

Abstract
Tissue factor (TF), the initiator of blood coagulation and thrombosis, is up-regulated after vascular injury and in atherosclerotic states. Systemic administration of recombinant TF pathway inhibitor (TFPI) has been reported to decrease intimal hyperplasia after vascular injury and also to suppress systemic mechanisms of blood coagulation and thrombosis. Here we report that, in heritable hyperlipidemic Watanabe rabbits, adenoviral gene transfer of TFPI to balloon-injured atherosclerotic arteries reduced the extent of intimal hyperplasia by 43% (P < 0.05) compared with a control vector used at identical titer (1 × 1010 plaque-forming units/ml). Platelet aggregation and coagulation studies performed 7 days after local gene transfer of TFPI failed to show any impairment in systemic hemostasis. At time of sacrifice, 4 weeks after vascular injury, the 10 Ad-TFPI treated carotid arteries were free of thrombi, whereas two control-treated arteries were occluded (P, not significant). These findings suggest that TFPI overexpressed in atherosclerotic arteries can regulate hyperplastic response to injury in the absence of changes in the hemostatic system, establishing a role for local TF regulation as target for gene transfer-based antirestenosis therapies.
Keywords: vascular smooth muscle cell, thrombosis, restenosis, hypercholesterolemia, atherosclerosis
Postinjury intimal hyperplasia and vascular stenosis remain unresolved problems in the treatment of vascular disorders. Catheter-based interventions, including balloon angioplasty and stenting, initially restore blood flow in obstructed arteries in more than 95% of patients. Within 6 months, however, vasospasm, thrombosis, and intimal hyperplasia cause clinically significant renarrowing of arteries in 15–40% of treated patients (1). The pathogenesis of intimal hyperplasia after vascular injury is believed to involve diverse signaling cascades that ultimately converge on vascular smooth muscle cells (VSMC), stimulating their proliferation and migration and their prolonged secretion of extracellular matrix (1, 2). Thrombus formation at the site of injury contributes to reobstruction of the vessel through its bulk and through the release of growth and chemotactic factors that further promote accumulation of intimal VSMC (1–3). Proteins involved in blood clotting, including the tissue factor (TF)/factor VIIa complex, factor Xa, and thrombin (4–6), exert chemotactic and mitogenic activity on VSMC and other cells.
TF, a member of the superfamily of cytokine receptors, plays a role in embryonic blood vessel formation (7) and is the initiator of blood coagulation and thrombosis (8). In healthy vessels, TF is present in the adventitia and in the deep tunica media (9, 10) but increases in the vascular intima and media after arterial injury (11) and in atherosclerotic states, where TF accumulates especially in lipid-laden plaques (10, 12, 13). In animal models of vascular injury, systemic administration of the inhibitor of TF (TF pathway inhibitor, or TFPI) (14–16), or of inhibitors of factor Xa (17) or thrombin (18), has suggested a role for the TF pathway of blood coagulation and thrombosis in the pathogenesis of postinjury intimal hyperplasia. However, these studies have not distinguished between the local influence of TF inhibition in the injured vessel wall and the systemic anticoagulant effects achieved with i.v. administration of antithrombotic agents. In addition, although local gene transfer of the thrombin inhibitor, hirudin, has been reported to reduce neointimal proliferation after balloon angioplasty in normal rat carotid arteries (19), few reports have evaluated the effects of local gene transfer in atherosclerotic injury models and none, to our knowledge, has examined the role of proteins engaged in blood coagulation and thrombosis.
Because TF is up-regulated in atherosclerotic plaques, which often rupture, triggering acute coronary thrombosis and infarction (12, 20), the effect of TFPI on the development of intimal hyperplasia may play a prominent role when vascular insult takes place in an atherosclerotic vessel. Accordingly, we examined regulation of intimal hyperplasia by local gene transfer of TFPI in balloon-injured atherosclerotic arteries of heritable hypercholesterolemic Watanabe rabbits (21, 22).
Construction and Amplification of a Replication-Deficient Adenovirus Encoding a Full-Length Human TFPI cDNA.
Construction of the replication-deficient adenoviral vector, which encodes a full-length human TFPI (23) driven by the cytomegalovirus immediate/early promoter/enhancer (Ad-TFPI), has been previously described (24). Ad-TFPI was plaque purified from 293 cells and further purified according to published procedures (24). Purified virions were suspended in sucrose (2% w/vol) and MgCl2 (2 mM) in PBS, desalted by Sepharose CL4B chromatography (Pharmacia & Upjohn), supplemented with 5% glycerol, and stored at −80°C. High-titer Ad-RR, a control vector identical to Ad-TFPI minus the foreign gene, was prepared in an identical fashion. The concentration of infectious viral particles was determined in 293 cells by plaque assay (25) and expressed as plaque-forming units (pfu).
Determination of TFPI Concentrations and Cell Culture.
