

Abstract
Huntington's disease (HD) is caused by a CAG repeat expansion in exon 1 of the HD gene resulting in a long polyglutamine tract in the N‐terminus of the protein huntingtin. Patients carrying the mutation display chorea in early stages followed by akinesia and sometimes dystonia in late stages. Other major symptoms include depression, anxiety, irritability or aggressive behavior, and apathy. Although many neuronal systems are affected, dysfunction and subsequent neurodegeneration in the basal ganglia and cortex are the most apparent pathologies. In HD, the primary hypothesis has been that there is an initial overactivity of glutamate neurotransmission that produces excitotoxicity followed by a series of complex changes that are different in the striatum and in the cortex. This review will focus on evidence for alterations in dopamine (DA)–glutamate interactions in HD, concentrating on the striatum and cortex. The most recent evidence points to decreases in DA and glutamate neurotransmission as the HD phenotype develops. However, there is some evidence for increased DA and glutamate functions that could be responsible for some of the early HD phenotype. Significant evidence indicates that glutamate and dopamine neurotransmission is affected in HD, compromising the fine balance in which DA modulates glutamate‐induced excitation in the basal ganglia and cortex. Restoring the balance between glutamate and dopamine could be helpful to treat HD symptoms.
Keywords: Cortex, Dopamine, Glutamate, Huntington's disease, Neuropathology, Striatum, Symptoms
Introduction
Huntington's disease (HD) is a neurodegenerative disorder caused by the mutation of a gene inducing expansion of the polyglutamine tract on the huntingtin (htt) protein [1]. Patients carrying the mutation display motor dysfunction, manifested as chorea in early stages, then as akinesia and sometimes dystonia in later stages. Other symptoms include depression, anxiety, irritability or aggressive behavior and apathy that typically precede the onset of motor abnormalities by many years and can be more devastating than movement disorders [2]. Brain MRIs indicate that abnormalities occur in the absence of overt motor signs in both cortical and subcortical regions and especially in the basal ganglia [3].
Although early studies stressed neuronal loss and degeneration, it remains unclear whether cell loss is “necessary” or if cell dysfunction alone or in combination with degeneration can lead to many of the symptoms in HD. We have proposed that a progressive disconnection between cortical and subcortical structures occurs as the disease becomes more severe [4]. This disconnection probably occurs because of abnormal neurotransmission due to imbalances among excitatory and inhibitory neurotransmitters like glutamate and gamma‐amino butyric acid (GABA), in both cortex and subcortical structures. This disconnection is especially apparent between the prefrontal cortex (PFC) and the striatum, interrupting the flow of information arising from the cerebral cortex to the basal ganglia. We also proposed that many of the symptoms of HD are associated with alterations in dopamine (DA) function and DA modulation of excitation and inhibition in cortex and basal ganglia nuclei. In this review, we will summarize findings concerning alterations in cortical and striatal glutamate and DA neurotransmission in HD, and in animal models of HD. We will show how these neurotransmitter systems interact and how their dysfunctions contribute to the progression of the disorder.
Animal Models of HD
Excitotoxic Lesions of the Striatum
Intrastriatal Injection of Glutamate Analogs
Testing the hypothesis that lesions of the striatum were responsible for HD, the first excitotoxic models examined the effects of intrastriatal injection of kainic acid in rats. These injections caused neurochemical, histological, and behavioral changes similar to those present in HD patients [5]. Lesions of the striatum can be induced by using other glutamate analogs like ibotenic acid, quisqualic acid, N‐methyl‐d‐aspartate (NMDA), and quinolinic acid [6, 7]. Use of quinolinic acid became a standard early excitotoxic HD model because this molecule was a powerful agonist of NMDA receptors [6] and led to the hypothesis that NMDA receptor function was abnormal in HD.
Alteration of Mitochondrial Function
The HD mutation alters mitochondrial function which in turn activates apoptotic pathways. The first mitochondrial HD models studied chronic blockade of succinate oxidation by systemic administration of the mitochondrial toxin 3‐nitro‐propionic acid (3NP). This model also replicated most of the clinical and pathophysiological hallmarks of HD, including spontaneous choreiform and dystonic movements, frontal‐type cognitive deficits, and progressive striatal neuronal degeneration [8].
Genetic Models
Homozygotic knock‐out (KO) of the mouse HD gene HDh is embryonically lethal in mice and mice heterozygous for HDh inactivation do not reproduce HD symptoms [9, 10] suggesting that HD is not caused by a simple loss of function. Therefore, transgenic mice with glutamine expansion in the coding region of the HD gene were created. Other genetic models include knock‐in and conditional models. Many reviews of the available HD mouse models have been published [11, 12, 13]. We therefore will provide only a short description of the mouse models referenced in this review. All transgenic mice with polyglutamine expansion in the HD gene present symptoms and neuropathology resembling the human condition, that change with the progression of the disease. However, depending on the length of repeats and/or on the length of htt, the progression of the neurological phenotype differs. For each model it is therefore important to determine behavioral and pathological changes corresponding to the early or late stages of the disease.
Transgenic Mouse Models
Studies in transgenic models have indicated that truncated portions of htt are more toxic than the full‐length protein. The R6 transgenic lines contain exon 1 of the human HD gene that generates a short N‐terminal fragment of htt [14]. R6/2 mice manifest a rapidly progressing form of HD while R6/1 mice display a more protracted time course. Onset ages of overt behavioral changes are approximately 4–5 weeks in R6/2 and 4–5 months in R6/1 mice. Reductions in brain weight are also present at 4–5 weeks, whereas decreased neostriatal volume and striatal neuron area, with a reduction in striatal and cortical neuron number, appear later, at 13 weeks in R6/2 mice [15, 16]. The mice display progressive weight loss and R6/2 mice typically die between 12 and 16 weeks of age while R6/1 mice survive for a longer time. The movement disorder includes irregular gait, stereotypic grooming movements, tremor and both hindlimb and forelimb clasping. N171‐82Q is another mouse model of HD with a N‐terminal truncation of htt that also shows rapid and aggressive progression of the disease [17]. It has been suggested that R6/2 mice recapitulate the juvenile form of the disease, questioning its validity as a model of adult HD. However, there are numerous commonalities between juvenile and adult‐onset HD that make the R6/2 a very reasonable model for both types.
HD46 mice and HD100 mice express an extended N‐terminal one‐third of htt with either 46 or 100 CAG repeats [18]. Both lines show neurological impairment and motor deficits starting between 3 and 10 months with severity increasing with age. In these mice striatal inclusions preceded the onset of the behavioral phenotype, whereas cortical changes, like the accumulation of htt in the nucleus and cytoplasm and the appearance of dysmorphic dendrites, predicted the onset and severity of the behavioral deficits.
Full‐length models have used yeast (YAC) or bacterial artificial chromosomes (BAC). YAC mice with 72 or 128 CAG repeats show a biphasic motor phenotype with hyperactivity at 3 months followed by hypoactivity in the open field at 12 months [19]. Modest striatal atrophy occurs at 9 months with both striatal and cortical atrophy at 12 months and loss of striatal neurons along with a decrease in striatal neuron area is apparent at 12 months. This suggests that symptoms appear well before the loss of striatal or cortical neurons. The BACHD model expresses full‐length human mutant htt with 97 CAG repeats. BACHD mice exhibit progressive motor deficits and neuronal synaptic dysfunction at 6 months, followed by late‐onset selective neuropathology at 12 months, which includes significant cortical and striatal atrophy and striatal dark neuron degeneration [20].
Knock‐in Mouse Models
CAG knock‐in lines express mutant htt with 50–150 glutamines under the endogenous mouse promoter in a manner comparable to the expression of mutant htt in HD patients [21]. Early abnormalities are mild, resembling the presymptomatic stages of the disease in which subtle motor anomalies occur. Knock‐in mice with 94 and 140 CAG repeats display biphasic changes in motor behavior, characterized by increased motor activity at 2 months of age, followed by hypoactivity at 4 months [22, 23]. A progressive gliosis and decreases in DARPP32 staining start at 12 months while a loss of striatal neurons is present at 2 years.
Glutamate and DA Pathways
Cortex
Prefrontal and motor cortices have important roles in motor and cognitive functions. Therefore, impairment of sensorimotor integration in motor areas would interfere with motor programs and contribute to the movement disorders in HD. The PFC receives major glutamatergic inputs from thalamic nuclei and a DA input from the ventral tegmental area (VTA) [24, 25]. Pyramidal neurons in superficial cortical layers project collaterals to other pyramidal neurons, while deep layers pyramidal neurons project to subcortical structures like the dorsal striatum involved in motor control and cognition, as well as the nucleus accumbens involved in motivated and cognitive behaviors [26, 27]. Thus, the corticostriatal pathway provides information used in motor planning and execution [28] as well in the control of cognition and motivation. Its disconnection, or abnormal input to the striatum and other subcortical pathways will have widespread effects.
Striatum
Afferent information to the striatum is filtered, integrated and transmitted to two target regions, the globus pallidus (GP) and the substantia nigra pars reticulata (SNr) via two primary output pathways originating from different subpopulations of medium‐sized spiny neurons (MSNs). MSNs comprise over 90% of striatal cells in rodents. The remainder of striatal neurons are different classes of interneurons. The two primary striatal output pathways are termed the direct and indirect pathways because of their connections. Direct pathway MSNs project preferentially to SNr and to the internal GP (GPi), while indirect pathway MSNs project to the external GP (GPe) which connects to the subthalamic nucleus (STN). STN projects to GPi and SNr [29, 30]. Direct and indirect pathway MSNs can be differentiated by the proteins they express. Direct pathway MSNs preferentially express DA D1‐like receptors, substance P (SP) and dynorphin while indirect pathway MSNs preferentially express D2‐like receptors and met‐enkephalin [31]. MSNs receive three main inputs: glutamate inputs from the cortex and the thalamus, GABA inputs from striatal interneurons and to a lesser extent from other MSNs, and DA inputs from substantia nigra pars compacta (SNc). In addition, MSNs receive intrastriatal cholinergic inputs from the large cholinergic interneurons.
Interestingly, MSNs expressing enkephalin and projecting to the GPe forming the indirect pathway appear to be particularly vulnerable in HD and markers such as enkephalin are lost in postmortem brains of fully symptomatic patients, but also in early symptomatic and presymptomatic brains [32, 33, 34]. In contrast, MSNs expressing SP, projecting to the GPi are relatively spared, although the SP‐containing projections to the SNr are more severely affected than the SP‐containing projections to the GPi and SNc. These results are consistent with the hypothesis that chorea results from preferential dysfunction and ultimate loss of indirect pathway MSNs and possibly that akinesia and dystonia in HD is a consequence of the additional dysfunction and loss of direct pathway MSNs [35].
