

Abstract
Generating cell aggregates is beneficial for various applications ranging from biotechnology to regenerative therapies. Previously, poly(ethylene glycol) (PEG) microwells have been demonstrated as a potentially useful method for generating controlled-size cell aggregates. In addition to controlling cell aggregate size and homogeneity, the ability to confine cell aggregates on glass adhesive substrates and subsequently retrieve aggregates from microwells for further experimentation and analysis could be beneficial for various applications. However, it is often difficult to retrieve cell aggregates from these microwells without the use of digestive enzymes. This study describes the stable formation of cell aggregates in responsive microwells with adhesive substrates and their further retrieval in a temperature dependent manner by exploiting the stimuli responsiveness of these microwells. The responsive polymer structure of the arrays can be used to thermally regulate the microwell diameters causing a mechanical force on the aggregates, subsequently facilitating the retrieval of cell aggregates from the microwells with high efficiency compared to PEG arrays. This approach can be potentially integrated into high-throughput systems and may become a versatile tool for various applications that require aggregate formation and experimentation, such as tissue engineering, drug discovery, and stem cell biology.
Introduction
Control of the cell microenvironment is important to better mimic native tissues by providing the appropriate conditions for cellular dynamics such as migration, spreading, differentiation, and proliferation. Microscale engineering approaches have been shown to be useful in controlling cellular behavior in vitro to fabricate modular tissues, to direct stem cell differentiation into desired cell types, and to perform high-throughput assays for drug toxicity and metabolism screening.1,2
Controlling the size and shape of cell aggregates can be used to form functional tissue units or to trigger embryonic stem (ES) cell differentiation.3–5 Particular cell types like hepatocytes and pancreatic cells require three dimensional (3D) environments to maintain their function.6–8 For example, hepatocyte spheroids drive bile canaliculi formation and enhance cell-cell interactions by increasing the degree of cell-cell contact.9–12 Furthermore, these spheroids better maintain hepatic activities such as production of albumin, urea and metabolic enzymes.9,13–15 Cell aggregates of differentiating ES cells, known as embryoid bodies (EBs) have also been shown to control the differentiation of ES cells.16–21 Formation of EBs with homogenous sizes and shapes is one approach to regulate ES cell differentiation.5,22 Suspension culture methods have been used to form EBs,20 but high rotation speeds generate shear forces which may influence ES cell proliferation, viability, and agglomeration.23 Another approach for EB formation is by the hanging drop method,21,24 though this method is cumbersome for generating large number of EBs and cannot be easily merged with high-throughput testing systems.
Microengineering systems have been used to form cellular aggregates and control the resulting microenvironmental signals. In one of these approaches, microwell structures have been fabricated from photocrosslinkable poly(ethylene glycol) (PEG) hydrogels5,25 by using soft lithography.26,27 The surface of PEG hydrogels is resistant to cell adhesion which enhances subsequent aggregate formation within the microwells.25 PEG microwells have also been used to form size and shape controlled EBs to direct the differentiation of ES cells5,28 and to immobilize stem cells within microfluidic devices.29 Microwells have also been used to generate controlled 3D co-cultures to control and analyze the interactions between of multiple cell types.2 All of these microwells have static environments such that their diameters and hydrogel properties cannot be controlled with external stimuli. To better mimic the temporally regulated aspects of the cellular microenvironment in the body, it may be important to fabricate microstructures with dynamic features like controllable microwell diameters and surface properties. Furthermore, aggregate harvesting from microwell arrays is a prerequisite for further encapsulation30 and biological analysis.5,31 In PEG bottomed arrays,22 the non-adhesive surface results in aggregates which do not attach to the substrate. These aggregates may be retrievable via agitation, gravitational forces or flow.28 The relative ease with which aggregates may be removed from non-adhesive, PEG bottomed arrays makes these systems non-ideal for high-throughput applications involving fluid flow or other agitations. On the other hand, aggregate retrieval is particularly difficult in experiments in which the underlying substrate is exposed (i.e. glass bottomed microwells),25 where cell aggregates often firmly adhere to the base of the microwells. Although an electrochemical method has been previously developed to harvest spheroids from microcavities, the technique requires a complicated substrate fabrication process and the need for applied electrical potential.32
To develop a method to form cell aggregates on adhesive substrates for high-throughput experimentation and to retrieve the resulting cell aggregates in a dynamic manner, we fabricated stimuli-responsive microwells with adhesive bottoms from photocrosslinkable poly(N-isopropyl-acrylamide) (PNIPAAm) by using soft lithography. PNIPAAm is a well-known stimuli responsive polymer which switches from hydrophobic to hydrophilic, and thus, swells under temperatures below its Lower Critical Solution Temperature (LCST) of 32 °C.33 Because of its temperature dependent switchable surface properties, PNIPAAm has been used in cell sheet tissue engineering,34–36 capillary network formation,37 drug delivery,38,39 and fabrication of hydrogel microstructures.40–42 Here dynamic microwells were fabricated with either shape varying or shape constant properties and were used to form cell aggregates. Furthermore, we used the temperature dependant properties of the microwells to drive retrieval of the aggregates. Given its tunable stimuli-responsive features, this microwell array system can be potentially useful in the formation and analysis of cell aggregates and can be integrated within high-throughput screening devices.
