

Abstract
Tissue injury due to acute and chronic alcohol consumption has extensive medical consequences, with the level and duration of alcohol exposure affecting both the magnitude of injury and the time frame to recovery. While the understanding of many of the molecular processes disrupted by alcohol has advanced, mechanisms of alcohol-induced tissue injury remain a subject of intensive research. Alcohol has multiple targets, since it affects diverse cellular and molecular processes. Some mechanisms of tissue damage due to alcohol may be common to many tissue types, while others are likely to be tissue-specific. Here we present a discussion of the alcohol-induced molecular and cellular disruptions associated with injury or recovery from injury in bone, muscle, skin and gastric mucosa. In every case, the goal of characterizing the sites of alcohol action is to devise potential measures for protection, prevention or therapeutic intervention.
Keywords: Acute alcohol, Alcoholic myopathy, Angiogenesis, Antioxidant, Binge alcohol, Bone fracture repair, Canonical Wnt signaling, Chronic alcohol, Cytoprotection, Extracellular matrix, Fracture non union, Gastric mucosa, Glutathione, Inflammation, Myopathy, Orthopaedic trauma, p34cdc2 kinase, Oxidative stress, Survivin, Tissue injury, Wound healing
OVERVIEW
Alcohol affects virtually every organ and tissue in the body, with multi-factorial actions on cellular and molecular functions. Alcohol itself alters biological function by direct interaction with cellular components, and also due to the direct effect of alcohol metabolism on the systemic oxidative and inflammatory state. Characterization of the cellular and molecular processes that are disrupted after exposure to alcohol is necessary to understanding and treating or preventing its pathophysiological effects. Throughout this report, the term alcohol refers to ethanol.
The metabolism of alcohol results in the generation of acetaldehyde and reactive oxygen (and other) species, biochemical moieties that damage healthy tissue. The oxidative stress resulting from these reactive oxygen and nitrogen species originates in many organs and tissues and varies in severity depending on the systemic inflammatory and oxidative state, and on systemic and local immune function. In addition, in different tissues, the magnitude and duration of oxidative stress depends on the metabolic state of the cells and on the ability of those tissues to metabolize and clear alcohol and its byproducts. Secondary sources of oxidative stress result from increased endotoxin leakage and increased release of pro-inflammatory cytokines from both immune and non-immune cells responding to alcohol. Tissue specific variation in local inflammatory/immune function and in the response to systemic factors may contribute to organ-specific differences in the pathological effects of alcohol. Overall, the presence of oxidative stress is balanced by cellular stress response systems, both those that prevent accumulation of oxidative stress and those that repair damage caused by oxidative species.
In addition to, and separate from, the consequences of alcohol metabolism, direct interactions of alcohol with molecular components affect physiological function. Alcohol has been shown to modify signal transduction at multiple sites through its interaction with cell membranes (Dolganiuc et al., 2006; Szabo et al., 2007) as well as with signaling proteins (Higashi et al., 1996; Resnicoff et al., 1994; Saso et al., 1997; Goral et al., 2005) and ion channels (Dopico, 2003). Modification of the function of receptors and other signaling molecules leads to altered function of multiple signaling pathways that mediate many essential processes. This review presents examples of the tissue-specific harm resulting from the alteration of basic signaling pathways by alcohol and by enhanced oxidative stress.
Both chronic and acute alcohol consumption have the potential to impair health and well-being. In addition to being a risk factor for the events that lead up to injuries and accidents, alcohol also leads to increased morbidity and mortality after those injuries. Alcohol-related diagnoses are associated with longer hospital stays, more complications, and greater medical expense. Nearly 50% of adult emergency room visits are associated with alcohol consumption, and, surprisingly, most of these patients are not chronic alcohol abusers, but rather those who consume alcohol on an acute or binge basis. As shown herein, acute alcohol has a negative impact on recovery after injury or illness. The cumulative tissue injury resulting from chronic alcohol exposure also results in significant detriment to overall health and to an impaired capacity to fend off illness.
With the understanding that some mechanisms of tissue damage due to alcohol may be common to multiple tissue types, while others are likely to be tissue-specific, we present a discussion of some of the organs systems and pathways affected by alcohol. This review focuses on mechanisms by which different levels of alcohol exposure yield tissue-specific injury and how the altered pathways affect repair processes in muscle, bone, gastric mucosa and skin.
Among the clinical implications of tissue-specific alcohol effects is the fact that injury is often not limited to a single tissue; for example a fractured bone may also be associated with skin and muscle or tendon damage. Moreover, devising therapeutic strategies to help accelerate the repair process in one tissue or organ system may result in a deleterious effect on others. Hence, gaining knowledge about how the response to alcohol-induced injury differs among these tissues and organs may aid in the design of treatments that will benefit the patient as a whole.
BINGE ALCOHOL EXPOSURE IMPAIRS BONE FRACTURE HEALING BY INHIBITION OF CANONICAL WNT SIGNALING
Social binge drinking of high alcohol content beverages by non-alcoholics is a commonly observed phenomenon in college aged and older adults in the USA. Approximately 40% of all orthopaedic trauma patients are intoxicated at the time of hospital admission (Levy et al., 1996), and delayed or incomplete development of the fracture callus is observed in these intoxicated patients (Frost, 1989a; Frost, 1989b). Thus, alcohol consumption increases the risk for incurring a traumatic injury (Savola et al., 2005), is associated with higher incidences of clinical complications following orthopaedic trauma (Levy et al., 1996) and also has an inhibitory effect on the fracture repair process (Janicke-Lorenz and Lorenz, 1984). Despite these statistics, little is known about the mechanisms underlying alcohol-induced effects on bone fracture repair. Early studies in both alcoholic patients and in rodent models of chronic alcohol consumption demonstrated an inhibition of bone fracture healing (Kristensson et al., 1980, Janicke-Lorenz and Lorenz, 1984). The lack of normal fracture callus formation in both intoxicated patients and rodents suggests that alcohol inhibits bone fracture repair at an early stage in the healing process. More recent studies demonstrate that production of inflammatory cytokines interleukin-1 (IL-1) and tumor necrosis factor alpha (TNFα) at the site of fracture injury may be associated with normal bone repair, that an alcohol-related modulation of this local inflammatory response may inhibit repair and that fracture healing in alcohol-exposed animals can be improved by administration of IL-1 and TNF antagonists (Perrien et al., 2004). Recent reports demonstrate that impairments in osteoinduction, a process by which an externally fixed fracture is stretched along the long axis of the bone in order to achieve greater limb length, may contribute to deficient bone repair in alcohol-exposed rodents. This hypothesis supports the supposition that bone formation is the primary target of alcohol during bone healing (Trevisiol et al., 2007).
