

Abstract
Cytosolic sulfotransferases (STs) catalyze the sulfation of hydroxyl containing compounds. Human phenol sulfotransferase (SULT1A1) is the major human ST that catalyzes the sulfation of simple phenols. Because of its broad substrate specificity and lack of endogenous substrates, the biological function of SULT1A1 is believed to be an important detoxification enzyme. In this report, amino acid modification, computer structure modeling, and site-directed mutagenesis were used for studies of Arg residues in the active site of SULT1A1. The Arg-specific modification reagent, 2,3-butanedione, inactivated SULT1A1 in an efficient, time- and concentration-dependent manner, suggesting Arg residues play an important role in the catalytic activity of SULT1A1. According to the computer model, Arg78, Arg130, and Arg257 may be important for SULT1A1 catalytic activity. Site-directed mutagenesis results demonstrated that the positive charge on Arg78 is not critical for SULT1A1 because R78A is still active. In contrast, a negative charge at this position, R78E, completely inactivated SULT1A1. Arg78 is in close proximity to the site of sulfuryl group transfer. Arg257 is located very close to the 3′-phosphate in adenosine 3′-phosphate 5′-phosphosulfate (PAPS). Site-directed mutagenesis demonstrated that Arg257 is critical for SULT1A1: both R257A and R257E are inactive. Although Arg130 is also located very close to the 3′-phosphate of PAPS, R130A and R130E are still active, suggesting that Arg130 is not a critical residue for the catalytic activity of SULT1A1. Computer modeling suggests that the ionic interaction between the positive charge on Arg257, and the negative charge on 3′-phosphate is the primary force stabilizing the specific binding of PAPS.
Sulfotransferases (STs)1 catalyze the sulfation of hydroxyl-containing molecules. The substrate specificities of STs are very broad. Most hydroxyl groups in phenols, alcohols, and N-substituted hydroxylamines are substrates for one of the ST isoforms. The co-substrate for sulfation of all STs is adenosine 3′-phosphate 5′-phosphosulfate (PAPS). Sulfation (sulfuryl transfer) is widely observed in various biological processes. Various biological signaling molecules, including hormones, neurotransmitters, peptides, and proteins, can be sulfated to alter biological activity. STs also catalyze the sulfation of a broad range of xenobiotics. Sulfation of drugs and xenobiotics is mainly associated with detoxification: biotransformation of a relatively hydrophobic xenobiotic into a more water-soluble sulfuric ester that is readily excreted. However, there are numerous important exceptions wherein the formation of chemically reactive sulfuric esters is an essential step in the metabolic pathways leading to toxic or carcinogenic responses. Detoxification or bioactivation is highly dependent on the electrophilic reactivity of the individual sulfuric ester products formed. Most sulfation products are stable enough for excretion, while other sulfuric ester products can be reactive toward nucleophilic sites on DNA, RNA, and protein, and so become involved in the initiation of carcinogenesis and other toxic responses.
Structure-activity relationship studies of STs started in the 1980s. Protein sequence alignments of the different STs have revealed two highly conserved regions, one (PKSGTTW) in the N-terminal region and one (RKGXXGDWK) in the C-terminal region (1). Since all STs use the same sulfuryl donor, it was speculated that these two regions were involved in PAPS binding (2, 3). Affinity labeling of rat liver aryl ST-IV identified a peptide sequence in the PAPS-binding site, which is in close proximity to the highly conserved sequence in the N-terminal region (4). Site-directed mutagenesis and [35S]PAPS affinity labeling studies of flavonol 3-ST have supported that the two regions mentioned above are involved in PAPS binding (5). Affinity chromatography and 31P NMR studies of flavonol 3-ST demonstrated the involvement of Lys59, Arg11, and Arg277 in PAPS binding (6). Point mutations and [35S]PAPS affinity labeling studies of guinea pig EST have also strongly supported the C-terminal conserved region mentioned above as being part of the PAPS-binding site (7). The x-ray crystal structure of mouse EST supports the idea that these two regions, residues 259–265 and 45–51 for mouse estrogen ST, are directly involved in PAPS binding (8). In addition to these two regions, Thr227, Trp53, and Phe229 of mouse EST have also been demonstrated to be involved in PAPS binding.
