

Abstract
This review examines the developments in optical biosensor technology, which uses the phenomenon of surface plasmon resonance, for the detection of paralytic shellfish poisoning (PSP) toxins. Optical biosensor technology measures the competitive biomolecular interaction of a specific biological recognition element or binder with a target toxin immobilised onto a sensor chip surface against toxin in a sample. Different binders such as receptors and antibodies previously employed in functional and immunological assays have been assessed. Highlighted are the difficulties in detecting this range of low molecular weight toxins, with analogues differing at four chemical substitution sites, using a single binder. The complications that arise with the toxicity factors of each toxin relative to the parent compound, saxitoxin, for the measurement of total toxicity relative to the mouse bioassay are also considered. For antibodies, the cross-reactivity profile does not always correlate to toxic potency, but rather to the toxin structure to which it was produced. Restrictions and availability of the toxins makes alternative chemical strategies for the synthesis of protein conjugate derivatives for antibody production a difficult task. However, when two antibodies with different cross-reactivity profiles are employed, with a toxin chip surface generic to both antibodies, it was demonstrated that the cross-reactivity profile of each could be combined into a single-assay format. Difficulties with receptors for optical biosensor analysis of low molecular weight compounds are discussed, as are the potential of alternative non-antibody-based binders for future assay development in this area.
Keywords: screening – biosensor, screening – immunoassays, toxicology, phycotoxins – PSP, seafood
Introduction
Paralytic shellfish poisoning (PSP) toxins are naturally occurring potent neurotoxins synthesised by microscopic dinoflagellates from the genii Alexandrium, Gymnodinium and Pyrodinium in marine and freshwater environments (Reis Costa et al. 2009). Increased temperatures, sunlight and nutrient-rich waters are considered to cause the rapid reproduction of dinoflagellate species and thereby lead to potential ‘harmful algal blooms’. Climate change, increased ocean eutrophication and commercial shipping are believed to contribute to the increasing frequency and occurrence of these blooms worldwide (Botana et al. 2009). Filterfeeding organisms, such as molluscan shellfish, consuming dinoflagellates may thereby accumulate the PSP toxins and potentially transfer them through the trophic chain (Deeds et al. 2008). The toxins do not appear to harm shellfish directly, but are potentially lethal to humans or other consumers such as marine mammals and birds (Huang et al. 1996).
No visible difference in appearance is observed between harmless and toxic shellfish. As the PSP toxins are heat stable they are not destroyed by cooking, but due to their solubility in water leaching into the cooking water occurs (Michalski 2007; Etheridge 2010), and canning procedures are reported to reduce toxin levels for this reason (Vieites et al. 1999). Following consumption the toxins bind with a high affinity to site 1 of the voltage-dependent sodium channel α-subunit of muscle and nerve cells, blocking the influx of sodium and preventing the generation and propagation of action potentials in these excitable cells (Cestele and Catterall 2000). Early symptoms following ingestion include tingling of the lips and tongue, which then progresses to the fingers and toes, followed by the loss of control of arms and legs. Difficulty in breathing ensues from paralysis of the muscles of the chest and abdomen, which results in death at high toxin doses. Fatal levels of less than 1 mg of the reference toxin saxitoxin (STX) have been reported (van Egmond et al. 2004). There is currently no antidote for intoxication with artificial respiration as the palliative care until the toxin is excreted from the body.
PSP has major implications to public health and thereby seriously threatening economic consequences in coastal communities and aquaculture industries worldwide. Therefore, globally shellfish destined for human consumption are routinely monitored for PSP toxins by regulatory bodies. The internationally accepted and European Union reference method of analysis for PSP toxins in shellfish is the Association of Analytical Communities (AOAC)-accredited biological method (AOAC 2005a) or more commonly referred to mouse bioassay. Using this method shellfish are deemed fit for consumption based on a regulatory limit, which in Europe is 800 μg of PSP toxins per kg of shellfish meat, a level which has been established for the prevention of acute poisoning (European Commission 2004). In some other countries this limit is lower (Campas et al. 2007). However, both ethical issues relating to the use of animals and technical concerns unfolding in relation to interference and sensitivity of the assay, particularly if European Food Safety Authority (EFSA) recommendations for lowering the regulatory limit are to be considered, is advancing the search for alternative methods of analysis (EFSA 2009).
As a family group, both the chemical and biological properties of the PSP toxins categorise the task of developing an alternative method as extremely challenging from any analytical perspective. Chemically, there are more than 20 common PSP toxins (Figure 1) that vary in structure at four main sites (R1–R4) of the parent compound STX (Llewellyn 2006a). The toxins are classified into groups based on the chemical substitutions at R4 as carbamate, decarbamoyl, sulfocarbamoyl and deoxydecarbamoyl. The carbamoyl and decarbamoyl toxins are the most toxic, whereas the sulfocarbamoyl toxins have a much lower potency. In contrast to the leaching of PSP toxins from shellfish to water it is interesting to note, however, that in the processing of canned seafood contaminated with PSP toxins, the sulfocarbamoyl toxins may be hydrolysed to the more toxic groups, which will increase the overall PSP toxicity (Christian and Luckas 2008). In the last number of years due to advances in mass spectrometry new structural analogues are also being identified (Dell'Aversano et al. 2008). Biologically, each analogue displays a different binding affinity to the sodium channel receptors which results in different toxicities relative to saxitoxin (Genenah and Shimizu 1981). Therefore, the CONTAM Panel of EFSA recommended that the regulatory limit to be expressed as μg STX-equivalents per kg of shellfish meat (EFSA 2009), which was accepted by the European Community. This method of expression of total toxicity or total level of contamination is therefore a simple concept for the biological methods whereby toxin levels correlate to toxicity and therefore the regulatory limit directly. However, this causes severe complications for both biochemical and analytical methodologies whereby conversion factors are required especially when certain toxin standards are limited or unavailable for all detectable PSP toxins in a contaminated sample.
Figure 1.
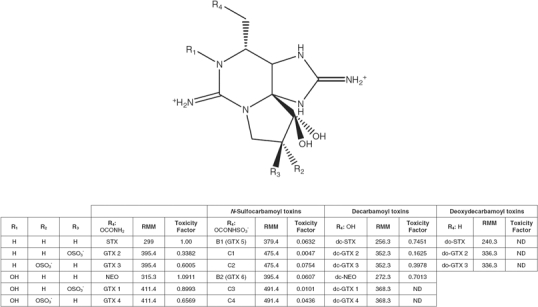
Structure, relative molecular mass (RMM) and toxicity factor of paralytic shellfish poisoning (PSP) toxins.