Global transcriptome analysis demonstrated that the canonical Wnt pathway, which regulates bone formation, is targeted by alcohol exposure in rat vertebral bone (Himes et al., 2008). Alcohol exposure decreased the expression of several genes in bone associated with canonical Wnt signaling including those coding for Lrp5 and β–catenin (Himes et al., 2008). As canonical Wnt signaling plays an important role in bone fracture repair (Chen et al., 2007), the effects of binge alcohol exposure on canonical Wnt signaling activity during the fracture repair process were evaluated. Using a binge alcohol exposure model (Callaci et al., 2004), male C57BL/6 mice were given alcohol for 3 days and then subjected to a mid-shaft, stabilized tibia fracture, shown previously to heal by both intramembranous and endochondral ossification, similar to stabilized human fractures (Le et al., 2001). The fracture callus was recovered from animals at 3, 6, 9 and 14 days post injury, and analyzed using histological, biomechanical and molecular analysis. Binge alcohol exposure prior to fracture injury decreased callus size at all time points examined. Callus composition was markedly different between control and alcohol-treated animals, with decreases in both cartilage and boney components of the callus at 6, 9 and 14 days post fracture. Material properties of the callus also differed between control and alcohol treated animals. Biomechanical 4-point bending analysis revealed a significant decrease in callus strength in alcohol-treated mice at 14 days post injury. Molecular analysis revealed that β-catenin protein expression in callus from alcohol-treated mice was significantly decreased at post fracture days 9 and 14. The alcohol-induced changes in Wnt/β-catenin signaling resulted in spatial and quantitative changes in downstream transcriptional activation of Wnt target gene expression in transgenic T-cell factor reporter mice (TCF transgenic mice).
The canonical Wnt signaling pathway plays a crucial role in bone formation (Gong et al., 2001), mesenchymal stem cell differentiation (Hill et al., 2005), and bone fracture repair (Chen et al., 2007). Recent data implicate the canonical Wnt signaling pathway as a cellular target in alcohol-induced bone loss (Himes et al., 2008) and the current data suggest deregulation of this pathway is also responsible for alcohol-induced deficient bone fracture repair (Figure 1). Alcohol exposure prior to fracture resulted in decreased callus size and altered cellular composition, indicating that alcohol may inhibit both cartilage and bone formation in the fracture callus. Osteoblast and chondrocyte cells share a common mesenchymal stem cell precursor, which requires tight regulation of β-catenin expression and activity for proper differentiation. These observations suggest that binge alcohol exposure prior to fracture injury may target the canonical Wnt pathway during the differentiation of mesenchymal precursors into bone and cartilage-forming cells, ultimately causing a decrease in the available pool of functional osteoblasts and chondrocytes available for normal fracture healing. Additionally, β-catenin signaling appears to be essential for normal osteoblast function and proliferation, which may also indicate that alcohol is capable of affecting the function of mature osteoblasts to produce osteoid in the callus. Evidence from TCF transgenic reporter mouse studies suggests that, in addition to alcohol’s detrimental effects on callus tissue constitution, the spatial organization of activated Wnt/β-catenin signaling in the callus is disrupted. At post-fracture day 6, active Wnt signaling was predominantly located in the marrow cavity of alcohol-treated animals rather than in the soft tissue comprising the callus as found in saline-treated controls, suggesting that alcohol may cause a delay in the differentiation of mesenchymal precursors into chondrocytes and osteoblasts following fracture injury. Quantification of β-catenin protein levels in the callus showed that an acute, three-day binge exposure prior to injury is sufficient to cause long-term effects on Wnt signaling, as noted by the sharp decline in β-catenin protein levels in callus tissue at days 9 and 14 post-fracture. This suggests that alcohol may not only have immediate consequences on the repair process by deregulating stem cell differentiation at the early stages of healing, but may also trigger significant downstream changes in osteoblast cells that persist long after alcohol exposure has ceased. Future studies directed toward the association between canonical Wnt signaling and alcohol exposure may reveal valuable therapeutic targets to alcohol-abusing patients sustaining orthopaedic injuries.
Figure 1. Alcohol targets Wnt signaling during bone fracture repair.
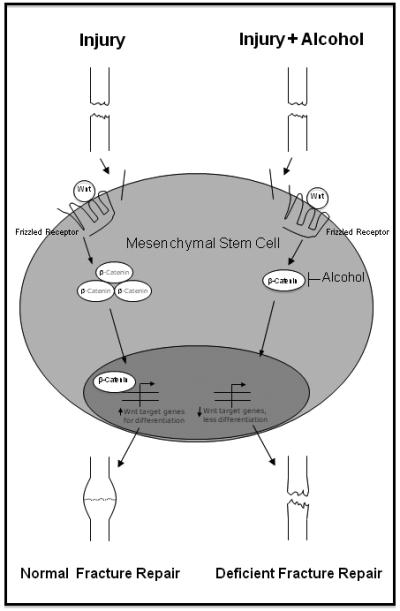
Bone fracture triggers the mobilization of mesenchymal stem cells from local and distant compartments to the site of injury, where they undergo differentiation into osteoblasts and chondrocytes under the control of canonical Wnt signaling. Wnt proteins bind to surface Frizzled receptors, leading to stabilization and translocation of cytosolic β-catenin to the nucleus. Nuclear β-catenin activates transcription of Wnt-related genes necessary for bone and cartilage formation, and its expression is tightly regulated throughout the repair process. Binge alcohol exposure prior to injury decreases protein levels of β-catenin and disrupts its precise pattern of expression, decreases downstream Wnt target gene expression, and ultimately causes decreased formation of osteoblasts and chondrocytes within the fracture callus. These effects lead to alcohol-induced deficient bone repair and fracture nonunion.