DA Inputs
DA is a major neuromodulator in the striatum as well as other neural areas and is involved in numerous functions, including motor activity, cognition, motivation, emotion, food intake, to name a few. The DA systems responsible for these functions have received much attention because their dysfunctions are involved in the etiology of numerous pathological conditions. Three major DA pathways innervate the forebrain and basal ganglia. The nigrostriatal DA pathway arises from the SNc and projects to the striatum. DA released from this pathway modulates activity of the direct and indirect striatal output pathways facilitating movement or inhibiting unwanted movement [36]. The mesocortical DA pathway arises from the VTA, projects to cingulate, entorhinal, and PFCs and plays a critical role in normal cognitive processes and neuropsychiatric pathologies [37]. The mesolimbic DA pathway arises from the VTA and projects to the limbic striatum (nucleus accumbens) and olfactory tubercle [38]. The mesolimbic DA system is part of the reward circuit involved in addiction and depression [39].
Roles of DA and Glutamate
DA Exerts Opposite Actions on Glutamate Ionotropic Receptors
DA exerts its actions through activation of multiple receptors. D1 and D2 receptors were initially identified [40, 41], followed by the D3 [42], the D4 [43], and the D5 [44] receptors. Based on their transduction mechanism and pharmacological properties, the five DA receptors are classified into D1‐like, encompassing D1 and D5 receptors, and D2‐like, encompassing D2, D3, and D4 receptors. In the striatum, DA modulates ionotropic glutamate receptors by altering glutamate currents and by modifying glutamate receptor surface localization. Postsynaptically, it is widely recognized that activation of D1‐like receptors increases striatal NMDA and α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid (AMPA) currents while activation of D2‐like receptors decrease them [45]. Activation of D1 receptors increases NMDA and AMPA receptor function through different signaling pathways. Among them, cAMP formation, NMDA receptor subunit phosphorylation and activation of voltage‐gated Ca2+ channels [46, 47, 48, 49, 50, 51, 52].
In the PFC, the D1 receptor family outnumbers the D2 family, and data show that signaling via the D1 receptor subtype plays a prominent role in modulating neuronal activity underlying working memory [53, 54]. In the cortex, DAs effects on glutamate currents are contradictory. Our group and others found that D1 receptor family activation facilitates postsynaptic glutamate (AMPA and NMDA) currents while D2 receptor family activation decreases these currents in rodents and in human cortex [55, 56, 57, 58, 59]. In contrast, other groups reported that DA or activation of D1 receptors decreased non‐NMDA excitatory postsynaptic currents [60, 61]. However, glutamate release probability is also modulated by DA and D1 agonists [60, 62] suggesting that DA could also modulate glutamate release through activation of presynaptic D1 receptors and this action could account for the contradictory findings.
DA Regulation of Glutamate and DA Release
The effects of DA on glutamate release are also contradictory, due to the complexity of the circuitry and the multiple neurotransmitters and modulators involved in controlling release. In the dorsal striatum, while D2 receptors are located on corticostriatal terminals and postsynaptically on MSN cell somata [63, 64], D1 receptors are preferentially located postsynaptically on MSN cell somata. Therefore, DA can modulate glutamate release from corticostriatal terminals directly by activating D2 receptors [65, 66]. In addition to DA's effect decreasing glutamate release through activation of D2 receptors on corticostriatal terminals, we and others also observed that DA can increase glutamate release through a combination of pre‐ and postsynaptic mechanisms [67, 68]. Glutamate release by corticostriatal and thalamostriatal terminals is regulated by other receptors located on presynaptic glutamatergic terminals. The metabotropic glutamate (mGlu) receptors, GABAB, serotonin, adenosine, endocannabinoids (CB1), and vanilloid (TRPV1) receptors can all modulate striatal glutamate transmission [69, 70]. These receptors ultimately may become targets to alleviate abnormal glutamate transmission in HD (see section on therapy).
Striatal DA is released by nigrostriatal terminals onto MSNs, corticostriatal terminals, and interneurons. D2 receptors are located on nigrostriatal DA neurons and act as autoreceptors, decreasing DA release when activated [71]. Initial studies showed that neuroleptics (D2 family receptor antagonists) increase whereas apomorphine (a DA receptor agonist) decreases evoked release of DA [72]. DA release is also influenced by TRPV1 receptors located on nigrostriatal terminals [73].
In the cortex, less is known about DA regulation of glutamate and DA release. In contrast to the striatum, cortical glutamate terminals contain D1 receptors and studies examining activation of presynaptic D1 receptors have yielded contradictory outcomes [60, 74]. Similar to the striatum, it is possible that DA regulation of glutamate release in the cortex depends on a combination of pre‐ and postsynaptic DA receptors as well as on other modulators. For example, recent studies report that cortical glutamate inputs are also modulated by CB1 via presynaptic CB1 receptors that decrease glutamate release when activated [75]. Cortical DA release is also modulated by presynaptic D2 receptors [76, 77].
Glutamate in HD
Symptomatic HD
Studies of glutamate in humans with HD have revealed a variety of alterations. While some studies report selective reductions of NMDA and AMPA receptors in patients in both caudate and cortex [78, 79], others found a nonsignificant loss of excitatory amino acid receptors in the caudate and no change in the cortex [80]. However, reduction of glutamate uptake was found in the PFC of HD patients, suggesting that impairment of glutamate uptake may also contribute to neuronal dysfunction and degeneration in HD [81]. In this study, the level of synaptic and astrocytic markers were unchanged suggesting that the loss of glutamate transporters was not due to neurodegeneration. In animal models, mostly decreases of striatal glutamate receptors and release have been reported. Studies in symptomatic animals show decreases of striatal NMDA mRNA in 8–12 week R6/2 mice and decreases of AMPA receptors in 12‐week R6/2 mice [82, 83]. In these studies, there was a loss of cortical glutamate receptors that occurred later than in the striatum. Basal striatal glutamate levels were also reduced by 43% in R6/1 mice at 16 weeks [84]. Li et al. report that decreases in [3H]glutamate release correlated with the increase in neuropil aggregates [85]. They observed that axons containing mutant htt aggregates had fewer synaptic vesicles and proposed that mutant htt binds tightly to synaptic vesicle membranes, inhibiting uptake and release of glutamate.
Presymptomatic HD
In contrast to studies reporting decreased striatal glutamate currents, we found that AMPA and NMDA currents were increased in presymptomatic R6/2, HD100, and YAC128 mice. These increases might have been overlooked because only subpopulation of cells demonstrated larger glutamate currents [86, 87, 88]. In addition, increases were transient, observed only in early stages while in later stages, NMDA and AMPA currents were reduced. Glutamate vulnerability also might depend on the receptor subunit composition. Expression of mutant htt has been shown to enhance currents and excitotoxicity mediated by predominantly NR2B‐type NMDA receptors suggesting that interaction between NR2B and mutant htt may contribute to selective neuronal dysfunction and degeneration [89]. Interestingly, endogenous quinolinic acid levels were elevated in the cortex and striatum of several mouse models showing neurodegeneration and motor and cognitive dysfunction, supporting the hypothesis that glutamate analogs play a role in neuropathology in HD [90]. Another study has shown decreased mRNA levels of the astroglial glutamate transporter in the striatum and cortex of R6 mice, accompanied by a concomitant decrease in glutamate uptake that could lead to neuronal damage [91]. In addition to increased glutamate receptors in presymptomatic animals, we found that glutamate neurotransmission was specifically increased in MSNs of the direct pathway in 50‐day old YAC128 [92]. Overactivation of direct pathway MSNs could lead to excessive inhibition of GPi and SNr neurons, to disinhibition of the thalamus and to abnormal movement [93]. In contrast, excessive glutamate could result in dysfunction and degeneration of the more vulnerable enkephalin‐containing MSNs forming the indirect pathway, which would disinhibit GPe, inhibit the STN and GPi and also induce abnormal movements (Figure 1). In support of this hypothesis, increased firing rate in GPe and decreased firing rates in GPi were found in an HD patient [94]. These findings may explain why deep brain stimulation of the GPi can improve abnormal movements in some HD patients [95, 96, 97].
Figure 1.
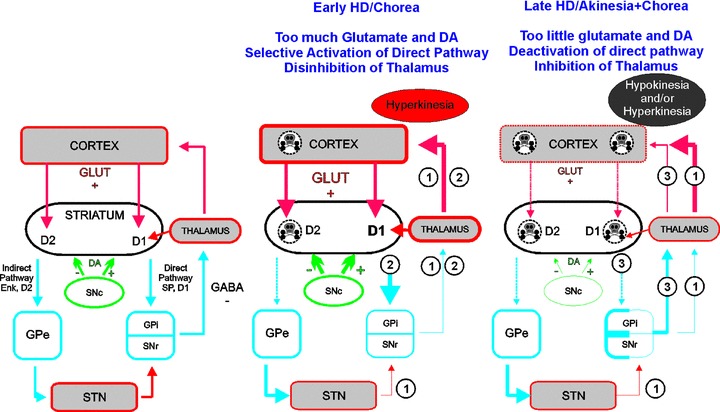
Glutamate and DA in basal ganglia function in intact brain and during HD. Left is a simplified schematic showing normal basal ganglia circuitry. Glutamate inputs are represented in red, GABA in blue and DA in green. Under normal conditions (left), the thalamus is inhibited by GPi and SNr GABA projections. In early HD (middle), cortical dysfunction induces excessive glutamate release in the striatum. Abnormally elevated striatal glutamate and DA levels trigger selective dysfunction of enkephalin‐containing MSNs. Imbalance between the direct and indirect pathways induces hyperkinesia via two pathways: (1) Selective dysfunction/degeneration (dashed lines) of enkephalin‐containing MSNs leads to decreased release of GABA in the GPe and to its disinhibition. In turn, overactivation of GABA neurons of the GPe leads to increased release of GABA and to inhibition of STN which decreases glutamate release and decreases activity of the GPi and SNr. (2) Overactivation of the direct pathway MSNs (by abnormal DA modulation and/or excessive glutamate) leads to increased release of GABA and to inhibition of GPi and SNr. Together, alterations in direct and indirect pathways in early HD induce inhibition of GPi and SNr GABA neurons and to a decreased release of GABA in their output structure, the thalamus. Disinhibition of the thalamus is responsible for abnormal movements. In late HD (right), corticostriatal and nigrostriatal inputs progressively degenerate, leading to decreased striatal glutamate and DA release. Low striatal glutamate and DA levels trigger dysfunction of both direct and indirect pathway MSNs. Imbalance between the direct and indirect pathways induces hyperkinesia and hypokinesia via two pathways: (1) Alterations in the indirect pathway are similar to early HD and lead to hyperkinetic movements. (3) Dysfunction/degeneration of direct pathway MSNs induces decreased release of GABA and disinhibition of GPi and SNr. Increased activity in GPi and SNr leads to inhibition of the thalamus and hypokinesia. Depending on the stage of dysfunction of direct and indirect pathway MSNs, activity in the basal ganglia could result in hypokinesia or hyperkinesia and would explain why some symptomatic patients display both chorea and akinesia. Enk = enkephalin; GLUT = glutamate; GPe = external segment of the globus pallidus; GPi = internal segment of the globus pallidus; SNc = substantia nigra pars compacta; SNr = substantia nigra pars reticulata; STN = subthalamic nucleus.