Materials and methods
Materials
N-isopropylacrylamide (NIPAAm), N,N-methylene-bis-acrylamide (MBAAm), dimethyl sulfoxide (DMSO), photo-initiator 2-hydroxy-2-methylpropiophenone (PI), 3-(tri-methoxysilyl)propyl-methacrylate (TMSPMA), and sodium Hydroxide (NaOH) were purchased from Sigma-Aldrich Chemical Company (St. Louis, MO). Poly(ethylene glycol) dimethacrylate (PEGDMA MW = 1000) was purchased from Polysciences Company (Warrington, PA). Irgacure-2959 was purchased from Ciba Specialty Chemical Corporation (Tarrytown, NY). Ethanol was purchased from Fisher Scientific (Fair Lawn, NJ). Silicon elastomer and curing agent were purchased from Dow Corning Corporation (Midland, MI). Dulbecco’s phosphate buffered saline (PBS), calcein-AM and ethidium homodimer, Dulbecco’s modified eagle medium (DMEM), fetal bovine serum (FBS), and penicillin streptomycin (Pen-strep) were purchased from Gibco Invitrogen (Carlsbad, CA).
PDMS molds and glass substrates
Silicon masters with 150 μm diameter cylindrical patterns were fabricated with SU-8 photolithography and used as templates to form poly(dimethylsiloxane) (PDMS) replicas. A mixture of 10 : 1 silicon elastomer and curing agent was cured at 70 °C for 2 h to generate PDMS stamps which were then detached from silicon masters. The resulting PDMS molds had cylindrical protruding patterns which were used to fabricate microwell structures of PNIPAAm and PEG.
Glass slides were first cleaned with 10 wt.% NaOH in deionized water for 12 h, then rinsed with deionized water and ethanol 3 times. After drying at ambient temperature, glass slides were treated with TMSPMA at 70 °C for 24 h. Subsequently, glass substrates were washed with ethanol 3 times and dried at room temperature for further usage.
Fabrication of PNIPAAm and PEG microwells
Prepolymer solutions to fabricate shape varying (PNIPAAm-1) and shape constant (PNIPAAm-2) microwell structures were prepared by mixing NIPAAm, MBAAm, DMSO, deionized water, and PI in the weight ratios of 1.3 : 0.04 : 1.87 : 1 : 0.09 and 2.18 : 0.12 : 3 : 1 : 0.15, respectively. These weight ratios for PNIPAAm-2 solution were previously used in the fabrication of adaptive liquid microlenses.43 The recipe of PNIPAAm-1 solution was designed with greater water and lower crosslinker (MBAAm) content than PNIPAAm-2 with the intention of creating a less densely crosslinked hydrogel. The PEG prepolymer solution was a mixture of PEGDMA-1000 and Irgacure as photoinitiator with weight ratios of 20 wt.% and 1 wt.%, respectively, dissolved in PBS.
Desired volumes of PNIPAAm prepolymer solutions were stirred overnight at ambient temperature and the photoinitiator was added to prepolymer solutions immediately before the photocrosslinking step. PEG prepolymer solutions were prepared on the day of fabrication. Microwell structures with glass bottom surfaces were fabricated with a micromolding approach by using PDMS mold substrates as shown in Fig. 1a. Prepolymer solution was put on a PDMS mold and a TMSPMA coated glass slide was gently placed on the PDMS substrate, then exposed to ultraviolet (UV) light to form microstructures. Both PNIPAAm-1 and PNIPAAm-2 microwells were crosslinked by UV light of 320–500 nm wavelength at an intensity of 4 mW cm−2 for 30 s using the OmniCure Series 2000 (EXFO, Mississauga, Canada). PEG microwells were formed by exposure to the same UV light source at an intensity of 52 mW cm−2 for 30 s. The depth and diameter of the microwells were adjusted to ~150 μm. Fabricated microwells were immersed in 70% ethanol solution to clean unreacted chemicals within the hydrogel structure, washed with PBS, and then kept in PBS until further experimentation.
Fig. 1.
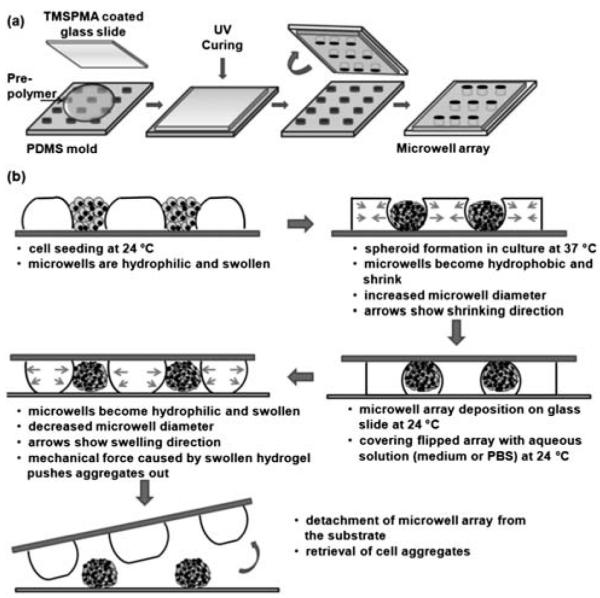
(a) Soft-lithographic fabrication of temperature responsive microwells. (b) Schematic of the steps of cell seeding, aggregate formation, and retrieval from glass bottomed temperature responsive microwells.