Toxicity-equivalence factors (TEFs) (Figure 1), originally determined by Oshima (1995) and provided as supplementary material for available toxin standards by the National Reference Centre in Canada (NRC), are used for the conversion of the individual toxin amounts to STXdiHCl equivalents. However, the EFSA working group on STX has recommended new TEFs that are due to be implemented later in 2010 in Europe with the major difference being that the TEF for dcSTX will double in value.
The use of TEFs has supported the development of analytical methods for PSP toxin analysis that can then be correlated to the mouse bioassay and the regulatory limits. Analytical methods such as high-performance liquid chromatography (HPLC) with fluorescence detection, using post-column (Oshima 1995; Asp et al. 2004) and pre-column oxidation methods (Lawrence and Niedzwiadek 2001; Lawrence et al. 2004, 2005), have been developed for the determination of PSP toxins. The method of Lawrence has now been AOAC accredited and accepted following rigorous validation studies by the European Union as a first action method for the toxins for which it is validated (AOAC 2005b). However, HPLC methods are slow, labour intensive and analytical standards are difficult to obtain. Oxidation of PSP toxins is required for HPLC with fluorescence detection because they lack a chromophore. For the AOAC HPLC method two oxidation steps (peroxide and periodate) are required resulting in two analytical runs. Lack of standard material, lengthy purification procedures and insufficient detectability has inhibited the progress in the development of other techniques such as mass spectrometry for routine monitoring. Though advances in mass spectrometry are being made through the use of a zwitterionic hydrophilic interaction chromatography column (Diener et al. 2007), nevertheless mass spectrometry methods are more suitable for confirmatory analysis rather than screening methods due to their high expenditure and the requirement of dedicated skilled scientists for processing the data for quantitative analysis. Hence, alternative screening methods are still sought that are effective and suitable for routine on-site monitoring by unskilled personnel. Different functional assays, biochemical and biosensor-based technologies employing various binding molecules have shown some potential as alternative screening methods, but surface plasmon resonance (SPR) optical biosensor technology has shown considerable promise in this field in a relatively short period of time. This review describes SPR optical biosensor technology and the progress and problems to date in developing an SPR assay for PSP toxin detection for full acceptance as a regulatory tool. Highlighted is a feasibility study of how combining binders has the potential to overcome certain issues and discussed are the possibilities of new novel binders with this technology.
Surface plasmon resonance (SPR) optical biosensor technology
SPR is a powerful physical technique that measures changes in refractive index and, therefore, in the resonance angle by which polarised light is reflected from a surface which is in turn related to a change in mass on that surface (Subrahmanyam et al. 2002). It is a phenomenon used to measure biomolecular interactions in real time in a label-free environment. These biomolecular interactions can be used to identify the binding of two or more interactants to each other to determine the affinity of the interactions, to measure the actual association and dissociation rates of the interaction, and for the purpose of PSP toxin detection can be exploited to quantify the concentration of one of the interactants (Karlsson 2004).
Optical biosensors based on SPR technology are now an established tool in biomedical research for exploring molecular interactions but are increasingly being investigated as monitoring tools by academic and industrial scientists interested in food safety for the detection of additives and contaminants.
As Biacore® biosensors had been proven to be a successful platform for detecting low-level chemical contaminants and toxins in food products, the Biacore Q platform was selected for assay development for the monitoring of PSP toxins (Campbell et al. 2007; Fonfria et al. 2007). This biosensor-based assay measured the real-time interaction of a specific biological recognition element or binder with a target toxin immobilised onto the sensor chip surface. A typical sensorgram, a measure of the binding response against time, is illustrated in Figure 2 (Abery 2001). The running buffer flowing over the surface established the baseline value and as the sample solution passed over the sensor surface, the profile of the molecular interaction appeared. During injection the observed binding rate was the difference between the molecules binding to the surface and molecules leaving the surface. Consequently, binding appears fastest at the beginning of the injection as the binder in the sample readily interacts with the high concentration of available toxin-binding partners on the sensor surface. As the injection continues, the observed rate of binding decreased as fewer toxin molecules are available on the surface. After the injection was complete, the binder dissociated and was washed away by the continuous flow of buffer. The sensor surface was regenerated back to the baseline value for the analysis of the next sample (Campbell et al. 2007).
Figure 2.
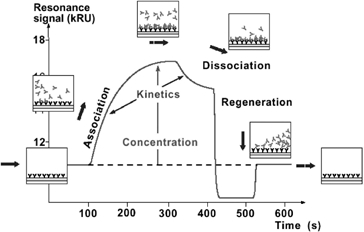
SPR optical biosensor sensorgram of the molecular interaction between the binder and immobilised surface. Source: Abery (2001). Courtesy: Biacore International AB, GE Healthcare.
The SPR optical biosensor method for PSP toxins was designed as an inhibition assay. A set amount of PSP binder was mixed with the sample and injected over the surface of a sensor chip to which toxin was immobilised. When no PSP toxins were present in the sample, the binder was then free to bind to the immobilised toxin producing a response (Figure 3a) (Campbell et al. 2007). When PSP toxins were present in the sample, these then bound to the binder, which inhibited the binder from binding to the immobilised toxin on the chip surface and a lower response was produced (Figure 3b) (Campbell et al. 2007). For most biomolecules the change in response is proportional to the mass of the material that has bound to the surface. Therefore, for binders to elicit a change in response from the baseline, they must be greater than 1000 daltons and hence this is why the low molecular weight toxins are immobilised to the surface. Using this assay format, the level of binding to the surface was inversely proportional to the concentration of PSP toxins present in the sample. Figure 4 shows a typical response against a concentration calibration curve.
Figure 3.
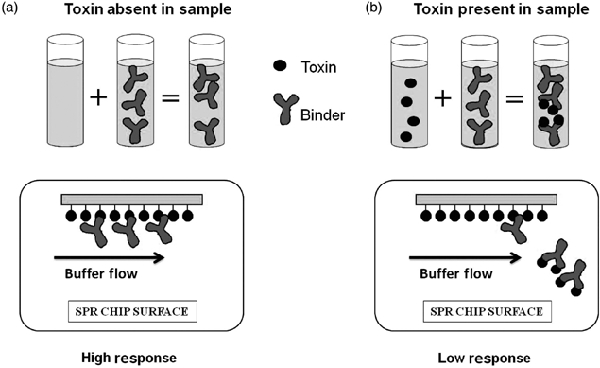
SPR optical biosensor inhibition assay format.
Figure 4.
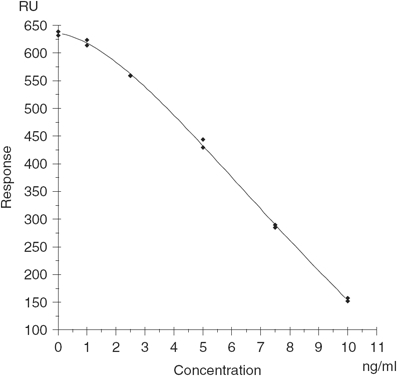
Typical response versus PSP toxin concentration calibration curve using a polyclonal antibody binder.