An ELISA for human TFPI (American Diagnostica, Greenwich, CT) measured TFPI, where TFPI is detected with a monoclonal antibody specific for the human TFPI Kunitz domain 1. Canine and rabbit VSMC were explanted, respectively, from femoral and carotid arteries as described (25), and cultured at passage 3–4 in DMEM supplemented with 10% FBS, streptomycin, and penicillin. Human VSMC were a gift from Tim Scott-Burden (deceased). Canine VSMC (2 × 103 cells/well) were infected for 6 h with Ad-TFPI at multiplicity of infection (moi) 20, 100, 200, and 500, or with Ad-RR (moi 500). The virus was then removed and the culture continued for 7 days. The conditioned medium was replaced daily. Additional wells of VSMC were infected at identical moi with Ad-TFPI and were daily counted to normalize TFPI secretion to 106 cells/24 h.
Determination of TFPI Activity.
TFPI activity was measured in a TF/factor VIIa inhibition assay (American Diagnostica), where the TF/factor VIIa activity remaining after incubation with the conditioned culture medium is assayed for its ability to convert factor X to Xa, as reflected by cleavage of a Xa-specific p-nitroaniline substrate. TFPI activity is expressed in TFPI units, whereby 1 TFPI unit corresponds to 55 ng of TFPI, the mean concentration of TFPI found in 1 ml of plasma in 300 human volunteers (American Diagnostica; data available on request).
Detection of TFPI by Immunohistochemistry.
No heparin was administered before the animals were killed (26). The carotid arteries were embedded in cryoprotective compound (Triangle Biomedical Sciences, Durham, NC) and transferred to −80°C. Five-micrometer tissue sections were fixed for 15 min in 4% formaldehyde in PBS, incubated in 3% H2O2, blocked in 2% horse serum, and exposed for 1 h to a monoclonal antibody to human TFPI (American Diagnostica, product no. 4903) or cytomegalovirus (Dako). Primary antibodies were diluted to 1:200. The antibody recognizing human TFPI used for immunohistochemistry is a mouse IgG1 raised against an epitope in the Kunitz I domain of human TFPI and is a neutralizing antibody. After incubation in a biotinylated anti-mouse antibody, the sections were treated with streptavidin–biotin horseradish peroxidase (Vector Laboratories). Antibody binding was visualized with diamino benzidine, and the sections were counterstained with 1% alcian blue/2% methyl green.
Anticoagulant Activity of Human TFPI in Rabbit Plasma.
To determine whether human TFPI produced by rabbit VSMC prolonged the clotting time of rabbit plasma, rabbit VSMC were infected with Ad-TFPI and the control vector, Ad-RR. After 24 h, the conditioned medium was added to rabbit plasma. The clotting time was determined after rabbit TF (Sigma) was added, as described in Results.
Adenovirus-Mediated TFPI Gene Transfer to Balloon-Injured Atherosclerotic Arteries.
Animal experiments were performed according to the institutional guidelines of the University of Texas–Houston Medical School. Twenty Watanabe rabbits of advanced age (32.2 ± 5 months; range 24–39 months) were used for gene-expression and dose-finding studies. Anesthesia was induced with xylazine and ketamine and maintained after endotracheal intubation with isofluorane in oxygen. A 5-F sheath was inserted into the right femoral artery, and heparin (150 units/kg) was administered. A balloon angioplasty catheter with a 20 × 2.5-mm balloon was introduced into the femoral sheath and advanced over a guide wire to the right common carotid artery. The balloon was inflated 5 times to 8 atm (1 atm = 101.3 kPa) (30 sec each inflation; 60-sec intervals between inflations). After the last inflation, the balloon was withdrawn to the caudal end of the injured carotid segment. This segment was temporarily isolated with umbilical tape to leave the catheter tip included in the isolated segment and rinsed with physiological saline solution. Ad-TFPI, the control vector Ad-RR, adenoviral vectors encoding unrelated genes, or viral buffer alone, were introduced into the isolated segment in an amount sufficient to barely distend the segment (250 ± 55 ml). Viral titers of 1 and 3.5 × 1010 pfu/ml were studied and removed after 30 min through the lumen of the catheter. The umbilical ties were released and the catheter withdrawn from the animals. The cut-down sites and skin wounds were repaired, and the rabbits were allowed to recover. Dalteparin, 60 units/kg (Fragmin, Pharmacia & Upjohn), was given 1 h after angioplasty and repeated once after 12 h. Of these older rabbits, one died during recovery and one within 36 h after surgery following a stroke. The rabbits were euthanized 4 days after surgery, and the carotid arteries were harvested to assess expression of human in the arterial wall. Tissues were frozen in cryoprotective agent and immunostained as described above.