GLUTATHIONE RESTORATION AS AN INTERVENTION FOR ALCOHOLIC MYOPATHY
The severity of skeletal muscle derangements due to alcohol abuse is directly proportional to the quantity and duration of alcohol consumption. Acute alcoholic myopathy, which may occur after single or multiple episodes of binge drinking, is an extremely rare condition that affects ~1% of alcoholics and may manifest in myalgia, muscle weakness and pain, renal impairment with myoglobinuria, and rhabdomyolysis. In contrast, chronic alcoholic myopathy has been estimated to occur in up to 70% of alcoholics and is reportedly more common than other alcohol-induced diseases, such as liver cirrhosis and cardiomyopathy. Chronic alcoholic myopathy is characterized by severe atrophy and muscle dysfunction, but appears to be a phenomenon specific to skeletal muscles with a predominantly glycolytic, fast-twitch phenotype such as the plantaris, while characteristically oxidative, slow-twitch muscles such as the soleus are largely spared. The fundamental cause(s) of the disease is unknown; however, the diverse toxicology of chronic alcohol consumption would suggest that the etiology and pathology are likely multi-factorial. Indeed, the development of alcoholic myopathy has been attributed to acetaldehyde protein adduct formation, gene dysregulation, altered growth hormone production, decreased muscle protein synthesis, increased muscle-derived and circulating catabolic factors, and oxidant stress (for comprehensive reviews see Lang et al., 2005; Fernandez-Solà et al., 2007).
Despite an alcoholic’s generally poor diet and high caloric content of alcohol in excess, nutritional status does not appear to play a leading role in the development of alcoholic myopathy. However, reduced plasma and muscle levels of several essential antioxidants have been reported in alcoholics with myopathy and likely contribute to the development of the disease (Fernández-Solà et al., 2002; Ward and Peters, 1992). Accordingly, research has used antioxidant or antioxidant co-factor supplementation to abate the surge in alcohol-induced reactive oxygen species, often with limited effectiveness. For example, providing alcoholics dietary supplements of zinc, an essential cofactor for superoxide dismutase and glutathione peroxidase, did not alleviate symptoms of alcoholic myopathy (Fernández-Solà et al., 1998). Further, α-tocopherol supplementation had little effect on the rates of protein synthesis or the total protein content in skeletal muscle (Koll et al., 2003). Moreover, precursors of the non-vitamin antioxidant glutathione have been used to rectify alcohol-induced derangements to lung and skeletal muscle tissue (Velasquez et al. 2002, Otis et al., 2007; Otis and Guidot, 2009).
Glutathione is a thiol-based, trimeric amino acid compound comprised of glycine, glutamate, and cysteine and is the principle nucleophilic scavenger of free radicals in cells. Additionally, glutathione stabilizes other antioxidants, maintains proteins in a reduced and, therefore, functional state, attenuates redox-sensitive catabolic factors, and preserves healthy biomembranes by limiting alcohol-induced lipid peroxidation (Wu et al., 2004). Glutathione levels can be depleted in certain disease states, but restoration is achievable with precursor compounds such as S-adenosyl methionine (SAMe), N-acetylcysteine (NAC), or L-2-oxothiazolidine-4-carboxylate (OTC or procysteine). Specifically, chronic alcohol ingestion decreases skeletal muscle levels of glutathione and cysteine, and decreases the enzyme activities of glutathione peroxidase and glutathione reductase (see Figure 2) (Fernández-Solà et al., 2002; Wu et al., 2004; Otis et al., 2007; Otis and Guidot, 2009).
Figure 2. Glutathione metabolism in alcoholic skeletal muscle.
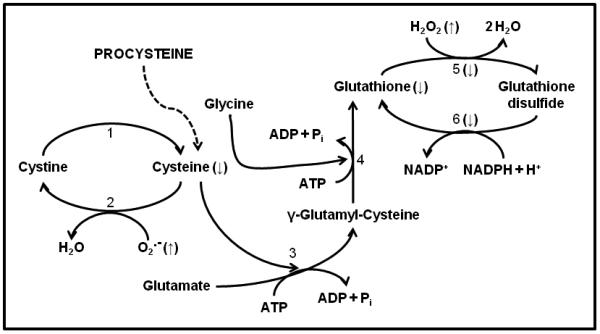
Alcoholic myopathy is characterized by increased formation of superoxide (O2.−), lipid and hydrogen peroxide (H2O2), and reactive oxygen species. Glutathione cycling is also altered suggesting a reduced capacity to sequester these alcohol-induced oxidants. Supplementing the diets of alcoholics with L-2-oxothiazolidine-4-carboxylate, a proform of L-cysteine (Procysteine), normalizes thiol levels of cysteine and glutathione. Enzymes that catalyze these reactions are: (1) glutathione dependent thiodisulfide, thioltransferase, and non-enzymatic reactions; (2) protein disulfide isomerase; (3) γ-glutamylcysteine synthetase; (4) glutathione synthetase; (5) glutathione peroxidase; and (6) glutathione reductase. Arrows (↑, ↓) denote direction of change as a result of chronic alcohol abuse. Other abbreviations used: ADP, adenosine diphosphate; ATP, adenosine triphosphate; H+, hydrogen ion; H2O, water; NADP+, nicotinamide adenine dinucleotide phosphate, NADPH, reduced form of NADP+; Pi, inorganic phosphate.