Further developments in optical SPR biosensor technology, particularly the progression of multichannel and multiplex instruments such as the Biorad Proteon XPR36 (http://www.bio-rad.com) or Biacore prototype developed as part of the European Union 6th Framework Project BioCop (http://www.biocop.org), provide additional options for dealing with toxicity factors of toxins and cross-reactivity issues of binders. Within the European Union 7th Framework programme the project CONffIDENCE is evaluating the BioCop multiplex optical SPR for developing a single assay for multiple toxin groups legislated by the European Union and emerging toxins to European Union waters (http://www.conffidence.eu). This multiplex prototype device is a four-channel instrument with four detection spots within each channel allowing for the simultaneous analysis of up to 16 different analytes in a sample. This increases the scope of binder interactions that could be employed to detect the different PSP analogues. An ideal scenario would be to have a highly specific binder for each of the main PSP toxin groups. Each binder assay for each group could be isolated in separate channels for simultaneous detection in a sample, and then not only would the analyst be able to determine if PSP toxin was present, but also to which group it belonged and a correlation made of the toxic potency level for that sample.
Binders for SPR analysis of PSP toxins
A number of different types of binder have been exploited in the development of various methodologies for the detection of PSP toxins. These include receptors such as the sodium channel receptor (SCR) and saxiphilin receptor, polyclonal and monoclonal antibodies raised against various toxin analogues and chemical binders (Llewellyn and Doyle 2001; Usleber et al. 2001; Gawley et al. 2007; van Dolah et al. 2009). The general advantages and disadvantages of binders that have been previously investigated in the development of assays for SPR analysis for PSP toxins are highlighted in Table 1.
Table 1.
General advantages and disadvantages of binders previously assessed for SPR analysis of PSP toxins.
| Binder | Advantages | Disadvantages |
|---|---|---|
| Receptors | Have a binding affinity that generally correlates with the biological response or toxicity of the toxins Can be isolated rapidly compared with antibody production Good sensitivity |
Wider scope of binding to all compounds that act on the same receptor Have to be isolated form biological tissues and in some cases the species of origin is difficult to obtain May display poor stability May be temperature sensitive for analytical purposes |
| Monoclonal antibodies | Constant renewable source of binder once produced with minimal batch to batch variation During production less immunizing agent is required compared with polyclonal production and it is possible to select for specific epitope specificities and generate antibodies against a wider range of antigens React with a single epitope on an antigen with high specificity Binding affinity for the immunizing antigen is generally better than a receptor Less background signal than polyclonal antibodies | For production, time, effort and commitment is high and use animals On average the binding affinity of a monoclonal antibody is lower than a polyclonal antibody As monoclonal antibodies are highly specific to a single epitope they may lose affinity to other antigens within a group with minor molecular modifications The binding affinity of an antibody does not correlate with biological response for a toxin group Antibodies sometimes display unexpected crossreactivity with unrelated antigens in biological matrices |
| Polyclonal antibodies | For production relatively quick and inexpensive to produce compared with monoclonal antibodies Polyclonals will recognize multiple epitopes on any one antigen and are therefore more tolerant of minor changes in the antigen than monoclonal antibodies providing better crossreactivity to structural antigens within a group The recognition of multiple epitopes generally provides more robust detection Binding affinity for the immunizing antigen is generally better than a monoclonal antibody or receptor |
Animals are required and are prone to batch to batch variability They produce large amounts of non-specific antibodies which can sometimes give background signal in some applications Multiple epitopes make it important to check for any cross-reactivity in biological matrixes The binding affinity of an antibody does not correlate with biological response for a toxin group |
Receptors
Receptors can be either cell membrane bound or cytoplasmic proteins that may be exploited for their ligand-binding capabilities for screening methodologies based on their functional response. SCRs from crude rat brain membrane preparations have been used in radioligand binding assays (Wiegele and Barchi 1978) and in recent years they have been configured into a microtitre plate format (Powell and Doucette 1999; Ruberu et al. 2003). This microtitre plate format has recently undergone successful single-laboratory validation (van Dolah et al. 2009). The fact that all STX analogues bind to SCRs with affinities that vary according to their toxic potency (Doucette et al. 1997) indicates that receptor-based competitive binding assays provide a measure of a sample's toxicity, irrespective of which PSP toxin(s) is present. The saxiphilin receptor, occurring naturally in various amphibians and terrestrial invertebrates, has also been integrated into a competitive binding assay using a microtitre plate format (Llewellyn and Doyle 2001). Llewellyn (2006b) studied the behaviour of mixtures of PSP toxins in these two unrelated competitive binding assays and found that the most potent toxins affect the toxicity of the mixture to the greatest extent with the less active toxins present required to be several orders of magnitude greater for the mixture to reflect their potency.
Receptor ligand binding assays using SPR technology had previously been investigated (Gestwicki et al. 2001; De Jong et al. 2005). As receptor binders for PSP toxins reflect toxicity in a similar manner to the mouse bioassay, it was envisaged that they could be an ideal candidate for the development of an SPR assay to replace the mouse bioassay. Campbell et al. (2007) examined crude extracts of SCR from rat brains using SPR analysis, but technical difficulties were encountered with the SCR in that this preparation appeared to be stable for only a short time at 4°C, which in effect was detrimental to reliability and reproducibility. Another issue with developing SPR assays for small molecular weight toxins is that when the toxin is immobilised onto the surface, the toxin may then be orientated in such a manner that it no longer locks into the binding site of the receptor pocket, making the toxin unrecognisable with minimal to no binding occurring. As SPR is dependent on changes in mass, for the receptor to be immobilised on the toxin surface the toxin binding in isolation is not of a large enough mass to achieve a change in response and the signal would need to be amplified. Amplification could be performed in a manner similar to a sandwich assay by using a secondary antibody. For small molecular weight compounds this is difficult to achieve due to a limited numbers of available epitopes that are not already encapsulated in the receptor pocket. A different approach that could be investigated is using a ligand of a high molecular weight that would complete and dissociate appropriately with the toxin for the receptor site when the receptor is immobilised on the surface (Karlsson 1994). This ligand would provide the molecular mass necessary for the SPR response, but sourcing a suitable competitor to PSP toxins maybe difficult. The limited availability of the saxiphillin receptor to only a few research groups has inhibited the characterisation of this binder in the role of PSP toxin detection by SPR. Research is on-going into the synthesis of cloned receptors that would prove beneficial in both functional assays and as more stable alternative binders to antibodies for sensor technologies.