Cortical Glutamate Changes
In contrast to the striatum, cortical NMDA and AMPA receptor‐induced currents were decreased in presymptomatic R6/2 mice and were unchanged in symptomatic R6/2 mice compared to controls [82, 98]. Decreased postsynaptic glutamate currents in pyramidal neurons could account for decreased vulnerability in cortical structures compared to the striatum. However, in symptomatic R6/2, YAC128 and CAG140 knock‐in mice, glutamate synaptic neurotransmission was increased and pyramidal neurons showed signs of hyperexcitability [99] suggesting that glutamate inputs are increased in cortex in models of HD while striatal glutamate inputs appear to be decreased [100]. The differential changes in glutamate currents observed in the striatum and cortex suggest that alterations induced by mutant htt are not solely cell dependent, but also depend on more complex interactions such as cell type, inputs and circuitry.
DA Receptors and DA Release in HD—Correlation with Neuropathology
Too Much or Too Little DA?
In patients with HD, the guiding hypothesis is that presynaptic activation of the nigrostriatal DA pathway induces chorea while loss of DA inputs induces akinesia [101, 102]. While there is no indication of an absolute increase in DA activity in chorea, relative overactivity in the nigrostriatal system might arise from a deficiency in GABA which normally inhibits DA release by activating GABA receptors on nigrostriatal cell somata and terminals [103, 104]. It is possible that an initial increase of DA release or abnormal extracellular levels of DA (along with elevated extracellular glutamate levels) induces excitotoxicity, loss of cortical and striatal neurons, and loss of DA terminals that could then lead to later symptoms such as akinesia or dystonia (Figure 1). However, it is not clear that DA levels are elevated in HD and bradykinesia (probably caused by low levels of DA in basal ganglia) is also observed in patients with chorea. In addition, some HD patients show dyskinesia that appears similar to l‐DOPA induced dyskinesia in Parkinson's disease patients (a result of too much DA or overactivation of DA receptors). Initial studies found that DA levels and activity of TH, the biosynthetic enzyme for DA were increased in the striatum of postmortem HD brains [102]. Also, binding for the presynaptic DA transporter was reduced in the caudate of HD patients [105, 106]. Although this could be interpreted as a loss of nigrostriatal terminals, another interpretation is that nigrostriatal DA transmission is altered, leading to choreatic movements (Figure 1) [101]. The neurochemical basis for this suggestion is based on the observations that antagonists of DA receptors and DA‐depleting agents reduce chorea, and that l‐DOPA exacerbates chorea in HD.
More recent imaging studies provide evidence supporting reduced DA function in HD patients. In presymptomatic and symptomatic patients, positron emission tomography for D1 and D2 ligands found that both striatal D1 and D2 receptor levels were reduced by 45–50% and that the loss progressed by 3–5% per year [107, 108]. Decreases in DA receptors also correlated with the duration of the disease, and with a decrease of GABA content, reflecting a loss neurons in the putamen [106].
Animal Studies
In contrast to human studies, in most animal models of HD, striatal DA receptors, DA release, and nigrostriatal terminals are reduced. The R6/2 mouse model displays decreases in D1 and D2 receptor binding at 6 weeks [14, 82]. Functionally, D1‐dependent modulation of AMPA currents was decreased in R6/2 mice at 6 weeks [109]. D1 receptor‐dependent striatal long‐term potentiation was also decreased in R6/2 mice compared to WT littermates [110]. Microdialysis studies showed that DA release was progressively reduced in R6/2 mice between 6 and 14 weeks [111, 112]. In the 3NP model of HD, striatal DA content was unchanged but evoked DA release was decreased [113]. Although DA receptor density was unchanged in 12‐month old YAC128 mice [114], this may represent a compensatory mechanism for decreased dopamine availability because the number of striatal neurons is reduced by 15% in this age group [19]. Our studies showed that in young YAC128 mice (1 month), D2 receptors are functional, but become much less effective at 12 months [115]. Interestingly, YAC128 mice exhibit initial hyperactivity, followed by the onset of a motor deficit and finally hypokinesis correlated with striatal neuronal loss [19]. In other models where lesions are more likely to be confined to the striatum, such as the quinolinic acid model, and the 3NP model, hyperkinesia occurred [116, 117]. It becomes important to determine loss of DA function relative to the general neuropathology in order to understand the role of DA in HD.
Glutamate, DA, and Glutamate‐DA Excitotoxicity
The excitotoxicity hypothesis of HD suggests that neurodegeneration in the striatum is caused by an excess of excitatory neurotransmitters such as glutamate or by overactivation of glutamate receptors, more probably the NMDA receptor. As pointed out earlier, this hypothesis was based on the observation that intrastriatal injections of glutamate receptor agonists induce neuronal loss and reproduce the symptoms similar to those seen in HD [6, 118]. In vitro, striatal and cortical neurons of HD mutant mice are more susceptible to NMDA‐induced cell death than control mice [86]. In mutant htt‐expressing neurons, elevated glutamate toxicity can be induced through elevation of intracellular Ca2+ concentrations and through proteolysis of caspases involved in apoptotic cascades [119, 120, 121].
High doses of DA also are able to directly induce cell death of striatal neurons in vitro[122, 123]. In vivo, striatal injection of DA induces loss of tyrosine hydroxylase (TH) immunoreactivity, a marker for DA terminals [124]. Increased DA release by methamphetamine has also been shown to be neurotoxic to nigrostriatal and mesolimbic projections [125]. DA can induce cell death through several mechanisms. The molecule itself, via receptor‐dependent mechanisms binds to D1 (or D2) receptors, which increases (or decreases) intracellular Ca2+ and activates (or inactivates) cascades leading to cell death [126]. DA also generates toxic metabolites via products of its metabolism or via autoxidation. Possible mechanisms include the generation of radical oxygen species and interactions with excitatory amino acids [127]. Another possibility is that DA plays a role in preferential vulnerability through interactions with mitochondria. Primary cultures of striatal neurons expressing mutant htt are vulnerable to DA, and this is correlated with a reduction in the level of complex II activity [128]. Interestingly, the same study found that the DA‐induced down‐regulation of complex II was mediated by D2 receptors. However, several lines of evidence suggest that DA toxicity is induced in part by activation of D1 receptors. D1 antagonists can limit cell death in cortical and striatal neuronal primary cultures incubated with DA [123]. In HD, NMDA receptors specifically potentiate the vulnerability of mutant htt cells (STHdhQ111) to DA toxicity as pretreatment with NMDA increased D1R‐induced cell death in mutant but not wild‐type cells [129].
Thus, abnormal DA/glutamate interactions could explain why the striatum and cortex are vulnerable in HD. Tang et al. showed that in primary MSN cultures from WT and YAC128 mice, DA in the presence of glutamate induced apoptosis in YAC128 cells and this effect was produced via glutamate‐induced Ca2+ signaling [130]. In vivo, they showed that MSN loss in 11‐month old YAC128 mice was increased by administration of l‐DOPA and reversed by tetrabenazine (TBZ), a monoamine depleting agent. Therefore, modifying glutamate or DA release modifies HD neuropathology. In agreement, hyperdopaminergia induced by knocking out the DA transporter (DAT) exacerbated mutant htt aggregates in the striatum and cortex of HdhQ92/Q92 mice as well as potentiating the emergence of behavioral symptoms [131]. In contrast, decreasing striatal DA or glutamate content by SN lesions or by decortication improved the neuropathology and behavioral performance in R6/2 mice [132].
Although the main interest of this review is about glutamate and DA, it is important to keep in mind that GABA is also a key player in the striatum and the cortex. There are important changes in the GABA system that are likely to play a role in the occurrence of symptoms and progression of HD. It has long been known that GABA content is decreased in the striatum of HD patients [102], due to the loss of MSNs. However, a recent study reported that GABAA and GABAB receptors are increased in output nuclei at all stages of the disease in HD patients, suggesting compensatory mechanisms to the loss of GABA terminals [133]. Our studies in animal models showing that striatal GABA currents are increased in HD also suggest compensatory mechanisms of the GABA system [134] that might be targeted for potential therapies.
Therapies
With the discovery of the gene in 1993 [1] and the ability to genetically test individuals at risk, various neuroprotective strategies designed to delay the progression of the disease have been examined. Drugs targeting glutamate and DA systems include typical and atypical neuroleptics, DA depleters, antidepressants, and antiglutamatergic drugs (for reviews, see [135, 136, 137]).
Targeting the DA System
TBZ causes depletion of brain DA, norepinephrine, and serotonin by binding to the presynaptic vesicular monoamine transporter, VMAT2. TBZ is the only FDA drug approved for treatment in HD and improves motor function in patients and in animals [130, 138, 139]. However, it can also produce side effects including depression and sleepiness in some patients, and has no effect on other items (total functional capacity, independence and behavioral scales, swallowing, speech and gait items of motor scale) of the Unified Huntington's Disease Rating Scale (UHDRS) (for review, see [137]). Beside its effect on striatal DA content, TBZ also has an effect on other monoamines [140] that could explain some of its side effects on cognition and depression. Although the trials assessing other anti‐dopaminergic agents targeting D2 (sulpiride, D2 receptor antagonist) and D4 receptors (clozapine, D4 receptor antagonist) were smaller, they report those two agents either had no effect, or improved chorea severity, but had too many adverse effects [141].
Since blocking DA release induces major side effects and may exacerbate psychiatric symptoms, more specific DA blockers have been investigated. Charvin et al. showed that early pharmacological blockade of D2 receptors by haloperidol attenuated aggregate formation and the loss of DARPP‐32 in a lentivirus‐based HD rat model, reinforcing the potential role of D2 antagonists in neuroprotection in HD [142]. Typical neuroleptics or typical antipsychotics also used in schizophrenia have a high affinity for D2 DA receptors and are used for the treatment of psychotic symptoms as well as chorea in HD. However, their potential side‐effects include parkinsonism, dystonias, tardive dyskinesia, and worsening of cognitive and affective symptoms, which can sometimes limit their therapeutic use. Haloperidol, tiapride, and sulpiride were generally well tolerated at low doses, but did not always reduce involuntary movements [135]. Clozapine is one of the only antipsychotics evaluated in clinical trials, and several studies show that the occurrence of adverse effects prevented an increase to a dose that might have produced a more potent antichoreic effect [143, 144]. Other antipsychotics like olanzapine, risperidone, and ziprasidone were reported to improve some motor and psychiatric symptoms in HD.