Muscle glutathione levels in rats fed alcohol for 6 weeks were replenished by procysteine administration, while oxidant stress and the expression of two catabolic factors, atrogin-1 and transforming growth factor β1 (TGFβ1), were decreased (Otis et al., 2007). Interestingly, oxidant stress and the induction of atrogin-1 and TGFβ1 occurred in the absence of clinically significant myopathy, suggesting that “pro-atrophy” programs may be remedied with glutathione restoration before the development of overt atrophy. Because the severity of myopathy progresses along a continuum as alcohol abuse persists, muscle alterations were also seen in rats that consumed alcohol for up to 35 weeks, a duration of abuse that produced overt muscle atrophy (Otis and Guidot, 2009). Long term alcohol ingestion created an overall catabolic state in atrophied rat plantaris muscles, as evidenced by oxidant stress and by the production of catabolic members of the interleukin-6 (IL-6) family (i.e., IL-6 and oncostatin M) and components of the ubiquitin proteasome system (i.e., atrogin-1 and muscle RING-finger protein-1 (MuRF1)). However, glutathione restoration was insufficient to attenuate these catabolic factors, but rather stimulated the production of several anabolic factors (i.e., insulin-like growth factor 1 (IGF-1), ciliary neurotrophic factor, and cardiotrophin-1) and ultimately led to increased plantaris fiber area in alcohol-fed rats. Taken together, these data reveal important temporal associations between early alcohol-induced oxidant stress and the induction of catabolic factors and resultant muscle atrophy. Further, if these salutary physiological responses to glutathione supplementation in animal models translate to the clinical setting, then glutathione replacement provided before the clinical onset of myopathy, and perhaps even after atrophy is established, could significantly improve muscle mass and function in chronic alcoholics.
IMPACT OF ACUTE ALCOHOL EXPOSURE ON DERMAL WOUND HEALING
The detrimental impact of alcohol exposure on tissue repair has been evident for decades, influencing multiple pathways across a wide range of tissues. These changes have been attributed to the metabolism of alcohol and production of toxic metabolites that directly impact normal cellular function required for efficient would repair. Intoxicated patients have a higher incidence of associated injuries, many of which sustain traumatic cutaneous wounds that demonstrate a correlation between alcohol intoxication and wound healing complications. For example, a blood alcohol level (BAL) of >200 mg/dl has been associated with a 2.6 fold increase in the incidence of wound-related infections (Gentilello et al., 1993). Although most studies have focused on chronic alcohol, the awareness of the frequency of acute alcohol abuse has led to the development of clinically relevant acute alcohol exposure models. Until recently, a comprehensive analysis identifying the effects of acute alcohol exposure on tissue repair was not established in vivo.
Wound repair is comprised of three overlapping phases that orchestrate a variety of cellular functions, with the endpoint being healed tissue up to 90% of its original integrity (Bernstein et al., 1996). The inflammatory phase initiates the healing process and involves the recruitment of proinflammatory cells into the wound site where they participate in host defense through secretion of chemokines and cytokines. This cascade subsequently activates the next phase, the proliferative phase, to increase cellular migration and proliferation to repopulate the wound bed that is devoid of epithelial, vascular, and matrix-producing cells. The final phase allows for remodeling and reorganization of newly synthesized matrix molecules to regain near normal functional capacity of the injured tissue. Thus, any disruption between the delicate balance of cellular recruitment and/or activation can lead to devastating defects in tissue regeneration.
The development of an acute alcohol model in mice in conjunction with an established wound healing model identified multiple defects in the wound repair process following one dose of acute alcohol thirty minutes prior to excisional injury. This model parallels a binge alcohol model yielding a blood alcohol concentration of 100 mg/dl that represents many of the clinical patients that frequent the emergency room with trauma injury. Wounds from alcohol-treated mice exhibited a significant reduction in the myeloperoxidase activity of neutrophils compared to saline controls, indicating a defect in neutrophil function. In addition, the levels of two major proinflammatory chemokines involved in macrophage recruitment, macrophage inflammatory protein-2 (MIP-2) and KC, the murine homolog of human interleukin-8, were significantly reduced compared to their saline-treated counterparts (Fitzgerald et al., 2007). Subsequent studies revealed that early defects in the inflammatory phase following acute alcohol exposure may have contributed to a delay in wound closure (Radek et al., 2005). Wounds from alcohol-treated mice were approximately 50% less re-epithelialized after two days following wounding compared to saline-treated mice. However, wounds from both groups were completely re-epithelialized after 5 days, indicating a transient effect of alcohol on keratinocyte migration.
Since angiogenesis is a critical element of the wound repair process, as it restores the underlying vasculature to restore oxygenation of the wound bed, an assessment of vascular density was made in wounds from alcohol-treated mice. Previous studies demonstrated that direct intragastric alcohol exposure (50% v/v) can actually promote vascular endothelial growth factor (VEGF)-induced angiogenesis in gastric epithelia (Jones et al., 1999). However, these concentrations are far beyond what would be available to cutaneous tissue following ingestion. In the presence of acute alcohol exposure, wound vascularity was significantly reduced up to 10 days following wounding compared to saline controls, despite near-normal levels of the pro-angiogenic cytokine, VEGF, and the wounds of alcohol-treated mice were notably more hypoxic. Subsequent in vitro studies revealed that the defect in vascularity seen with acute alcohol in vivo is attributed, in part, to reduced phosphorylation of the VEGF receptor, critical to a signaling pathway involved in endothelial cell proliferation and differentiation into capillaries (Radek et al., 2008). Markers of matrix integrity were also assessed and revealed that wounds from alcohol treated animals had significantly less collagen, while exhibiting an increase in matrix proteolytic activity. Stimulation of fibroblasts, the matrix producing cells of the skin, with alcohol (100 mg/dl) in vitro also reduced the gene expression of collagen type I (Radek et al., 2007). Previously, alcohol exposure (>5% v/v) diminished the ability of fibroblasts to proliferate and sufficiently produce collagen type I in the presence of TGF-β1 in vitro (Stephens et al., 1996). Together, these data demonstrate that acute alcohol exposure renders fibroblasts unable to properly synthesize the required matrix molecules to re-establish dermal stability. This defect can reduce the capacity of endothelial cells to properly migrate to form capillary networks within the newly synthesized matrix, culminating in a functionally defective wound bed that is more vulnerable to wound dehiscence and infection.
One of the major difficulties involved in deciphering the direct effects of alcohol from indirect effects on the various cell populations involved in wound repair is due to the promiscuity of alcohol and its metabolites. Collectively, the prolonged consequences of alcohol exposure on dermal tissue repair (Figure 3) evoke concern for intoxicated trauma patients who may succumb to opportunistic infections after injury and/or surgical intervention. Ultimately, more detailed insight into the specific alcohol mediated changes on cell signaling pathways that occur during tissue repair will be essential to develop more effective treatment strategies to limit physiologic perturbations in intoxicated patients.