Antibodies
Antibodies unlike receptors are biochemical binders and bind selectively to chemical moieties on the structure of the toxin to which they are produced. These binders do not show a similar correlation between the toxicity of a toxin and binding affinity in the same manner as receptors for PSP toxins. The difficulty in trying to detect such small molecular weight compounds using antibodies is that there are limited functional groups to conjugate a protein for antibody production based on site specificity and, therefore, few chemical reactions can be utilised for immunogen synthesis. Restricted and limited availability of toxins makes the synthesis of alternative derivatives impractical unless sufficient yield can be achieved for immunogen production. The formation of the toxin protein conjugate must also reduce the toxicity of the toxin or when immunised the animal may be affected with PSP symptoms. The first antibodies produced against PSP toxins were reported by Johnston et al. (1964). For most PSP toxin antibodies produced since then either a modification of the Mannich reaction or reductive alkylation chemistry (Bürk et al. 1995) has been employed for the synthesis of the protein conjugate. As highlighted by Usleber et al. (2001) during these reactions there are three potential reactive sites for the carbamate toxins. These include the amino, guanidine and imine groups, but it is difficult to characterise which is the predominant orientation of the toxin to the protein conjugate or the ratio of protein conjugation to all three sites.
Immunochemical methods are frequently used for qualitative, semi-quantitative and quantitative measurements of many chemical substances with high biological impact. These methods are well established in clinical chemistry and endocrinology and have become in recent years more sophisticated in their use for food and environmental analysis and toxicology screening due to their ability to function effectively in complex biological matrices without major purification. From the production of the first antibodies for PSP toxins, it was decades later when immunoassays were actually utilised to monitor PSP toxins in shellfish. Immunoassay methods for PSP toxins have been reviewed in the past (Usleber et al. 2001) and several antibody-based approaches for detecting these toxins have been described since that time. These include radioimmunoassay (Carlson et al. 1984), immunoaffinity columns (Dietrich et al. 1996), lateral flow devices (Jellett et al. 1998; Laycock et al. 2010) and numerous competitive enzyme immunoassays (EIAs). Indirect EIAs have been developed for STX (Chu and Fan 1985; Cembella et al. 1990; Micheli et al. 2002), neosaxitoxin (NEO) (Chu et al. 1992; Bürk et al. 1995) and gonyautoxin (GTX) 2/3 (Kawatsu et al. 2002) and direct EIAs for STX and NEO (Usleber et al. 1991; Schneider et al. 1995; Chu et al. 1996; Huang et al. 1996; Dubois et al. 2010) and GTX 2/3 (Kawatsu et al. 2002). These assays all utilise antibodies produced using the previously mentioned protein conjugation chemistries with the primary modification being the PSP toxin to which the protein is conjugated. Although several EIAs and lateral flow test kits are commercially available (Laycock et al. 2010; http://www.abraxiskits.com; http://www.biosense.com; http://www.r-biopharm.com), one shortcoming is that they are highly specific to certain toxins. This specificity, corresponding mainly to chemical structure but not proportionately to toxicity, limits an accurate measure of the total toxicity in a sample as required by regulatory authorities. For such diverse potency within this relatively large toxin group, whose small molecular weight structures are highly similar, it is extremely difficult to match the cross-reactivity profile of an antibody to the potency of each toxin for similar comparability as a receptor assay to the mouse bioassay. Usleber et al. (2001) state that from an immunological point of view the PSP toxin family must be subdivided into STX (non-hydroxylated) and NEO (hydroxylated) groups. Previous ELISA studies have demonstrated that when the antibodies are specific to either group they are less specific to the other. Therefore, to incorporate the full spectra of PSP toxins to an equivalent extent, antibodies produced to both the non-hydroxylated (STX) or hydroxylated toxins (NEO) will have to be utilised. Other drawbacks to these commercial kits are the high costs, primarily due to the availability and quantity of toxin required for producing either coating antigen or enzyme labels, and the potentially subjective interpretation of results.
Current progress of SPR analysis of PSP toxins using antibody binders
To date although antibodies may have specificity issues relating to the analysis of such a large toxin group they are the most successful binder in terms of sensitivity for the detection of PSP toxins. For this reason antibodies were chosen as the model binder to demonstrate the feasibility of SPR optical biosensor technology for PSP toxin analysis. The applicability of SPR analysis to this task was demonstrated using both a monoclonal antibody (GT-13A) raised to gonyautoxin 2/3 and a polyclonal antibody (R895) raised to STX (Campbell et al. 2007). When an STX surface was employed, which can be regenerated over 1000 times for reuse conserving toxin supplies and reducing cost, these two antibodies displayed vast differences in their cross-reactivity profiles. The GT-13A binder showed a high degree of segregation between non-hydroxylated and hydroxylated toxins indicating that when samples are analysed and compared with the mouse bioassay using this binder the toxin content of a sample would be underestimated if the toxin composition is predominately the hydroxylated toxins, NEO or GTX1/4 (Campbell et al. 2007). However, PSP toxins such as C1/C2 and GTX5 displayed high specificity, although their toxic potency is relatively low to STX. Therefore, samples containing levels of these toxins will overestimate compared with the mouse bioassay. For regulatory purposes, underestimation could have severe health implications to the consumer whereby contaminated material is declared safe for consumption, whereas overestimation could cause detrimental economic losses to the industry through the unnecessary closure of harvesting beds. This GT-13A binder was further assessed by SPR to determine its tolerance capabilities to various shellfish matrices and PSP toxin extraction solvents (Fonfria et al. 2007). Fonfria et al. applied different extraction procedures, previously employed for PSP toxin analysis, to different shellfish species and they highlighted that by either using an ethanolic extraction procedure reported by Garthwaite et al. (2001) or the AOAC HPLC acetic acid extraction (AOAC 2005b), the determination of PSP toxins in shellfish could be performed by SPR. The Garthwaite method was recommended for further evaluation based on analysis time, but only 50% recovery of STX was achieved using this extraction procedure. However, samples containing C1/C2 toxins overestimated compared with HPLC and mouse bioassay results due to the low toxic potency but high specificity of this toxin. As a proof of concept this monoclonal antibody was also employed to demonstrate that SPR biosensor-based biochemical analysis could be combined with state-of-the-art mass spectrometry chemical analysis for joint screening and confirmatory determination of PSP toxins (Marchesini et al. 2009).
The R895 binder showed a much more diverse but narrower pattern in cross-reactivity profile with improved sensitivities to all toxins compared with GT-13A, but the hydroxylated toxins dcNEO and GTX1/4 will require further improvement. Rawn et al. (2009) performed an evaluation of both binders by SPR using the Garthwaite extraction procedure for analysing 88 natural samples and compared the results with the AOAC HPLC method (AOAC 2005b). This study demonstrated that when using this extraction procedure, different shellfish matrix effects were considerable compared with a buffer curve, and if a buffer curve were used for analysis, this would cause an overestimation in results of 50%. Therefore, natural samples were evaluated using an extracted mussel calibration curve for better correlation in recovery and matrix effects. In general, the polyclonal antibody correlated better with HPLC than the monoclonal antibody due to the differences in cross-reactivity profiles. However, it was recommended that improved antibody response to the hydroxylated toxins would be necessary if SPR were to be a replacement for the mouse bioassay for regulatory testing. As a screening tool, it was suggested that this SPR method using either antibody could eliminate greater than 80% of samples from the mouse bioassay at the current regulatory limit. In relation to the three Rs of replace, reduce and refine at this stage, the SPR assay may not be a replacement tool but a reduction tool for the number of animal assays performed for regulatory purposes.
