

Abstract
Transforming growth factor β (TGFβ) has both tumor suppressive and oncogenic roles in cancer development. We previously showed that SB431542 (SB) a small molecule inhibitor of the TGFβ type I receptor (ALK5) kinase suppressed benign epidermal tumor formation but enhanced malignant conversion. Here we show that SB treatment of primary K5rTA x tetORASV12G bitransgenic keratinocytes did not alter HRASV12G- induced keratinocyte hyperproliferation. However, continuous SB treatment significantly enhanced HRASV12G-induced cornified envelope formation and cell death linked to increased expression of enzymes transglutaminase 1 (TGM1) and 3 (TGM3) and constituents of the cornified envelope small proline-rich protein 1A (SPR1A) and 2H (SPR2H). In contrast, TGFβ1 suppressed cornified envelope formation by HRASV12G keratinocytes. Similar results were obtained in HRASV12G transgenic mice treated topically with SB or by co-expressing TGFβ1 and HRASV12G in the epidermis. Despite significant cell death, SB resistant HRASV12G keratinocytes repopulated the primary culture that had overcome HRas-induced senescence. These cells expressed reduced levels of p16ink4a and were growth stimulated by SB but remained sensitive to a calcium-induced growth arrest. Together these results suggest that differential responsiveness to cornification may represent a mechanism by which pharmacological blockade of TGFβ signaling can inhibit the outgrowth of preneoplastic lesions but may cause a more progressed phenotype in a separate keratinocyte population.
Keywords: ALK5, cornified envelope, SB431542, skin, skin carcinogenesis
Introduction
Transforming growth factor β1 (TGFβ1) is a member of a large family of regulatory molecules that play both positive and negative roles in epithelial cancers. Genetic studies in the 2-stage skin carcinogenesis model have shown that overexpression of TGFβ (1) or related family members (2) suppresses benign tumor formation while inactivation of TGFβ type I (3, 4) and/or II receptor (5) signaling increases tumor development and malignant conversion. Similarly, Ferguson-Smith disease in humans which results in rapidly growing locally invasive squamous carcinoma-like skin tumors that spontaneously heal are a result of inactivating mutations in the TGFβ type I receptor (6). However overexpression of TGFβ in mice also leads to outgrowth of a few poorly differentiated spindle cell carcinomas (7) and increased metastasis of benign papillomas (8).
In contrast to these genetic models we have recently shown that pharmacological inhibition of the TGFβ type I receptor (ALK5) during skin tumor promotion with SB431542 (SB) suppresses outgrowth of benign lesions although this also enhances malignant conversion (9). While it is generally accepted that the tumor suppressive function of TGFβ is linked to its role as a negative regulator of epithelial cell proliferation (10), this result suggests that the function of TGFβ signaling in the premalignant keratinocyte is more complex. In the 2-stage carcinogenesis model, cancer initiation occurs through mutational activation of the HRas gene (11). Keratinocytes expressing activated HRas are hyperproliferative in vitro (12) and exhibit abnormal responses to differentiation inducing signals, with suppression of the early differentiation program such as induction of keratin 1 and 10 but overexpression of late differentiation genes (13). However, previous studies have shown that these cells both produce and remain growth inhibited by exogenous TGFβ1 (12). To identify mechanisms underlying suppression of tumor formation by ALK5 inhibition we examined the response of primary keratinocytes expressing an activated HRas oncogene to SB using bitrangenic primary keratinocytes from a doxycyline (Dox) inducible K5rtTA/tetORASV12G line. Here we show that pharmacological inhibition of ALK5 in HRASV12G-expressing primary keratinocytes causes cornification and cell death in the majority of keratinocytes, but also the secondary outgrowth of a cornification resistant non-senescent subpopulation. These results suggest that the ability of SB to inhibit benign tumor formation but enhance malignant conversion in epidermal carcinogenesis is linked to distinct effects on different populations of initiated keratinocytes.
Materials and Methods
Cell culture
Primary mouse keratinocytes were isolated and cultured according to a standard protocol (14) from newborn transgenic littermates. tetORASV12G (15) mice that express HRASV12G when induced by the bovine keratin 5 promoter (K5rtTA) (16) were obtained from the NCI mouse repository and newborn mice of each genotype identified by PCR were obtained from FVB/n background crosses of heterozygous K5rtTA and homozygous tetORASV12G mice. HRASV12G expression was induced by addition of 1μg/ml Dox to culture media. The v-HRas retrovirus was used as described (12). Unless otherwise indicated 0.5μM SB431542 (Sigma, St. Louis, MO) was used to block TGFβ1 signaling. Keratinocytes were treated with 1ng/ml TGFβ1 for indicated times (R&D systems, Minneapolis, MN). For all long-term experiments media was changed every other day. Photomicrographs of keratinocytes were made using an inverted Olympus CKV41 microscope, UPlan Fl objectives, SPOT RT-KE Mono-IR camera and SPOT Software 4.5 (Diagnostics Instruments, Inc., Sterling Heights, MI)
Measurement of Cell Proliferation
For cell growth curves, K5rtTA/tetORASV12G keratinocytes were pre-attached plated in high calcium (1.4mM) for 2 hours to remove contaminating fibroblasts which attach faster than keratinocytes, and then seeded at a density of 200,000 cells in 24-well culture trays. On day 2 post-plating, keratinocytes were treated with Dox and/or SB in 0.05 mM Ca2+ media and media was replaced every other day. Triplicate samples of each treatment group were counted twice at indicated time points using a Z1 Coulter particle counter (Beckman Coulter, Hialeah, FL). For 2-color analysis of cell cycle keratinocytes were pulsed with 40μM 5-bromo-2-deoxyuridine (BrdU, Becton Dickson, Franklin Lakes, NJ) 1 hour before harvesting, fixed in 70% ethanol, and then stained with anti-BrdU-FITC antibody (ABFM-18, Phoenix Flow Systems, San Diego, CA) and propidium iodide (PI, Invitrogen, Carlsbad, CA). Cells were analyzed using an EPICS-XL-MCL flow cytometer (Beckman Coulter) and the percentage of cells at each phase of the cell cycle determined with FlowJo Flow Cytometry analysis software.