Limited information is available on the structural determinants of the ST substrate-binding site. Of the four regions proposed by Varin’s group for flavonol STs, regions I and IV are the same two regions mentioned above for the PAPS-binding site, while domain II, spanning amino acids 92–194 of the flavonol 3-ST sequence, is believed to contain all the determinants for substrate binding (2, 9). Subsequent mutational analysis of domain II of flavonol 3-ST did not identify amino acids critical for substrate binding; however, Leu95 may be in direct contact with the flavonol B ring (10). N-Bromoacetyl-4-hydroxyphenylamine has been used for affinity labeling of the substrate-binding site in rat liver aryl ST-IV (11). Four modified amino acids, Cys232, Cys283, Lys286, and Cys289 have been identified. Sakakibara et al. (3) reported localization and a functional analysis of the substrate specificity and catalytic domains of the human M-PST and P-PST-1. By comparing the kinetic parameters of a series of expressed chimeric PSTs, where the M-form and P-form coding regions were exchanged, it was concluded that amino acid residues 84–148 contain the structural determinants for substrate specificity in both forms. They also demonstrated the differential roles of the two highly variable regions (amino acid residues 84–89 and 143–148) in substrate binding, catalysis, and sensitivity to inhibition by 2,6-dichloro-4-nitrophenol. Similar studies on human aryl-STs (HAST1, HAST3, and HAST4) suggested that the two highly divergent regions, region A (amino acids 44–107) and region B (amino acids 132–164), determine the substrate specificity of human aryl STs (12, 13). A single amino acid change in HAST1 (A146E) altered its activity for p-nitrophenol (PNP) to that of HAST3. Studies on guinea pig 3-hydroxysteroid ST isoforms revealed that the amino acid residue at position 51 plays a fundamental role in determining the stereospecificity exhibited by the α- and β-isoforms. Mutational studies on mouse EST demonstrated that Tyr81 determines the substrate specificity of this enzyme (14). The x-ray crystal structure of mouse EST showed that residues Phe142, Ile146, Tyr149 contribute to binding of steroid 17β-estradiol. Asn86 is believed to be in a position to form a hydrogen bond with the 17β-hydroxyl group, while Lys106 and His108 are within hydrogen bonding distance of the 3α-phenol group of the steroid (8).
Studies of structure-function relationships on human STs are relatively limited. The work on human phenol STs has revealed two regions and one specific amino acid (position 146) as important for the determination of substrate specificity, as mentioned above (3, 12, 13). To our knowledge, studies on the identification of specific residues in the active site of STs by amino acid modification reagents are limited. Borchardt et al. (16, 17) used 2,3-butanedione and phenylglyoxal to identify the arginyl residues in the active site of rat liver PST. N-Ethylmaleimide has also been used for the identification of essential sulfhydryl residues in rat PST (17). Ribonucleotide dialdehydes (ATPDA, ADPDA, AMPDA, and APSDA) have been characterized as affinity labeling reagents for rat liver PST, and it was speculated that the dialdehydes inactivated the ST by possible formation of a Schiff’s base adduct with an active site lysine residue (17, 18). ATPDA has been used as an affinity labeling reagent for the identification of a peptide sequence in the PAPS-binding site of rat liver AST-IV (4). We have studied the carboxyl amino acid residues in the active site of SULT1A1 (19). Our results indicated that the conserved residues Asp134 and Asp263 are critical for the binding of PAPS. Glu83 and Glu246 may be important for the binding of substrate, and Glu83 may be involved in the catalysis reaction.
Since the crystallization of the mouse estrogen ST (8), a few human STs have also been crystallized and their crystal structures partially solved. They include human monoamine ST (M-PST, SULT1A3) (20, 21), human estrogen ST (EST, SULT1E1) (22), and hydroxysteroid ST (DHEA-ST, SULT2B1) (23). The crystal structure of the human simple phenol ST (P-PST, SULT1A1) has not been shown. We built the computer model of SULT1A1 based on the other known ST crystal structures (19). Even with the known crystal structure, the roles of amino acid residues in the active site still need to be studied using experimental methods.
In this report, we studied Arg residues in the active site of human phenol sulfotransferase (SULT1A1) using amino acid modification, computer modeling, and site-directed mutagenesis. Arginine is one of the two amino acids that carry a positive charge. It may be very important for the catalytic activity of STs. Arg-specific modification reagents have been used to study the importance of Arg for the catalytic activity of SULT1A1. Our results demonstrate that the positive charge of Arg78 does not contribute to the catalytic activity of SULT1A1. A negative charge at this position inactivated SULT1A1. Although both Arg257 and Arg130 are in close proximity to the 3′-phosphate on PAPS, only Arg257 appears to be critical for the catalytic activity of SULT1A1.
EXPERIMENTAL PROCEDURES
Material
2,3-Butanedione (BD), 4-phenylphenol (4PP), 2-naphthol (2NP), 3′-phosphoadenosine-5′-phosphate (PAP), PAPS, isopropyl-1-thio-β-D-galactopyranoside, ampicillin, and dithiothreitol were purchased from Sigma. Amylose affinity resin and maltose were purchased from New England Biolabs (Beverly, MA). All other chemicals and solvents were of the highest grade available.
Enzymatic Assay
Methylene Blue Assay
This method is used for determining activity of SULT1A1 and its mutants and for SULT1A1 mutant protein kinetic parameter determinations. This assay method was developed by Nose and Lipmann (24). SULT1A1 activity was determined in the assay mixture containing 0.125 M phosphate buffer, pH 6.2, 8 μM PAPS, 2–10 μg mutant protein, and different concentrations of substrate in a total volume of 400 μl. After incubating at 37.0 °C for 30 min, the reaction was stopped by adding 0.5 ml of methylene blue solution (250 mg of methylene blue, 10 ml of H2SO4, 50 g of Na2SO4 in 1 liter of water) and 1 ml of chloroform. After vortexing, the tubes were centrifuged in a benchtop centrifuge for 1 min. The chloroform phase was then transferred to tubes containing 20–50 mg of dehydrated Na2SO4 and read at 651 nm. Kinetic constants were calculated according to the Michaelis-Menten equation.
p-Nitrophenyl Sulfate (PNPS) Assay
This method utilizes PNPS to regenerate PAPS from product PAP. At the same time, the color reagent PNP is generated for colorimetric measurement (25–27). This method was used for the determination of time- and concentration-dependent inactivation, kinetic parameter determination for partially inactivated SULT1A1, and PAP and substrate protection for BD inactivation of SULT1A1.