Further work was then performed on the extraction procedure in order to calibrate the SPR assay using a buffer curve to eliminate the requirement of sourcing uncontaminated material and using relatively excessive quantities of toxin in the preparation of a calibration curve (Campbell et al. 2009). This was achieved in a comparative study using a second polyclonal binder raised to STX (BC67) for SPR analysis compared with EIA, HPLC and mouse bioassay (Campbell et al. 2009). This study demonstrated that the key for immunological assay development was the binder and highlighted that the advantages of using SPR over all the other methods were simplicity, ease of use and speed of analysis. This new extraction procedure using pH 5 sodium acetate buffer followed by centrifugation followed by dilution and analysis was then applied to the STX antibody and a single laboratory validation of this SPR method performed highly satisfactorily (Campbell et al. 2010). An inter-laboratory study between seven international laboratories is currently underway. Threshold values for the assay have been discussed for regulatory purposes, but this level could vary depending on geographical location and the risk-management strategies that have to be considered whether by industrial or by regulatory monitoring laboratories. To date there has been no reports of PSP toxins occurring in isolation, but if GTX1/4 were to do so, this method could then become invalid even as a screening tool due to the lack of sensitivity of this binder to this toxin. Therefore, alternative binders are still being sought with improved sensitivity towards the hydroxylated toxins and different heterologous assay formats are being investigated that could improve the overall toxicity determination of PSP toxins in a sample.
A combination of two binders, one raised to STX (Campbell et al. 2007), the other to NEO (Bürk et al. 1995), were assessed on a NEO chip surface to determine the feasibility of combining the cross-reactivity profiles of each in order to be able to detect the full range of PSP toxins in isolation at the regulatory level (Campbell et al., unpublished data). The sensitivity and specificity of each binder relative to STX and its analogues were assessed. The cross-reactivity profile of the STX and NEO antibodies on the NEO surface was favoured to the non-hydroxylated and hydroxylated toxins at the R1 position respectively (Table 2). The percentage cross-reactivity of STX antibody in relation to STXdiHCl was variable in relation to toxin structure with the NEO surface. Toxins with modifications in the R4 position (Figure 1) displayed the highest percentage cross-reactivity followed by those with modifications to the R2 and R3 positions and then those toxins hydroxylated in the R1 position. Combinations of modifications showed an additive decrease in percentage cross-reactivity with the outcome for GTX1/4, which is modified at the R1, R2 and R3 positions, displaying the lowest percentage cross-reactivity at ≤ 1.0%. For the NEO antibody binder the chemical reaction used to prepare the chip surfaces was different from the reaction used to prepare the toxin protein conjugate used as the immunogen. A change in orientation of the toxin conjugated to the surface could explain the relatively low antibody titres required to produce a response on this NEO chip surface in relation to the titre of this antibody reported in the ELISA study (Bürk et al. 1995). This antibody was significantly more sensitive to the R1 hydroxylated toxins compared with the corresponding non-hydroxylated toxins as in agreement with the ELISA study.
Table 2.
Sensitivity and specificity data for both STX and NEO antibody on a NEO chip surface (Campbell et al., unpublished data).
| Chip surface | Neosaxitoxin chip surface | |||||
| Antibody | STX | NEO | ||||
| Antibody dilution in HBS-EP | 1/250 | 1/50 | ||||
| Antibody ratio to standard | 1:1 | 1:1 | ||||
| Flow rate (μl min−1) | 25 | 25 | ||||
| Contact time (min) | 2 | 2 | ||||
| PSP toxin analogue | IC50 (ng ml−1) | Percentage cross-reactivity | Dynamic range, IC2o to IC80 (ng ml−1) | IC50 (ng ml−1) | Percentage cross-reactivity | Dynamic range, IC2o to IC80 (ng ml−1) |
| Saxitoxin dihydrochloride | 4.8 | 100 | 2.7–8.1 | 41.8 | 100 | 6.6–203 |
| Neosaxitoxin | 13.4 | 35.8 | 3.7–84.8 | 2.4 | 1742 | 1.1–5.5 |
| Gonyautoxin 1/4 | 474b | 1.0b | 77–2780b | 4.1 | 1032 | 1.6–14.9 |
| Gonyautoxin 2/3 | 7.0 | 68.7 | 2.1–22.0 | 19.6 | 213 | 4.1–91.6 |
| Decarbamoyl saxitoxin | 2.9 | 165.1 | 1.3–6.3 | >1000 | <4.2 | n.d.a |
| Decarbamoyl neosaxitoxin | 100 | 4.8 | 14.4–200 | 192.1 | 21.8 | 41.9–512 |
| Decarbamoyl gonyautoxin 2/3 | 18.7 | 25.8 | 4.4–62.3 | >1000 | <4.2 | n.d. |
| C1/C2 | 14.8 | 32.6 | 3.3–53.6 | >1000 | <4.2 | n.d. |
| Gonyautoxin 5 | 4.1 | 117 | 2.7–7.7 | >1000 | <4.2 | n.d. |
| C3/C4 | 206b | 2.3b | 27–590b | 100 | 41.3 | n.d. |
Notes: an.d., Not determined due to lack of the standard.
bValues calculated based on extrapolation of the four-parameter fit applied to the standard curve due to availability of the standard.
Initially for the carbamate toxins different ratios of STX antibody to NEO antibody titres were assessed with the most suitable for the mix study being 1/500 and 1/50, respectively. Different percentage ratios of STX to NEO antibody at these dilutions were assessed using the neosaxitoxin chip surface and each of the carbamate toxins at a concentration of 10 ng ml−−1(Figure 5). The full cross-reactivity profile was determined using an optimum 20% STX and 80% NEO antibody (Table 3) as this ratio showed the lowest differential in cross-reactivity for the carbamate toxins. On comparison with the cross-reactivity profile of STX and NEO antibody on their own with this surface, the cross-reactivity profile has been amalgamated to produce a potential single-assay format for all PSP toxins. With the exceptions of GTX5 and dcSTX, which will overestimate based on this cross-reactivity profile, this antibody mix correlates much better than either binder individually with the mouse bioassay toxicity factors (Figure 1) for each of the toxins. This is illustrated in Table 3 as the cross-reactivity profile in STXdiHCl equivalents.