Previous studies of adenoviral TFPI gene transfer in domestic pigs (24) and normal rabbits (27) detected no impairment of hemostatic variables at a time when antithrombotic protection was observed (respectively 5 and 6 days after injury). We further investigated the integrity of the hemostatic system after local adenoviral gene transfer of TFPI. At baseline, before balloon injury and virus delivery, blood was drawn from an ear vein of the Watanabe rabbits and platelet aggregation, activated partial thrombin times, prothrombin times, and activated clotting times were measured with standard procedures (see Results). Hemostatic variables and platelet aggregation were measured again 7 days after angioplasty and infection of the injured arteries with Ad-TFPI (n = 10) and Ad-RR (n = 9), after which the rabbits were killed to prevent blood loss and hemodilution from interfering with thrombosis.
Neointima formation, vascular stenosis, and incidence of thrombosis were evaluated by histological examination 4 weeks after injury in a group of 12- to 16-month-old Watanabe rabbits of either sex. Ten rabbits were analyzed in each group. One additional Ad-RR-treated rabbit was killed 3 days after injury, because of development of a stroke.
Computer-Assisted Histomorphometric Evaluation of Intimal Hyperplasia.
Four weeks after injury, the rabbits were killed with sodium pentobarbital overdose. The carotid arteries were pressure perfusion-fixed with 10%-buffered formaldehyde at 100 mmHg (1 mmHg = 133 Pa) and filled gently with a latex solution to minimize collapse of the vessel walls during processing of the artery (25). Because of the multiple steps of tissue processing and handling, the latex filling is often lost during the final washing steps. This has in general little, if any, importance, given that at this stage the arteries are fixed and hard. In addition, in this study, many arteries were relatively hardened before the fixation step.
The injured carotid artery and a segment of the contralateral uninjured carotid artery of each rabbit (Ad-TFPI, n = 10; Ad-RR, n = 10) were collected into 10%-buffered formaldehyde, cut in 2-mm rings, and embedded in paraffin. Seven to nine arterial rings were obtained from each injured carotid segment and stained with the Verhoeft–Van Gieson elastic stain. Magnified images were captured by using an Axiophot microscope (Zeiss) and digital camera (Leaf Systems Lumina, Southboro, MA). Images were processed with software from Optima Imaging Analysis Systems (Version 6.5, Silver Spring, MD). A technician unaware of the treatment the rabbits had received performed measurements.
Plasma Cholesterol Measurement.
At time of surgery, total plasma cholesterol was measured with a Cholestech LDX apparatus (Cholestech, Haywood, CA). To obtain cholesterol levels, 35–60 μl of whole blood (collected from an ear vein) were transferred to the sample cassette and placed in the apparatus, which prints out a cholesterol level within 5 min.
Statistical Analysis.
In studies evaluating the extent neointima formation after experimental treatments, multiple sections from each injured artery (divided in rings) have been measured to compute a single mean for each artery (or “N”). On the basis of these individual means, a mean ± SD is then calculated for each group. This method does not take into account that, (i) repeated measurements performed to compute a single value for each “N”, i.e., the mean value for each artery; (ii) the variability in the number of microscopic sections (or rings) evaluated per artery; (iii) the variability in the extent of underlying atherosclerotic or other preexisting lesions (where present); (iv) the variability in neointima formation between individual sections (rings) of an individual artery; (v) the dependence of within-subject observations (i.e., the correlation between sections from the same artery). To integrate these variables into the final statistical analysis, we analyzed the data and calculated statistical differences of histomorphometric variables between the two treatment groups by using the general estimation equation for repeated site-specific measurements (30–32) (SAS Institute, Cary, NC, Ver. 8.0). Statistical significance was defined by a P value <0.05. Values are indicated as mean ± SD.
Results
TPFI Secretion by Cultured VSMC.
After infection of the canine VSMC with Ad-TFPI as described above, TFPI secretion was nearly 1 μg/106 cells within 24 h after infection at moi 500, peaked at 4 days, and declined thereafter (Fig. 1a). A nonlinear viral-dose response was seen with Ad-TFPI from moi 20–500 (Fig. 1b), whereas no TFPI was detected in the conditioned medium of the canine VSMC infected with the Ad-RR control vector. Three days after infection of the human VSMC with Ad-TFPI at moi 100, the TFPI concentration in the conditioned medium of cultured human VSMC was 315 ± 99 ng/106 cells/24 h, whereas no TFPI was detected in the conditioned medium of uninfected human VSMC or human VSMC infected with Ad-RR. TFPI secreted by the transduced canine VSMC inhibited factor X activation by the catalytic TF/factor VIIa complex. No anti-TF/VIIa activity was detected in control-treated (Ad-RR) VSMC (Fig. 1c).
Figure 1.