Cornified envelope assay
A protocol adapted from published methods (17) was used to quantify cornified envelope formation. Floating/cornified cells were boiled at 90°C for 10 minutes in 2% sodium dodecyl sulfate/20mM DTT as cornified cells are resistant to this treatment. Phase contrast microscopy was used to view and count cornified cells under a hemocytometer which appear as “ghost cells” (18). The attached cells were counted using a Z1 Coulter particle counter (Beckman Coulter).
Measurement of Senescence
Keratinocytes were plated in 12-well tissue culture plates and on day 3 post-plating treated with Dox and/or SB for 11 days. Senescence associated-β-galactosidase (SA-βgal) staining was performed as described (12) and an inverted Olympus CKX41 microscope (20x microscope frame) was used. Positive cells were expressed as a percentage of total cells for each treatment group. Three different fields from each well were counted and triplicate samples were analyzed for each treatment group.
Calcium resistance
To determine responsiveness to increased calcium concentrations, passaged/immortalized K5rtTA/tetORASV12G keratinocytes at 5000 cells/60mm dish were seeded in 0.05 mM Ca2+ media with 1μg/ml Dox and 0.5μM SB. After 48 hours the calcium concentration was increased to 0.5mM and treatment continued for 9 days. Calcium resistant colonies were counted after the dishes were fixed and stained in hematoxylin.
RNA and protein analysis
Whole skin was homogenized using a Qiagen Tissuelyzer (Qiagen, Valencia, CA) in Trizol (Invitrogen). Quantitative RT-PCR (qPCR) was done for the indicated genes and normalized to 18s rRNA expression using the MyIQ system (BioRad Laboratories, Hercules, CA) and PerfeCTa SYBR Green SuperMix for iQ Quanta Biosciences, Gaithersburg, MD). All PCR samples were normalized to GAPDH. Primer sequences were obtained from published studies or using Primer 3 (19) software with Genebank sequence information. Protein was isolated with 0.5% NP-40 lysis buffer (0.5% IGEPAL CA-630, 250mM NaCl, 50mM Tris HCl, pH 7.4) with protease and phosphatase inhibitors. Antibodies directed against Smad 2/3, p-Smad2, p-ERK, ERK, p15, GAPDH (Cell Signaling Technology, Inc., Danvers, MA); HRAS, p-38, p16, p21 (Santa Cruz Biotechnology, Santa Cruz, CA); p-p38 (New England Biolabs, Ipswich, MA); and β-actin (Millipore, Billerica, MA) were used.
Animal studies
Crosses of Involucrin tTA (20) and tetORASV12G mice were maintained on 10 μg/ml Dox water, which was switched to water alone to induce HRASV12G expression. Seven-week-old mice were treated with 200 μL of acetone and/or 200 μL of 10.0 μM SB431542 every other day for 5 days, and skin was harvested 24h after the last SB treatment. Non-transgenic, K5rtTA x tetOTGFβ1 (21), K5rtTA x tetORASV12G, and K5rtTA x tetORASV12G/tetOTGFβ1 mice were treated with Dox for 2 days in chow (1gm/kg) to induce expression of HRASV12G and/or TGFβ1. All transgenic mice are on an FVB/n background. All animals were kept under a controlled environment of temperature and humidity and a 12h light/dark cycle. Animal studies were conducted under approved IACUC protocols.
Statistical Analysis
One-way ANOVA and Tukey’s Multiple Comparison post-test were used to test significance of multiple groups within an experiment. Student’s t test was used to compare two groups only, and the significance of the difference was described.
Results
ALK5 inhibition induces and TGFβ1 suppresses cornification of keratinocytes expressing an HRAS oncogene
We first tested if SB could suppress endogenous TGFβ1 signaling in keratinocytes expressing human oncogenic HRASV12G. Primary keratinocytes were isolated from bitransgenic K5rtTA/tetORASV12G mice and treated with Dox to induce HRASV12G in the presence or absence of SB. Figure 1 shows that 24 hours after the addition of Dox there was a large increase in expression of HRAS as detected by immunoblotting and this was accompanied by a corresponding increase in levels of p-Erk1/2, but expression of p-p38 MAPK was not detected. Although there was no major increase in total or pSmad2 levels after induction of HRASV12G, addition of SB caused an immediate reduction of pSmad2 and this was maintained with continuous treatment.
Figure 1.

SB431542 inhibits Smad2 phosphorylation in bitransgenic keratinocytes. A, Immunoblot showing induction of HRASV12G and Erk1/2 phosphorylation with Dox and reduction in p-Smad2 levels in HRASV12G-expressing and control keratinocytes treated with SB for indicated times. SB does not inhibit phosphorylation of ERK and no increase in p38 MAPK phosphorylation was observed with HRASV12G induction. Protein isolation and analysis as described in Materials and Methods, with GAPDH used as a loading control. N=2. C, Control; R, HRASV12G expression induced by Dox.
Since TGFβ1 is a potent growth inhibitor for keratinocytes expressing v-HRas (12) it was anticipated that inhibition of autocrine TGFβ signaling would increase proliferation. However, Figure 2A shows that there was no effect of SB on the fraction of HRASV12G-expressing keratinocytes in G1, S, or G2/M phases of the cell cycle, as measured by 2-color flow cytometry. Additionally, SB did not enhance proliferation of keratinocytes expressing reduced levels of HRASV12G produced using a suboptimal concentration of Dox (data not shown). Consistent with these results induction of HRASV12G in keratinocytes caused an increase in cell number over time relative to control keratinocytes but SB caused no additional increase in cell number within the first 5 days. However, after 5 days of continuous exposure to SB the number of attached HRASV12G keratinocytes rapidly decreased below that of control keratinocytes with a 50% decrease in attached cell number compared to HRASV12G alone (Fig. 2B). HRASV12G keratinocytes that remained attached to the culture dish had a more spindle shaped morphology compared to HRASV12G alone (Fig. 2C). Importantly, there was no effect of SB on normal keratinocytes that did not express HRASV12G.