Purification of SULT1A1
Human SULT1A1 cDNA (and its mutants) was expressed using the pMAL-c2 expression system as described previously (26, 28). Briefly, the maltose-binding fusion protein (MBSULT1A1 and its mutants) was expressed in XL1-Blue cells containing the pMAL-SULT1A1 vector. The cytosol of fusion protein was loaded onto an amylose affinity column (New England Biolabs) pre-washed with 5 mM phosphate buffer, pH 7.4. After washing out other proteins, the fusion protein (mutants or wild type) was eluted using a 0–1 mM maltose gradient in the same buffer. The final purified SULT1A1 was apparent homogenous according to SDS-PAGE analysis.
SULT1A1 Inactivation by 2,3-Butanedione
SULT1A1 (0.1 mg/ml) was incubated in 0.05 M borate, pH 7.5, containing 1 M NaCl at room temperature. Different concentrations of 2,3-butanedione (in ethanol) were added to start the inactivation reaction. Aliquots (10 μl) were taken at different times for standard assay of SULT1A1 activity. Arginine (0.5 M) was included in the enzyme assay reaction mixture (for time- and concentration-dependent inactivation) or used to stop the reaction (for partially inactivation of SULT1A1). For substrate or PAP protection experiments, phenol substrates (2-naph-thol or 4-phenylphenol) or PAP were added to the SULT1A1 solution before the addition of 2,3-butanedione.
Modeling of the SULT1A1 Structure
The molecular coordinates of mSULT1E1 were obtained from the Protein Data Bank file 1AQU. A Silicon Graphics Iris work station was used for modeling. Since the primary sequence of SULT1A1 and mSULT1E1 could be aligned with no gaps, a significant amino acid similarity (93%) existed between the two proteins, and each had the same number of amino acids. Biosym’s Homology program was used to build the backbone of SULT1A1 based on the coordinates of mSULT1E1. Side chain bumps of 0.1% overlap of the Van der Walls radii were relieved manually. The structure was then minimized using steepest descents (2000 steps) followed by conjugate gradients (1000 steps) using Biosym’s Discover program, version 2.9. No constraints were used. The program RasMol, version 2.6, was used for display of the SULT1A1 structure.
Site-directed Mutagenesis of the cDNA Encoding SULT1A1
The cDNA encoding SULT1A1 in the pKK233-3 vector was from Dr. Charles Falany, and this construct has been described previously (29, 30). All mutant cDNAs were created with the QuikChange mutagenesis kit (Stratagene, La Jolla, CA), and all primers for mutagenesis were obtained from Integrated DNA Technologies Inc. (Coralville, IA). The primers used for the mutations are list in Table I. The primers were designed using the Gene Fisher primer designing and Multialignment software. Mutant cDNA was generated and selected through a series of three steps consisting of primer extension/thermocycling, digestion of parental DNA, and transformation as outlined in the manufacturer’s protocol. Colonies resulting from the transformation were grown in sterile medium by standard protocols and plasmid was isolated from cells with the QIAprep spin mini-prep kit (Qiagen Inc., Valencia, CA). The presence of the desired mutations was confirmed by DNA sequencing which was performed by The Oklahoma State University, Department of Biochemistry and Molecular Biology, Recombinant DNA/Protein Resource Facility.
Table I.
Primer pairs for SULT1A1 mutants
| Mutantsa | Primers |
|---|---|
| R78A | 5′-GCTCCCATCTTCATGGCCGTGCCCTTCC-3′ |
| R78E | 5′-GCTCCCATCTTCATGGAGGTGCCCTTCC-3′ |
| R78r | 5′-GGAAACTGCCACATCCTTTGCGTTGCGG-3′ |
| R130A | 5′-GGTCTATGTTGCCGCCAACGCAAAGGATGTGC-3′ |
| R130E | 5′-GGTCTATGTTGCCGAGAACGCAAAGGATGTGC-3′ |
| R130r | 5′-GGTAGAGAACAGGGTGGGTCCGGCTCAGCTCC-3′ |
| R257A | 5′-GCATCTCCCCCTTCATGGCCAAAGGCATGGCT-3′ |
| R257E | 5′-GCATCTCCCCCTTCATGGAGAAAGGCATGGCT-3′ |
| R257r | 5′-CGCTTGGTCAGGTTTGATTCGCACACTCCCTC-3′ |
| R213E | 5′-CCTGGAGTTTGTGGGGCAATCCCTGCCAGAC-3′ |
| R213r | 5′-GGACCTCAAACACCCCGTTAGGGACGGTCTC-3′ |
Note: two different mutants in the same position used the same reverse primer (r).
RESULTS AND DISCUSSION
Chemical Inactivation of SULT1A1 by Arginine Residue-specific Modification Reagent 2,3-Butadione
BD is a reagent specifically reactive toward the Arg residues of a protein (Fig. 1). BD has been used for the characterization of Arg residues in various enzymes (31–34). Because borate is necessary to form a stable adduct (Fig. 1), the inactivation reactions were done in 50 mM borate buffer, pH 7.5.
Fig. 1.

Chemical reaction for 2,3-butanedione inactivation of Arg residues in a protein.