Targeting the Glutamate System
Antiglutamatergic Drugs
Antiglutamatergic drugs (receptor antagonists or release blockers), such as amantadine, remacemide, riluzole, and ketamine have been tested in clinical trials. Although some of these drugs improved chorea and motor scores, most also had adverse effects. Amantadine proved to have beneficial effects on the choreic dyskinesias in some patients [145, 146]. Contrasting results were found in other trials that reported no improvement in the UHDRS chorea scores [147] suggesting that amantadine may not be a potent antichorea medication at a dose of less than 400 mg/day [135]. Remacemide, a nonselective NMDA receptor antagonist, showed a trend toward improvement in chorea but did not impact functional decline and induced adverse effects such as psychiatric disturbances, gastrointestinal upset, and dizziness [148, 149]. Riluzole, an inhibitor of glutamate release had no significant effect on chorea, behavioral, cognitive, independence, and functional scores in a 3‐year study in Europe [150]. The NMDA blocker ketamine induced deteriorations in the total motor score and eye movements measured by the UHDRS [137].
Ampakines
Ampakines are benzamide drugs that increase excitatory transmission in the forebrain via allosteric modulation of AMPA‐type glutamate receptors [151]. There is evidence that in vivo, ampakine treatment reduces neuronal death in animal models of Parkinson's disease and reduces excitotoxic brain damage [152, 153]. In the CAG140 model of HD, the ampakine CX929 rescued long‐term potentiation and reduced learning dysfunction in 8 and 16‐week old animals, while having no effect on locomotor activity [154] suggesting that, given that ampakines are well tolerated in clinical trials and are effective in this study after brief exposures, they could be used as a novel strategy for chronic treatment of the cognitive difficulties in the early stages of HD. One of the mechanisms by which ampakines might exert its neuroprotective effect is through the increase of brain derived neurotrophic factor that modulates MSN activity and has been shown to provide neuroprotection [155, 156].
Targeting Glutamate Release
Cannabinoids
Cannabinoid receptors CB1 modulate neurotransmitter release in virtually all brain regions [157, 158]. Cannabinoid ligands and receptors are particularly abundant in basal ganglia structures (especially nuclei that receive striatal efferents, like the GPi, GPe, and SNr) compared with other brain regions [159, 160]. The cannabinoid system plays a prominent role in basal ganglia function by modulating release of neurotransmitters that operate in the basal ganglia circuits [161, 162]. In addition, cannabinoids that activate CB1 receptors have an important influence on motor responses [163]. Activation of CB1 receptors decrease locomotion while antagonizing CB1 receptors causes hyperlocomotion [164, 165]. CB1 also bind to TRPV1 receptors located on DA terminals and can reduce hyperdopaminergia‐related hyperactivity [166]. CB1 may potentially be useful to reduce the effects of neurodegeneration [167]. They were shown to protect neurons via CB1 receptor‐mediated inhibition of glutamate exocytosis, closing of voltage sensitive calcium channels, and reducing antioxidant activity [168, 169]. Therefore, cannabinoid‐based treatment might be beneficial in basal ganglia disorders [170]. Although cannabinoid receptors are decreased in HD, CB1 could still have beneficial effects by decreasing glutamate release without inducing deleterious NMDA antagonist side effects. AM404, an inhibitor of the endocannabinoid reuptake process has been shown to reduce hyperlocomotion in an animal model of HD via activation of TRPV1 receptors [171]. Earlier studies reported no beneficial effect of cannabidiol in HD [172, 173]. However, much has to be discovered about endocannabinoid ligands and receptors, in both healthy and pathological conditions to determine the potential therapeutic benefits of targeting this system in HD or other basal ganglia disorders. Another potentially beneficial effect of CB1 could take place through activation of CB2 receptors present on microglia which would in turn decrease microglia activation. Indeed, activation of CB2 receptors has been demonstrated to decrease neuroinflammation and motor symptoms in R6/2 mice [174].
Adenosine
Indirect pathway MSNs selectively express high levels of the neuromodulator adenosine A2A subytpe of receptor [175]. A2A receptors activate the cAMP/PKA pathway as opposed to D2 receptors, which inhibit adenylyl cyclase and decrease cAMP [176]. Another consequence of A2A receptor activation is an increase in glutamate release via activation of A2A receptors located on presynaptic glutamate terminals [177]. Therefore, high concentrations of adenosine and overactivation of A2A receptors could lead to toxicity [178, 179]. A transient increase in A2A receptor density has been reported in presymptomatic (1–2 weeks) R6/2 mice while large reductions were found in later stages [180, 181], suggesting adenosine could indeed exacerbate excitotoxicity in early stages of HD.
It has been proposed that A2A receptor agonists could have beneficial effects in late HD, when A2A receptors are downregulated. One study found that A2A receptor agonists significantly attenuated the progressive deterioration of motor coordination and reduced brain atrophy [180]. However, another study showed that despite the neuroprotective effect on NMDA‐induced toxicity in symptomatic R6/2 mice, treatment with an A2A receptor agonist did not rescue the deficit in motor coordination and even worsened the performance of R6/2 mice [182]. It is possible that the motor effects of A2A receptor agonists depend on mechanisms independent from those involved in neuroprotection. Several lines of evidence suggest that blocking presynaptic A2A receptors can be beneficial because it decreases excitotoxicity while the blockade of the postsynaptic receptors can be deleterious [183]. Finally, since in HD brains, a dramatic loss of A2A receptor binding was observed in grade 0 cases (34–35% of controls) and more advanced cases showed no detectable A2A receptor binding [184], it remains unknown whether therapy targeting A2A receptors will be beneficial in HD patients.
Conclusions
It is now clear that neuronal dysfunction occurs in both striatum and cortex well before a clinical diagnosis of HD is made. Because the cortex and striatum are interconnected, it is difficult to determine if cortical changes precede or follow striatal changes. However, the fact that HD patients experience cognitive and emotional disturbances before the occurrence of sensorimotor symptoms provides evidence that cortical dysfunction might precede striatal pathology. Activation of both D1 and D2 receptors can lead to toxic changes in cortical and striatal neurons as well as their terminals. It is therefore probable that diverse and complex interactions among glutamate and DA neurotransmission will contribute to the selective dysfunction of neurons in the cortex and basal ganglia. Subsequent and progressive dysfunction and degeneration are likely to be responsible for the complex and heterogeneous clinical phenotype in HD. The complexity of mutant htt‐induced alterations throughout these systems renders the development of therapies difficult, and disease‐modifying therapies preventing the onset of clinical symptoms in individuals at risk are still not available. While existing antidopaminergic or antiglutamatergic drugs seem to have a positive effect on motor symptoms, their use can be limited due to the side‐effects they trigger. Therefore, it will be necessary to develop more specific drugs with selective targets. Other glutamate and DA release modulators such as adenosine or CB1 could have potential therapeutic benefits in HD.
Conflict of Interest
The authors have no conflict of interest.
References
- 1. The Huntington's Disease Collaborative Research Group . A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 1993;72:971–983. [DOI] [PubMed] [Google Scholar]
- 2. Bonelli RM, Hofmann P. A systematic review of the treatment studies in Huntington's disease since 1990. Expert Opin Pharmacother 2007;8:141–153. [DOI] [PubMed] [Google Scholar]
- 3. Rosas HD, Salat DH, Lee SY, Zaleta AK, Pappu V, Fischl B, Greve D. Cerebral cortex and the clinical expression of Huntington's disease: Complexity and heterogeneity. Brain 2008;131:1057–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cepeda C, Wu N, Andre VM, Cummings DM, Levine MS. The corticostriatal pathway in Huntington's disease. Prog Neurobiol 2007;81:253–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Coyle JT, Schwarcz R. Lesion of striatal neurones with kainic acid provides a model for Huntington's chorea. Nature 1976;263:244–246. [DOI] [PubMed] [Google Scholar]
- 6. Beal MF, Kowall NW, Ellison DW, Mazurek MF, Swartz KJ, Martin JB. Replication of the neurochemical characteristics of Huntington's disease by quinolinic acid. Nature 1986;321:168–171. [DOI] [PubMed] [Google Scholar]
- 7. McGeer EG, McGeer PL, Singh K. Kainate‐induced degeneration of neostriatal neurons: Dependency upon corticostriatal tract. Brain Res 1978;139:381–383. [DOI] [PubMed] [Google Scholar]
- 8. Brouillet E, Conde F, Beal MF, Hantraye P. Replicating Huntington's disease phenotype in experimental animals. Prog Neurobiol 1999;59:427–468. [DOI] [PubMed] [Google Scholar]
- 9. Nasir J, Floresco SB, O’Kusky JR, Diewert VM, Richman JM, Zeisler J, Borowski A. Targeted disruption of the Huntington's disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell 1995;81:811–823. [DOI] [PubMed] [Google Scholar]