Responsiveness tests for fabricated microwells
Responsiveness tests for all microwell arrays were conducted in ambient and cell-culture conditions to observe and quantify microwell behaviors under temperature stimuli. Microwells were washed with 70% ethanol solution and PBS to rinse off unreacted chemicals and kept in PBS. Time lapse images were taken with an inverted microscope (Nikon Eclipse TE2000-U) for all microwells at 24 °C with 1 h time intervals for 3 h. Arrays were subsequently moved to 37 °C and kept for 6 h; time-lapse images were taken every 1 h. Finally, all microwell arrays were moved back to 24 °C for 6 h and time-lapse images were taken every 1 h. There were 3 samples for each microwell type (n = 3) and 6 single microwells per microscope image taken on each sample. To quantify the dynamic behavior under different temperatures, the approximate top-view surface area of each microwell was measured by Spot Advanced software. The responsiveness of each microwell type at corresponding time points was represented by a mean value and standard deviation.
Scanning electron microscopy
To remove the water, PNIPAAm microwells were dried at room temperature for 24 h. The samples were subsequently mounted onto aluminium stages, sputter coated with gold, and analyzed under scanning electron microscopy (SEM) (JEOL JSM 6060) at a working distance of 20 mm.
Cell culture and cell seeding on the microwells
Human hepatoblastoma cells, HepG2, were cultured at 37 °C in a 5% CO2 humidified incubator in culture medium containing 89% DMEM, 10% FBS, and 1% Penicillin-Streptomycin.
All microwell types were placed in 6 well culture plates after fabrication, rinsed with ethanol, and kept in PBS until cell seeding. HepG2 cells were trypsinized and prepared in culture medium at a density of 4 × 106 cells/mL. PNIPAAm microwells were kept at room temperature for at least 1 h to make the hydrogel surface hydrophilic for a better cell-seeding condition at 24 °C. PEG microwells were also subjected to the same conditions. After aspirating PBS from each 6 well plate, 200 μL of a solution containing 4 × 106 cells/mL was seeded on each microwell array and kept at ambient temperature for 20 min to drive spreading of the cell suspension on the hydrophilic microwell array surface. Subsequently, microwell arrays were gently washed with PBS to remove cells on the hydrogel surface and immersed in fresh culture medium (Fig. 1b). Cell seeding onto PNIPAAm-1 arrays was also performed at physiological temperature (37 °C) while microwells were open to their full capacity. Seeded microwell arrays were kept in a 5% CO2 humidified incubator at 37 °C for 3 days. Microscope images were taken daily to analyze aggregate formation in the microwell structures.
Cell aggregate retrieval from the microwells
After culturing cell-seeded microwell arrays for 3 days, cell aggregates were retrieved from the microwells by deposition of the arrays on glass slides as shown in Fig. 1b. For release experiments at 24 °C, microwell arrays were gently placed on a glass slide and immersed in 24 °C PBS with the wells facing down. Control experiments with the same method were conducted at 37 °C to test whether the temperature was the main driving force in releasing the aggregates from the microwells. For control experiments at 37 °C, microwells were covered with 37 °C PBS. The duration at the experimental temperature while wells were face down was defined as the retrieval time. Different retrieval times (5, 10, and 15 min) were tested in order to analyze the controlled release behavior of responsive microwells. Release experiments with the same retrieval times at 24 °C and 37 °C were also done for PEG microwells to compare the aggregate release characteristics of static microwells with those of dynamic microwell arrays. Three samples of each microwell type (n = 3) were analyzed for each retrieval time and incubation temperature. Retrieved aggregates from each sample were counted with Image-J software after taking low magnification (2×) pictures from 3 different parts of each glass slide. The percentage of aggregates that were released per array was calculated by dividing the total number of aggregates released from one microwell array by the total number of microwells on the array. Approximate diameters of retrieved aggregates were measured with Spot Advanced software to determine the frequency of diameters of aggregates formed within dynamic microstructures.
Live/dead staining
Live/dead assay was performed by using a solution containing 2 μM of calcein-AM and 4 μM of ethidium homodimer in PBS. For live and dead evaluation, each microwell array was placed in live/dead solution during deposition on glass substrate for a maximum of 15 min at 24 °C or 37 °C. Calcein AM stained live cells with fluorescent green color while homodimer caused fluorescent red staining in dead cells. Cells were visualized under an inverted microscope (Nikon Eclipse TE2000-U).
Statistical analysis
Data was shown as the mean and ± standard deviation (±sd). Statistical analysis was performed with an unpaired student’s t-test and p < 0.05 was considered as significant.