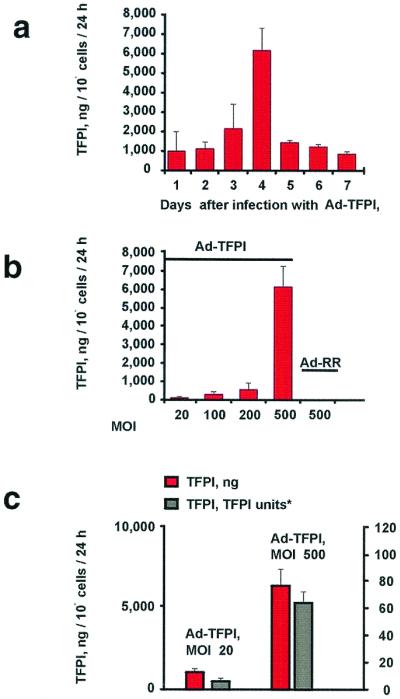
TFPI secretion in canine VSMC. Human TFPI was measured by the ELISA in the conditioned medium of dog VSMC. (a) Time course of TFPI (in ng/106 cells/24 h) secreted by VSMC infected for 6 h with Ad-TFPI or Ad-RR (identical construct minus the foreign gene) at moi 500 (see Methods). (b) Viral dose response of TFPI secretion in VSMC (ng/106 cells/24 h) 4 days after a 6-h infection with Ad-TFPI. No TFPI was detected in VSMC infected with Ad-RR. (c) Anti-TF/factor VIIa activity of human TFPI secreted by dog VSMC. TFPI activity in the conditioned medium is expressed in TFPI units (where 1 unit corresponds to about 55 ng TFPI in human plasma; see Methods).
Rabbit VSMC were treated with mock (buffer alone), Ad-TFPI, and Ad-RR at moi 100. After 24 h, the TFPI concentration in the conditioned medium was 1,238 ± 151 ng/ml in Ad-TFPI-infected cells but was undetectable in control-treated cells. When rabbit TF was used as initiator, the clotting time of rabbit plasma was 15.1 ± 0.6, 15 ± 0.5, and 20.5 ± 0.5 sec after addition of the conditioned medium from VSMC treated, respectively, with no virus (mock), the Ad-RR control, and Ad-TFPI (P < 0.05 compared with mock and Ad-RR).
Adenovirus-Mediated TFPI Gene Transfer to Balloon-Injured Atherosclerotic Arteries.
To determine whether TFPI can be transferred to severely atherosclerotic rabbit arteries, Ad-TFPI, Ad-RR, viral buffer alone, and adenoviral constructs, encoding cyclooxygenase-1 or enhanced green fluorescent protein, were delivered at 1 and 3.5 × 1010 pfu/ml to the carotid arteries of 20 Watanabe rabbits of advanced age (32.2 ± 5.1 months, mean ± SD). Similar to humans with familial hypercholesterolemia, these rabbits do not express low-density lipoprotein receptors and develop systemic atherosclerosis at about 6 months of age (21, 22).
By using a neutralizing monoclonal antibody against an epitope in the Kunitz I domain of human TFPI (see Methods), expression of TFPI was observed after local delivery of Ad-TFPI at 1 × 1010 pfu/ml to severely atherosclerotic plaques (Fig. 2 a and b) and in lesions of lesser severity (data not shown). Injured carotid arteries of animals treated with virus suspension buffer alone, with Ad-RR, with Ad-COX-1, or with Ad-eGFP did not show immunostaining for TFPI (results not shown). In addition, no immunostaining for endogenous rabbit TFPI was observed in the uninjured endothelium of rabbit carotid arteries and s.c. microvessels of rabbits, which had not been injured or infected with Ad-TFPI (Fig. 2 c and d). Accordingly, this lower viral titer was used to examine whether gene transfer of TFPI reduces intimal hyperplasia in balloon-injured carotid arteries of atherosclerotic Watanabe rabbits.
Figure 2.

TFPI gene transfer in balloon-injured atherosclerotic rabbit arteries and TFPI immunoreactivity in normal rabbit endothelium. Atherosclerotic carotid plaque of a Watanabe rabbit killed 4 days after infection with Ad-TFPI at 1 × 1010 pfu/ml (see Methods). (a) Immunostain for human TFPI. (b) The TFPI antibody used in a is replaced with an unrelated (cytomegalovirus) control antibody in an adjacent section; c and d show endothelium, respectively, of the carotid artery and s.c. microvasculature of a normal New Zealand White rabbit killed without prior injury. Tissue sections were immunostained with the antibody recognizing human TFPI. a and b, ×200; c, ×400; d, ×100.
Severe injury was induced by transluminal balloon angioplasty of carotid arteries of 12- to 16-month-old Watanabe rabbits of either sex. Immediately after injury, the damaged arterial segments were isolated externally, and Ad-TFPI or the control vector, Ad-RR, was instilled into the isolated arterial segments as described. One rabbit treated with Ad-RR was prematurely killed and excluded from analysis after developing a stroke 3 days after injury. The total plasma cholesterol concentrations at time of surgery were 403 ± 101 and 406 ± 73, respectively, in Ad-RR- and Ad-TFPI-treated rabbits.