BD inactivated SULT1A1 in an efficient, time- and concentration-dependent manner (Fig. 2A). In the concentration range from 0.0625 to 2 mM, BD effectively inactivated SULT1A1. At 2 mM, BD inactivated 95% of SULT1A1 activity within 30 s. At all BD concentrations, log(percent activity) is linear versus time. These facts indicate that the inactivation is specific through active site Arg residues. According to the equation d[E]/dt = Kapp[E] or log[E] = 2.3Kappt (where E represents active SULT1A1, t is time, Kapp is apparent first order rate constant), Kapp can be calculated from the slopes of Fig. 2A. According to equation Kapp = K[BD]n or logKapp = logK + nlog[BD] (where K is the rate constant and n is the reaction order for BD), when logKapp is plotted against the log[BD], a straight line results (Fig. 2B). From the data in Fig. 2B, n = 1.07 and K = 450 min−1 M−1. This further demonstrates that BD is a specific reagent for the inactivation of critical Arg residues in the active site of SULT1A1. The BD reaction order, n, is ~1. This suggests that there may be only one Arg residue in the active site whose modification could lead to complete inactivation of SULT1A1.
Fig. 2. Time- and concentration-dependent inactivation of SULT1A1 by 2,3-butanedione.

A, log(percent activity) versus time. SULT1A1 (0.1 mg/ml) was incubated with different concentrations of 2,3-butanedione at room temperature in 50 mM borate, 1 M NaCl, pH 7.5. Aliquots were taken at different time points. SULT1A1 remaining activity was assayed using the PNPS assay method in a mixture containing 0.5 M arginine. B, log(Kapp) versus log(BD).
SULT1A1 was partially inactivated using 0.25 mM BD in 50 mM borate buffer, pH 7.5, containing 1 M NaCl for 1 min at room temperature. The inactivation was stopped by addition of an equal volume of 1 M arginine solution at pH 7.5. The partially inactivated SULT1A1 was used to determine kinetic parameter changes by varying either PAPS or substrate 4PP concentrations. Ethanol (BD stock solution was prepared in ethanol) was used for the control. Fig. 3, A and B, demonstrate that the partially inactivated SULT1A1 did not change Km for either PAPS or substrate 4PP. This suggests that the SULT1A1 completely lost its catalytic activity when BD modified the active site Arg residues. If any of the BD modifications to SULT1A1 had lead to partial loss of catalytic activity, the above partially inactivated SULT1A1 would have changed in Km for either PAPS or 4PP.
Fig. 3. Kinetic parameter determination of partially inactivated SULT1A1.

The PNPS assay method was used for the determination of enzyme activity. A, varied substrate 4-phenylphenol concentration. B, varied PAPS concentration.
Fig. 4 demonstrates that both PAP and phenol substrates (4-phenylphenol and 2-naphthol) protected the BD inactivation of SULT1A1. This further suggests that the inactivation is due to the modification of Arg residues in the active site. Because the inactivation is irreversible and substrate or PAP binding is reversible, the protection cannot be 100%.
Fig. 4. PAP and substrate protection of 2,3-butanedione inactivation of SULT1A1.

The PNPS assay method was used to determine the remaining enzyme activity.
Computer Modeling Structure of SULT1A1
The computer model of human SULT1A1 was generated (19) based on the published crystal structure of mouse estrogen ST (mSULT1E1) (Protein Data Bank file 1AQU) (15).
According to the computer SULT1A1 model, Arg78, Arg130, and Arg257 may be important for the catalytic activity of SULT1A1 (Fig. 5). Arg257 and Arg130 are in close proximity to the 3′-phosphate group on PAPS. Arg78 is in close proximity to both the 5′-phosphosulfate on PAPS and the hydroxyl group on substrate, 2-naphthol. Arg78 may affect the catalytic activity of SULT1A1. All other Arg residues are located on the surface of the protein structure. Table II lists the shortest calculated distances of these Arg residues to different functional groups.
Fig. 5. Computer modeling structure of SULT1A1.

The structure was depicted by RasMol 2.6. The whole structure was displaced by “Backbone.” PAPS, 2-naphthol, Arg78, Arg130, Arg257, and Arg213 were displaced by “Spacefill.”
Table II.
Calculated shortest distance between the related residues to the functional groups
| Arg78 | Arg78 | Arg130 | Arg213 | Arg213 | Arg257 | Lys106 | Lys106 | |
|---|---|---|---|---|---|---|---|---|
| Functional group | PAPS (5′-sulfate) | 2NP (OH) | PAPS (3′-phosphate) | PAPS (5′-sulfate) | 2NP (OH) | PAPS (3′-phosphate) | PAPS (5′-sulfate) | 2NP (OH) |
| Distance (Å) | 11.3 | 11.1 | 2.8 | 23.8 | 23.3 | 2.0 | 3.7 | 2.0 |
Site-directed Mutagenesis of Important Arg Residues
According to the BD inactivation results and computer modeling structure, Arg78, Arg130, and Arg257 were selected for site-directed mutagenesis studies. Arg213 (located on the surface of SULT1A1) was selected for control mutation. Arg78, Arg130, and Arg257 were mutated to either the neutral amino acid alanine or the negatively charged amino acid glutamic, acid. Arg213 was only mutated to glutamic acid. All the mutated SULT1A1s were purified. Pure proteins of the mutants were used for kinetic enzyme activity measurements. Fig. 6 shows the SDS-PAGE for the purified mutant proteins. All mutant proteins were pure based on the SDS-PAGE. Fig. 7 shows the enzymatic specific activities of different mutants at 100 μM 2-naphthol, 8 μM PAPS, pH 6.2. Fig. 7 demonstrates that R213E (control mutant) did not change SULT1A1 catalytic activity. R257A and R257E almost completely inactivated SULT1A1. This suggests that a positive charge at the Arg257 position is critical for the catalytic activity (or PAPS binding) of SULT1A1. Although R130 is also in close proximity to the 3′-phosphate of PAPS, neither R130A nor R130E significantly changed the catalytic activity of SULT1A1. Arg130 may not play a critical role for the binding of PAPS or the catalytic activity of SULT1A1.