Figure 3. Detrimental effects of acute alcohol exposure on cutaneous wound repair.

Acute alcohol exposure disrupts the balance of cellular processes to favor diminished cellular function leading to matrix degradation. Impaired immune cell function, vascularity, and matrix regeneration contribute to the increase in susceptibility to wound infection. EC= endothelial cell, VEGF= Vascular Endothelial Growth Factor.
ROLE OF SURVIVIN IN ADAPTIVE CYTOPROTECTION AGAINST ALCOHOL INDUCED GASTRIC INJURY
In 1983, Andre Robert and co-workers first described the phenomenon they termed “adaptive cytoprotection” whereby pre-ingestion (or experimental pre-administration in animal models) of “mild-irritant” alcohol concentrations (~10-20% v/v) resulted in the preservation of gastric mucosal (stomach) integrity against the damaging effects of subsequent ingestion (administration) of strong (≥50% v/v) alcohol (Robert et al., 1983). They and numerous other groups have since verified this phenomenon and attributed it to an increase in the production of prostaglandins. Although there is no doubt that prostaglandins (particularly prostaglandin E2 and prostacyclin) play important contributory roles in gastric adaptive cytoprotection, several studies suggest that other, prostaglandin-independent, factors also participate in this phenomenon. These include nitric oxide, vagal innervation, sensory nerves, blood flow, calcium (Ca2+) influx, heat shock proteins and a physical barrier resulting from mucosal surface exfoliation (Jones et al., 2008).
Survivin is a recently discovered protein that possesses a dual function as both a regulator of cell division and cell survival. This protein is highly expressed in most organ tissues during embryonic development where it plays an essential role in organ tissue remodeling through the regulation of cell proliferation and apoptosis. Following the completion of development, survivin expression disappears in most adult differentiated organ tissues where its function is presumably no longer needed. Interestingly, however, survivin is highly expressed in all known forms of human cancer thus classifying it as a “universal tumor antigen” (Andersen et al., 2007). This has opened a floodgate of investigation into the most efficacious means by which to universally target/inhibit survivin expression as a component of cancer therapy. Nevertheless, survivin has also been shown to be a factor in liver regeneration (Deguchi et al., 2002), angiogenesis (O’Connor et al., 2000) and vascular injury and repair (Simosa et al., 2005; Conte and Altieri, 2006). In at least some normal adult organ tissues, therefore, survivin may be a crucial player in maintaining the physiologically important balance between cellular proliferation and apoptosis.
Survivin is expressed in normal, non-cancerous, adult differentiated gastric mucosa (Chiou et al., 2003). In addition, in response to exposure of gastric epithelial cells to cytoprotective alcohol concentrations, survivin accumulation is required for the full cytoprotective effect against subsequent exposure to cytotoxic alcohol concentrations (Jones et al., 2008). The latter was demonstrated by showing that: a) the cytoprotection produced by pre-exposure to “mild irritant” alcohol against subsequent exposure to cytotoxic alcohol was blocked by inhibiting survivin accumulation/expression using small interfering RNA (an experimental strategy used to attenuate targeted protein expression); and, b) forced overexpression of survivin by transfection, or gene transfer, produced substantial cytoprotection against exposure to a cytotoxic alcohol concentration even in the absence of pre-exposure to “mild irritant” alcohol.
How do survivin expression levels accumulate in the gastric mucosa in response to cytoprotective alcohol ingestion or exposure? The complete answer to this question, at present, remains moot since the mechanism(s) by which survivin expression levels accumulate in gastric epithelial cells grown in the laboratory and the mechanism(s) by which survivin expression levels accumulate in the mammalian gastric mucosa, including that of humans, is likely to differ somewhat. For instance, exposure of gastric epithelial cells to cytoprotective alcohol results in an increase in survivin protein levels by virtue of being stabilized against normal degradation via phosphorylation of amino acid residue, threonine-34. Evidence suggests that this modification occurs or is enhanced, following cytoprotective alcohol exposure, by the induction/activation of one or more regulatory kinases, including the cell cycle-dependent kinase, p34cdc2 (Jones et al., 2008). The same modification has been observed in the gastric mucosa from experimental animals administered mild irritant alcohol; further, preliminary data also suggest that de novo transcription of the survivin gene may be involved in gastric cytoprotection to whole animals.
A key question remaining is how cytoprotective alcohol exposure enhances the activation of the kinase(s) that lead(s) to survivin stabilization/accumulation in gastric epithelial cells. One possibility, supported by as yet unpublished data, is that this may occur via Ca2+ mobilization and the resulting activation of calmodulin-dependent kinase, CaM kinase II. Nevertheless, an alternative possibility, that the activities of upstream membrane-associated kinases (e.g. receptor kinases) are influenced by physical perturbations in the surface membranes of cells exposed to cytoprotective alcohol, cannot be ruled out.
SUMMARY
The examples presented here attest to the multifactorial and multisystemic mechanisms by which, even after a single acute or binge exposure, alcohol leads to tissue damage. In gastric mucosa and in bone, alcohol exposure alters basic signaling processes. In the repair of skin damage following injury or surgery, alcohol disrupts signaling in a broader context, in multiple tissue types. The derangement of the inflammatory response by alcohol leads to altered cytokine and chemokine production by multiple cell types and this, in turn, influences the responses of other cell types. Disruption of multiple molecular processes by alcohol contributes to myopathy and muscle atrophy, with the accumulation of oxidative stress playing a major role.
In every case, the goal of characterizing the sites of alcohol action is to identify potential targets for intervention, either preventative or therapeutic. Recovery from bone injury after alcohol exposure is adversely affected by alterations in the Wnt signaling pathway, which is essential to the repair process. Undisturbed Wnt signaling is essential to bone formation, since disruption of mesenchymal stem cell differentiation compromises both bone and callus formation. Restoration or protection of Wnt signaling may improve the prognosis for recovery from bone fractures sustained with alcohol exposure.