Twenty-eight days after balloon injury, both carotid arteries were harvested, and arterial sections were analyzed by quantitative histomorphometry. Despite marked variability, intima/media ratios of individual arterial sections clustered toward lower values in Ad-TFPI- compared with Ad-RR-treated rabbits (Fig. 3a). The mean intima/media ratio of Ad-TFPI-treated injured carotid arteries was reduced by 43% (P < 0.05) compared with that of Ad.RR-treated vessels (Fig. 3b), whereas the reduction in absolute intima area (0.41 ± 0.25 mm2, Ad-RR; 0.26 ± 17 mm2, Ad-TFPI) and stenosis (42 ± 20%, Ad-RR; 26 ± 15%, Ad-TFPI) was of borderline statistical significance (Fig. 3 b and c). The average outer diameter and thickness of the tunica media were virtually identical between the two groups, indicating that a trend toward a larger lumen size was caused exclusively by a reduction in neointima formation (Fig. 3d).
Figure 3.

Histomorphometric observations in balloon-injured carotid arteries of Watanabe rabbits treated with Ad-TFPI. The carotid arteries of 12- to 16-month-old Watanabe rabbits of either sex were treated at the time of severe balloon injury with Ad-TFPI, encoding a human TFPI cDNA, or Ad-RR, an identical vector minus the transgene. Both vectors were locally delivered at a titer of 1 × 1010 pfu/ml. Animals were killed 4 weeks after injury, and carotid arteries were processed as described in Methods. Quantitative histomorphometry was performed in blinded fashion. Shown are (a) individual intima/media ratios observed in injured and adenovirally treated arterial sections; (b) mean areas of intima and media and mean intima/media ratios; (c) the degree of stenosis in the injured arteries; (d) the average outer diameter of the media, computed as average diameter between opposite sides of the media/adventitia border (a measure of external vessel size independent of dissection artifacts of the adventitia) and the average lumen area; and (e) intima/media ratio and stenosis of the uninjured contralateral carotid arteries.
To compare the extent of atherosclerosis between both groups, histomorphometry was performed on the contralateral uninjured carotid artery of each rabbit. Fig. 3e illustrates the degree of variability in the uninjured atherosclerotic carotid arteries, which showed a marginal trend toward more severe lesions in the rabbits assigned to treatment with Ad-TFPI. Histological examples of adenovirus treated arteries are shown in Fig. 4.
Figure 4.

Examples of carotid arteries of Watanabe rabbits. (a and b) Verhoeft-Van Gieson stain of sections from Watanabe rabbit carotid arteries treated with Ad-TFPI (a) and Ad-RR (b). (c) Plaque in the noninjured contralateral carotid artery of an Ad-TFPI-treated rabbit. (d) Section adjacent to that shown in c and stained with hematoxylin-eosin to illustrate the difference in texture between arterial tissue and latex filled arterial lumen. I, intima; L, lumen filled with residual latex to prevent postmortem distortion of lumen and vessel wall; P, atherosclerotic plaque. →, internal elastic membrane. (×40.)
Carotid thrombi at the site of injury were observed in two arteries treated with the Ad-RR control vector and in none treated with Ad-TFPI (P, not significant). No prolongation in coagulation variables or decrease in platelet aggregation was detected 7 days after viral delivery to the balloon-injured arteries (Tables 1 and 2), indicating that, at this time, the hemostatic system was not affected by local TFPI gene transfer.
Table 1.
Hemostatic variables in Watanabe rabbits treated locally with Ad-TFPI (n = 10) and Ad-RR (n = 9)
| ACT, s | PT, s | aPTT, s | BT, s | |
|---|---|---|---|---|
| Ad-TFPI | ||||
| Day 0 | 116 ± 7 | 9 ± 0 | 28 ± 5 | 114 ± 18 |
| Day 7 | 110 ± 13 | 9 ± 0 | 28 ± 7 | 108 ± 22 |
| Ad-RR | ||||
| Day 0 | 14 ± 18 | 9 ± 1 | 30 ± 9 | 108 ± 17 |
| Day 7 | 113 ± 7 | 9 ± 0 | 29 ± 8 | 110 ± 16 |
To measure activated coagulation times (ACT), prothrombin times (PT), and activated partial thromboplastin times (aPTT), blood was collected from an earvein before injury (day 0) and from the femoral vein on day 7. Variables were measured in a Hemochron-80 Dual Coagulation System (International Technidyne, Edison, NJ). Bleeding times (BT) were determined as ear-skin bleeding time, using a Simplex II device, as described in Methods.
Table 2.
