

Abstract
Introduction
Pendred syndrome, a combination of sensorineural deafness, impaired organification of iodide in the thyroid and goitre, results from biallelic defects in pendrin (encoded by SLC26A4), which transports chloride and iodide in the inner ear and thyroid respectively. Recently, pendrin has also been identified in the kidneys, where it is found in the apical plasma membrane of non-α-type intercalated cells of the cortical collecting duct. Here, it functions as a chloride–bicarbonate exchanger, capable of secreting bicarbonate into the urine. Despite this function, patients with Pendred syndrome have not been reported to develop any significant acid–base disturbances, except a single previous reported case of metabolic alkalosis in the context of Pendred syndrome in a child started on a diuretic.
Case report
We describe a 46-year-old female with sensorineural deafness and hypothyroidism, who presented with severe hypokalaemic metabolic alkalosis during inter-current illnesses on two occasions, and who was found to be homozygous for a loss-of-function mutation (V138F) in SLC26A4. Her acid–base status and electrolytes were unremarkable when she was well.
Conclusion
This case illustrates that, although pendrin is not usually required to maintain acid–base homeostasis under ambient condition, loss of renal bicarbonate excretion by pendrin during a metabolic alkalotic challenge may contribute to life-threatening acid–base disturbances in patients with Pendred syndrome.
Introduction
First reported in 1896 (1), Pendred syndrome is an autosomal recessive disorder due to biallelic mutations in SLC26A4 (2). This gene encodes pendrin, a member of the SLC26 family of multi-functional membrane-spanning anion transporters. Pendrin was initially shown to be expressed in the inner ear and thyroid. In the cochlea, it acts as a chloride/bicarbonate exchanger, where it contributes to endolymph homeostasis, whereas in the thyroid, it mediates iodide transport at the apical membrane of thyrocytes (3) (Fig. 1). Recently, pendrin has also been identified in the kidney (4, 5). Here, it localises to the apical plasma membrane of non-α-type intercalated cells of the cortical collecting duct, where it functions as a chloride–bicarbonate exchanger, secreting bicarbonate into the luminal fluid in alkalotic states. However, the exact physiological role of renal pendrin in humans has remained uncertain. In this study, we describe a woman with Pendred syndrome that was only recognised after two episodes of life-threatening metabolic alkalosis. Our case illustrates that, although not required under normal conditions, pendrin expressed in the kidney does play a critical role in humans in protecting against metabolic alkalosis.
Figure 1.

Cellular functions of pendrin. In the thyroid, pendrin is involved in apical iodide transport (see ref. (3) for discussion); in the inner ear it transports bicarbonate into endolymph in exchange for chloride; and in renal collecting duct β-intercalated cells, it participates in urinary bicarbonate excretion with tubular chloride reabsorption. NIS, sodium iodide symporter; I, iodide; Cl, chloride; HCO3, bicarbonate. Full colour version of this figure available via http://dx.doi.org/10.1530/EJE-11-0101.
Case report
A 46-year-old Caucasian female was admitted to hospital after being found on the floor of her home, confused and with rigid limbs. The medical history elicited from her family included childhood-onset sensorineural hearing loss and mild hypothyroidism that had been diagnosed at the time of cochlear implantation 1 year earlier. There was a long history of alcohol excess. Her only medication was levothyroxine 50 μg daily. Initial examination revealed a small goitre, a sinus tachycardia (120 beats/min) and reduced oxygen saturation (91% on air). Tone in all limbs was increased with bilateral carpal spasms and rightward tonic deviation of the neck.
Initial arterial blood gas measurements indicated a severe metabolic alkalosis with hypoventilation: pH 7.59, pO2 7.08 kPa, pCO2 6.39 kPa, bicarbonate (HCO3) 45 mmol/l and base excess +20.4 (normal ranges 7.36–7.44; 9.3–13.3 kPa; 4.5–6.0 kPa; 22–26 mmol/l and −2 to +2 respectively). Venous biochemistry (Table 1) showed severe hypokalaemia and hypochloraemia. In addition, her deranged liver function tests, low platelets, severe hypomagnesaemia and raised mean cell volume (MCV) (Table 1) were consistent with chronic excessive alcohol intake. Inflammatory markers and lactate level were elevated, suggestive of infection. Her thyroid function tests (Table 1) were indicative of either under-replacement or non-compliance.
Table 1.