Figure 2.

SB431542 enhances terminal differentiation in HRASV12G-expressing keratinocytes but does not affect cell proliferation. A, 2-color flow cytometric analysis of cell proliferation after 4 days of SB treatment shows no significant effect on cell cycle of either control or HRASV12G keratinocytes. Cells were stained with BrdU/PI and analyzed according to Materials and Methods. n=2, N=2. B, Attached cell number of HRASV12G keratinocytes is significantly reduced after 5 days of continuous treatment with SB. n=3, N=2. C, Photomicrographs showing effect of SB on bitransgenic cell monolayers after 5 days. HRASV12G keratinocytes alone (left) and HRASV12G + SB (right) on day 5. 100x magnification. D, Photomicrograph of floating cornified cells from SB treated HRASV12G keratinocytes. 200x magnification, scale bar represents 50μm. E, Enhanced formation of cornified envelopes in keratinocytes expressing HRASV12G treated with SB. Floating cells counted in triplicate by cornified envelope assay and expressed relative to attached cells on day 5. n=3, N=3. F, TGFβ1 blocks cornified envelope formation in HRASV12G- expressing keratinocytes. HRASV12G was induced for 3 days followed by an additional 2 days in the presence or absence of 1ng/ml TGFβ1. Cornified cells were measured as described in Materials in Methods and expressed relative to total attached cells. All keratinocytes were cultured and treated in 0.05mM Ca2+ media. n=3, N=2. C, Control; HRAS, HRASV12G expression induced by Dox. Values with * are significantly different at p <0.05. Error bars represent standard error of the mean.
Although flow cytometric analysis did not show a significant increase in the sub-G1 fraction prior to 5 days of SB treatment there was a significant increase in the number of floating cells with a morphology consistent with that of a cornified envelope, the insoluble product of keratinocyte terminal differentiation characterized by extensive protein crosslinking (22) (Fig. 2D). Since cornified envelopes are resistant to sodium dodecyl sulfate/DTT and boiling, we used this method to quantify the fraction of cornified envelope in cultures of HRASV12G keratinocytes treated with SB. Consistent with earlier studies showing aberrant terminal differentiation in v-HRas expressing mouse keratinocytes (13), expression of HRASG12V caused an increase in cornified envelopes relative to control keratinocytes. Treatment with SB significantly increased the number of cornified envelopes produced by HRASV12G keratinocytes but had no effect on control keratinocytes (Fig. 2E). Figure 2F shows that TGFβ1 treatment of HRASV12G keratinocytes significantly reduced the level of cornified envelopes indicating that the effect of SB was due to inhibition of TGFβ signaling rather than an off-target effect. Together these results suggest that autocrine TGFβ signaling does not act as a negative regulator of cell proliferation in these premalignant keratinocytes but is directly linked to terminal differentiation.
TGFβ signaling regulates expression of cornified envelope genes
We examined expression of genes encoding specific cornified envelope proteins and transglutaminases (TGM1, TGM3) that cause crosslinking to form the cornified envelope. TGFβ1 inhibited the induction of TGM1 and reduced baseline TGM3 expression by HRASV12G (Fig. 3A) but there was no effect on expression of Small Proline Rich (SPR) genes 1A and 2H (data not shown). Conversely, inhibition of TGFβ signaling with SB increased expression of TGM1 and TGM3 and significantly increased the expression of SPR1A and SPR2H (Fig. 3B). No change in keratin 1 or involucrin expression was detected between these treatment groups. Figure 3C shows that similar to TGFβ1 treatment, infection of HRASV12G keratinocytes with a constitutively active ALK5 adenovirus reduced TGM1 gene expression. Conversely, infection with an adenovirus expressing a dominant-negative TGFβ type II receptor increases TGM1 expression. To further test the ability of TGFβ1 to suppress aberrant differentiation in HRASV12G-expressing keratinocytes we used a different model in which primary keratinoyctes were transduced with a replication defective retrovirus expressing v-HRas. Previous studies have shown that v-HRas suppresses calcium-mediated induction of early differentiation markers such as keratin 1 but increases expression of late differentiation markers (13). Figure 3D shows that TGFβ1 reduced expression of the cornifed envelope protein filaggrin in v-HRas expressing keratinocytes in both proliferative (0.05 mM Ca2+) and differentiation inducing media (0.5 mM Ca2+).
Figure 3.

TGFβ signaling regulates expression of terminal differentiation genes in preneoplastic keratinocytes. A, Reduced expression of TGM1 and TGM3 in HRASV12G keratinocytes treated with TGFβ1. Bitransgenic keratinocytes were treated with Dox for 24h and then a further 12h with Dox ±TGFβ1 prior to isolation of RNA. n=4, N=2. B, SB increases expression of terminal differentiation genes in HRASV12G keratinocytes. Bitransgenic keratinocytes were treated for 4 days with Dox±SB prior to isolation of RNA. n=4, N=3. C, Infection of HRASV12G-expressing keratinocytes with adenoviruses expressing either a constitutively activated TGFβ type I receptor (ALK5) or a dominant negative TGFβ type II receptor inhibits and induces TGM1 expression, respectively. RNA was isolated from cells 24h after adenoviral infection. n=3, N=2. D, TGFβ1 blocks elevated Ca2+ induced expression of filaggrin in v-HRas infected keratinocytes. Protein extracts were isolated 24h after switching v-HRas infected keratinocytes to 0.5mM Ca2+ in the presence or absence of 1ng/ml TGFβ1, and immunoblotted for the indicated proteins. β-actin was used as a loading control. N=1. C, control; HRAS, HRASV12G expression induced by Dox. Values with * are significantly different at p <0.05. Error bars represent standard error of the mean.