Results and discussion
Fabrication of temperature responsive microwells
Microwells were fabricated by micromolding of photocrosslinkable hydrogels as illustrated in Fig. 1a. Two distinct types of dynamic microwell structures were formed with shape varying (PNIPAAm-1) and shape constant (PNIPAAm-2) properties. The diameter of both types of PNIPAAm microwells changed in a temperature dependent manner by swelling or shrinking (Fig. 2a). Within 1h after being placed at 24 °C, PNIPAAm-1 microwells not only changed their diameter but also changed their shape (Supplementary Movie-1) whereas PNIPAAm-2 microwells maintained their rounded shape and only changed their diameter (Supplementary Movie-2). The shape varying feature of PNIPAAm-1 microwells may be a result of less chemical crosslinker (MBAAm) concentration in the prepolymer solution. In contrast, PNIPAAm-2 prepolymer solution has a higher amount of crosslinker, resulting in more controlled shape structures. Interestingly, PNIPAAm-1 structures changed their microwell diameters non-uniformly under temperature variation, whereas PNIPAAm-2 structures responded to external stimuli by changing their diameters uniformly (Fig. 2a). PNIPAAm-2 structures were also more transparent than those made of PNIPAAm-1. Furthermore, SEM images and time-lapse microscopy of the microwell array swelling show that PNIPAAm-2 microwells (Fig. 3b, d, Supplementary Movie-2) were smooth in comparison to PNIPAAm-1 (Fig. 3a, c, Supplementary Movie-1) which qualitatively suggests that the higher crosslinker ratio in PNIPAAm-2 solution results in higher pattern transfer fidelity. It is important to note that the microwells were dehydrated for SEM analysis.
Fig. 2.
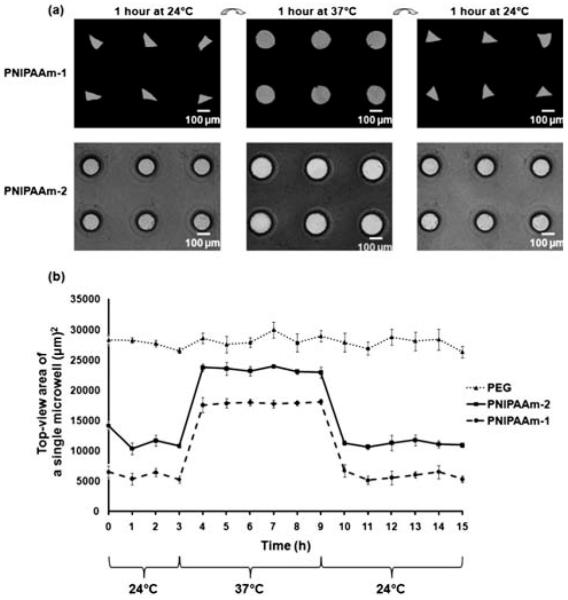
(a) Temperature responsiveness of shape varying (PNIPAAm-1) and shape constant (PNIPAAm-2) microwells. Time lapse images were taken in 1h time intervals at 24 °C and 37 °C. (b) Responsiveness of different microwell types was analyzed by measuring the top-view area of a single microwell. The microwells were initially kept at 24 °C for 3 h, then at 37 °C and 24 °C for 6 h each. Data represents average ± sd for n = 3 samples.
Fig. 3.

Scanning electron micrographs of fabricated PNIPAAm-1 (a, c) and PNIPAAm-2 (b, d) microwells. High resolution images of single microwells for PNIPAAm-1 (c) and PNIPAAm-2 (d).
Temperature responsiveness test for microwell arrays
To quantify the dynamics of responsive microwells, we kept arrays at ambient (24 °C) and physiological (37 °C) temperatures for different time intervals. Fig. 2b shows a plot of the average microwell area as a function of time, for all microwell types. Within 1 h after changing the temperature from 24 °C to 37 °C, the free space of the PNIPAAm microwells expanded as a result of hydrogel contraction, presumably caused by the increase in temperature above the expected LCST point (~32 °C) of PNIPAAm. Microwell areas returned to their initial values within 1 h when temperature was decreased to 24 °C, demonstrating that both microwell types possess reversible shape changing property. PNIPAAm-1 microwells exhibited smaller initial microwell areas as a result of lower crosslinker to monomer ratio compared to PNIPAAm-2. Interestingly, the absolute change in microwell areas due to the temperature transition was similar for both PNIPAAm-1 and PNIPAAm-2 microwells. Control experiments with PEG microwells demonstrated that PEG microwells were non-responsive to temperature stimuli. Thus, PNIPAAm microwells have a potential advantage over PEG arrays with their tunable thermo-responsive characteristics.