Venous biochemical and haematological parameters at presentation.
| Results | Reference range | |
|---|---|---|
| Sodium (mmol/l) | 137 | 135–145 |
| Potassium (mmol/l) | 1.4 | 3.4–5.0 |
| Calcium (mmol/l) | 2.07 | 2.1–2.5 |
| Magnesium (mmol/l) | 0.19 | 0.7–1.0 |
| Phosphate (mmol/l) | 0.62 | 0.8–1.4 |
| Chloride (mmol/l) | 86 | 95–106 |
| Bicarbonate (mmol/l) | 45 | 21–32 |
| Glucose (mmol/l) | 10.5 | 3.5–5.5 |
| Creatinine (μmol/l) | 126 | 35–125 |
| Urea (mmol/l) | 9.3 | 0–7.5 |
| Lactate (mmol/l) | 2.6 | Up to 2.0 |
| White cells (×109/l) | 24.2 | 4.0–11 |
| Neutrophils (×109/l) | 20.69 | 2.0–8.0 |
| C-reactive protein (mg/l) | 81 | 0–6 |
| Haemoglobin (g/dl) | 15.7 | 11.5–16.0 |
| MCV (fl) | 112.3 | 80–100 |
| Platelets (×109/l) | 91 | 150–400 |
| Alanine aminotransferase (U/l) | 177 | 0–50 |
| Alkaline phosphatase (U/l) | 141 | 30–135 |
| Bilirubin (μmol/l) | 51 | 0–17 |
| Albumin (g/l) | 32 | 30–35 |
| TSH (mU/l) | 6.30 | 0.35–5.5 |
| Free thyroxine (pmol/l) | 8.4 | 11.5–22.7 |
Central venous access was obtained for fluid and electrolyte resuscitation, with close monitoring of fluid balance. She desaturated and developed a severe respiratory acidosis 24 h after admission, culminating in respiratory arrest that required intubation and ventilation. In the ICU, she received a continuous infusion (20–30 mmol/h) of potassium with hourly electrolyte measurement, but also required several infusions of magnesium (each 20 mmol/2 h), together with i.v. boluses of calcium gluconate. A ventricular fibrillation (VF) cardiac arrest responded to DC cardioversion and i.v. amiodarone 36 h post-admission, but she experienced several further VF arrests despite improvements in her venous K and Mg levels.
Her electrolyte balance stabilised by day 4, but her recovery after extubation was complicated by a chest infection requiring antibiotic treatment. Her potassium and calcium levels eventually normalised without ongoing replacement. Her elevated random blood glucose level normalised without treatment, suggesting that this was a stress response. Oral magnesium supplementation on discharge (day 37) was discontinued 4 weeks later, after which her electrolyte balance remained normal. Her blood pressure was 130/80 mmHg when she was well.
Review of previous charts indicated that the patient had been hospitalised 1 year earlier, following 3 days of persistent vomiting, with similarly deranged biochemistry. During admission, her biochemical disturbances normalised after appropriate treatment, without undue complications. Retrospective analysis of investigations at that time revealed inappropriately high urinary potassium excretion in the face of hypokalaemia (urine K 41.3 mmol/l; serum K 1.4 mmol/l; urine osmolality 490 mOsm) and raised fractional excretion of both magnesium and chloride (5.8 and 4.75% respectively, normal ranges <4.0 and <0.8%). Aldosterone levels were appropriately suppressed. In particular, between the current and the previous hospital admissions, her plasma electrolytes had not required supplementation. Her thyroid dysfunction at the time of diagnosis of hypothyroidism in 2006 was mild (TSH 6.9 mU/l; free thyroxine 9.7 pmol/l; reference ranges as in Table 1) and anti-thyroid peroxidase antibodies were normal.
Review of inner ear imaging showed bilaterally enlarged vestibular aqueducts (EVA; Fig. 2A) and single-cavity cochleae (Mondini defect; Fig. 2B) (6). The combination of bilateral sensorineural hearing loss, goitrous non-autoimmune hypothyroidism and cochlear defects suggested a unifying diagnosis of Pendred syndrome. Sequencing of SLC26A4 confirmed this, revealing that the patient is homozygous for a missense valine to phenylalanine mutation (V138F) in pendrin, which has previously been described in this disorder (7).
Figure 2.

Otologic features of Pendred syndrome in patient. (A) Enlarged vestibular aqueduct of patient compared with normal individual (white arrows). (B) Mondini defect: absence of middle turn of the cochlea and smaller cochlea in our patient compared with normal individual (narrow white arrow – apical turn, wide white arrow – middle turn, black arrow – basal turn of the cochlea).