Fig. 6. SDS-PAGE of the purified SULT1A1 mutant proteins.
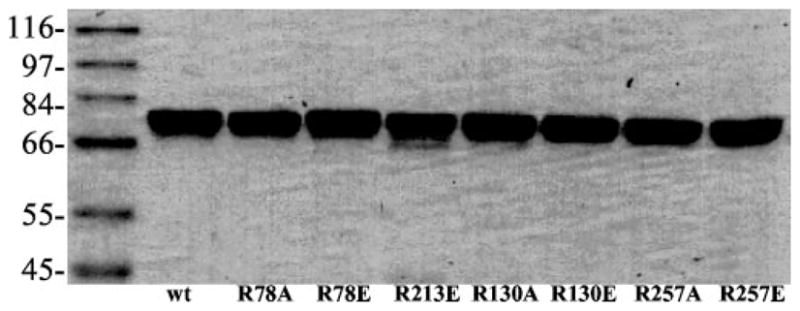
12% acrylamide gel was used for the separation of proteins. The gel was run at 200 V.
Fig. 7. 2-Naphthol sulfation activity of SULT1A1 mutant proteins.

The methylene blue assay method was used for enzyme activity determinations. 0.1 M 2-naphthol was used as substrate for the assay at 8 μM PAPS, pH 6.2.
R78A retained activity (Fig. 7 and Table III). This suggests that the positive charge on Arg78 is not critical for the catalytic activity of SULT1A1. R78E was inactive, suggesting that a negative charge at position 78 will inactivate SULT1A1. Based on the computer model of SULT1A1 (Fig. 8), Lys106 (the other amino acid carrying a positive charge on its side chain) is in closer proximity than Arg78 to the location of sulfuryl group transfer (Table II). The positive charge on Arg78 is neutralized by Asp59 (negatively charged residue). Therefore the positive charge at position 78 is not critical for catalytic activity of SULT1A1. If position 78 is mutated to a negatively charge amino acid, Lys106 may be affected, thus inactivating SULT1A1. We are currently investigating the role of Lys residues on the catalytic activity of SULT1A1.
Table III.
2-Naphthol sulfation kinetic parameters for SULT1A1 mutants
| wt | R213E | R78A | R130A | R130E | |
|---|---|---|---|---|---|
| Vmax (nmol/min/mg) | 13.2 | 14.4 | 13.2 | 14.0 | 16.4 |
| Km (mM) | 16.0 | 20.9 | 18.2 | 18.4 | 18.4 |
| Vmax/Km | 0.84 | 0.68 | 0.72 | 0.76 | 0.88 |
Fig. 8. Computer modeling structure of SULT1A1 showing Arg78 and related residues and ligands.
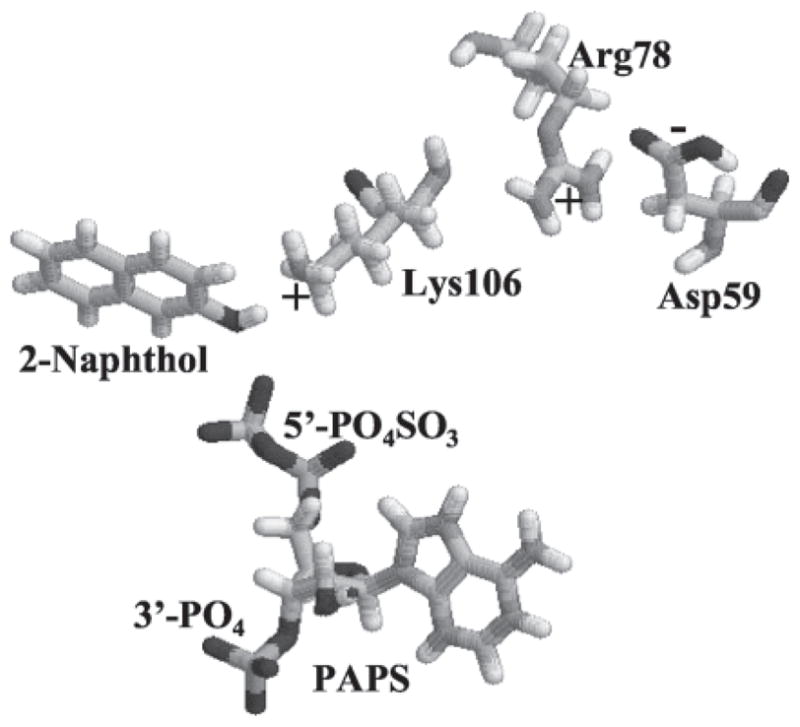
The model structure of SULT1A1 was depicted by RasMol 2.6. Only Arg78, PAPS, 2-naphthol, Lys106, and Asp59 are displaced.