Preclinical testing of methods for correcting alcoholic myopathy has generated promising results. The causes of alcohol-induced myopathy are multifaceted, with the disruption of oxidative balance playing a significant role. Alcohol affects levels of antioxidant compounds as well as the activity of enzymes involved in oxidative balance. Significantly, restoration of the antioxidant compound glutathione by precursor supplementation restores oxidative balance. Supplementation of the glutathione precursor after binge alcohol consumption, prior to the appearance of muscle atrophy, has the potential to prevent or reduce muscle damage. Further, replenishing glutathione by precursor supplementation even in the presence of full-blown atrophy after chronic alcohol consumption restores some measures of metabolic balance in an animal model, suggesting the potential for correction of cumulative tissue injury.
The derangements of the inflammatory response in the presence of alcohol consumption adversely affect the process of wound healing. Many aspects of the inflammatory response are essential to proper healing of dermal wounds, potentially providing multiple therapeutic targets. The many cytokines and chemokines released in response to tissue damage and the many cell types that are mobilized during inflammation all contribute to the process of wound healing. Normalization of the inflammatory response in the presence of alcohol would improve the outlook for recovery after dermal injury, particularly in the context of wound infections.
The fact that mechanisms of injury and of repair processes differ among tissues brings to the fore the potential for complications in patients with multiple injuries. For example, targeting survivin in tumor tissue has the potential to lead to unanticipated or undesired elimination of necessary survivin functions, including protection of gastric mucosa. Survivin protein function is essential to the development of cytoprotection. Maintenance of survivin levels result from modification of the kinase activity of p34cdc2 after modest alcohol exposure. Other evidence suggests modification at the expression level may also be occurring.
The effects of acute alcohol exposure on the healing process persist many days out in wound healing, in recovery from bone fracture (both reported here), as well as after burn injury (Messingham et al., 2002), and other forms of traumatic injury. While the sustained disruption of healing is discouraging, it presents multiple potential approaches for intervention. The response to a short term alcohol state suggests that a seminal process is altered, leading to branching downstream effects. Identifying and targeting the primary disruption would be ideal, but if missed, the potential to intervene to restore downstream functions may also be considered.
Acknowledgements
This research was supported by NIH grants R01AA016138 (JJC), T32AA013527 (KLL), K01AA017190 (JSO), R01AA014946 (MKJ), P30AA019373 (EJK, KAR), R01AA012034 (EJK), the Ralph and Marian C Falk Medical Research Trust (EJK), the Margaret A. Baima Endowment Fund for Alcohol Research (EJK), and the VA Biomedical Laboratory Research & Development Service (MKJ).
Footnotes
Current address: Alcohol Research Program, Burn & Shock Trauma Institute, Department of Surgery, Loyola University Stritch School of Medicine, Maywood, IL
Contributor Information
M. Katherine Jung, Division of Metabolism and Health Effects, National Institute on Alcohol Abuse and Alcoholism, Bethesda, MD.
John J. Callaci, Alcohol Research Program, Burn & Shock Trauma Institute, Department of Orthopaedic Surgery, Loyola University Stritch School of Medicine, Maywood, IL.
Kristen L. Lauing, Alcohol Research Program, Burn & Shock Trauma Institute, Department of Orthopaedic Surgery, Loyola University Stritch School of Medicine, Maywood, IL.
Jeffrey S. Otis, Division of Pulmonary, Allergy, and Critical Care Medicine, Emory University School of Medicine, Atlanta, GA.
Katherine A. Radek, VA San Diego Healthcare System, San Diego, CA, and Division of Dermatology, Department of Medicine, University of California, San Diego, CA.
Michael K. Jones, Research Healthcare Group, VA Long Beach Healthcare System, Long Beach, CA, and Department of Medicine, University of California, Irvine, CA.
Elizabeth J. Kovacs, Alcohol Research Program, Burn & Shock Trauma Institute, Department of Surgery, Loyola University Stritch School of Medicine, Maywood, IL.
REFERENCES
- Andersen MH, Svane IM, Becker JC, Straten PT. The universal character of the tumor-associated antigen survivin. Clin Cancer Res. 2007;13:5991–5994. doi: 10.1158/1078-0432.CCR-07-0686. [DOI] [PubMed] [Google Scholar]
- Bernstein E, Bolten LL, Mauviel A, Frank T, McGrath JA, Uitto J. Principles of Cutaneous Surgery. McGraw Hill; New York: 1996. [Google Scholar]
- Callaci JJ, Juknelis D, Patwardhan A, Sartori M, Frost N, Wezeman FH. The effects of binge alcohol exposure on bone resorption and biomechanical and structural properties are offset by concurrent bisphosphonate treatment. Alcohol Clin Exp Res. 2004;28:182–191. doi: 10.1097/01.ALC.0000108661.41560.BF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Whetstone HC, Lin AC, Nadesan P, Wei Q, Poon R, Alman BA. Beta-catenin signaling plays a disparate role in different phases of fracture repair: implications for therapy to improve bone healing. PLoS Med. 2007;4:e249. doi: 10.1371/journal.pmed.0040249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou SK, Moon WS, Jones MK, Tarnawski AS. Survivin expression in the stomach: implications for mucosal integrity and protection. Biochem Biophys Res Commun. 2003;305:374–379. doi: 10.1016/s0006-291x(03)00724-1. [DOI] [PubMed] [Google Scholar]