Discussion
About 150 diverse mutations in SLC26A4, predominantly missense, have been reported in Pendred syndrome (8). The V138F change found in our patient has been shown to result in loss of function via retention of the mutant protein in the endoplasmic reticulum (9). Failure of chloride transport in the inner ear alters endolymphatic potential, leading to endolymphatic hydrops manifesting as an EVA, as seen in our patient. The late-onset hypothyroidism is attributable to defective iodide transport and organification in the thyroid (10). We suggest that her propensity to profound metabolic alkalosis in particular clinical contexts is mediated by loss of pendrin function in the kidney.
Under normal conditions, the human omnivorous diet generates a net gain of acid of about 1 mmol H+/kg body weight/day, such that there is little need for renal bicarbonate-excretory function. Instead, the kidney usually acts to conserve bicarbonate, with 90% of filtered bicarbonate being reabsorbed in the proximal convoluted tubule and the residual 10% in the cortical collecting duct via the chloride–bicarbonate exchanger AE1 (known in its red cell isoform as Band-3) at the basolateral surface of α-intercalated cells. In contrast, the pendrin-expressing β-intercalated cells in the collecting duct are bicarbonate secreting, but this function is likely to be less active in humans given our diet, and few β-intercalated cells are observed on renal microscopy (11). Thus, under ambient conditions, individuals with Pendred syndrome can maintain normal acid–base homeostasis.
However, our patient illustrates the failure of normal compensatory mechanisms that operate during metabolic alkalosis. Vomiting and alcohol excess were likely causative factors that initially disturbed acid–base homeostasis in our patient (12), which spiralled in a vicious cycle resulting in severe metabolic alkalosis because renal β-intercalated cell function could not be up-regulated. In support of this, murine studies during metabolic alkalosis have shown significant up-regulation of renal pendrin mediating enhanced bicarbonate excretion in the cortical collecting duct, together with down-regulation of proximal tubular bicarbonate reabsorption via other transporters, as normal compensatory mechanisms. Furthermore, pendrin-null mice exhibit higher serum bicarbonate levels and impaired excretion of hydroxide equivalents following dietary electrolyte or pharmacological manipulation (13), confirming its role in acid–base homeostasis. By analogy, we suggest that the marked metabolic alkalosis in our patient reflected failure of a similar compensatory mechanism, with deficiency of pendrin in the kidney impairing bicarbonate excretion. In addition, hypokalaemia due to insufficient oral intake, transient renal tubular loss or a combination of both probably further contributed to metabolic alkalosis by enhancing cellular H+ ion uptake (14).
Only a single previous case of metabolic alkalosis in the context of Pendred syndrome has been reported (15). In that report, a child with the disorder developed severe metabolic alkalosis and hypokalaemia, but in the context of commencement of thiazide diuretic therapy for endolymphatic hydrops, presumably being unable to compensate for drug-induced renal chloride and potassium losses, leading to alkalosis.
The severity of our patient's metabolic abnormalities, particularly her hypomagnesaemia and apparent resistance to corrective measures, did prompt us to consider alternative underlying predisposing causes. However, the Gitelman/Bartter spectrum or a primary hypomagnesaemia was ruled out by her recovery without long-term requirement for electrolyte supplements.
In summary, this patient's unusual clinical presentation highlights that in the kidney, pendrin does play a part in maintaining acid–base homeostasis in humans, with its absence and consequent failure of renal bicarbonate excretion leading to potentially life-threatening metabolic alkalosis in the context of inter-current illness.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This study did not receive any specific grant from any funding agency in the commercial or not-for-profit sector. It was supported by the NIHR Cambridge Biomedical Research Centre. F Karet and K Chatterjee are supported by the Wellcome Trust.