- 10. Zeitlin S, Liu JP, Chapman DL, Papaioannou VE, Efstratiadis A. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington's disease gene homologue. Nat Genet 1995;11:155–163. [DOI] [PubMed] [Google Scholar]
- 11. Heng MY, Detloff PJ, Albin RL. Rodent genetic models of Huntington disease. Neurobiol Dis 2008;32:1–9. [DOI] [PubMed] [Google Scholar]
- 12. Levine MS, Cepeda C, Hickey MA, Fleming SM, Chesselet MF. Genetic mouse models of Huntington's and Parkinson's diseases: Illuminating but imperfect. Trends Neurosci 2004;27:691–697. [DOI] [PubMed] [Google Scholar]
- 13. Ramaswamy S, McBride JL, Kordower JH. Animal models of Huntington's disease. Ilar J 2007;48:356–373. [DOI] [PubMed] [Google Scholar]
- 14. Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 1996;87:493–506. [DOI] [PubMed] [Google Scholar]
- 15. Stack EC, Kubilus JK, Smith K, Cormier K, Del Signore SJ, Guelin E, Ryu H. Chronology of behavioral symptoms and neuropathological sequela in R6/2 Huntington's disease transgenic mice. J Comp Neurol 2005;490:354–370. [DOI] [PubMed] [Google Scholar]
- 16. Turmaine M, Raza A, Mahal A, Mangiarini L, Bates GP, Davies SW. Nonapoptotic neurodegeneration in a transgenic mouse model of Huntington's disease. Proc Natl Acad Sci U S A 2000;97:8093–8097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schilling G, Becher MW, Sharp AH, Jinnah HA, Duan K, Kotzuk JA, Slunt HH. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N‐terminal fragment of huntingtin. Hum Mol Genet 1999;8:397–407. [DOI] [PubMed] [Google Scholar]
- 18. Laforet GA, Sapp E, Chase K, McIntyre C, Boyce FM, Campbell M, Cadigan BA. Changes in cortical and striatal neurons predict behavioral and electrophysiological abnormalities in a transgenic murine model of Huntington's disease. J Neurosci 2001;21:9112–9123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Slow EJ, Van Raamsdonk J, Rogers D, Coleman SH, Graham RK, Deng Y, Oh R. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet 2003;12:1555–1567. [DOI] [PubMed] [Google Scholar]
- 20. Gray M, Shirasaki DI, Cepeda C, Andre VM, Wilburn B, Lu XH, Tao J. Full‐length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J Neurosci 2008;28:6182–6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Menalled LB. Knock‐in mouse models of Huntington's disease. NeuroRx 2005;2:465–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hickey MA, Kosmalska A, Enayati J, Cohen R, Zeitlin S, Levine MS, Chesselet MF. Extensive early motor and non‐motor behavioral deficits are followed by striatal neuronal loss in knock‐in Huntington's disease mice. Neuroscience 2008;157:280–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Menalled LB, Sison JD, Dragatsis I, Zeitlin S, Chesselet MF. Time course of early motor and neuropathological anomalies in a knock‐in mouse model of Huntington's disease with 140 CAG repeats. J Comp Neurol 2003;465:11–26. [DOI] [PubMed] [Google Scholar]
- 24. Bannon MJ, Wolf ME, Roth RH. Pharmacology of dopamine neurons innervating the prefrontal, cingulate and piriform cortices. Eur J Pharmacol 1983;92:119–125. [DOI] [PubMed] [Google Scholar]
- 25. Groenewegen HJ, Wright CI, Uylings HB. The anatomical relationships of the prefrontal cortex with limbic structures and the basal ganglia. J Psychopharmacol 1997;11:99–106. [DOI] [PubMed] [Google Scholar]
- 26. Carr DB, O’Donnell P, Card JP, Sesack SR. Dopamine terminals in the rat prefrontal cortex synapse on pyramidal cells that project to the nucleus accumbens. J Neurosci 1999;19:11049–11060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. O’Donnell P, Grace AA. Tonic D2‐mediated attenuation of cortical excitation in nucleus accumbens neurons recorded in vitro . Brain Res 1994;634:105–112. [DOI] [PubMed] [Google Scholar]
- 28. Graybiel AM, Aosaki T, Flaherty AW, Kimura M. The basal ganglia and adaptive motor control. Science 1994;265:1826–1831. [DOI] [PubMed] [Google Scholar]
- 29. Bolam JP, Hanley JJ, Booth PA, Bevan MD. Synaptic organisation of the basal ganglia. J Anat 2000;196(Pt 4):527–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gerfen CR. The neostriatal mosaic: Multiple levels of compartmental organization. Trends Neurosci 1992;15:133–139. [DOI] [PubMed] [Google Scholar]
- 31. Haber SN, Nauta WJ. Ramifications of the globus pallidus in the rat as indicated by patterns of immunohistochemistry. Neuroscience 1983;9:245–260. [DOI] [PubMed] [Google Scholar]
- 32. Albin RL, Reiner A, Anderson KD, et al Preferential loss of striato‐external pallidal projection neurons in presymptomatic Huntington's disease. Ann Neurol 1992;31:425–430. [DOI] [PubMed] [Google Scholar]
- 33. Reiner A, Albin RL, Anderson KD, D’Amato CJ, Penney JB, Young AB. Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci U S A 1988;85:5733–5737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Richfield EK, Maguire‐Zeiss KA, Vonkeman HE, Voorn P. Preferential loss of preproenkephalin versus preprotachykinin neurons from the striatum of Huntington's disease patients. Ann Neurol 1995;38:852–861. [DOI] [PubMed] [Google Scholar]
- 35. Albin RL, Reiner A, Anderson KD, Penney JB, Young AB. Striatal and nigral neuron subpopulations in rigid Huntington's disease: Implications for the functional anatomy of chorea and rigidity‐akinesia. Ann Neurol 1990;27:357–365. [DOI] [PubMed] [Google Scholar]
- 36. Crossman AR. Functional anatomy of movement disorders. J Anat 2000;196(Pt 4):519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tzschentke TM. Pharmacology and behavioral pharmacology of the mesocortical dopamine system. Prog Neurobiol 2001;63:241–320. [DOI] [PubMed] [Google Scholar]
- 38. Seamans JK, Yang CR. The principal features and mechanisms of dopamine modulation in the prefrontal cortex. Prog Neurobiol 2004;74:1–58. [DOI] [PubMed] [Google Scholar]
- 39. Niehaus JL, Cruz‐Bermudez ND, Kauer JA. Plasticity of addiction: A mesolimbic dopamine short‐circuit? Am J Addict 2009;18:259–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kebabian JW, Calne DB. Multiple receptors for dopamine. Nature 1979;277:93–96. [DOI] [PubMed] [Google Scholar]
- 41. Spano PF, Govoni S, Trabucchi M. Studies on the pharmacological properties of dopamine receptors in various areas of the central nervous system. Adv Biochem Psychopharmacol 1978;19:155–165. [PubMed] [Google Scholar]
- 42. Sokoloff P, Giros B, Martres MP, Bouthenet ML, Schwartz JC. Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature 1990;347:146–151. [DOI] [PubMed] [Google Scholar]
- 43. Van Tol HH, Bunzow JR, Guan HC, Sunahara RK, Seeman P, Niznik HB, Civelli O. Cloning of the gene for a human dopamine D4 receptor with high affinity for the antipsychotic clozapine. Nature 1991;350:610–614. [DOI] [PubMed] [Google Scholar]
- 44. Sunahara RK, Guan HC, O’Dowd BF, Seeman P, Laurier LG, Ng G, George SR. Cloning of the gene for a human dopamine D5 receptor with higher affinity for dopamine than D1. Nature 1991;350:614–619. [DOI] [PubMed] [Google Scholar]
- 45. Cepeda C, Buchwald NA, Levine MS. Neuromodulatory actions of dopamine in the neostriatum are dependent upon the excitatory amino acid receptor subtypes activated. Proc Natl Acad Sci U S A 1993;90:9576–9580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Blank T, Nijholt I, Teichert U, Kugler H, Behrsing H, Fienberg A, Greengard P. The phosphoprotein DARPP‐32 mediates cAMP‐dependent potentiation of striatal N‐methyl‐d‐aspartate responses. Proc Natl Acad Sci U S A 1997;94:14859–14864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cepeda C, Levine MS. Dopamine and N‐methyl‐d‐aspartate receptor interactions in the neostriatum. Dev Neurosci 1998;20:1–18. [DOI] [PubMed] [Google Scholar]
- 48. Dunah AW, Standaert DG. Dopamine D1 receptor‐dependent trafficking of striatal NMDA glutamate receptors to the postsynaptic membrane. J Neurosci 2001;21:5546–5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Flores‐Hernandez J, Cepeda C, Hernandez‐Echeagaray E, Calvert CR, Jokel ES, Fienberg AA, Greengard P. Dopamine enhancement of NMDA currents in dissociated medium‐sized striatal neurons: Role of D1 receptors and DARPP‐32. J Neurophysiol 2002;88:3010–3020. [DOI] [PubMed] [Google Scholar]
- 50. Liu JC, DeFazio RA, Espinosa‐Jeffrey A, Cepeda C, De Vellis J, Levine MS. Calcium modulates dopamine potentiation of N‐methyl‐d‐aspartate responses: Electrophysiological and imaging evidence. J Neurosci Res 2004;76:315–322. [DOI] [PubMed] [Google Scholar]
- 51. Price CJ, Kim P, Raymond LA. D1 dopamine receptor‐induced cyclic AMP‐dependent protein kinase phosphorylation and potentiation of striatal glutamate receptors. J Neurochem 1999;73:2441–2446. [DOI] [PubMed] [Google Scholar]
- 52. Snyder GL, Fienberg AA, Huganir RL, Greengard P. A dopamine/D1 receptor/protein kinase A/dopamine‐ and cAMP‐regulated phosphoprotein (Mr 32 kDa)/protein phosphatase‐1 pathway regulates dephosphorylation of the NMDA receptor. J Neurosci 1998;18:10297–10303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Castner SA, Williams GV. Tuning the engine of cognition: A focus on NMDA/D1 receptor interactions in prefrontal cortex. Brain Cogn 2007;63:94–122. [DOI] [PubMed] [Google Scholar]
- 54. Sawaguchi T, Goldman‐Rakic PS. D1 dopamine receptors in prefrontal cortex: Involvement in working memory. Science 1991;251:947–950. [DOI] [PubMed] [Google Scholar]
- 55. Cepeda C, Li Z, Cromwell HC, Altemus KL, Crawford CA, Nansen EA, Ariano MA. Electrophysiological and morphological analyses of cortical neurons obtained from children with catastrophic epilepsy: Dopamine receptor modulation of glutamatergic responses. Dev Neurosci 1999;21:223–235. [DOI] [PubMed] [Google Scholar]