To determine if inhibition of the TGFβ pathway could enhance terminal differentiation of premalignant keratinocytes in vivo, we used bitransgenic Involucrin tTA/tetORASV12G mice in part due to rapid mortality of mice expressing HRASV12G in the basal layer. Bitransgenic mice were taken off Dox to induce HRASV12G and were then treated topically with SB or acetone for 5 days. SB treatment caused a significant increase in the thickness of cornified layers compared to acetone alone (Fig. 4A–B), and this correlated with a significant increase in TGM1 and SPR1A mRNA levels (Fig. 4C). Although it is unclear why expression of HRASV12G causes an increase in TGM3 in vivo but reduced expression in vitro, SB treatment caused a similar but not statistically significant increase in TGM3 and SPR2H. To test the opposite response we co-induced HRASV12G and TGFβ1 in triple transgenic mice containing tetORASV12G and tetOTGFβ1 transgenes. TGFβ1 reduced expression of TGM3 with a similar trend for TGM1 (Fig. 4D). Expression of TGFβ1 alone also inhibited baseline TGM1 expression in vivo (Fig. 4D) similar to its effects on TGM1 expression in vitro (Fig. 3A). Although oncogenic HRASV12G induced SPR expression, TGFβ1 did not alter this response and no change was detected with TGFβ1 expression alone compared to control (data not shown). Taken together these results indicate that TGFβ1 signaling suppresses cornification of keratinocytes expressing oncogenic HRAS while pharmacological disruption of the TGFβ pathway with SB enhances cornification.
Figure 4.

TGFβ signaling pathway regulates expression of terminal differentiation markers in vivo. A, Photomicrographs showing increased thickness of the cornified layer in bitransgenic Involucrin tTA/tetORASV12G mice treated with SB (10.0μM in 200μl acetone) for 5 days. Mice were removed from Dox to induce HRASV12G expression. Magnification 200x, scale bar represents 50 μm. n=5. C, Cornified layer; E, Epidermis. B, Quantification of cornified layer thickness, measured at magnification 200x, 30 measurements/slide. n=5. C, Topical SB (10.0μM) treatment of InvtTA/tetORASV12G mice significantly increases TGM1 and SPR1A gene expression, with similar trends for TGM3 and SPR2H. n=5. D, Co-induction of TGFβ1 with HRASV12G in triple transgenic mice reduces expression of TGM1 and TGM3 compared to HRASV12G alone. n=5. A, Acetone; HRAS, HRASV12G expression induced by Dox removal; TGFβ, tetOTGFβ. Values with * are significantly different at p <0.05. Error bars represent standard error of the mean.
HRASV12G keratinocytes resistant to SB-induced cornification have an altered senescence phenotype
While there was a significant induction of cornification in the SB treated HRASV12G keratinocytes, some remained which were resistant to this effect and these cells reconstituted the culture over time in the presence of continuous SB and Dox treatment. Figure 5A–B shows that the large drop in attached cells which occurred in the SB treated HRASV12G keratinocytes at 5–6 days was followed by a rapid recovery such that by 11 days the culture became nearly confluent with no significant difference in cell number between HRASV12G treated with and without SB. These cells were keratinocyte in origin since they expressed keratin 1 (data not shown). A similar increase in cell number did not occur in SB treated control keratinocytes indicating that the effect was linked to expression of oncogenic HRAS.
Figure 5.

Continued proliferation of HRASV12G keratinocytes resistant to SB431542-induced cornification. A, Growth curve showing expansion of SB resistant cells and recovery of attached cell numbers after 11 days continuous treatment. n=3, N=2. B, Photomicrographs of bitransgenic monolayers cultured for 11 days with and without HRASV12G expression and SB, showing loss of attached cells at day 6, re-growth by day 11 and lack of effect of SB on keratinocytes not expressing oncogenic HRas. C, Control, bitransgenic keratinocytes without Dox or SB; HRAS, HRASV12G expression induced by Dox.
Since v-HRas expression in mouse keratinocytes causes hyperproliferation followed by senescence, and this is dependent on autocrine TGFβ signaling (12), we tested whether SB altered the senescence response in cornification resistant HRASV12G keratinocytes. Figure 6A shows that 11 days after HRASV12G induction, 65% of the keratinocytes expressed senescence associated-β-galactosidase (SA-βgal) a commonly used marker for cellular senescence (12), while cells that did not undergo cornification and continued to proliferate in the presence of SB were only 30% SA-βgal positive. Similarly, when v-HRas transduced Balb/c keratinocytes, which have a pronounced senescence response (23) were treated with SB for 11 days, there was a reduced senescence response as measured by SA-βgal (Fig. 6A). The senescence-associated induction of the tumor suppressor p16ink4a was reduced in HRASV12G keratinocytes treated with SB (Fig. 6B), but there was little effect on expression of the cyclin-dependent kinase inhibitor p21waf1. Similarly, when v-HRas keratinocytes were treated continuously with SB there was a dose dependent reduction in p16ink4a relative to untreated senescent cultures with no change in p21waf1 (Fig. 6C). Treatment of day 11 senescent keratinocytes with SB for an additional 4 days did not cause a significant reduction in p16ink4a levels suggesting that the effect of SB on expression of this tumor suppressor was indirect (data not shown), or once activated its expression was independent of TGFβ signaling. These non-senescent keratinocytes were still sensitive to SB as pSmad2 levels were reduced relative to control and expression of PAI-1, a well-characterized TGFβ1 responsive gene (24) was inhibited by SB to the same extent in senescent resistant cells as primary bitransgenic cells after a 24 hr treatment (Supplementary Fig. 1A–B). To test if the cornification and senescence resistant cells represented a subpopulation of HRASV12G-expressing keratinocytes we plated primary bitransgenic keratinocytes at low density before switching to Dox media with or without SB for 11 days. Figure 7A shows that after 11 days of SB treatment large colonies of densely packed cells formed only in the SB treated HRASV12G keratinocytes in contrast to a thin lawn of cells with the other treatment groups. To determine the growth properties of these cells we subcultured a mass population following 14 days in Dox and SB and passaged twice. Cells were grown in culture media without Dox or SB for 2 days and then tested for responsiveness to TGFβ1, and SB with and without HRASV12G. Figure 7B shows TGFβ1 inhibited the growth of these keratinocytes indicating no gross selection for resistance to this growth factor. As expected, induction of HRASV12G with Dox stimulated proliferation as with the initial primary culture, but in contrast to primary cultures, SB now stimulated proliferation of these cells with and without HRASV12G. To test if these senescence resistant keratinocytes had undergone malignant conversion we determined their ability to form colonies under differentiation-inducing conditions of elevated calcium, a hallmark of malignant conversion of primary keratinocytes expressing oncogenic Ras (25, 26). When plated at clonal density and cultured with Dox and SB in low calcium proliferation media (0.05 mM Ca2+), these keratinocytes readily formed colonies (Fig. 7C). No colonies formed in the absence of either Dox or SB indicating continued dependence on oncogenic HRas and blocked TGFβ signaling (not shown). When cells were switched from proliferation to differentiation media (0.5mM Ca2+) containing Dox and SB, colony formation did not occur, even though induction of HRASV12G in these cells was sufficient to suppress keratin 1 induction (not shown). In addition, when senescence-resistant keratinocytes were skin grafted onto athymic nude mice (14) predosed with Dox, 0/7 grafts formed tumors, even with topical SB treatment. Thus these cells do not have characteristics of fully malignant keratinocytes but exhibit altered growth and differentiation responses to inhibition of TGFβ1 signaling.