References
- Pendred V. Deaf-mutism and goitre. Lancet. 1896;2:532. doi: 10.1016/S0140-6736(01)74403-0. [DOI] [Google Scholar]
- Everett LA, Glaser B, Beck JC, Idol JR, Buchs A, Heyman M, Adawi F, Hazani E, Nassir E, Baxevanis AD, Sheffield VC, Green ED. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS) Nature Genetics. 1997;17:411–422. doi: 10.1038/ng1297-411. [DOI] [PubMed] [Google Scholar]
- Kopp P, Pesce L, Solis-S JC. Pendred syndrome and iodide transport in the thyroid. Trends in Endocrinology and Metabolism. 2008;19:260–268. doi: 10.1016/j.tem.2008.07.001. [DOI] [PubMed] [Google Scholar]
- Royaux IE, Wall SM, Karniski LP, Everett LA, Suzuki K, Knepper MA, Green ED. Pendrin, encoded by the Pendred syndrome gene, resides in the apical region of renal intercalated cells and mediates bicarbonate secretion. PNAS. 2001;98:4221–4226. doi: 10.1073/pnas.071516798. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
Soleimani M, Greeley T, Petrovic S, Wang Z, Amlal H, Kopp P, Burnham CE. Pendrin: an apical
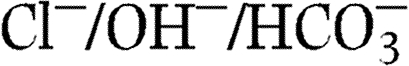 exchanger in the kidney cortex. American Journal of Physiology. Renal Physiology. 2001;280:F356–F364. doi: 10.1152/ajprenal.2001.280.2.F356. [DOI] [PubMed] [Google Scholar]
exchanger in the kidney cortex. American Journal of Physiology. Renal Physiology. 2001;280:F356–F364. doi: 10.1152/ajprenal.2001.280.2.F356. [DOI] [PubMed] [Google Scholar] - Illum P. Mondini type of cochlear malformation – survey of literature. Archives of Otolaryngology. 1972;96:305–311. doi: 10.1001/archotol.1972.00770090481002. [DOI] [PubMed] [Google Scholar]
- Borck G, Roth C, Martine U, Wildhardt G, Pohlenz J. Mutations in the PDS gene in German families with Pendred's syndrome: V138F is a founder mutation. Journal of Clinical Endocrinology and Metabolism. 2003;88:2916–2921. doi: 10.1210/jc.2002-021334. [DOI] [PubMed] [Google Scholar]
- Palos F, Garcia-Rendueles MER, Araujo-Vilar D, Obregon MJ, Calvo RM, Cameselle-Teijeiro J, Bravo SB, Perez-Guerra O, Loidi L, Czarnocka B, Alvarez P, Refetoff S, Dominguez-Gerpe L, Alvarez CV, Lado-Abeal J. Pendred syndrome in two galician families: insights into clinical phenotypes through cellular, genetic, and molecular studies. Journal of Clinical Endocrinology and Metabolism. 2008;93:267–277. doi: 10.1210/jc.2007-0539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JP, Metcalfe RA, Watson PF, Weetman AP, Trembath RC. Mutations of the PDS gene, encoding pendrin, are associated with protein mislocalization and loss of iodide efflux: implications for thyroid dysfunction in Pendred syndrome. Journal of Clinical Endocrinology and Metabolism. 2002;87:1778–1784. doi: 10.1210/jc.87.4.1778. [DOI] [PubMed] [Google Scholar]
- Vaidya B, Coffey R, Coyle B, Trembath R, San Lazaro C, Reardon W, Kendall-Taylor P. Concurrence of Pendred syndrome, autoimmune thyroiditis, and simple goiter in one family. Journal of Clinical Endocrinology and Metabolism. 1999;84:2736–2738. doi: 10.1210/jc.84.8.2736. [DOI] [PubMed] [Google Scholar]
- Smith AN, Skaug J, Choate KA, Nayir A, Bakkaloglu A, Ozen S, Hulton SA, Sanjad SA, Al-Sabban EA, Lifton RP, Scherer SW, Karet FE. Mutations in ATP6N1B, encoding a new kidney vacuolar proton pump 116-kD subunit, cause recessive distal renal tubular acidosis with preserved hearing. Nature Genetics. 2000;26:71–75. doi: 10.1038/82492. [DOI] [PubMed] [Google Scholar]
- Clarke RSJ. Nausea and vomiting. British Journal of Anaesthesia. 1984;56:19–27. doi: 10.1093/bja/56.1.19. [DOI] [PubMed] [Google Scholar]
- Kim YH, Pech V, Spencer KB, Beierwaltes WH, Everett LA, Green ED, Shin W, Verlander JW, Sutliff RL, Wall SM. Reduced ENaC protein abundance contributes to the lower blood pressure observed in pendrin-null mice. American Journal of Physiology. Renal Physiology. 2007;293:F1314–F1324. doi: 10.1152/ajprenal.00155.2007. [DOI] [PubMed] [Google Scholar]
- Galla JH. Metabolic alkalosis. Journal of the American Society of Nephrology. 2000;11:369–375. doi: 10.1681/ASN.V112369. [DOI] [PubMed] [Google Scholar]
- Pela I, Bigozzi M, Bianchi B. Profound hypokalemia and hypochloremic metabolic alkalosis during thiazide therapy in a child with Pendred syndrome. Clinical Nephrology. 2008;69:450–453. doi: 10.5414/cnp69450. [DOI] [PubMed] [Google Scholar]