- 56. Chen G, Greengard P, Yan Z. Potentiation of NMDA receptor currents by dopamine D1 receptors in prefrontal cortex. Proc Natl Acad Sci U S A 2004;101:2596–2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rubinstein M, Cepeda C, Hurst RS, Flores‐Hernandez J, Ariano MA, Falzone TL, Kozell LB. Dopamine D4 receptor‐deficient mice display cortical hyperexcitability. J Neurosci 2001;21:3756–3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tseng KY, O’Donnell P. Dopamine‐glutamate interactions controlling prefrontal cortical pyramidal cell excitability involve multiple signaling mechanisms. J Neurosci 2004;24:5131–5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zheng P, Zhang XX, Bunney BS, Shi WX. Opposite modulation of cortical N‐methyl‐d‐aspartate receptor‐mediated responses by low and high concentrations of dopamine. Neuroscience 1999;91:527–535. [DOI] [PubMed] [Google Scholar]
- 60. Seamans JK, Durstewitz D, Christie BR, Stevens CF, Sejnowski TJ. Dopamine D1/D5 receptor modulation of excitatory synaptic inputs to layer V prefrontal cortex neurons. Proc Natl Acad Sci U S A 2001;98:301–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Urban NN, Gonzalez‐Burgos G, Henze DA, Lewis DA, Barrionuevo G. Selective reduction by dopamine of excitatory synaptic inputs to pyramidal neurons in primate prefrontal cortex. J Physiol 2002;539:707–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gao WJ, Krimer LS, Goldman‐Rakic PS. Presynaptic regulation of recurrent excitation by D1 receptors in prefrontal circuits. Proc Natl Acad Sci U S A 2001;98:295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Dumartin B, Doudnikoff E, Gonon F, Bloch B. Differences in ultrastructural localization of dopaminergic D1 receptors between dorsal striatum and nucleus accumbens in the rat. Neurosci Lett 2007;419:273–277. [DOI] [PubMed] [Google Scholar]
- 64. Hersch SM, Ciliax BJ, Gutekunst CA, Rees HD, Heilman CJ, Yung KK, Bolam JO. Electron microscopic analysis of D1 and D2 dopamine receptor proteins in the dorsal striatum and their synaptic relationships with motor corticostriatal afferents. J Neurosci 1995;15:5222–5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bamford NS, Zhang H, Schmitz Y, Wu NP, Cepeda C, Levine MS, Schmauss C. Heterosynaptic dopamine neurotransmission selects sets of corticostriatal terminals. Neuron 2004;42:653–663. [DOI] [PubMed] [Google Scholar]
- 66. Flores‐Hernandez J, Galarraga E, Bargas J. Dopamine selects glutamatergic inputs to neostriatal neurons. Synapse 1997;25:185–195. [DOI] [PubMed] [Google Scholar]
- 67. Andre VM, Cepeda C, Cummings DM, Jocoy EL, Fisher YE, Yang XW, Levine MS. Dopamine modulation of excitatory currents in striatum is dictated by the expression of D1 or D2 receptors and modified by endocannabinoids. Eur J Neurosci 2010;31:14–28. [DOI] [PubMed] [Google Scholar]
- 68. Hernandez A, Sierra A, Valdiosera R, Floran B, Erlij D, Aceves J. Presynaptic D1 dopamine receptors facilitate glutamatergic neurotransmission in the rat globus pallidus. Neurosci Lett 2007;425:188–191. [DOI] [PubMed] [Google Scholar]
- 69. Lovinger DM, McCool BA. Metabotropic glutamate receptor‐mediated presynaptic depression at corticostriatal synapses involves mGLuR2 or 3. J Neurophysiol 1995;73:1076–1083. [DOI] [PubMed] [Google Scholar]
- 70. Nisenbaum ES, Berger TW, Grace AA. Presynaptic modulation by GABAB receptors of glutamatergic excitation and GABAergic inhibition of neostriatal neurons. J Neurophysiol 1992;67:477–481. [DOI] [PubMed] [Google Scholar]
- 71. Arbilla S, Langer SZ. Stereoselectivity of presynaptic autoreceptors modulating dopamine release. Eur J Pharmacol 1981;76:345–351. [DOI] [PubMed] [Google Scholar]
- 72. Farnebo LO, Hamberger B. Regulation of (3H)5‐hydroxytryptamine release from rat brain slices. J Pharm Pharmacol 1974;26:642–644. [DOI] [PubMed] [Google Scholar]
- 73. De Lago E, De Miguel R, Lastres‐Becker I, Ramos JA, Fernandez‐Ruiz J. Involvement of vanilloid‐like receptors in the effects of anandamide on motor behavior and nigrostriatal dopaminergic activity: In vivo and in vitro evidence. Brain Res 2004;1007:152–159. [DOI] [PubMed] [Google Scholar]
- 74. Wang Z, Feng XQ, Zheng P. Activation of presynaptic D1 dopamine receptors by dopamine increases the frequency of spontaneous excitatory postsynaptic currents through protein kinase A and protein kinase C in pyramidal cells of rat prelimbic cortex. Neuroscience 2002;112:499–508. [DOI] [PubMed] [Google Scholar]
- 75. Schlicker E, Kathmann M. Modulation of transmitter release via presynaptic cannabinoid receptors. Trends Pharmacol Sci 2001;22:565–572. [DOI] [PubMed] [Google Scholar]
- 76. Ohmori T, Koyama T, Yamashita I. Measurement of endogenous dopamine and norepinephrine release from superfused slices of rat prefrontal cortex in vitro: Modulation by D2 and alpha‐2 presynaptic receptors. Life Sci 1991;48:283–289. [DOI] [PubMed] [Google Scholar]
- 77. Schmitz Y, Benoit‐Marand M, Gonon F, Sulzer D. Presynaptic regulation of dopaminergic neurotransmission. J Neurochem 2003;87:273–289. [DOI] [PubMed] [Google Scholar]
- 78. Young AB, Greenamyre JT, Hollingsworth Z, Albin R, D’Amato C, Shoulson I, Penney JB. NMDA receptor losses in putamen from patients with Huntington's disease. Science 1988;241:981–983. [DOI] [PubMed] [Google Scholar]
- 79. Wagster MV, Hedreen JC, Peyser CE, Folstein SE, Ross CA. Selective loss of [3H]kainic acid and [3H]AMPA binding in layer VI of frontal cortex in Huntington's disease. Exp Neurol 1994;127:70–75. [DOI] [PubMed] [Google Scholar]
- 80. Dure LS IV, Young AB, Penney JB. Excitatory amino acid binding sites in the caudate nucleus and frontal cortex of Huntington's disease. Ann Neurol 1991;30:785–793. [DOI] [PubMed] [Google Scholar]
- 81. Hassel B, Tessler S, Faull RL, Emson PC. Glutamate uptake is reduced in prefrontal cortex in Huntington's disease. Neurochem Res 2008;33:232–237. [DOI] [PubMed] [Google Scholar]
- 82. Cha JH, Kosinski CM, Kerner JA, Alsdorf SA, Mangiarini L, Davies SW, Penney JB. Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human huntington disease gene. Proc Natl Acad Sci U S A 1998;95:6480–6485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Cha JH, Frey AS, Alsdorf SA, Kerner JA, Kosinski CM, Mangiarini L, Penney JB Jr., Altered neurotransmitter receptor expression in transgenic mouse models of Huntington's disease. Philos Trans R Soc Lond B Biol Sci 1999;354:981–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Nicniocaill B, Haraldsson B, Hansson O, O’Connor WT, Brundin P. Altered striatal amino acid neurotransmitter release monitored using microdialysis in R6/1 Huntington transgenic mice. Eur J Neurosci 2001;13:206–210. [DOI] [PubMed] [Google Scholar]
- 85. Li H, Wyman T, Yu ZX, Li SH, Li XJ. Abnormal association of mutant huntingtin with synaptic vesicles inhibits glutamate release. Hum Mol Genet 2003;12:2021–2030. [DOI] [PubMed] [Google Scholar]
- 86. Levine MS, Klapstein GJ, Koppel A, Gruen E, Cepeda C, Vargas ME, Jokel ES. Enhanced sensitivity to N‐methyl‐d‐aspartate receptor activation in transgenic and knockin mouse models of Huntington's disease. J Neurosci Res 1999;58:515–532. [PubMed] [Google Scholar]
- 87. Cepeda C, Ariano MA, Calvert CR, Flores‐Hernández J, Chandler SH, Leavitt BR, Hayden MR. NMDA receptor function in mouse models of Huntington disease. J Neurosci Res 2001;66:525–539. [DOI] [PubMed] [Google Scholar]
- 88. Starling AJ, Andre VM, Cepeda C, De Lima M, Chandler SH, Levine MS. Alterations in N‐methyl‐d‐aspartate receptor sensitivity and magnesium blockade occur early in development in the R6/2 mouse model of Huntington's disease. J Neurosci Res 2005;82:377–386. [DOI] [PubMed] [Google Scholar]
- 89. Chen N, Luo T, Wellington C, Metzler M, McCutcheon K, Hayden MR, Raymond LA. Subtype‐specific enhancement of NMDA receptor currents by mutant huntingtin. J Neurochem 1999;72:1890–1898. [DOI] [PubMed] [Google Scholar]
- 90. Guidetti P, Bates GP, Graham RK, Hayden MR, Leavitt BR, MacDonald ME, Slow EJ. Elevated brain 3‐hydroxykynurenine and quinolinate levels in Huntington disease mice. Neurobiol Dis 2006;23:190–197. [DOI] [PubMed] [Google Scholar]
- 91. Lievens JC, Woodman B, Mahal A, Spasic‐Boscovic O, Samuel D, Kerkerian‐Le Goff L, Bates JP. Impaired glutamate uptake in the R6 Huntington's disease transgenic mice. Neurobiol Dis 2001;8:807–821. [DOI] [PubMed] [Google Scholar]
- 92. Andre VM, Cummings DM, Cepeda C, Leavitt BR, Hayden MR, Levine MS. Glutamate inputs and dopaminergic modulation are differentially altered in D1 and D2 receptor‐expressing striatal neurons in the YAC128 mouse model of Huntington's disease. Soc Neurosc Abstr 2008;443.10. [Google Scholar]
- 93. Chevalier G, Deniau JM. Disinhibition as a basic process in the expression of striatal functions. Trends Neurosci 1990;13:277–280. [DOI] [PubMed] [Google Scholar]
- 94. Starr PA, Kang GA, Heath S, Shimamoto S, Turner RS. Pallidal neuronal discharge in Huntington's disease: Support for selective loss of striatal cells originating the indirect pathway. Exp Neurol 2008;211:227–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Biolsi B, Cif L, Fertit HE, Robles SG, Coubes P. Long‐term follow‐up of Huntington disease treated by bilateral deep brain stimulation of the internal globus pallidus. J Neurosurg 2008;109:130–132. [DOI] [PubMed] [Google Scholar]
- 96. Fasano A, Mazzone P, Piano C, Quaranta D, Soleti F, Bentivoglio AR. GPi‐DBS in Huntington's disease: Results on motor function and cognition in a 72‐year‐old case. Mov Disord 2008;23:1289–1292. [DOI] [PubMed] [Google Scholar]
- 97. Moro E, Lang AE, Strafella AP, Poon YY, Arango PM, Dagher A, Hutchison WR. Bilateral globus pallidus stimulation for Huntington's disease. Ann Neurol 2004;56:290–294. [DOI] [PubMed] [Google Scholar]