Cell-seeding and formation of cell aggregates within microwells
Microwell arrays were previously shown to be useful tools for 3D cell culture and aggregate formation.2,11,22,25,28,29 The responsive microwell structures exhibit switchable swelling properties, a potentially useful feature in controlling the microwell surface adhesiveness for cell aggregation and retrieval. After fabrication, microwells were usually kept at room temperature for 1 h to form a swollen gel and a smaller exposed area before cell-seeding at 24 °C. In some cases, PNIPAAm-1 arrays were kept at 37 °C until microwells were in an open state with a maximum area of ~18000 μm2. HepG2 cells were used as the model cell type due to their relevance in drug discovery and their ability to form spheroids. To generate cell aggregates in the microwells, a suspension of HepG2 cells was pipetted onto a glass slide containing microwells at a density of ~2.7 × 105 cells cm−2. After 20 min, arrays were gently washed to remove cells that were not in the microwells. Cell seeded microwell arrays were kept in the incubator for 3 days to form cell aggregates. Fig. 4 illustrates that the cell-seeding efficiency and spheroid formation in responsive microwells were comparable to those formed in glass-bottomed PEG microwells (all samples were seeded with cells at 24 °C). After one day, cell aggregates could be observed in many microwells and by day 3 tightly packed cell clusters were visible in majority of the wells. At 37 °C, the microwells were hydrophobic which may increase the level of protein adsorption from the serum and subsequent cell adhesion. Thus after several days, some aggregates in PNIPAAm microwells appeared to adhere to the surrounding hydrogel matrix (Fig. 4). This contact may increase the stability of cell aggregates in the PNIPAAm microwells in comparison to their PEG counterparts.
Fig. 4.

Light-microscopy images of spheroid formation within PNIPAAm-1, PNIPAAm-2, and PEG microwells over a 3 day incubation period.
To further analyze the effect of stimuli-responsiveness of microstructures on aggregates, microwells were removed from 37 °C and placed at 24 °C for 1 h. We observed that the diameter change of responsive microwells applied a mechanical force on the aggregates formed in PNIPAAm-1 (Supplementary Movie-3) and PNIPAAm-2 microwells (Supplementary Movie-4). Taken together our cell seeding and aggregate formation experiments demonstrate that responsive microwell structures are potentially useful templates for 3D culture conditions and, with their switchable and tunable properties, offer potential advantages over static microstructures.
Controlled retrieval of spheroids from microwell arrays
Aggregate retrieval with high-efficiency is a desirable property of microwell array culture systems. Successful retrieval allows for further experimentation, such as molecular biology analysis of EBs5,31 or encapsulation of spheroids within biomaterials30 for developing functional tissue constructs. We developed a temperature responsive strategy for aggregate harvesting from microfabricated stimuli-responsive devices by exploiting the switchable swelling/deswelling properties of PNIPAAm-1. PNIPAAm-2 microwells were not tested due to the frequent detachment of the array from the TMSPMA coated surface during long-term culture. For aggregate retrieval, bare glass slides were gently placed on the microwell arrays, and the entire structures were inverted to initiate the release of cell aggregates, as shown in Fig. 1b. To keep the microwells in an aqueous environment, inverted microwell arrays were covered with PBS. The experiment was performed at 24 °C and 37 °C for PNIPAAm-1 and PEG microwells. Here we defined the retrieval time as the duration in which microwell arrays were face down on a glass slide at a particular temperature. Aggregate release experiments were conducted with 3 different retrieval times of 5, 10, and 15 min. After the retrieval period, excess solution was removed from the periphery and the microwell array was gently detached from the deposition surface as shown in Fig. 1b.
When the aggregate retrieval experiment was performed at 37 °C, with the PNIPAAm-1 arrays in a hydrophobic/contracted state, limited aggregate release was observed for both the PNIPAAm-1 and PEG microwells (Fig. 5a, c). However, when the procedure was conducted at 24 °C, the PNIPAAm microwells demonstrated a dramatic increase in aggregate release, while the PEG microwells showed no change (Fig. 5b, d). Time course experiments for PNIPAAm-1 at 24 °C showed a moderate increase in aggregate retrieval at 5 min, followed by a significantly greater increase at 10 min (Fig. 5e). Furthermore, we observed high cell viability levels based on live/dead staining images for all time points in the PNIPAAm-1 experiments (Fig. 5e), qualitatively suggesting that the PNIPAAm-1 microwells did not adversely affect the vitality of the cells. Due to the potential lack of dye penetration into cell aggregates, cell viability was not estimated quantitatively.
Fig. 5.

Phase contrast and fluorescent images of released aggregates from (a) PNIPAAm-1 microwells at 37 °C for 10 min retrieval time (control experiment) (b) PNIPAAm-1 microwells at 24 °C for 10 min retrieval time (c) PEG microwells at 37 °C for 10 min retrieval time (d) PEG microwells at 24 °C for 10 min retrieval time (e) PNIPAAm-1 microwells at 24 °C for 5, 10, and 15 min retrieval times. (f, g) Retrieval efficiency from microwells and size distribution of released cell aggregates. (f) Cell aggregates were formed within microwells for 3 days. On day 3, microwell arrays were inverted on the glass slides with the wells facing down for 5, 10, and 15 min time scales to release the aggregates. PNIPAAm-1 microwells were tested at 24 °C to show that aggregate release percentage is more than previously developed PEG microwells and gravity is less effective in the retrieval. Release experiments were also conducted at 37 °C as control experiments to show that temperature is the main driving force for the aggregate release from the stimuli-responsive microwells. Data for percentage aggregate release per microwell array were shown with average ± sd (n = 3). * shows from=PNIPAAm-1 statistically significant difference in variance (p < 0.05). (g) Cumulative frequency of spheroid diameters for released aggregates microwells at 24 °C for all retrieval times.