Figure 5.
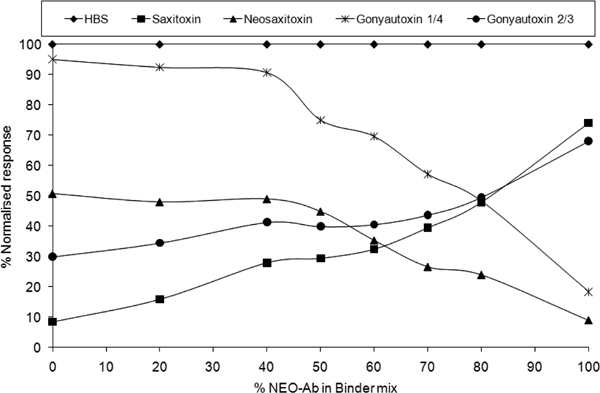
Trends in carbamate toxin response at a fixed concentration of 10 ng ml−−1 with increasing percentage of NEO antibody in STX/NEO antibody binder mix. Source: Campbell et al, unpublished data.
Table 3.
Sensitivity and specificity of the STX/NEO antibody mix for each PSP toxin (Campbell et al., unpublished data).
| STX |
1/500 |
|||||
| Antibody dilution in HBS-EP | NEO | 1/50 | ||||
| Antibody mix | 20% STX/80% NEO | |||||
| Antibody ratio to standard | 1:1 | |||||
| Flow rate (μl min−−1) | 12 | |||||
| Injection volume (μl) | 60 | |||||
| Contact time (min) | 5 | |||||
| Toxin concentration (ng ml−−1) | Toxin concentratioas STXdiHCl equivalents (ng ml−−1 | |||||
| PSP toxin analogue | IC50 (ng ml−−1) | Percentage cross-reactivity | Dynamic range, IC20 to IC80 (ng ml−−1) | IC50 (ng ml−−1) | Percentage cross-reactivity | Dynamic range, IC20 to IC80 (ng ml−−1) |
| Saxitoxin dihydrochloride | 2.8 | 100 | 0.2–68.8 | 2.8 | 100 | 0.20–68.8 |
| Neosaxitoxin | 2.8 | 100 | 0.8–9.9 | 3.1 | 90.3 | 0.87–10.8 |
| Gonyautoxin 1/4 | 4.4 | 63.6 | 0.8–25.3 | 4.0 | 70 | 0.72–22.8 |
| Gonyautoxin 2/3 | 4.9 | 57.1 | 0.9–28.0 | 2.9 | 96.6 | 0.54–16.8 |
| Decarbamoyl saxitoxin | 0.4 | 700 | 0.1–1.2 | 0.3 | 933 | 0.07–0.9 |
| Decarbamoyl neosaxitoxin | 9.1 | 30.7 | 1.7–48.7 | 6.4 | 43.8 | 1.19–34.2 |
| Decarbamoyl gonyautoxin 2/3 | 17.1 | 16.4 | 3.1–94.7 | 6.8 | 41.1 | 1.23–37.7 |
| C1/C2 | 14.5 | 19.3 | 2.0–102.7 | 1.1 | 255 | 0.15–7.7 |
| Gonyautoxin 5 | 0.4 | 700 | 0.1–1.5 | 0.03 | 9333 | 0.01–0.1 |
| C3/C4 | 91.8 | 3.0 | 24.0–198 | 4.0 | 70 | 1.03–8.5 |
Consequently, by using optical SPR biosensor analysis, there is the potential to resolve cross-reactivity issues that arise using a single binder. Combining the two polyclonal antibodies STX and NEO with a common denominator of the NEO surface enabled the design of a single-assay format for both hydroxylated and non-hydroxylated toxins. In addition, as the cross-reactivity is more in line with the toxicity profile for the mouse bioassay, this is an advantage in having one screening test for all PSP toxins compared with two ELISA tests. This approach of mixing antibodies raised in the same species could be used for other families of chemical contaminants or toxins where there is significant variability in the cross-reactivity profile as long as there is a common denominator in the family for chip surface immobilisation.
Potential binders for future optical SPR analysis of PSP toxins
A wide range of non-antibody-based binders have been reviewed previously as alternative molecules for the detection of pathogens (Ngundi et al. 2006). The most promising non-antibody binders worth investigating in their role for PSP toxin analysis by SPR are chemosensors (Bell and Hext 2004), molecular imprinted polymers (MIPs) (Hall et al. 2005) and aptamers (Tombelli et al. 2005). The advantages and disadvantages of these particular binders are highlighted in Table 4.
Table 4.
Advantages and disadvantages of potential binders for SPR analysis of PSP toxins.
| Binder | Advantages | Disadvantages |
|---|---|---|
| Chemosensors | Non-animal-based binders Can be incorporated into self-assembly monolayers for sensing on a surface |
Lower binding affinity than antibodies No correlation between the binding intensity and biological response or toxicity |
| Molecular imprinted polymers (MIPS) | Non-animal-based binder Target analyte defines its own recognition site Stable at extreme temperature and pH Show specificity in natural systems Adaptable and flexible in their use Low cost and ease of manufacture May be used in non-aqueous media or aggressive environments |
May be sensitive to small alterations in target analyte structure and properties Show non-specific binding Diversity of binding sites Template bleeding requires suitable template analogue for imprinting and affects quantitative applications Generally lower binding affinities than antibodies |
| Aptamers | Nucleic acid aptamers are non-animalbased binders produced by chemical synthesis resulting in limited batch to batch variation Selectively bind to low molecular weight compounds Binding affinities of aptamers are in the μM to nM range and kinetic parameters can be altered on demand Toxins that do not illicit a good immune response can be used to generate high-affinity aptamers Aptamers are stable for long-term storage and can be transported at ambient temperature Selection conditions can be manipulated to obtain aptamers with properties desirable for in vitro assays, e.g. non-physiological buffers, solvents, etc. Reporter molecules can be attached to aptamers at precise locations so as not to affect binding |
Aptamers are costly to generate and long aptamer sequences are difficult to achieve DNA aptamers have a smaller range of three-dimensional structures obtainable compared with RNA aptamers but they can bind their target to the entire sequence DNA and RNA aptamers are susceptible to enzymatic degradation by nucleases, thus requiring highly pure environments. This can be overcome Peptide aptamers are constrained to the scaffold protein so they are less flexible, which may affect their effectiveness. Only the variable range is used for binding Peptide aptamers require biological systems for selection purposes |
Chemosensors
Chemists have used a variety of approaches to design artificial receptors or chemosensors capable of selectively binding analytes. Chemosensors are molecules specifically designed for the qualitative and quantitative monitoring of analytes and are being researched in the areas of biological and analytical chemistry, medicine and environmental sciences (Bell and Hext 2004; Minkin et al. 2008). These molecules bind to the target analyte through non-covalent interactions and produce a change in light absorption or fluorescence. Molecular structures that have displayed some potential as chemosensors are polyalcohols, crown ethers, calixarenes, helicenes, sterically geared tripods, metal complexes, pinwheels, porphyrins and fused ring heterocycles. Most chemosensors described do not display sufficient sensitivity or selectivity to their targets for practical applications, and they usually require synthetic modification for immobilisation onto a sensor surface. However, chemosensors such as crown ether sensors like calix[4]arene have been investigated as alternative binders for STX detection (Chen et al. 2007). These binders are showing considerable promise in detecting STX using fluorescence, but as yet the sensitivity is five orders of magnitude lower than that achievable using the mouse bioassay (Gawley et al. 2002, 2007). For naturally contaminated shellfish samples there was no correlation between the intensity of absorption and toxicity of the sample. However, SPR spectroscopic detection using these binders increased the sensitivity up to 1000-fold (Chen et al. 2007). Shellfish matrices have not as yet been evaluated.