Figure 6.

Reduced Senescence in HRas-expressing keratinocytes treated long term with SB431542. A, HRASV12G and v-HRas expressing Balb/c keratinocytes treated for 11 days with SB have reduced percentage of senescent cells compared to untreated HRas expressing controls. n=3, N=2. B, Senescence-associated increase in p16ink4a is blocked in HRASV12G keratinocytes cultured in SB. Blot was probed for p21waf1 followed by p16ink4a without stripping. N=2. C, Dose dependent inhibition of p16ink4a protein expression by SB in v-HRas infected keratinocytes. Retrovirally infected Balb/c keratinocytes were treated with the indicated dose of SB continuously for 11 days prior to isolation of cell extract for immunoblotting. No change in p15ink4b was observed (not shown). β-actin was used as a loading control. N=2. C, Control; R, HRASV12G expression induced by Dox. Values with * are significantly different at p <0.05. Error bars represent standard error of the mean.
Figure 7.
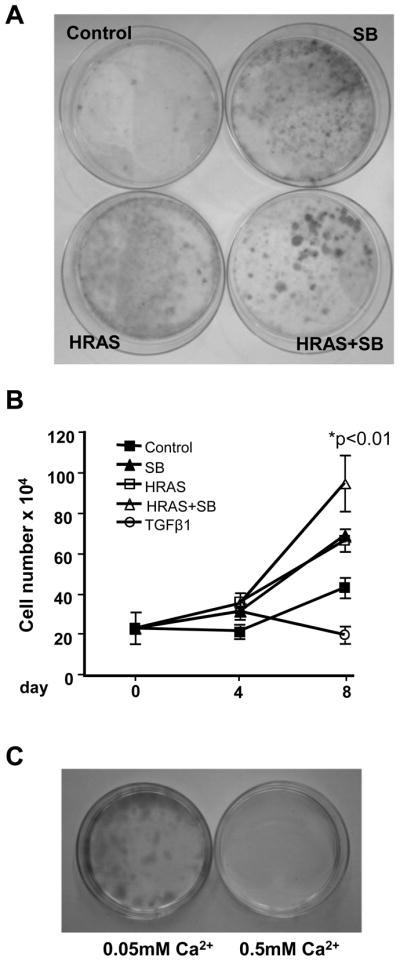
SB431542 resistant cells arise from a subpopulation of HRASV12G keratinocytes stimulated to proliferate by SB. A, HRASV12G keratinocytes plated at low density and treated with SB (2.0 μM) for 11 days form large colonies while cells expressing HRASV12G or treated with SB alone do not. N=3. B, Passaged resistant cells are growth stimulated by re-expression of HRASV12G and SB. A mass culture of SB resistant cells was passaged twice before plating in media without supplements for 2 days followed by treatment as indicated, and attached cell number was determined at the indicated days. There was a significant increase in cell number in cells expressing HRASV12G+SB compared to HRASV12G expression alone. n=3. N=2. C, Passaged HRASV12G+SB resistant keratinocytes can grow clonally in proliferation media (0.05 mM Ca2+) with continued Dox and SB treatment (left) but are unable to form colonies in differentiation media (0.5mM Ca2+ media, right). No colonies formed in either growth condition when cells were cultured with Dox or SB alone or without any treatment. Passaged cells were seeded at clonal density in the presence of Dox and SB to allow attachment and then switched to the indicated conditions. N=4. HRAS, HRASV12G expression induced by Dox; TGFβ1, 1ng/ml. Values with * are significantly different at p <0.05. Error bars represent standard error of the mean.
Discussion
TGFβ1 signaling has a well-characterized role in chemical carcinogenesis of the mouse epidermis. Recently we demonstrated that, in contrast to the paradigm of TGFβ as a tumor suppressor derived from genetic models, pharmacological inhibition of TGFβ signaling with the small molecule ALK5 inhibitor SB431542 suppressed benign tumor formation and enhanced malignant conversion. Here we have used primary keratinocytes expressing inducible oncogenic HRASV12G to study potential mechanisms through which ALK5 inhibition could produce these dual responses in vivo. Our study provides support for the novel hypothesis that subpopulations of HRASV12G-expressing keratinocytes have distinct responses to inhibition of TGFβ signaling and these dual responses are linked to tumorigenic potential.