Qualitative aggregate release efficiencies were confirmed by quantification of the percentage of aggregates that were released from a microwell array (Fig. 5f). PEG microwells exhibited a maximum aggregate release of 15.4 ± 3.8% for any time point or temperature. Similarly, we observed PNIPAAm-1 microwells at 37 °C to have a maximum aggregate release of 19.7 ± 4%. Alternatively, PNIPAAm-1 microwells at 24 °C showed a marked increase in aggregate release at 5 min (41.7 ± 3.8%) and a maximal aggregate release at 10 min (74.8 ± 10.4%). No further significant increase in aggregate retrieval was observed at 15 min compared to 10 min (p > 0.1), suggesting that 10 min at 24 °C is sufficient to achieve the maximum recovery of cell aggregates from PNIPAAm-1 microwell arrays.
Comparing the results of control experiments at 37 °C for PNIPAAm-1 microwells with the results of release experiments at 24 °C for PNIPAAm-1 microwells, we infer that temperature is the main triggering force in aggregate retrieval from PNIPAAm-1 microwell arrays. We hypothesize that the swollen state of these temperature responsive PNIPAAm-1 microwells at 24 °C generates a mechanical force on the aggregates which drives their release from the microwells. In contrast, static PEG microwells provided no mechanical force to overcome the adhesion of cell aggregates to the glass bottomed substrate.
Aggregate retrieval from previously developed static PEG bottomed microwells has been achieved by flowing an aqueous solution over the microwell arrays or by agitation of the arrays22,28 both of which may lead to damage of the spheroids. The significant improvement in aggregate retrieval in the PNIPAAm-1 arrays suggests an obvious advantage compared to their PEG counterparts. Furthermore, the use of enzymes to detach the aggregates from substrates also disrupts cell-cell contacts and may not be suitable for these applications.
It should be noted that all microwell arrays in our experiments had a glass bottom, which after the adhesion of the serum proteins, provided adhesive regions for the cells25 making the mechanical force caused by the swollen state of the responsive microwells more effective than gravity in aggregate retrieval. Thus, stimuli-responsive microwells possess a prominent advantage over previous aggregate retrieval methods from static microwell structures22,28 with their controllable aggregate release property. However, it should be noted that although live/dead images suggest that mechanical detachment of cell aggregates from the glass bottomed microwells did not adversely affect cell viability, the possibility of undesirable stress-induced effects resulting from forced detachment from the glass surface has not been ruled out.
To characterize the uniformity of the retrieved aggregates, the diameters of released aggregates from PNIPAAm-1 microwells at 24 °C were quantified. As shown in Fig. 5g, a wide distribution in the frequency of aggregate diameter ranges was observed. This may be due to the effects of mechanical forces from the swelling of the microwell walls. Another reason could be that the smaller diameters of PNIPAAm-1 microwells during cell seeding at 24 °C, prevented a high initial number of cells to dock in the microwells. As shown in Fig. 2b, during the cell seeding process at 24 °C, PNIPAAm-1 microwells held a maximum area of ~6000 μm2 whereas PEG microwells had a maximum area of ~28000 μm2. This may explain the fact that spheroids formed in PEG microwells appeared to be more packed than those formed in PNIPAAm microwells as shown in Fig. 4. We also seeded cells onto PNIPAAm-1 arrays at 37 °C while microwells were in an open state with a maximum area of ~18000 μm2. It was shown that spheroids formed in PNIPAAm-1 microwells were larger than those formed when cell seeding was performed at 24 °C (Supplementary Fig. 1). However, aggregate release was less than 10% for any retrieval time or temperature (data not shown). In further studies, it may be possible to form homogenous aggregates with more shape constant dynamic microwells or by better controlling cell-seeding efficiency on the responsive microwells. We also observed that removing the responsive microwells from the glass surface after aggregate retrieval sometimes led to detachment from TMSPMA surfaces at 24 °C, which may also cause aggregate deformation if the microwell array is not removed gently from the deposition surface. In further investigations, applying new surface chemistry methods on glass substrates may alleviate stability limitations of temperature responsive microwell structures in a variety of aqueous conditions.
Conclusions
Here we show that microfabricated PNIPAAm based microwell arrays with different shape changing characteristics can be used to form cell aggregates on adhesive glass substrates and enable their subsequent release in a controllable manner. These dynamic microwells have potential advantages over static microwell structures due to their high aggregate retrieval efficiency and tunable stimuli-responsiveness. These microwells may also be used to generate EBs and direct ES cell differentiation in a controllable manner, generate micro tissues, and can easily be integrated into high-throughput screening systems.
Supplementary Material
Acknowledgements
We would like to thank Dr. Seunghwan Lee for helpful discussions. This research was supported by the U.S. Army Research Office through the Institute for Soldier Nanotechnologies at MIT under the project DAAD-19-02-D-002, the Draper Laboratory and the NIH (DE01323, DE016516, HL092836, DE019024, EB007249).
Footnotes
Published as part of a special issue dedicated to Emerging Investigators: Guest Editors: Aaron Wheeler & Amy Herr.