The kinetic parameters for the active mutants were determined (Table III). The results in Table III indicate that R213E, R78A, R130A, and R130E have catalytic characteristics similar to wild type SULT1A1. Km and Vmax for these mutants did not change significantly compared with the wild type.
CONCLUSION
The effect of specific Arg residues on SULT1A1 activity has not been previously studied. To the best of our knowledge, the only report on the study of Arg residues in STs was published by Borchardt and Schasteen in 1977 (16). They studied 2,3-butandione inactivation of a rat liver phenol ST. Their results indicated that BD inactivated this rat enzyme in a time- and concentration-dependent manner. They suggested a possible role for arginyl residues as anionic recognition sites for sulfate transfer reactions. In this work, we focused on the study of Arg residues in the active site of human SULT1A1 using the combination of amino acid modification, computer modeling, and site-directed mutagenesis methods. Our results consistently suggest that among the Arg residues, only Arg257 is critical for the catalytic activity of SULT1A1. The inactivation reaction order for BD (n) is ~1. This suggests that there is only one Arg modification that can completely inactivate SULT1A1. Because R78A is active, the BD modification of R78 should not inactivate SULT1A1. Because both R130A and R130E are active, the BD modification of Arg130 also should not inactivate SULT1A1. BD partially inactivated SULT1A1 did not change the Km values for either substrate or PAPS (Fig. 3), suggesting that when BD modified Arg257, SULT1A1 would completely lose its activity. When other Arg residues were modified, SULT1A1 was still active. These findings agree with the site-directed mutagenesis results.
Mutational analysis of substrate-binding domains in SULT1A1 (P-PST) and SULT1A3 (M-PST) demonstrated that the amino acid at position 146 (Ala146 in SULT1A1 and Glu146 in SULT1A3) determine the substrate specificity of SULT1A1 and SULT1A3 (12, 13, 35). Our SULT1A1 model indicates that Ala146 is in direct contact with substrate 2-naphthol. Mutation of His108 in both SULT1A1 and SULT1A3 resulted in complete loss of activity suggesting His108 is a critical residue in SULT1A1 and SULT1A3 (35). Our recent amino acid modification experimental results demonstrated that His108 is critical for catalysis in both SULT1A1 and SULT1A3 but not for the binding of substrate or PAPS.2 According to our SULT1A1 model, His108 is located at the site of sulfuryl group transfer. The imidazole group in His108 may act as a base catalyst for the sulfuryl transfer. These results demonstrate critical residues for catalysis and substrate binding.
In this work, Arg257 was found to be critical for SULT1A1 catalytic activity. Arg257 is located near the 3′-phosphate but not the site of sulfuryl group transfer. This suggests that Arg residues in SULT1A1 do not contribute to the transfer of the sulfuryl group but do contribute to the specific binding of PAPS. A mutational study on rat liver phenol ST demonstrated that Ser134 plays a key role in PAPS binding but not in catalysis (15). This work also demonstrated the roles of Arg126 and Arg253 (corresponding to Arg130 and Arg257, respectively, in human SULT1A1) in the specific binding of PAP or AMP. Our work demonstrates only Arg257, and not Arg130, to be critical for human SULT1A1 activity. This is in agreement with the commonly recognized fact that the two regions, PKSGTTK (47–53 for SULT1A1) and RKGXXGDWKXXFT (257–269 for SULT1A1), are the primary sequences in the specific binding of PAPS to STs (1–8, 15). Arg257 is in the latter conserved region, whereas Arg130 is in neither. Arg130 in SULT1A1 and Arg126 in rat phenol ST may have different roles in the two different enzymes.
Fig. 9 shows PAPS and both positively and negatively charged residues to be within 5 Å of 3′-phosphate. Both Arg257 and Arg130 are in close proximity to the 3′-phosphate. Lys258 is also close to the 3′-phosphate (within 2 Å), but its positively charged ammonium ion lies more than 8 Å away. There are two negatively charged residues, Asp134 and Asp263, within 5 Å distance. Asp134 is near Arg130 (2 Å), neutralizing its positive charge. Asp263 is near Lys258 (2 Å), neutralizing its positive charge. Arg257 is not neutralized by any neighboring residues. The ionic interaction between the positive charge on Arg257 and the negative charge on 3′-phosphate is the primary force stabilizing the specific binding of PAPS. Arg257 is conserved in all STs and is one of the most important residues for specific binding of PAPS.
Fig. 9. Computer modeling structure of SULT1A1 showing charged residues in close proximity to 3′-phosphate of PAPS.