Whole blood aggregation of Watanabe rabbits treated locally with Ad-TPFI (n = 10) and Ad-RR (n = 9)
| Aggregation, %
|
|||||
|---|---|---|---|---|---|
| Collagen,
μ/ml
|
Thrombin, units/ml
|
||||
| 5 | 10 | .125 | .25 | .5 | |
| Ad-RR | |||||
| Day 0 | 33 ± 12 | 52 ± 9 | 30 ± 29 | 65 ± 21 | 92 ± 16 |
| Day 7 | 38 ± 16 | 50 ± 24 | 21 ± 28 | 86 ± 22 | 86 ± 23 |
| Ad-TFP | |||||
| Day 0 | 27 ± 8 | 44 ± 15 | 10 ± 14 | 57 ± 25 | 89 ± 15 |
| Day 7 | 37 ± 10 | 47 ± 20 | 18 ± 2 | 92 ± 34 | 102 ± 27 |
Whole blood aggregation (%) was measured as electrical impedance (ohms) in a Chrono-Log aggregometer (Chrono-Log Corporation, Havertown, PA) before injury (day 0) and after 7 days. Blood was drawn into heparin (2 units/ml) for aggregation to collagen (Hormon Chemie, Munich) or into 3.8% sodium citrate (1:9 = vol/vol) for aggregation to α/β thrombin (Fibrindex, Ortho-McNeil Pharmaceutical, Raritan, NJ).
Discussion
We examined the role of TF in the intimal hyperplastic response to vascular damage in atherosclerotic arteries of heritable hyperlipidemic Watanabe rabbits. We found that overexpression of TFPI markedly inhibited intimal hyperplasia, as reflected by a 43% reduction in the intima/media ratio of carotid segments examined 4 weeks after balloon injury. No impairment of systemic hemostasis or excess bleeding was observed in this or previous studies of adenoviral TFPI gene transfer in vivo (24, 27), indicating the potential of locally overexpressed TFPI to inhibit neointima formation in the absence of detectable hemorrhagic risk. We found that a trend toward an increased lumen size in Ad-TFPI-treated vessels was associated with outer diameter and thickness of the tunica media virtually identical to that of Ad-RR controls. Thus, in contrast to previous observations with locally overexpressed COX-1 (25) and eNOS (31), adenoviral gene transfer of TFPI does not appear to result in persistent dilatation or “remodeling” of the injured vessel.
All arteries treated with Ad-TFPI were patent at the time of sacrifice, whereas occlusive thrombi were present on histological examination in 2 of the 10 Watanabe rabbits treated with the control vector, Ad-RR. Carotid blood flow was not monitored during the 4-week observation period in this study. Thus, the absence of thrombi 4 weeks after injury does not exclude in either group the presence of thrombi that may have dissolved or were intermittently embolizing, generating cyclic flow reductions in the injured artery (3). However, previous studies (25, 27) have shown that adenoviral gene transfer of TFPI exerts significant protection against the development of cyclic flow reductions, in accordance with reports that administration of recombinant TFPI markedly reduced the procoagulant properties of balloon-injured rabbit arteries (32, 33). We therefore speculate that local gene transfer of TFPI may prevent intimal hyperplasia, at least in part, by decreasing the local burden of thrombus-dependent growth factors (3, 4).
The antithrombotic properties of local TFPI gene transfer may play an important role as a potential therapeutic approach in circumstances where thrombus formation is prominent. On the other hand, given the low incidence of thrombi on postmortem examination of rabbits in both groups (2/10, Ad-RR control; 0/10, Ad-TFPI), the efficacy of overexpressed TFPI in reducing neointima formation at sites of vascular injury suggests that TF and its downstream proteases, factor Xa and thrombin, may function in vivo much like cytokines—promoting intimal hyperplasia and vessel obstruction through signals activating migration and proliferation of VSMC (4–6). Of note, in a study examining the effect of systemically administered TFPI on neointima formation in repeatedly injured but nonatherosclerotic rabbit arteries, the protective effect of systemically administered TFPI was found to be significant only after deep arterial injury but was not observed after mild endothelial denudation alone (34). Thus, atherosclerotic vessels undergoing severe injury, in principle, may be a promising clinical target for TFPI gene transfer.
Drug treatments developed to reduce intimal hyperplasia and restenosis include systemic administration of antithrombotic drugs, such as inhibitors of thrombin and platelet aggregation. However, these therapies have not been consistently effective, and their dosing is limited given their associated hemorrhagic risk (1, 35, 36). After local gene transfer of TFPI, we observed no derangements in hemostasis. This finding agrees with short-term studies in thrombosis models indicating a normally functioning hemostatic systemic at 5 and 6 days after local TFPI gene therapy (24, 27). Taken together, these findings warrant reconsideration of systemic anticoagulant and antithrombotic approaches to the problem of recurrent thrombosis and restenosis. With one exception (35), such strategies have failed to reduce the need for repeated revascularization after percutaneous vascular interventions, leading to a quest for more potent and prolonged antithrombotic treatment, especially in the secondary prevention of acute coronary syndromes (37, 38). Although we cannot exclude that intense systemic anticoagulation, added to local inhibition of TF, may provide additional protection against postinjury intimal hyperplasia and thrombotic occlusion, in situ expression of TFPI alone is shown in our study to markedly inhibit neointima formation in damaged atherosclerotic arteries without the bleeding risks associated with intense prolonged systemic antithrombotic therapies. Ongoing searches for improved gene transfer vectors associated with less immune-inflammatory potential and longer gene expression may facilitate further preclinical evaluation of vascular TFPI gene transfer.