- Conte MS, Altieri DC. Survivin regulation of vascular injury. Trends Cardiovasc Med. 2006;16:114–117. doi: 10.1016/j.tcm.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Deguchi M, Shiraki K, Inoue H, Okano H, Ito T, Yamanaka T, Sugimoto K, Sakai T, Ohmori S, Murata K, Furusaka A, Hisatomi H, Nakano T. Expression of survivin during liver regeneration. Biochem Biophys Res Commun. 2002;297:59–64. doi: 10.1016/s0006-291x(02)02128-9. [DOI] [PubMed] [Google Scholar]
- Dolganiuc A, Bakis G, Kodys K, Mandrekar P, Szabo G. Acute ethanol treatment modulates toll-like receptor-4 association with lipid rafts. Alcohol Clin Exp Res. 2006;30:76–85. doi: 10.1111/j.1530-0277.2006.00003.x. [DOI] [PubMed] [Google Scholar]
- Dopico AM. Ethanol sensitivity of BK(Ca) channels from arterial smooth muscle does not require the presence of the beta 1-subunit. Am J Physiol Cell Physiol. 2003;284:C1468–C1480. doi: 10.1152/ajpcell.00421.2002. [DOI] [PubMed] [Google Scholar]
- Fernández-Solà J, Villegas E, Nicolas JM, Deulofeu R, Antunez E, Sacanella E, Estruch R, Urbano-Márquez A. Serum and muscle levels of alpha-tocopherol, ascorbic acid, and retinol are normal in chronic alcoholic myopathy. Alcohol Clin Exp Res. 1998;22:422–427. [PubMed] [Google Scholar]
- Fernández-Solà J, Garcia G, Elena M, Tobias E, Sacanella E, Estruch R, Nicolas JM. Muscle anti-oxidant status in chronic alcoholism. Alcohol Clin Exp Res. 2002;26:1858–1862. [PubMed] [Google Scholar]
- Fernández-Solà J, Preedy VR, Lang CH, Gonzalez-Reimers E, Arno M, Lin JC, Wiseman H, Zhou S, Emery PW, Nakahara T, Hashimoto K, Hirano M, Santolaria-Fernández F, González-Hernández T, Fatjó F, Sacanella E, Estruch R, Nicolás JM, Urbano-Márquez A. Molecular and cellular events in alcohol-induced muscle disease. Alcohol Clin Exp Res. 2007;31:1953–1962. doi: 10.1111/j.1530-0277.2007.00530.x. [DOI] [PubMed] [Google Scholar]
- Fitzgerald DJ, Radek KA, Chaar M, Faunce D, DiPietro LA, Kovacs EJ. Effects of acute ethanol exposure on the early inflammatory response after excisional injury. Alcohol Clin Exp Res. 2007;31:317–323. doi: 10.1111/j.1530-0277.2006.00307.x. [DOI] [PubMed] [Google Scholar]
- Frost HM. The biology of fracture healing. An overview for clinicians. Part I. Clin Orthop Relat Res. 1989;248:283–293. [PubMed] [Google Scholar]
- Frost HM. The biology of fracture healing. An overview for clinicians. Part II. Clin Orthop Relat Res. 1989;248:294–309. [PubMed] [Google Scholar]
- Gentilello LM, Cobean RA, Walker AP, Moore EE, Wertz MJ, Dellinger EP. Acute ethanol intoxication increases the risk of infection following penetrating abdominal trauma. J Trauma. 1993;34:669–674. doi: 10.1097/00005373-199305000-00009. [DOI] [PubMed] [Google Scholar]
- Gong Y, et al. Osteoporosis-Pseudoglioma Syndrome Collaborative Group LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell. 2001;107(4):513–23. doi: 10.1016/s0092-8674(01)00571-2. [DOI] [PubMed] [Google Scholar]
- Goral J, Kovacs EJ. In vivo ethanol exposure down-regulates TLR2-, TLR4-, and TLR9-mediated macrophage inflammatory response by limiting p38 and ERK1/2 activation. J Immunol. 2005;174:456–463. doi: 10.4049/jimmunol.174.1.456. [DOI] [PubMed] [Google Scholar]
- Higashi K, Hoshino M, Nomura T, Saso K, Ito M, Hoek JB. Interaction of protein phosphatases and ethanol on phospholipase C-mediated intracellular signal transduction processes in rat hepatocytes: role of protein kinase A. Alcohol Clin Exp Res. 1996;20:320A–324A. [PubMed] [Google Scholar]
- Hill TP, Taketo MN, Birchmeier W, Hartmann C. Canonical Wnt/β-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev Cell. 2005;8:727–738. doi: 10.1016/j.devcel.2005.02.013. [DOI] [PubMed] [Google Scholar]
- Himes R, Wezeman FH, Callaci JJ. Identification of novel bone-specific molecular targets of binge alcohol and ibandronate by transcriptome analysis. Alcohol Clin Exp Res. 2008;32(7):1167–1180. doi: 10.1111/j.1530-0277.2008.00736.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janicke-Lorenz J, Lorenz R. Alcoholism and fracture healing. A radiological study in the rat. Arch Orthop Trauma Surg. 1984;103:286–289. doi: 10.1007/BF00387336. [DOI] [PubMed] [Google Scholar]
- Jones MK, Itani RM, Wang H, Tomikawa M, Sarfeh IJ, Szabo S, Tarnawski AS. Activation of VEGF and Ras genes in gastric mucosa during angiogenic response to ethanol injury. Am J Physiol. 1999;276:G1345–1355. doi: 10.1152/ajpgi.1999.276.6.G1345. [DOI] [PubMed] [Google Scholar]
- Jones MK, Padilla OR, Webb NA, Norng M. The anti-apoptosis protein, survivin, mediates gastric epithelial cell cytoprotection against ethanol-induced injury via activation of the p34cdc2 cyclin dependent kinase. J Cell Physiol. 2008;215:750–764. doi: 10.1002/jcp.21358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koll M, Beeso JA, Kelly FJ, Simanowski UA, Seitz HK, Peters TJ, Preedy VR. Chronic alpha-tocopherol supplementation in rats does not ameliorate either chronic or acute alcohol-induced changes in muscle protein metabolism. Clin Sci (Lond) 2003;104:287–294. doi: 10.1042/CS20020312. [DOI] [PubMed] [Google Scholar]