Molecular imprinted polymers (MIPs)
MIPs have received controversial exposure over the past 10 years from researchers and industries in their potential role in the environmental and food analysis of chemical contaminants and toxins (Lotierzo et al. 2004; Henry et al. 2005; Baggiani et al. 2008). They are artificially created ligand binding sites that can selectively recognise a target molecule. The synthesis involves the polymerisation of functional and cross-linking monomers around the target of interest or a structural mimic template through non-covalent interactions or reversible covalent bonds or both with the functional groups of the template. These three-dimensional polymer-based systems can be optimised to yield recognition sites highly specific and selective to the target of interest in the same manner as either a polyclonal or a monoclonal antibody. However, the mechanism of interaction or recognition of a particular analyte to the polymer is still not completely comprehended. Unlike antibodies, these are fairly rigid macroscopic structures are insoluble in any solvent. MIPs therefore are more commonly marketed and used for selectively separating molecules for clean-up and pre-concentration of analytes from complex samples, but they have also been demonstrated in detection assays (Ramstrom et al. 2001; Chianella et al. 2003). The applicability of MIPs as a surface template in the SPR analysis of domoic acid has also been demonstrated (Lotierzo et al. 2004). Mahony et al. (2005) stated that combinatorial chemistry and computational molecular modelling are the most promising concepts for the development of MIPs for enhanced recognition properties. Using this technology with advances in polymeric materials, in the future there is the potential to develop and design binders with the highest selectivity to the PSP toxin family as a whole. However, the elimination of problems associated with template leaching for quantitative analysis would have to occur.
Aptamers
Aptamers, first described by Tuerk and Gold (1990), are creating a new wave of enthusiasm as alternative binders to antibodies due to progress in aptamer screening methods (Wang and Jia 2009) and their potential in the field of aptasensors (Wilner and Zayats 2007). Aptamers can be divided into those created from polymers of nucleic acids (RNA and DNA) or amino acids (peptides). Nucleic acid aptamers are small synthetic oligonucleotides of up to 100 base pairs that can bind specifically to a variety of targets, such as whole cells, proteins, drugs, toxins and low molecular weight compounds, with affinities in the nanomolar and sub-nanomolar ranges. They can bind with a high affinity and specificity to a target molecule through complementary shape interactions as a result of their three-dimensional shape. Nucleic acid aptamers are produced by an in vitro process called systematic evolution of ligands by exponential enrichment (SELEX) (Stoltenburg et al. 2007). These aptamers can provide high molecular discrimination in being able to distinguish differences in a methyl group between two compounds (Jenison et al. 1994). Aptamers offer several distinct advantages compared with antibodies or receptors. They are isolated in vitro, and the targets are of a broad spectrum. Following aptamer selection, DNA aptamers with a long shelf life can be prepared with high batch consistency and low cost by automated DNA synthesis, and RNA aptamers can be prepared by simple in vitro transcription. Aptamers are also simple to modify and introduce various reactive groups, affinity tags or reporting moieties essential for biosensor applications. Nucleic acid aptamers have a limited number of structures compared with peptide aptamers, but offer better structural stability. Protein scaffolds are an adaptation of peptide aptamers that may overcome stability issues. Peptide aptamers still require biological systems for selection purposes. Aptamers have been demonstrated in therapeutic applications, in the field of separation chemistry, in environmental and food analysis of chemical contaminants and toxins, and in a number of aptasensor applications (Tombelli et al. 2007; Wilner and Zayats 2007; Stead et al. 2010). These binders are now developing rapidly and may be the one binder group that in future replaces antibodies for diagnostic and detection applications. To date, they offer the best opportunity of finding a single binder for the detection of the whole PSP toxin family or individual binders for each of the varying toxic groups within this family.
Conclusion
Optical SPR biosensor analysis for PSP toxin detection has been demonstrated over the past five years to be a highly effective rapid screening method that has the potential to reduce the vast number of mouse bioassays performed worldwide. This review has highlighted the key problems associated with antibody cross-reactivity in over- and underestimating total toxicity and how, by using a dual antibody binder system with a generic surface, there is the potential to adjust the cross-reactivity profile to help overcome this problem. Alternative non-antibody-based binders, however, may offer a complete non-animal-based methodology for the detection of PSP toxins by SPR. In addition, advances in SPR technology, with the development of multiplexing multichannel instruments, could help resolve some of the difficulties in binder specificity not relating to toxicity. Multiplex multichannel SPR biosensors with highly designed binders such as nucleic acid aptamers (Mok and Li 2008) may be the way forward not only for monitoring phycotoxins, but also for a wide variety of food additives and contaminants.
Acknowledgements
This study was funded by the European Commission as part of the 6th Framework Programme Integrated Project BioCop (Contract Number FOOD-CT-2004-06988) and the 7th Framewok Project CONffIDENCE (Contract Number 211326).