In chemical carcinogenesis studies in the mouse skin, mutational activation of HRas is an essential first step in tumor formation. Keratinocytes expressing oncogenic HRas have abnormal responses to terminal differentiation signals (27, 28). The end product of keratinocyte differentiation is the cornified envelope, a highly insoluble complex of crosslinked proteins that is essential for the barrier function of the epidermis. As keratinocytes leave the basement membrane and withdraw from the cell cycle, they express specific proteins that are involved in formation of the cornified envelope including involucrin, filaggrin, specific suprabasal keratins, and small proline-rich proteins as well as transglutaminases that cause crosslinking of these structural proteins (22). In vitro studies show that oncogenic HRas blocks calcium-mediated induction of the early differentiation markers keratin 1 and 10, but causes overexpression of proteins associated with later stages of cornification including loricrin, filaggrin and transglutaminase (13). We observed increased cornified envelope formation in HRASV12G keratinocytes but this also was balanced by hyperproliferation such that cell loss was not observed. While it is generally accepted that exogenous TGFβ1 acts as a growth inhibitor for normal and preneoplastic keratinocytes and other epithelial cells, we found that inhibition of endogenous TGFβ signaling with SB had no effect on cell cycle or proliferation of HRASV12G keratinocytes. Rather, there was a significant increase in cornified envelope formation accompanied by loss of attached cells and increased expression of genes associated with cornified envelope formation. This indicates that the majority of HRASV12G-expressing keratinocytes were being driven towards cornification without self-renewal when ALK5 signaling was blocked. Conversely, our in vitro and in vivo studies show that TGFβ1 signaling can block cornified envelope formation and genes associated with formation of the cornified envelope in HRASV12G-expressing keratinocytes. Although some studies have not demonstrated any effects of TGFβ1 on terminal differentiation in normal human keratinocytes (29), our results are consistent with other studies showing that TGFβ1 can suppress squamous differentiation in normal and transformed human and mouse keratinocytes (30–33) and indicate that at this stage TGFβ effects on differentiation rather than proliferation predominate. Taken together our studies suggest that one mechanism underlying the ability of SB to suppress benign papilloma formation in 2-stage skin carcinogenesis is through enhanced terminal differentiation of epidermal keratinocytes containing a mutated HRas gene.
Although SB enhanced terminal differentiation and cell death in keratinocytes expressing HRASV12G, a subpopulation of these cells were resistant to this effect and continued to proliferate. Further analysis showed that the growth of these cells was stimulated when TGFβ1 signaling was blocked with SB suggesting that TGFβ signaling was now linked to negative regulation of the cell cycle. These cells also had reduced expression of the senescence marker SA-βgal and the tumor suppressor p16ink4a suggesting that they were also resistant to the senescence phenotype associated with oncogenic Ras expression in primary mouse keratinocytes. These results are consistent with previous studies which showed that TGFβ1−/−, Smad3−/− or DNTβRII expressing primary keratinocytes could overcome a v-HRas-induced senescence response (12, 34) and that TGFβ1 can induce p16ink4a expression in v-HRas keratinocytes (35). In this study however, treatment of senescent keratinocytes with SB did not reduce p16ink4a levels indicating that the effects following long-term treatment are indirect. We did not observe a significant effect of SB on the induction of Smad7 by HRASV12G at early timepoints although Smad7 expression was downregulated in the resistant cells (data not shown). The significance of this downregulation to the phenotype of these cells remains to be determined. The observation that SB stimulated outgrowth of discrete large colonies of bitransgenic keratinocytes expressing HRASV12G and that SB stimulated proliferation of passaged, resistant keratinocytes while no effect was observed in the initial primary cultures suggests that cornification and senescence resistant keratinocytes represent a distinct population of primary keratinocytes from those that undergo cornification in response to SB. No difference in HRAS protein levels were seen in attached and cornified cells 5 days after SB treatment (data not shown) so this is unlikely to represent the underlying mechanism. It is well established that subpopulations of primary epidermal keratinocytes exist with differential responsiveness to external stimuli such as TPA: the majority form cornified envelopes, while a small minority proliferate instead (36). Although speculative, one possibility is that resistant cells are derived from hair follicle stem cells present in the newborn mouse keratinocyte preparation that upon induction of oncogenic Ras and inhibition of ALK5 proliferate rather than undergo terminal differentiation. Studies showing that genetic inactivation of the Bone Morphogenic Protein Receptor 1A (BMPR1A) in the stem cell niche causes proliferation and blocked differentiation of previously quiescent stem cells (37, 38) support this concept.
While evasion of oncogene-induced senescence is associated with malignant progression in cells with TGFβ1 and other pathway defects (39), the keratinocytes that grew out resistant to SB-induced cornification and HRASV12G senescence did not form tumors when grafted onto athymic nude mice, remained dependent on continued HRASV12G expression and ALK5 inhibition for proliferation, and could not proliferate under differentiation inducing conditions indicating that they were not fully transformed. Nevertheless proliferation and reduced senescence following long-term SB treatment of keratinocytes harboring activated HRASV12G provides a mechanism for a more progressed phenotype that may be dependent on interaction with the tumor microenvironment or further mutations for full malignancy.
While a number of different small molecule ALK5 inhibitors can suppress the malignant phenotype including attenuated epithelial to mesenchymal transition, cell motility, migration and invasion in vitro and can reduce tumor growth, metastasis (9, 40), and enhanced tumor immunogenicity in vivo (41), this is the first study to demonstrate potential suppressive effects of ALK5 inhibition through induction of terminal differentiation in a premalignant epithelial cell as well as bidirectional responses to ALK5 inhibition within a primary cell population. While this expands the potential therapeutic potential for ALK5 inhibitors it also highlights the potential difficulties associated with long term use.
Supplementary Material
Acknowledgments
Grant Support: This work was supported by the National Institutes of Health [RO1 CA117957, RO1 CA122109 to A.B.G]; and the authors would also like to thank the Dermal Toxicology Specialty Section of the National Society of Toxicology for partial funding of this project through the Battelle Student Research Award to L.M.M.