- 98. Andre VM, Cepeda C, Venegas A, Gomez Y, Levine MS. Altered cortical glutamate receptor function in the R6/2 model of Huntington's disease. J Neurophysiol 2006;95:2108–2119. [DOI] [PubMed] [Google Scholar]
- 99. Cummings DM, Andre VM, Uzgil BO, Gee SM, Fisher YE, Cepeda C, Levine MS. Alterations in cortical excitation and inhibition in genetic mouse models of Huntington's disease. J Neurosci 2009;29:10371–10386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Cepeda C, Hurst RS, Calvert CR, Hernández‐Echeagaray E, Nguyen OK, Jocoy E, Christian LJ. Transient and progressive electrophysiological alterations in the corticostriatal pathway in a mouse model of Huntington's disease. J Neurosci 2003;23:961–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Bird ED. Chemical pathology of Huntington's disease. Annu Rev Pharmacol Toxicol 1980;20:533–551. [DOI] [PubMed] [Google Scholar]
- 102. Spokes EGS. The neurochemistry of Huntington's chorea. TINS 1981;4:115–118. [Google Scholar]
- 103. Bolam JP, Smith Y. The GABA and substance P input to dopaminergic neurones in the substantia nigra of the rat. Brain Res 1990;529:57–78. [DOI] [PubMed] [Google Scholar]
- 104. Paladini CA, Tepper JM. GABA(A) and GABA(B) antagonists differentially affect the firing pattern of substantia nigra dopaminergic neurons in vivo . Synapse 1999;32:165–176. [DOI] [PubMed] [Google Scholar]
- 105. Backman L, Robins‐Wahlin TB, Lundin A, Ginovart N, Farde L. Cognitive deficits in Huntington's disease are predicted by dopaminergic PET markers and brain volumes. Brain 1997;120(Pt 12):2207–2217. [DOI] [PubMed] [Google Scholar]
- 106. Ginovart N, Lundin A, Farde L, Halldin C, Backman L, Swahn CG, Pauli S. PET study of the pre‐ and post‐synaptic dopaminergic markers for the neurodegenerative process in Huntington's disease. Brain 1997;120(Pt 3):503–514. [DOI] [PubMed] [Google Scholar]
- 107. Andrews TC, Weeks RA, Turjanski N, Gunn RN, Watkins LH, Sahakian B, Hoddges JR. Huntington's disease progression. PET and clinical observations. Brain 1999;122(Pt 12):2353–2363. [DOI] [PubMed] [Google Scholar]
- 108. Van Oostrom JC, Dekker M, Willemsen AT, De Jong BM, Roos RA, Leenders KL. Changes in striatal dopamine D2 receptor binding in pre‐clinical Huntington's disease. Eur J Neurol 2009;16:226–231. [DOI] [PubMed] [Google Scholar]
- 109. Bibb JA, Yan Z, Svenningsson P, Snyder GL, Pieribone VA, Horiuchi A, Nairn AC. Severe deficiences in dopamine signaling in presymptomatic Huntington's disease mice. Proc Natl Acad Sci U S A 2000;97:6809–6814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Kung VW, Hassam R, Morton AJ, Jones S. Dopamine‐dependent long term potentiation in the dorsal striatum is reduced in the R6/2 mouse model of Huntington's disease. Neuroscience 2007;146:1571–1580. [DOI] [PubMed] [Google Scholar]
- 111. Hickey MA, Reynolds GP, Morton AJ. The role of dopamine in motor symptoms in the R6/2 transgenic mouse model of Huntington's disease. J Neurochem 2002;81:46–59. [DOI] [PubMed] [Google Scholar]
- 112. Johnson MA, Rajan V, Miller CE, Wightman RM. Dopamine release is severely compromised in the R6/2 mouse model of Huntington's disease. J Neurochem 2006;97:737–746. [DOI] [PubMed] [Google Scholar]
- 113. Kraft JC, Osterhaus GL, Ortiz AN, Garris PA, Johnson MA. In vivo dopamine release and uptake impairments in rats treated with 3‐nitropropionic acid. Neuroscience 2009;161:940–949 [DOI] [PubMed] [Google Scholar]
- 114. Benn CL, Slow EJ, Farrell LA, Graham R, Deng Y, Hayden MR, Cha JH. Glutamate receptor abnormalities in the YAC128 transgenic mouse model of Huntington's disease. Neuroscience 2007;147:354–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Joshi PR, Wu NP, Andre VM, Cummings DM, Cepeda C, Joyce JA, Carroll JB. Age‐dependent alterations of corticostriatal activity in the YAC128 mouse model of Huntington disease. J Neurosci 2009;29:2414–2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Brouillet E, Hantraye P, Ferrante RJ, Dolan R, Leroy‐Willig A, Kowall NW, Beal MF. Chronic mitochondrial energy impairment produces selective striatal degeneration and abnormal choreiform movements in primates. Proc Natl Acad Sci U S A 1995;92:7105–7109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Sanberg PR, Calderon SF, Giordano M, Tew JM, Norman AB. The quinolinic acid model of Huntington's disease: Locomotor abnormalities. Exp Neurol 1989;105:45–53. [DOI] [PubMed] [Google Scholar]
- 118. McGeer EG, McGeer PL. Duplication of biochemical changes of Huntington's chorea by intrastriatal injections of glutamic and kainic acids. Nature 1976;263:517–519. [DOI] [PubMed] [Google Scholar]
- 119. Fernandes HB, Baimbridge KG, Church J, Hayden MR, Raymond LA. Mitochondrial sensitivity and altered calcium handling underlie enhanced NMDA‐induced apoptosis in YAC128 model of Huntington's disease. J Neurosci 2007;27:13614–13623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Graham RK, Deng Y, Slow EJ, Haigh B, Bissada N, Lu G, Pearson J. Cleavage at the caspase‐6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell 2006;125:1179–1191. [DOI] [PubMed] [Google Scholar]
- 121. Oliveira JM, Chen S, Almeida S, Riley R, Goncalves J, Oliveira CR, Hayden MR. Mitochondrial‐dependent Ca2+ handling in Huntington's disease striatal cells: Effect of histone deacetylase inhibitors. J Neurosci 2006;26:11174–11186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Cheng N, Maeda T, Kume T, Kaneko S, Kochiyama H, Akaike A, Goshima Y. Differential neurotoxicity induced by l‐DOPA and dopamine in cultured striatal neurons. Brain Res 1996;743:278–283. [DOI] [PubMed] [Google Scholar]
- 123. McLaughlin BA, Nelson D, Erecinska M, Chesselet MF. Toxicity of dopamine to striatal neurons in vitro and potentiation of cell death by a mitochondrial inhibitor. J Neurochem 1998;70:2406–2415. [DOI] [PubMed] [Google Scholar]
- 124. Hastings TG, Lewis DA, Zigmond MJ. Role of oxidation in the neurotoxic effects of intrastriatal dopamine injections. Proc Natl Acad Sci U S A 1996;93:1956–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Seiden LS, Commins DL, Vosmer G, Axt K, Marek G. Neurotoxicity in dopamine and 5‐hydroxytryptamine terminal fields: A regional analysis in nigrostriatal and mesolimbic projections. Ann N Y Acad Sci 1988;537:161–172. [DOI] [PubMed] [Google Scholar]
- 126. Bozzi Y, Borrelli E. Dopamine in neurotoxicity and neuroprotection: What do D2 receptors have to do with it? Trends Neurosci 2006;29:167–174. [DOI] [PubMed] [Google Scholar]
- 127. Jakel RJ, Maragos WF. Neuronal cell death in Huntington's disease: A potential role for dopamine. Trends Neurosci 2000;23:239–245. [DOI] [PubMed] [Google Scholar]
- 128. Benchoua A, Trioulier Y, Diguet E, Malgorn C, Gaillard MC, Dufour N, Elalouf JM. Dopamine determines the vulnerability of striatal neurons to the N‐terminal fragment of mutant huntingtin through the regulation of mitochondrial complex II. Hum Mol Genet 2008;17:1446–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Paoletti P, Vila I, Rife M, Lizcano JM, Alberch J, Gines S. Dopaminergic and glutamatergic signaling crosstalk in Huntington's disease neurodegeneration: The role of p25/cyclin‐dependent kinase 5. J Neurosci 2008;28:10090–10101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Tang TS, Chen X, Liu J, Bezprozvanny I. Dopaminergic signaling and striatal neurodegeneration in Huntington's disease. J Neurosci 2007;27:7899–7910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Cyr M, Sotnikova TD, Gainetdinov RR, Caron MG. Dopamine enhances motor and neuropathological consequences of polyglutamine expanded huntingtin. Faseb J 2006;20:2541–2543. [DOI] [PubMed] [Google Scholar]
- 132. Stack EC, Dedeoglu A, Smith KM, Cormier K, Kubilus JK, Bogdanov M, Matson WR. Neuroprotective effects of synaptic modulation in Huntington's disease R6/2 mice. J Neurosci 2007;27:12908–12915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Allen KL, Waldvogel HJ, Glass M, Faull RL. Cannabinoid (CB(1)), GABA(A) and GABA(B) receptor subunit changes in the globus pallidus in Huntington's disease. J Chem Neuroanat 2009;37:266–281. [DOI] [PubMed] [Google Scholar]
- 134. Cepeda C, Starling AJ, Wu N, Nguyen OK, Uzgil B, Soda T, Andre VM. Increased GABAergic function in mouse models of Huntington's disease: Reversal by BDNF. J Neurosci Res 2004;78:855–867. [DOI] [PubMed] [Google Scholar]
- 135. Adam OR, Jankovic J. Symptomatic treatment of Huntington disease. Neurotherapeutics 2008;5:181–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Jankovic J. Treatment of hyperkinetic movement disorders. Lancet Neurol 2009;8:844–856. [DOI] [PubMed] [Google Scholar]
- 137. Mestre T, Ferreira J, Coelho MM, Rosa M, Sampaio C. Therapeutic interventions for symptomatic treatment in Huntington's disease. Cochrane Database Syst Rev 2009. doi: 10.1002/14651858.CD006456.pub2. [DOI] [PubMed] [Google Scholar]
- 138. Huntington Study Group . Tetrabenazine as antichorea therapy in Huntington disease: A randomized controlled trial. Neurology 2006;66:366–372. [DOI] [PubMed] [Google Scholar]
- 139. Pakkenberg H. The effect of tetrabenazine in some hyperkinetic syndromes. Acta Neurol Scand 1968;44:391–393. [DOI] [PubMed] [Google Scholar]
- 140. Pettibone DJ, Totaro JA, Pflueger AB. Tetrabenazine‐induced depletion of brain monoamines: Characterization and interaction with selected antidepressants. Eur J Pharmacol 1984;102:425–430. [DOI] [PubMed] [Google Scholar]
- 141. Quinn N, Marsden CD. A double blind trial of sulpiride in Huntington's disease and tardive dyskinesia. J Neurol Neurosurg Psychiatry 1984;47:844–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Charvin D, Roze E, Perrin V, Deyts C, Betuing S, Pages C, Regulier E. Haloperidol protects striatal neurons from dysfunction induced by mutated huntingtin in vivo . Neurobiol Dis 2008;29:22–29. [DOI] [PubMed] [Google Scholar]