Electronic Supplementary Information (ESI) available: Supplementary movies for thermo responsive behavior of microwells and supplementary movies for dynamic manipulation of cell aggregates. Supplementary figure for the frequency of aggregate diameter ranges. See DOI: 10.1039/c004732e/
References
- 1.Khademhosseini A, Langer R, Borenstein J, Vacanti JP. Proc. Natl. Acad. Sci. U. S. A. 2006;103:2480–2487. doi: 10.1073/pnas.0507681102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fukuda J, Khademhosseini A, Yeo Y, Yang XY, Yeh J, Eng G, Blumling J, Wang CF, Kohane DS, Langer R. Biomaterials. 2006;27:5259–5267. doi: 10.1016/j.biomaterials.2006.05.044. [DOI] [PubMed] [Google Scholar]
- 3.Mohr JC, de Pablo JJ, Palecek SP. Biomaterials. 2006;27:6032–6042. doi: 10.1016/j.biomaterials.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 4.Mohr JC, Zhang J, Azarin SM, Soerens AG, de Pablo JJ, Thomson JA, Lyons GE, Palecek SP, Kamp TJ. Biomaterials. 31:1885–1893. doi: 10.1016/j.biomaterials.2009.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hwang YS, Chung BG, Ortmann D, Hattori N, Moeller HC, Khademhosseini A. Proc. Natl. Acad. Sci. U. S. A. 2009;106:16978–16983. doi: 10.1073/pnas.0905550106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koide N, Shinji T, Tanabe T, Asano K, Kawaguchi M, Sakaguchi K, Koide Y, Mori M, Tsuji T. Biochem. Biophys. Res. Commun. 1989;161:385–391. doi: 10.1016/0006-291x(89)91609-4. [DOI] [PubMed] [Google Scholar]
- 7.Fukuda J, Mizumoto H, Nakazawa K, Kajiwara T, Funatsu K. International Journal of Artificial Organs. 2004;27:1091–1099. doi: 10.1177/039139880402701213. [DOI] [PubMed] [Google Scholar]
- 8.Hober C, Benhamou PY, Watt PC, Watanabe Y, Nomura Y, Stein E, Brunicardi FC, Mullen Y. Pancreas. 1997;14:199–204. doi: 10.1097/00006676-199703000-00014. [DOI] [PubMed] [Google Scholar]
- 9.Koide N, Sakaguchi K, Koide Y, Asano K, Kawaguchi M, Matsushima H, Takenami T, Shinji T, Mori M, Tsuji T. Exp. Cell Res. 1990;186:227–235. doi: 10.1016/0014-4827(90)90300-y. [DOI] [PubMed] [Google Scholar]
- 10.Abu-Absi SF, Friend JR, Hansen LK, Hu WS. Exp. Cell Res. 2002;274:56–67. doi: 10.1006/excr.2001.5467. [DOI] [PubMed] [Google Scholar]
- 11.Fukuda J, Sakai Y, Nakazawa K. Biomaterials. 2006;27:1061–1070. doi: 10.1016/j.biomaterials.2005.07.031. [DOI] [PubMed] [Google Scholar]
- 12.Lee PJ, Hung PJ, Lee LP. Biotechnol. Bioeng. 2007;97:1340–1346. doi: 10.1002/bit.21360. [DOI] [PubMed] [Google Scholar]
- 13.Fukuda J, Okamura K, Nakazawa K, Ijima H, Yamashita Y, Shimada M, Shirabe K, Tsujita E, Sugimachi K, Funatsu K. Cell Transplant. 2003;12:51–58. doi: 10.3727/000000003783985151. [DOI] [PubMed] [Google Scholar]
- 14.Matsushita T, Ijima H, Koide N, Funatsu K. Appl. Microbiol. Biotechnol. 1991;36:324–326. doi: 10.1007/BF00208150. [DOI] [PubMed] [Google Scholar]
- 15.Wu FJ, Friend JR, Remmel RP, Cerra FB, Hu WS. Cell Transplantation. 1999;8:233–246. doi: 10.1177/096368979900800304. [DOI] [PubMed] [Google Scholar]
- 16.Watt FM, Hogan BLM. Science. 2000;287:1427–1430. doi: 10.1126/science.287.5457.1427. [DOI] [PubMed] [Google Scholar]
- 17.Falconnet D, Csucs G, Grandin HM, Textor M. Biomaterials. 2006;27:3044–3063. doi: 10.1016/j.biomaterials.2005.12.024. [DOI] [PubMed] [Google Scholar]
- 18.Ng ES, Davis RP, Azzola L, Stanley EG, Elefanty AG. Blood. 2005;106:1601–1603. doi: 10.1182/blood-2005-03-0987. [DOI] [PubMed] [Google Scholar]
- 19.Doetschman TC, Eistetter H, Katz M, Schmidt W, Kemler R. Journal of Embryology and Experimental Morphology. 1985;87:27. [PubMed] [Google Scholar]