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
References
- 1.Fowlis DJ, Cui W, Johnson SA, Balmain A, Akhurst RJ. Altered epidermal cell growth control in vivo by inducible expression of transforming growth factor β1 in the skin of transgenic mice. Cell Growth Differ. 1996;7:679–87. [PubMed] [Google Scholar]
- 2.Blessing M, Nanney LB, King LE, Hogan BL. Chemical skin carcinogenesis is prevented in mice by the induced expression of a TGF-β related transgene. Teratog Carcinog Mutagen. 1995;15:11–21. doi: 10.1002/tcm.1770150103. [DOI] [PubMed] [Google Scholar]
- 3.Honjo Y, Bian Y, Kawakami K, Molinolo A, Longenecker G, Boppana R, et al. TGF-beta receptor I conditional knockout mice develop spontaneous squamous cell carcinoma. Cell Cycle. 2007;6:1360–6. doi: 10.4161/cc.6.11.4268. [DOI] [PubMed] [Google Scholar]
- 4.Bian Y, Terse A, Du J, Hall B, Molinolo A, Zhang P, et al. Progressive tumor formation in mice with conditional deletion of TGF-beta signaling in head and neck epithelia is associated with activation of the PI3K/Akt pathway. Cancer Res. 2009;69:5918–26. doi: 10.1158/0008-5472.CAN-08-4623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amendt C, Schirmacher P, Weber H, Blessing M. Expression of a dominant negative type II TGF-β receptor in mouse skin results in an increase in carcinoma incidence and an acceleration of carcinoma development. Oncogene. 1998;17:25–34. doi: 10.1038/sj.onc.1202161. [DOI] [PubMed] [Google Scholar]
- 6.Goudie DR, D’Alessandro M, Merriman B, Lee H, Szeverenyi I, Avery S, et al. Multiple self-healing squamous epithelioma is caused by a disease-specific spectrum of mutations in TGFBR1. Nat Genet. 2011;43:365–9. doi: 10.1038/ng.780. [DOI] [PubMed] [Google Scholar]
- 7.Cui W, Fowlis DJ, Bryson S, Duffie E, Ireland H, Balmain A, et al. TGFβ1 inhibits the formation of benign skin tumors, but enhances progression to invasive spindle carcinomas in transgenic mice. Cell. 1996;86:531–42. doi: 10.1016/s0092-8674(00)80127-0. [DOI] [PubMed] [Google Scholar]
- 8.Weeks BH, He W, Olson KL, Wang XJ. Inducible expression of transforming growth factor beta1 in papillomas causes rapid metastasis. Cancer Res. 2001;61:7435–43. [PubMed] [Google Scholar]
- 9.Mordasky Markell L, Perez-Lorenzo R, Masiuk KE, Kennett MJ, Glick AB. Use of a TGFbeta type I receptor inhibitor in mouse skin carcinogenesis reveals a dual role for TGFbeta signaling in tumor promotion and progression. Carcinogenesis. 2010;31:2127–35. doi: 10.1093/carcin/bgq191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glick AB. TGFbeta1, Back to the Future: Revisiting its Role as a Transforming Growth Factor. Cancer Biol Ther. 2004;3:276–83. doi: 10.4161/cbt.3.3.849. [DOI] [PubMed] [Google Scholar]
- 11.Yuspa SH. The pathogenesis of squamous cell cancer: lessons learned from studies of skin carcinogenesis--Thirty-third G.H.A. Clowes Memorial Award Lecture. Cancer Res. 1994;54:1178–89. [PubMed] [Google Scholar]
- 12.Tremain R, Marko M, Kinnimulki V, Ueno H, Bottinger E, Glick A. Defects in TGFβ signaling overcome senescence of mouse keratinocytes expressing v-ras. Oncogene. 2000;19:1698–709. doi: 10.1038/sj.onc.1203471. [DOI] [PubMed] [Google Scholar]
- 13.Dlugosz AA, Cheng C, Williams EK, Dharia AG, Denning MF, Yuspa SH. Alterations in murine keratinocyte differentiation induced by activated rasHa genes are mediated by protein kinase C-alpha. Cancer Res. 1994;54:6413–20. [PubMed] [Google Scholar]
- 14.Dlugosz AA, Glick AB, Tennenbaum T, Weinberg WC, Yuspa SH. Isolation and utilization of epidermal keratinocytes for oncogene research. In: Vogt PK, Verma IM, editors. Methods in Enzymology. New York: Academic Press; 1995. pp. 3–20. [DOI] [PubMed] [Google Scholar]
- 15.Chin L, Tam A, Pomerantz J, Wong M, Holash J, Bardeesy N, et al. Essential role for oncogenic Ras in tumour maintenance. Nature. 1999;400:468–72. doi: 10.1038/22788. [DOI] [PubMed] [Google Scholar]
- 16.Diamond I, Owolabi T, Marco M, Lam C, Glick A. Conditional gene expression in the epidermis of transgenic mice using the tetracycline regulated transactivators tTA and rTA linked to the keratin 5 promoter. J Invest Dermatol. 2000;115:788–94. doi: 10.1046/j.1523-1747.2000.00144.x. [DOI] [PubMed] [Google Scholar]
- 17.Hough-Monroe L, Milstone LM. Quantitation of cross-linked protein: an alternative to counting cornified envelopes as an index of keratinocyte differentiation. Anal Biochem. 1991;199:25–8. doi: 10.1016/0003-2697(91)90264-t. [DOI] [PubMed] [Google Scholar]
- 18.Nagae S, Lichti U, De Luca LM, Yuspa SH. Effect of retinoic acid on cornified envelope formation: difference between spontaneous envelope formation in vivo or in vitro and expression of envelope competence. J Invest Dermatol. 1987;89:51–8. doi: 10.1111/1523-1747.ep12580383. [DOI] [PubMed] [Google Scholar]
- 19.Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–86. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- 20.Jaubert J, Patel S, Cheng J, Segre JA. Tetracycline-regulated transactivators driven by the involucrin promoter to achieve epidermal conditional gene expression. J Invest Dermatol. 2004;123:313–8. doi: 10.1111/j.0022-202X.2004.23203.x. [DOI] [PubMed] [Google Scholar]