Acknowledgments
This work was supported in part by National Institutes of Health Grants R01-HL50179 and R01-HL54839, an American Heart Association National Center Scientist Development Award, and a DREAMS Grant of the U.S. Armed Forces. We thank Dr. Tze-Chein Wun for the full-length TFPI cDNA, Dr. Robert D. Gerard for the pACpLpA plasmid, Ad-RR control vector and 293 cells, and gratefully acknowledge the help of Dr. Seung-Ho Kang with the statistical calculations.
Abbreviations
- TF
tissue factor
- TFPI
TF pathway inhibitor
- VSMC
vascular smooth muscle cells
- pfu
plaque-forming unit
- moi
multiplicity of infection
References
- 1.Lefkovits J, Topol E. Prog Cardiovasc Dis. 1997;40:141–158. doi: 10.1016/s0033-0620(97)80006-0. [DOI] [PubMed] [Google Scholar]
- 2.Schwartz S. Circ Res. 1999;85:877–879. doi: 10.1161/01.res.85.10.877. [DOI] [PubMed] [Google Scholar]
- 3.Willerson J, Yao S, McNatt J, Benedict C, Anderson H, Golino P, Murphree S, Buja L. Proc Natl Acad Sci USA. 1991;88:10624–10628. doi: 10.1073/pnas.88.23.10624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sato Y, Asada Y, Marutsuka K, Hatakeyama K, Kamikubo Y, Sumiyoshi A. Thromb Haemostasis. 1997;78:1138–1141. [PubMed] [Google Scholar]
- 5.Feng N, Yang Y, Huang S, Ou J. J Clin Invest. 1996;98:1493–1501. doi: 10.1172/JCI118938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McNamara C, Sarembock I, Gimple L, Fenton J, Coughlin S, Owens G. J Clin Invest. 1993;91:94–98. doi: 10.1172/JCI116206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carmeliet P, Mackman N, Moons L, Luther T, Gressens P, Van Vlaenderen I, Demunck H, Kasper M, Breier G, Evrard, et al. Nature (London) 1996;383:73–75. doi: 10.1038/383073a0. [DOI] [PubMed] [Google Scholar]
- 8.Rappaport S, Rao L. Arterioscler Thromb. 1992;12:1111–1121. doi: 10.1161/01.atv.12.10.1111. [DOI] [PubMed] [Google Scholar]
- 9.Drake T, Morrissey J, Edgington T. Am J Pathol. 1989;34:1087. [PMC free article] [PubMed] [Google Scholar]
- 10.Wilcox J, Smith K, Schwartz S, Gordon D. Proc Natl Acad Sci USA. 1989;86:2839–2843. doi: 10.1073/pnas.86.8.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marmur J, Rossikhina M, Guha A, Fyfe B, Friedrich V, Mendlowitz M, Nemerson Y, Taubman M. J Clin Invest. 1993;91:2253–2259. doi: 10.1172/JCI116452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Annex B, Denning S, Keith M, Sketch M, Stack R, Morissey J, Peters K. Circulation. 1995;91:619–622. doi: 10.1161/01.cir.91.3.619. [DOI] [PubMed] [Google Scholar]
- 13.Toschi V, Gallo R, Letino M, Fallon J, Gallo R, Ferndez-Ortiz A, Badimon L, Chesebro J, Nemerson Y, Fuster F, et al. Circulation. 1997;95:594–599. doi: 10.1161/01.cir.95.3.594. [DOI] [PubMed] [Google Scholar]
- 14.Jang Y, Guzman L, Lincoff M, Topol E. Circulation. 1995;92:3041–3050. doi: 10.1161/01.cir.92.10.3041. [DOI] [PubMed] [Google Scholar]
- 15.Oltrona L, Speidel C, Recchia D, Wickline S, Eisenberg P, Abendschein D. Circulation. 1997;96:646–652. doi: 10.1161/01.cir.96.2.646. [DOI] [PubMed] [Google Scholar]
- 16.Han X, Girard T J, Baum P, Abendschein D R, Broze G J., Jr Arterioscler Thromb Vasc Biol. 1999;19:2563–2567. doi: 10.1161/01.atv.19.10.2563. [DOI] [PubMed] [Google Scholar]