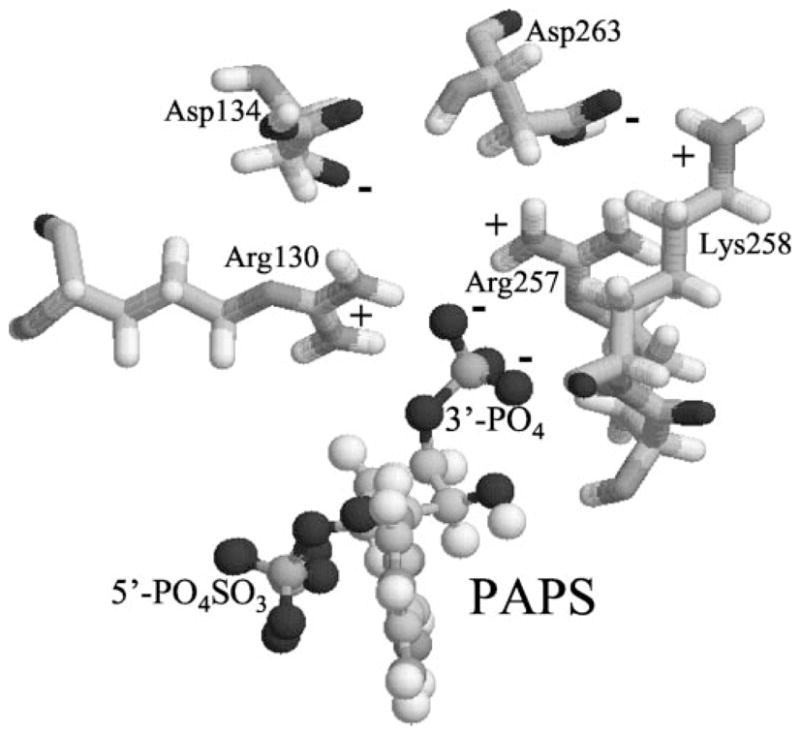
The model structure of SULT1A1 was depicted by RasMol 2.6. Only PAPS and charged residues within 5 Å to 3′-phosphate are displaced.
Acknowledgments
We thank Sharon Baker for her evaluation of the manuscript. We are grateful to Dr. Anna Radominska-Pandya (University of Arkansas for Medical Sciences) for her extensive help with this research project, the generous gift of SULT1A1 E. coli strain from Dr. Charles N. Falany (University of Alabama at Birmingham), and help by Dr. J. Lyndal York (University of Arkansas for Medical Sciences) with generation of the SULT1A1 computer model are deeply appreciated.
Footnotes
This work was supported in part by National Institutes of Health Grant GM59873 (to G. C.).
The abbreviations used are: ST, sulfotransferase; PAP, 3′-phosphoadenosine-5′-phosphate; PAPS, 3′-phosphate 5′-phosphosulfate; EST, expressed sequence tag; DA, dialdehyde; BD, 2,3-butadione; 4PP, 4-phenylphenol; 2NP, 2-naphthol; PNPS, p-nitrophenyl sulfate; PNP, p-nitrophenol.
G. Chen, manuscript in preparation.
Note Added in Proof—The authors sincerely apologize that our statement in the introduction “The crystal structure of the human simple phenol ST (P-PST, SULT1A1) has not been shown” is not true. This crystal structure complexed with PAP and p-nitrophenol (Protein Data Bank entry 1LS6) has been solved and published (Gamage, N. U., Duggleby, R. G., Barnett, A. C., Tresillian, M., Latham, C. F., Liyou, N. E., McManus, M. E., and Martin, J. L. (2003) J. Biol. Chem. 278, 7655–7662).
References
- 1.Weinshilboum R, Aksoy I. Chem Biol Interact. 1994;92:233–246. doi: 10.1016/0009-2797(94)90066-3. [DOI] [PubMed] [Google Scholar]
- 2.Varin L, Marsolais F, Richard M, Rouleau M. FASEB J. 1997;11:517–525. doi: 10.1096/fasebj.11.7.9212075. [DOI] [PubMed] [Google Scholar]
- 3.Sakakibara Y, Takami Y, Nakayama T, Suiko M, Liu C. J Biol Chem. 1998;273:6242–6247. doi: 10.1074/jbc.273.11.6242. [DOI] [PubMed] [Google Scholar]
- 4.Zheng Y, Bergold A, Duffel MW. J Biol Chem. 1994;269:30313–30319. [PubMed] [Google Scholar]
- 5.Marsolais F, Varin L. J Biol Chem. 1995;270:30458–30463. doi: 10.1074/jbc.270.51.30458. [DOI] [PubMed] [Google Scholar]
- 6.Marsolais F, Laviolette M, Kakuta Y, Negishi M, Pedersen LC, Auger M, Varin L. Biochemistry. 1999;38:4066–4071. doi: 10.1021/bi982239m. [DOI] [PubMed] [Google Scholar]
- 7.Komatsu K, Driscoll WJ, Koh YC, Strott CA. Biochem Biophys Res Commun. 1994;204:1178–1185. doi: 10.1006/bbrc.1994.2587. [DOI] [PubMed] [Google Scholar]
- 8.Kakuta Y, Pedersen LG, Carter CW, Negishi M, Pedersen C. Nat Struct Biol. 1997;4:904–908. doi: 10.1038/nsb1197-904. [DOI] [PubMed] [Google Scholar]
- 9.Varin L, Marsolais F, Brisson N. J Biol Chem. 1995;270:12498–12502. doi: 10.1074/jbc.270.21.12498. [DOI] [PubMed] [Google Scholar]
- 10.Marsolais F, Varin L. Eur J Biochem. 1997;247:1056–1062. doi: 10.1111/j.1432-1033.1997.01056.x. [DOI] [PubMed] [Google Scholar]
- 11.Duffel MW, Chen G, Sharma V. Chem Biol Interact. 1998;109:81–92. doi: 10.1016/s0009-2797(97)00122-1. [DOI] [PubMed] [Google Scholar]
- 12.Brix LA, Barnett AC, Duggleby RG, Leggett B, McManus ME. Biochemistry. 1999;38:10474–10479. doi: 10.1021/bi990795q. [DOI] [PubMed] [Google Scholar]