- 21.Liu X, Alexander V, Vijayachandra K, Bhogte E, Diamond I, Glick A. Conditional epidermal expression of TGFbeta 1 blocks neonatal lethality but causes a reversible hyperplasia and alopecia. Proc Natl Acad Sci U S A. 2001;98:9139–44. doi: 10.1073/pnas.161016098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eckert RL, Crish JF, Robinson NA. The epidermal keratinocyte as a model for the study of gene regulation and cell differentiation. Physiol Rev. 1997;77:397–424. doi: 10.1152/physrev.1997.77.2.397. [DOI] [PubMed] [Google Scholar]
- 23.Woodworth CD, Michael E, Smith L, Vijayachandra K, Glick A, Hennings H, et al. Strain-dependent differences in malignant conversion of mouse skin tumors is an inherent property of the epidermal keratinocyte. Carcinogenesis. 2004;25:1771–8. doi: 10.1093/carcin/bgh170. [DOI] [PubMed] [Google Scholar]
- 24.Wikner NE, Elder JT, Persichitte KA, Mink P, Clark RA. Transforming growth factor-beta modulates plasminogen activator activity and plasminogen activator inhibitor type-1 expression in human keratinocytes in vitro. J Invest Dermatol. 1990;95:607–13. doi: 10.1111/1523-1747.ep12505603. [DOI] [PubMed] [Google Scholar]
- 25.Yuspa SH, Hawley-Nelson P, Koehler B, Stanley JR. A survey of transformation markers in differentiating epidermal cell lines in culture. Cancer Res. 1980;40:4694–703. [PubMed] [Google Scholar]
- 26.Kulesz-Martin M, Kilkenny AE, Holbrook KA, Digernes V, Yuspa SH. Properties of carcinogen altered mouse epidermal cells resistant to calcium-induced terminal differentiation. Carcinogenesis. 1983;4:1367–77. doi: 10.1093/carcin/4.11.1367. [DOI] [PubMed] [Google Scholar]
- 27.Yuspa SH, Kilkenny AE, Stanley J, Lichti U. Keratinocytes blocked in phorbol ester-responsive early stage of terminal differentiation by sarcoma viruses. Nature. 1985;314:459–62. doi: 10.1038/314459a0. [DOI] [PubMed] [Google Scholar]
- 28.Yuspa SH, Vass W, Scolnick E. Altered growth and differentiation of cultured mouse epidermal cells infected with oncogenic retrovirus: contrasting effects of viruses and chemicals. Cancer Res. 1983;43:6021–30. [PubMed] [Google Scholar]
- 29.George MD, Vollberg TM, Floyd EE, Stein JP, Jetten AM. Regulation of transglutaminase type II by transforming growth factor-β 1 in normal and transformed human epidermal keratinocytes. J Biol Chem. 1990;265:11098–104. [PubMed] [Google Scholar]
- 30.Reiss M, Sartorelli AC. Regulation of growth and differentiation of human keratinocytes by Type β transforming growth factor and epidermal growth factor. Cancer Res. 1987;47:6705–9. [PubMed] [Google Scholar]
- 31.Saunders NA, Jetten AM. Control of growth regulatory and differentiation-specific genes in human epidermal keratinocytes by interferon gamma. Antagonism by retinoic acid and transforming growth factor beta 1. J Biol Chem. 1994;269:2016–22. [PubMed] [Google Scholar]
- 32.Dahler AL, Cavanagh LL, Saunders NA. Suppression of keratinocyte growth and differentiation by transforming growth factor beta1 involves multiple signaling pathways. J Invest Dermatol. 2001;116:266–74. doi: 10.1046/j.1523-1747.2001.01243.x. [DOI] [PubMed] [Google Scholar]
- 33.Reiss M, Zhou ZL. Uncoupling of the calcium-induced terminal differentiation and the activation of membrane-associated transglutaminase in murine keratinocytes by type-beta transforming growth factor. Exp Cell Res. 1989;183:101–11. doi: 10.1016/0014-4827(89)90421-7. [DOI] [PubMed] [Google Scholar]
- 34.Vijayachandra K, Lee J, Glick AB. Smad3 regulates senescence and malignant conversion in a mouse multistage skin carcinogenesis model. Cancer Res. 2003;63:3447–52. [PubMed] [Google Scholar]
- 35.Vijayachandra K, Higgins W, Lee J, Glick A. Induction of p16(ink4a) and p19(ARF) by TGFbeta1 contributes to growth arrest and senescence response in mouse keratinocytes. Mol Carcinog. 2008 doi: 10.1002/mc.20472. [DOI] [PubMed] [Google Scholar]
- 36.Yuspa SH, Ben T, Hennings H, Lichti U. Divergent responses in epidermal basal cells exposed to the tumor promoter 12-O-tetradecanoylphorbol-13-acetate. Cancer Res. 1982;42:2344–9. [PubMed] [Google Scholar]
- 37.Kobielak K, Pasolli HA, Alonso L, Polak L, Fuchs E. Defining BMP functions in the hair follicle by conditional ablation of BMP receptor IA. J Cell Biol. 2003;163:609–23. doi: 10.1083/jcb.200309042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kobielak K, Stokes N, de la CJ, Polak L, Fuchs E. Loss of a quiescent niche but not follicle stem cells in the absence of bone morphogenetic protein signaling. Proc Natl Acad Sci U S A. 2007;104:10063–8. doi: 10.1073/pnas.0703004104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Collado M, Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer. 2010;10:51–7. doi: 10.1038/nrc2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsuchida K, Nakatani M, Uezumi A, Murakami T, Cui X. Signal transduction pathway through activin receptors as a therapeutic target of musculoskeletal diseases and cancer. Endocr J. 2008;55:11–21. doi: 10.1507/endocrj.kr-110. [DOI] [PubMed] [Google Scholar]
- 41.Uhl M, Aulwurm S, Wischhusen J, Weiler M, Ma JY, Almirez R, et al. SD-208, a novel transforming growth factor beta receptor I kinase inhibitor, inhibits growth and invasiveness and enhances immunogenicity of murine and human glioma cells in vitro and in vivo. Cancer Res. 2004;64:7954–61. doi: 10.1158/0008-5472.CAN-04-1013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.