- 17.Ragosta M, Gimple L, Gertz S, Durewiddie C, Vlasuk G, Haber H, Powers E, Roberts W, Sarembock U. Circulation. 1994;89:1262–1271. doi: 10.1161/01.cir.89.3.1262. [DOI] [PubMed] [Google Scholar]
- 18.Sarembock I, Gertz S, Gimple L, Owen R, Powers E, Roberts W. Circulation. 1991;84:232–243. doi: 10.1161/01.cir.84.1.232. [DOI] [PubMed] [Google Scholar]
- 19.Rade J, Schulick A, Virmani R, Dichek D A. Nat Med. 1996;2:293–298. doi: 10.1038/nm0396-293. [DOI] [PubMed] [Google Scholar]
- 20.Marmur J, Thiruvikraman S, Fyfe B, Guha A, Sharma S, Ambrose J, Falloon J, Nemerson Y, Taubman M. Circulation. 1996;94:1226–1232. doi: 10.1161/01.cir.94.6.1226. [DOI] [PubMed] [Google Scholar]
- 21.Buja L, Clubb F, Bilheimer D, Willerson J. Eur Heart J. 1990;11:41–52. doi: 10.1093/eurheartj/11.suppl_e.41. [DOI] [PubMed] [Google Scholar]
- 22.Aliev G, Burnstock G. Histol Histopathol. 1998;13:797–817. doi: 10.14670/HH-13.797. [DOI] [PubMed] [Google Scholar]
- 23.Wun T, Kretzmer K, Girard T, Miletich J, Broze G. J Biol Chem. 1998;263:6001–6004. [PubMed] [Google Scholar]
- 24.Zoldhelyi P, McNatt J, Shelat H, Yamamoto Y, Chen Z-Q, Willerson J T. Circulation. 2000;101:289–295. doi: 10.1161/01.cir.101.3.289. [DOI] [PubMed] [Google Scholar]
- 25.Zoldhelyi P, McNatt J, Xu X-M, Meidell R, Loose-Mitchell D, Willerson J T, Wu K K. Circulation. 1996;93:10–17. doi: 10.1161/01.cir.93.1.10. [DOI] [PubMed] [Google Scholar]
- 26.Hansen J, Sandset P, Huseby K, Huseby N, Nordøy A. Thromb Haemostasis. 1996;76:703–709. [PubMed] [Google Scholar]
- 27.Nishida T, Ueno H, Atsuchi N, Kawano R, Asada Y, Nakahara Y, Kamikubo Y, Takeshita A, Yasui H. Circ Res. 1999;84:1446–1452. doi: 10.1161/01.res.84.12.1446. [DOI] [PubMed] [Google Scholar]
- 28.Liang K Y, Zeger S L. Biometrika. 1986;73:13–22. [Google Scholar]
- 29.Zeger S L, Liang K Y, Albert P S. Biometrics. 1989;44:1049–1060. , and erratum (1989) 45, 347. [PubMed] [Google Scholar]
- 30.Park T. Stat Med. 1993;12:1723–1732. doi: 10.1002/sim.4780121807. [DOI] [PubMed] [Google Scholar]
- 31.Varenne O, Pislaru S, Gillijns H, Van Pelt N, Gerard R D, Zoldhelyi P, Van de Werf F, Collen D, Janssens S P. Circulation. 1998;98:919–926. doi: 10.1161/01.cir.98.9.919. [DOI] [PubMed] [Google Scholar]
- 32.Speidel C, Eisenberg P, Ruf W, Edgington T, Abendschein D. Circulation. 1995;92:3323–3330. doi: 10.1161/01.cir.92.11.3323. [DOI] [PubMed] [Google Scholar]
- 33.St. Pierre J, Yang L Y, Tamirisa K, Scherrer D, De Ciechi P, Eisenberg P, Tolunay E, Abendschein D. Arterioscler Thromb Vasc Biol. 1999;19:2263–2268. doi: 10.1161/01.atv.19.9.2263. [DOI] [PubMed] [Google Scholar]
- 34.Asada Y, Hara S, Tsuneyoshi A, Hatakeyama K, Kisanuki A, Marutsuka K, Sato Y, Kamikubo Y, Sumiyoshi A. Thromb Haemostasis. 1998;80:506–511. [PubMed] [Google Scholar]
- 35.Topol E J, Califf R M, Weisman H F, Ellis S G, Tcheng J E, Worley S, Ivanhoe R, George B S, Fintel D, Weston M, et al. Lancet. 1994;343:881–886. doi: 10.1016/s0140-6736(94)90007-8. [DOI] [PubMed] [Google Scholar]
- 36.Welsch D, Novotny W, Wun T. Thromb Res. 1991;64:213–222. doi: 10.1016/0049-3848(91)90120-l. [DOI] [PubMed] [Google Scholar]
- 37.The symphony Investigators. Lancet. 2000;355:337–345. [PubMed] [Google Scholar]
- 38.Brower M A, Van den Bergh P J, Vromans R P J W, Aengevaeren W R M, Gerrit Veen G, Luijten J E. Circulation. 2000;102 (Supplement II):600. [Google Scholar]