- 143. Caine ED, Polinsky RJ, Kartzinel R, Ebert MH. The trial use of clozapine for abnormal involuntary movement disorders. Am J Psychiatry 1979;136:317–320. [DOI] [PubMed] [Google Scholar]
- 144. Van Vugt JP, Siesling S, Vergeer M, Van Der Velde EA, Roos RA. Clozapine versus placebo in Huntington's disease: A double blind randomised comparative study. J Neurol Neurosurg Psychiatry 1997;63:35–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Lucetti C, Del Dotto P, Gambaccini G, Dell’ Agnello G, Bernardini S, Rossi G, Murri L. IV amantadine improves chorea in Huntington's disease: An acute randomized, controlled study. Neurology 2003;60:1995–1997. [DOI] [PubMed] [Google Scholar]
- 146. Verhagen Metman L, Morris MJ, Farmer C, Gillespie M, Mosby K, Wuu J, Chase TN. Huntington's disease: A randomized, controlled trial using the NMDA‐antagonist amantadine. Neurology 2002;59:694–699. [DOI] [PubMed] [Google Scholar]
- 147. O'Suilleabhain P, Dewey RB Jr. A randomized trial of amantadine in Huntington disease. Arch Neurol 2003;60:996–998. [DOI] [PubMed] [Google Scholar]
- 148. Huntington Study Group . A randomized, placebo‐controlled trial of coenzyme Q10 and remacemide in Huntington's disease. Neurology 2001;57:397–404. [DOI] [PubMed] [Google Scholar]
- 149. Kieburtz K, Feigin A, McDermott M, Como P, Abwender D, Zimmerman C, Hickey L. A controlled trial of remacemide hydrochloride in Huntington's disease. Mov Disord 1996;11:273–277. [DOI] [PubMed] [Google Scholar]
- 150. Landwehrmeyer GB, Dubois B, De Yebenes JG, Kremer B, Gaus W, Kraus PH, Przuntek K. Riluzole in Huntington's disease: A 3‐year, randomized controlled study. Ann Neurol 2007;62:262–272. [DOI] [PubMed] [Google Scholar]
- 151. Lauterborn JC, Lynch G, Vanderklish P, Arai A, Gall CM. Positive modulation of AMPA receptors increases neurotrophin expression by hippocampal and cortical neurons. J Neurosci 2000;20:8–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Bahr BA, Bendiske J, Brown QB, Munirathinam S, Caba E, Rudin M, Urwyler S. Survival signaling and selective neuroprotection through glutamatergic transmission. Exp Neurol 2002;174:37–47. [DOI] [PubMed] [Google Scholar]
- 153. Dicou E, Rangon CM, Guimiot F, Spedding M, Gressens P. Positive allosteric modulators of AMPA receptors are neuroprotective against lesions induced by an NMDA agonist in neonatal mouse brain. Brain Res 2003;970:221–225. [DOI] [PubMed] [Google Scholar]
- 154. Simmons DA, Rex CS, Palmer L, Pandyarajan V, Fedulov V, Gall CM, Lynch G. Up‐regulating BDNF with an ampakine rescues synaptic plasticity and memory in Huntington's disease knockin mice. Proc Natl Acad Sci U S A 2009;106:4906–4911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Altar CA, Cai N, Bliven T, Juhasz M, Conner JM, Acheson AL, Lindsay RM. Anterograde transport of brain‐derived neurotrophic factor and its role in the brain. Nature 1997;389:856–860. [DOI] [PubMed] [Google Scholar]
- 156. Baquet ZC, Gorski JA, Jones KR. Early striatal dendrite deficits followed by neuron loss with advanced age in the absence of anterograde cortical brain‐derived neurotrophic factor. J Neurosci 2004;24:4250–4258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 1990;346:561–564. [DOI] [PubMed] [Google Scholar]
- 158. Munro S, Thomas KL, Abu‐Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993;365:61–65. [DOI] [PubMed] [Google Scholar]
- 159. Herkenham M, Lynn AB, De Costa BR, Richfield EK. Neuronal localization of cannabinoid receptors in the basal ganglia of the rat. Brain Res 1991;547:267–274. [DOI] [PubMed] [Google Scholar]
- 160. Bisogno T, Delton‐Vandenbroucke I, Milone A, Lagarde M, Di Marzo V. Biosynthesis and inactivation of N‐arachidonoylethanolamine (anandamide) and N‐docosahexaenoylethanolamine in bovine retina. Arch Biochem Biophys 1999;370:300–307. [DOI] [PubMed] [Google Scholar]
- 161. Lovinger DM. Presynaptic modulation by endocannabinoids In: Sudhoh TC, Starke K, editors. Handbook of experimental pharmacology. Berlin , Heidelberg : Springer‐Verlag, 2008;435–477. [DOI] [PubMed] [Google Scholar]
- 162. Kano M, Ohno‐Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M. Endocannabinoid‐mediated control of synaptic transmission. Physiol Rev 2009;89:309–380. [DOI] [PubMed] [Google Scholar]
- 163. Fernandez‐Ruiz J, Gonzales S. Cannabinoid control of motor function at the basal ganglia. Handb Exp Pharmacol 2005;168:479–507. [DOI] [PubMed] [Google Scholar]
- 164. Compton DR, Aceto MD, Lowe J, Martin BR. In vivo characterization of a specific cannabinoid receptor antagonist (SR141716A): Inhibition of delta 9‐tetrahydrocannabinol‐induced responses and apparent agonist activity. J Pharmacol Exp Ther 1996;277:586–594. [PubMed] [Google Scholar]
- 165. Ledent C, Valverde O, Cossu G, Petitet F, Aubert JF, Beslot F, Bohme GA. Unresponsiveness to cannabinoids and reduced addictive effects of opiates in CB1 receptor knockout mice. Science 1999;283:401–404. [DOI] [PubMed] [Google Scholar]
- 166. Tzavara ET, Li DL, Moutsimilli L, Bisogno T, Di Marzo V, Phebus LA, Nomikos GG. Endocannabinoids activate transient receptor potential vanilloid 1 receptors to reduce hyperdopaminergia‐related hyperactivity: Therapeutic implications. Biol Psychiatry 2006;59:508–515. [DOI] [PubMed] [Google Scholar]
- 167. Van Der Stelt M, Fox SH, Hill M, Crossman AR, Petrosino S, Di Marzo V, Brotchie JM. A role for endocannabinoids in the generation of parkinsonism and levodopa‐induced dyskinesia in MPTP‐lesioned non‐human primate models of Parkinson's disease. Faseb J 2005;19:1140–1142. [DOI] [PubMed] [Google Scholar]
- 168. Abood ME, Rizvi G, Sallapudi N, McAllister SD. Activation of the CB1 cannabinoid receptor protects cultured mouse spinal neurons against excitotoxicity. Neurosci Lett 2001;309:197–201. [DOI] [PubMed] [Google Scholar]
- 169. Shen M, Thayer SA. Cannabinoid receptor agonists protect cultured rat hippocampal neurons from excitotoxicity. Mol Pharmacol 1998;54:459–462. [DOI] [PubMed] [Google Scholar]
- 170. Fernandez‐Ruiz J. The endocannabinoid system as a target for the treatment of motor dysfunction. Br J Pharmacol 2009;156:1029–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Lastres‐Becker I, De Miguel R, De Petrocellis L, Makriyannis A, Di Marzo V, Fernandez‐Ruiz J. Compounds acting at the endocannabinoid and/or endovanilloid systems reduce hyperkinesia in a rat model of Huntington's disease. J Neurochem 2003;84:1097–1109. [DOI] [PubMed] [Google Scholar]
- 172. Consroe P, Laguna J, Allender J, Snider S, Stern L, Sandyk R, Kennedy K. Controlled clinical trial of cannabidiol in Huntington's disease. Pharmacol Biochem Behav 1991;40:701–708. [DOI] [PubMed] [Google Scholar]
- 173. Muller‐Vahl KR, Kolbe H, Schneider U, Emrich HM. Cannabis in movement disorders. Forsch Komplementarmed 1999;6(Suppl 3):23–27. [DOI] [PubMed] [Google Scholar]
- 174. Palazuelos J, Aguado T, Pazos MR, Julien B, Carrasco C, Resel E, Sagredo O. Microglial CB2 cannabinoid receptors are neuroprotective in Huntington's disease excitotoxicity. Brain 2009;132:3152–3164. [DOI] [PubMed] [Google Scholar]
- 175. Fink JS, Weaver DR, Rivkees SA, Peterfreund RA, Pollack AE, Adler EM, Reppert SM. Molecular cloning of the rat A2 adenosine receptor: Selective co‐expression with D2 dopamine receptors in rat striatum. Brain Res Mol Brain Res 1992;14:186–195. [DOI] [PubMed] [Google Scholar]
- 176. Fisone G, Hakansson K, Borgkvist A, Santini E. Signaling in the basal ganglia: Postsynaptic and presynaptic mechanisms. Physiol Behav 2007;92:8–14. [DOI] [PubMed] [Google Scholar]
- 177. Popoli P, Betto P, Reggio R, Ricciarello G. Adenosine A2A receptor stimulation enhances striatal extracellular glutamate levels in rats. Eur J Pharmacol 1995;287:215–217. [DOI] [PubMed] [Google Scholar]
- 178. Jacobson KA, Hoffmann C, Cattabeni F, Abbracchio MP. Adenosine‐induced cell death: Evidence for receptor‐mediated signalling. Apoptosis 1999;4:197–211. [DOI] [PubMed] [Google Scholar]
- 179. Popoli P, Blum D, Domenici MR, Burnouf S, Chern Y. A critical evaluation of adenosine A2A receptors as potentially “druggable” targets in Huntington's disease. Curr Pharm Des 2008;14:1500–1511. [DOI] [PubMed] [Google Scholar]
- 180. Chou SY, Lee YC, Chen HM, Chiang MC, Lai HL, Chang HH, Wu YC. CGS21680 attenuates symptoms of Huntington's disease in a transgenic mouse model. J Neurochem 2005;93:310–320. [DOI] [PubMed] [Google Scholar]
- 181. Tarditi A, Camurri A, Varani K, Borea PA, Woodman B, Bates G, Cattaneo E. Early and transient alteration of adenosine A2A receptor signaling in a mouse model of Huntington disease. Neurobiol Dis 2006;23:44–53. [DOI] [PubMed] [Google Scholar]
- 182. Domenici MR, Scattoni ML, Martire A, Lastoria G, Potenza RL, Borioni A, Venerosi A. Behavioral and electrophysiological effects of the adenosine A2A receptor antagonist SCH 58261 in R6/2 Huntington's disease mice. Neurobiol Dis 2007;28:197–205. [DOI] [PubMed] [Google Scholar]
- 183. Tebano MT, Pintor A, Frank C, Domenici MR, Martire A, Pepponi R, Potenza RL. Adenosine A2A receptor blockade differentially influences excitotoxic mechanisms at pre‐ and postsynaptic sites in the rat striatum. J Neurosci Res 2004;77:100–107. [DOI] [PubMed] [Google Scholar]
- 184. Glass M, Dragunow M, Faull RL. The pattern of neurodegeneration in Huntington's disease: A comparative study of cannabinoid, dopamine, adenosine and GABA(A) receptor alterations in the human basal ganglia in Huntington's disease. Neuroscience 2000;97:505–519. [DOI] [PubMed] [Google Scholar]