- Kristensson H, Lunden A, Nilsson BE. Fracture incidence and diagnostic roentgen in alcoholics. Acta Orthop Scand. 1980;51:205–207. doi: 10.3109/17453678008990787. [DOI] [PubMed] [Google Scholar]
- Lang CH, Frost RA, Summer AD, Vary TC. Molecular mechanisms responsible for alcohol-induced myopathy in skeletal muscle and heart. Int J Biochem Cell Biol. 2005;37:2180–2195. doi: 10.1016/j.biocel.2005.04.013. [DOI] [PubMed] [Google Scholar]
- Le AX, Miclau T, Hu D, Helms JA. Molecular aspects of healing in stabilized and non-stabilized fractures. J Ortho Res. 2001;19:78–84. doi: 10.1016/S0736-0266(00)00006-1. [DOI] [PubMed] [Google Scholar]
- Levy RS, Hebert CK, Munn BG, Barrack RL. Drug and alcohol use in orthopedic trauma patients: a prospective study. J Orthop Trauma. 1996;10:21–27. doi: 10.1097/00005131-199601000-00004. [DOI] [PubMed] [Google Scholar]
- Messingham KAN, Faunce DE, Kovacs EJ. Alcohol, injury and cellular immunity. Alcohol. 2002;28:137–149. doi: 10.1016/s0741-8329(02)00278-1. [DOI] [PubMed] [Google Scholar]
- O’Connor DS, Schechner JS, Adida C, Mesri M, Rothermel AL, Li F, Nath AK, Pober JS, Altieri DC. Control of apoptosis during angiogenesis by survivin expression in endothelial cells. Am J Pathol. 2000;156:393–398. doi: 10.1016/S0002-9440(10)64742-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otis JS, Brown LAS, Guidot DM. Oxidant-induced atrogin-1 and TGFβ1 precede alcoholic myopathy in rats. Muscle Nerve. 2007;36:842–848. doi: 10.1002/mus.20883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otis JS, Guidot DM. Procysteine stimulates expression of key anabolic factors and reduces plantaris atrophy in alcohol-fed rats. Alcohol Clin Exp Res. 2009;33:1450–1459. doi: 10.1111/j.1530-0277.2009.00975.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrien DS, Wahl EC, Hogue WR, Feige U, Aronson J, Ronis MJJ, Badger TM, Lumpkin CK. IL-1 and TNF antagonists prevent inhibition of fracture healing by ethanol in rats. Toxicological Sci. 2004;82:656–660. doi: 10.1093/toxsci/kfi002. [DOI] [PubMed] [Google Scholar]
- Radek KA, Matthies AM, Burns AL, Heinrich SA, Kovacs EJ, Dipietro LA. Acute ethanol exposure impairs angiogenesis and the proliferative phase of wound healing. Am J Physiol Heart Circ Physiol. 2005;289:H1084–1090. doi: 10.1152/ajpheart.00080.2005. [DOI] [PubMed] [Google Scholar]
- Radek KA, Kovacs EJ, DiPietro LA. Matrix proteolytic activity during wound healing: modulation by acute ethanol exposure. Alcohol Clin Exp Res. 2007;31:1045–1052. doi: 10.1111/j.1530-0277.2007.00386.x. [DOI] [PubMed] [Google Scholar]
- Radek KA, Kovacs EJ, Gallo RL, DiPietro LA. Acute ethanol exposure disrupts VEGF receptor cell signaling in endothelial cells. Am J Physiol Heart Circ Physiol. 2008;295:H174–184. doi: 10.1152/ajpheart.00699.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnicoff M, Rubini M, Baserga R, Rubin R. Ethanol inhibits insulin-like growth factor-1-mediated signalling and proliferation of C6 rat glioblastoma cells. Lab Invest. 1994;71:657–62. [PubMed] [Google Scholar]
- Robert A, Nezamis JE, Lancaster C, Davis JP, Field SO, Hanchar AJ. Mild irritants prevent gastric necrosis through “adaptive cytoprotection” mediated by prostaglandins. Am J Physiol. 1983;245:G113–G121. doi: 10.1152/ajpgi.1983.245.1.G113. [DOI] [PubMed] [Google Scholar]
- Saso K, Moehren G, Higashi K, Hoek JB. Differential inhibition of epidermal growth factor signaling pathways in rat hepatocytes by long-term ethanol treatment. Gastroenterology. 1997;112:2073–88. doi: 10.1053/gast.1997.v112.pm9178701. [DOI] [PubMed] [Google Scholar]
- Savola O, Niemela O, Hillbom M. Alcohol intake and the pattern of trauma in young adults and working aged people admitted after trauma. Alcohol Alcohol. 2005;40:269–73. doi: 10.1093/alcalc/agh159. [DOI] [PubMed] [Google Scholar]
- Simosa HF, Wang G, Sui X, Peterson T, Narra V, Altieri DC, Conte MS. Survivin expression is up-regulated in vascular injury and identifies a distinct cellular phenotype. J Vasc Surg. 2005;41:682–690. doi: 10.1016/j.jvs.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Stephens P, al-Khateeb T, Davies KJ, Shepherd JP, Thomas DW. An investigation of the interaction between alcohol and fibroblasts in wound healing. Int J Oral Maxillofac Surg. 1996;25:161–164. doi: 10.1016/s0901-5027(96)80065-8. [DOI] [PubMed] [Google Scholar]
- Szabo G, Dolganiuc A, Dai Q, Pruett SB. TLR4, ethanol, and lipid rafts: a new mechanism of ethanol action with implications for other receptor-mediated effects. J Immunol. 2007;178:1243–1249. doi: 10.4049/jimmunol.178.3.1243. [DOI] [PubMed] [Google Scholar]
- Trevisiol CH, Turner RT, Pfaff JE, Hunter JC, Nemagh PG, Hardin K, Ho E, Iwaniec UT. Impaired osteoinduction in a rat model for chronic alcohol abuse. Bone. 2007;41:175–180. doi: 10.1016/j.bone.2007.04.189. [DOI] [PubMed] [Google Scholar]
- Velasquez A, Bechara RI, Lewis JF, Malloy J, McCaig L, Brown LA, Guidot DM. Glutathione replacement preserves the functional surfactant phospholipid pool size and decreases sepsis-mediated lung dysfunction in ethanol-fed rats. Alcohol Clin Exp Res. 2002;26:1245–1251. doi: 10.1097/01.ALC.0000024269.05402.97. [DOI] [PubMed] [Google Scholar]
- Ward RJ, Peters TJ. The anti-oxidant status of patients with either alcohol-induced liver damage or myopathy. Alcohol Alcohol. 1992;27:359–365. [PubMed] [Google Scholar]
- Wu G, Fang Y-Z, Yang S, Lupton JR, Turner ND. Glutathione metabolism and its implications for health. J Nutr. 2004;134:489–492. doi: 10.1093/jn/134.3.489. [DOI] [PubMed] [Google Scholar]