- 13.Brix LA, Duggleby RG, Gaedigk A, McManus ME. Biochem J. 1999;337:337–343. [PMC free article] [PubMed] [Google Scholar]
- 14.Petrotchenko EV, Doerflein ME, Kakuta Y, Pedersen LC, Negishi M. J Biol Chem. 1999;274:30019–30022. doi: 10.1074/jbc.274.42.30019. [DOI] [PubMed] [Google Scholar]
- 15.Hsiao YS, Yang YS. Biochemistry. 2002;41:12959–12966. doi: 10.1021/bi0261239. [DOI] [PubMed] [Google Scholar]
- 16.Borchardt RT, Schasteen CS. Biochem Biophys Res Commun. 1977;78:1067–1073. doi: 10.1016/0006-291x(77)90529-0. [DOI] [PubMed] [Google Scholar]
- 17.Borchardt RT, Schasteen CS, Wu SE. Biochim Biophys Acta. 1982;708:280–293. doi: 10.1016/0167-4838(82)90438-1. [DOI] [PubMed] [Google Scholar]
- 18.Borchardt RT, Wu SE, Schasteen CS. Biochem Biophys Res Commun. 1978;81:841–849. doi: 10.1016/0006-291x(78)91428-6. [DOI] [PubMed] [Google Scholar]
- 19.Chen G, Rabjohn PA, York JL, Wooldridge C, Zhang D, Falany CN, Radominska-Pandya A. Biochemistry. 2000;39:16000–16007. doi: 10.1021/bi0021479. [DOI] [PubMed] [Google Scholar]
- 20.Bidwell LM, McManus ME, Gaedigk A, Kakuta Y, Negishi M, Pedersen L. J Mol Biol. 1999;293:521–530. doi: 10.1006/jmbi.1999.3153. [DOI] [PubMed] [Google Scholar]
- 21.Dajani R, Cleasby A, Neu M, Wonacott AJ, Jhoti H, Hood AM, Modi S, Hersey A, Taskinen J, Cooke RM, Manchee GR, Coughtrie MW. J Biol Chem. 1999;274:37862–37868. doi: 10.1074/jbc.274.53.37862. [DOI] [PubMed] [Google Scholar]
- 22.Pedersen LC, Petrotchenko E, Shevtsov S, Negishi M. J Biol Chem. 2002;277:17928–17932. doi: 10.1074/jbc.M111651200. [DOI] [PubMed] [Google Scholar]
- 23.Pedersen LC, Petrotchenko EV, Negishi M. FEBS Lett. 2000;475:61–64. doi: 10.1016/s0014-5793(00)01479-4. [DOI] [PubMed] [Google Scholar]
- 24.Nose Y, Lipmann F. J Biol Chem. 1958;233:1348–1351. [PubMed] [Google Scholar]
- 25.Frame LT, Ozawa S, Nowell SA, Chou HC, DeLongchamp RR, Doerge DR, Lang NP, Kadlubar FF. Drug Metab Dispos. 2000;28:1063–1068. [PubMed] [Google Scholar]
- 26.Chen G, Battaglia E, Senay C, Falany CN, Radominska-Pandya A. Protein Sci. 1999;8:2151–2157. doi: 10.1110/ps.8.10.2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen G, Yin S, Maiti S, Shao X. J Biochem Mol Toxicol. 2002;16:279–285. doi: 10.1002/jbt.10048. [DOI] [PubMed] [Google Scholar]
- 28.Falany CN, Comer KA, Dooley TP, Glatt H. Ann N Y Acad Sci. 1995;774:59–72. doi: 10.1111/j.1749-6632.1995.tb17372.x. [DOI] [PubMed] [Google Scholar]
- 29.Wilborn TW, Comer KA, Dooley TP, Reardon IM, Heinrikson RL, Falany CN. Mol Pharmacol. 1993;43:70–77. [PubMed] [Google Scholar]
- 30.Falany CN, Zhuang W, Falany JL. Chem Biol Interact. 1994;92:57–66. doi: 10.1016/0009-2797(94)90053-1. [DOI] [PubMed] [Google Scholar]
- 31.Yankeelov JA, Jr, Mitchell CD, Crawford TH. J Am Chem Soc. 1968;90:1664–1666. doi: 10.1021/ja01008a056. [DOI] [PubMed] [Google Scholar]
- 32.Riordan JF. Biochemistry. 1973;12:3915–3923. doi: 10.1021/bi00744a020. [DOI] [PubMed] [Google Scholar]
- 33.Eriksson O, Fontaine E, Bernardi P. J Biol Chem. 1998;273:12669–12674. doi: 10.1074/jbc.273.20.12669. [DOI] [PubMed] [Google Scholar]
- 34.Alkema WB, Prins AK, de Vries E, Janssen DB. Biochem J. 2002;365:303–309. doi: 10.1042/BJ20011468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu MC, Suiko M, Sakakibara Y. J Biol Chem. 2000;275:13460–13464. doi: 10.1074/jbc.275.18.13460. [DOI] [PubMed] [Google Scholar]