- 20.Itskovitz-Eldor J, Schuldiner M, Karsenti D, Eden A, Yanuka O, Amit M, Soreq H, Benvenisty N. Molecular Medicine. 2000;6:88–95. [PMC free article] [PubMed] [Google Scholar]
- 21.Keller GM. Curr. Opin. Cell Biol. 1995;7:862–869. doi: 10.1016/0955-0674(95)80071-9. [DOI] [PubMed] [Google Scholar]
- 22.Moeller HC, Mian MK, Shrivastava S, Chung BG, Khademhosseini A. Biomaterials. 2008;29:752–763. doi: 10.1016/j.biomaterials.2007.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schroeder M, Niebruegge S, Werner A, Willbold E, Burg M, Ruediger M, Field LJ, Lehmann J, Zweigerdt R. Biotechnol. Bioeng. 2005;92:920–933. doi: 10.1002/bit.20668. [DOI] [PubMed] [Google Scholar]
- 24.Yamada T, Yoshikawa M, Kanda S, Kato Y, Nakajima Y, Ishizaka S, Tsunoda Y. Stem Cells. 2002;20:146–154. doi: 10.1634/stemcells.20-2-146. [DOI] [PubMed] [Google Scholar]
- 25.Khademhosseini A, Yeh J, Jon S, Eng G, Suh KY, Burdick JA, Langer R. Lab Chip. 2004;4:425–430. doi: 10.1039/b404842c. [DOI] [PubMed] [Google Scholar]
- 26.Revzin A, Sekine K, Sin A, Tompkins RG, Toner M. Lab Chip. 5:30–37. doi: 10.1039/b405557h. [DOI] [PubMed] [Google Scholar]
- 27.Kim YS, Suh KY, Lee HH. Appl. Phys. Lett. 2001;79:2285–2287. [Google Scholar]
- 28.Karp JM, Yeh J, Eng G, Fukuda J, Blumling J, Suh KY, Cheng J, Mahdavi A, Borenstein J, Langer R, Khademhosseini A. Lab Chip. 2007;7:786–794. doi: 10.1039/b705085m. [DOI] [PubMed] [Google Scholar]
- 29.Khademhosseini A, Yeh J, Eng G, Karp J, Kaji H, Borenstein J, Farokhzad OC, Langer R. Lab Chip. 2005;5:1380–1386. doi: 10.1039/b508096g. [DOI] [PubMed] [Google Scholar]
- 30.Chia SM, Leong KW, Li J, Xu X, Zeng KY, Er PN, Gao SJ, Yu H. Tissue Eng. 2000;6:481–495. doi: 10.1089/107632700750022134. [DOI] [PubMed] [Google Scholar]
- 31.Carpenedo RL, Sargent CY, McDevitt TC. Stem Cells. 2007;25:2224–2234. doi: 10.1634/stemcells.2006-0523. [DOI] [PubMed] [Google Scholar]
- 32.Inaba R, Khademhosseini A, Suzuki H, Fukuda J. Biomaterials. 2009;30:3573–3579. doi: 10.1016/j.biomaterials.2009.03.045. [DOI] [PubMed] [Google Scholar]
- 33.Alarcon CDH, Pennadam S, Alexander C. Chem. Soc. Rev. 2005;34:276–285. doi: 10.1039/b406727d. [DOI] [PubMed] [Google Scholar]
- 34.Shimizu H, Ohashi K, Utoh R, Ise K, Gotoh M, Yamato M, Okano T. Biomaterials. 2009;30:5943–5949. doi: 10.1016/j.biomaterials.2009.07.042. [DOI] [PubMed] [Google Scholar]
- 35.Tsuda Y, Kikuchi A, Yamato M, Nakao A, Sakurai Y, Umezu M, Okano T. Biomaterials. 2005;26:1885–1893. doi: 10.1016/j.biomaterials.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 36.Matsuda N, Shimizu T, Yamato M, Okano T. Adv. Mater. 2007;19:3089–3099. [Google Scholar]
- 37.Tsuda Y, Yamato M, Kikuchi A, Watanabe M, Chen GP, Takahashi Y, Okano T. Adv. Mater. 2007;19:3633. [Google Scholar]
- 38.Hsiue GH, Hsu SH, Yang CC, Lee SH, Yang IK. Biomaterials. 2002;23:457–462. doi: 10.1016/s0142-9612(01)00127-2. [DOI] [PubMed] [Google Scholar]
- 39.Schmaljohann D. Adv. Drug Delivery Rev. 2006;58:1655–1670. doi: 10.1016/j.addr.2006.09.020. [DOI] [PubMed] [Google Scholar]
- 40.van der Linden H, Olthuis W, Bergveld P. Lab Chip. 2004;4:619–624. doi: 10.1039/b406247g. [DOI] [PubMed] [Google Scholar]
- 41.Sershen SR, Mensing GA, Ng M, Halas NJ, Beebe DJ, West JL. Adv. Mater. 2005;17:1366. doi: 10.1002/adma.200401239. [DOI] [PubMed] [Google Scholar]
- 42.Kim D, Beebe DJ. Lab Chip. 2007;7:193–198. doi: 10.1039/b612995a. [DOI] [PubMed] [Google Scholar]
- 43.Dong L, Agarwal AK, Beebe DJ, Jiang HR. Nature. 2006;442:551–554. doi: 10.1038/nature05024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.