References
- Abery J. Detecting the molecular ties that bind. Modern Drug Disc. 2001;4:34–36. [Google Scholar]
- Association of Analytical Communities (AOAC) Official methods of analysis of AOAC International. 18th ed. Gaithersburg (MD): AOAC International; 2005a. AOAC Official Method 959.08. Paralytic shellfish poison. Biological method. Section 49.10.01. [Google Scholar]
- Association of Analytical Communities (AOAC) Official methods of analysis of AOAC International. 18th ed. Gaithersburg (MD): AOAC International; 2005b. AOAC Official Method 2005.06. Paralytic shellfish poisoning toxins in shellfish. Section 49.10.03. [Google Scholar]
- Asp TN, Larsen S, Aune T. Analysis of PSP toxins in Norwegian mussels by a post-column derivatization HPLC method. Toxicon. 2004;43(3):319–327. doi: 10.1016/j.toxicon.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Baggiani C, Anfossi L, Giovannoli C. Molecular imprinted polymers as synthetic receptors for the analysis of myco- and phyco-toxins. Analyst. 2008;133:719–730. doi: 10.1039/b711352h. [DOI] [PubMed] [Google Scholar]
- Bell TW, Hext NM. Supramolecular optical chemo-sensors for organic analytes. Chem Soc Rev. 2004;33:589–598. doi: 10.1039/b207182g. [DOI] [PubMed] [Google Scholar]
- Botana LM, Alfonso A, Botana A, Vieytes MR, Vale C, Vilarino N, Louzao C. Functional assays for marine toxins as an alternative, high-throughput screening solution to animal tests. Trends in Anal Chem. 2009;28(5):603–611. [Google Scholar]
- Bürk C, Usleber E, Dietrich R, Märtlbauer E. Production and characterisation of antibodies against neosaxitoxin utilizing a novel immunogen synthesis procedure. Food Agr Immunol. 1995;7:315–322. [Google Scholar]
- Campas C, Prieto-Simon B, Marty JL. Biosensors to detect marine toxins: assessing seafood safety. Talanta. 2007;72:884–895. doi: 10.1016/j.talanta.2006.12.036. [DOI] [PubMed] [Google Scholar]
- Campbell K, Bürk C, Dietrich R, Elliott CT. Use of two antibodies to enhance cross-reactivity for paralytic shellfish poisoning toxins using optical biosensor technology. Unpublished data.
- Campbell K, Haughey SA, van den Top H, van Egmond H, Vilarino N, Botana L, Elliott CT. Single laboratory validation of a surface plasmon resonance biosensor screening method for paralytic shellfish poisoning toxins. Anal Chem. 2010;82(7):2977–2988. doi: 10.1021/ac1000338. [DOI] [PubMed] [Google Scholar]
- Campbell K, Huet AC, Charlier C, Higgins C, Delahaut P, Elliott CT. Comparison of ELISA and SPR biosensor technology for the detection of paralytic shellfish poisoning toxins. J Chrom B. 2009;877:4079–4089. doi: 10.1016/j.jchromb.2009.10.023. [DOI] [PubMed] [Google Scholar]
- Campbell K, Stewart LD, Fodey TL, Haughey SA, Doucette GJ, Kawatsu K, Elliott CT. Assessment of specific binding proteins suitable for the detection of paralytic shellfish poisons using optical biosensor technology. Anal Chem. 2007;79:5906–5914. doi: 10.1021/ac070342o. [DOI] [PubMed] [Google Scholar]
- Carlson RE, Lever ML, Lee BW, Guire PE. Development of immunoassays for paralytic shellfish poisoning. A radio-immunoassay for saxitoxin. In: Ragelis EP, editor. Seafood toxins. Washington (DC): American Chemical Society; 1984. pp. 181–192. [Google Scholar]
- Cembella A, Parent Y, Jones D, Lamoureux G. Specificity and cross-reactivity of an absorption inhibition enzyme linked immunoassay for the detection of paralytic shellfish toxins. In: Granéli E, Sundstrom B, Edler L, Anderson DM, editors. Toxic marine phytoplankton. New York (NY): Elsevier; 1990. pp. 339–344. [Google Scholar]
- Cestele S, Catterall WA. Molecular mechanisms of neurotoxin action on voltage-gated sodium channels. Biochimie. 2000;82:883–892. doi: 10.1016/s0300-9084(00)01174-3. [DOI] [PubMed] [Google Scholar]
- Chen H, Kim YS, Keum SR, Kim SH, Choi HJ, Lee J, An WG, Koh K. Surface plasmon spectroscopic detection of saxitoxin. Sensors. 2007;7:1216–1223. [Google Scholar]
- Chianella I, Piletsky SA, Tothill IE, Chen B, Turner APF. MIP based solid phase extraction cartridges combined with MIP based sensors for the detection of microcystin-LR. Biosens Biolectron. 2003;18:119–127. doi: 10.1016/s0956-5663(02)00165-3. [DOI] [PubMed] [Google Scholar]
- Christian B, Luckas B. Determination of marine biotoxins relevant for regulations: from the mouse bioassay to coupled LC-MS methods. Anal Bioanal Chem. 2008;391:117–134. doi: 10.1007/s00216-007-1778-x. [DOI] [PubMed] [Google Scholar]
- Chu FS, Fan TSL. Indirect enzyme-linked immuno-sorbent assay for saxitoxin in shellfish. J AOAC Int. 1985;68:13–16. [PubMed] [Google Scholar]
- Chu FS, Hsu KH, Huang X, Barrett R, Allison C. Screening of paralytic shellfish poisoning toxins in naturally contaminated samples with three different direct competitive enzyme linked immunosorbent assays. J Agric Food Chem. 1996;44:4043–4047. [Google Scholar]
- Chu FS, Huang X, Hall S. Production and characterisation of antibodies against neosaxitoxin. J AOAC Int. 1992;75:341–345. [Google Scholar]
- De Jong LAA, Uges DRA, Franke JP, Bischoff R. Receptor ligand binding assays: Technologies and applications. J Chrom B. 2005;829:1–25. doi: 10.1016/j.jchromb.2005.10.002. [DOI] [PubMed] [Google Scholar]
- Deeds JR, Landsberg JH, Etheridge SM, Pitcher GC, Longan SW. Non-traditional vectors for paralytic shellfish poisoning. Mar Drugs. 2008;6:308–348. doi: 10.3390/md20080015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell'Aversano C, Walter JA, Burton IW, Stirling DJ, Fattorusso E, Quilliam MA. Isolation and structure elucidation of new and unusual saxitoxin analogues from mussels. J Nat Prod. 2008;71(9):1518–1523. doi: 10.1021/np800066r. [DOI] [PubMed] [Google Scholar]
- Diener M, Erler K, Christian B, Luckas B. Application of a new zwitterionic hydrophilic interaction chromatography column for the determination of paralytic shellfish poisoning toxins. J Sep Sci. 2007;30:1821–1826. doi: 10.1002/jssc.200700025. [DOI] [PubMed] [Google Scholar]
- Dietrich R, Usleber E, Burk C, Martlbauer E. In: Immunoassays for residue analysis: food safety. Beier RC, Stanker LH, editors. Washington, DC: American Chemical Society; 1996. pp. 395–403. [Google Scholar]
- Doucette GJ, Logan ML, Ramsdell JS, van Dolah FM. Development and preliminary validation of a microtiter plate-based receptor binding assay for paralytic shellfish poisoning toxins. Toxicon. 1997;35:625–636. doi: 10.1016/s0041-0101(96)00189-4. [DOI] [PubMed] [Google Scholar]
- Dubois M, Demoulin L, Charlier C, Singh G, Godefroy SB, Campbell K, Elliott CT, Delahaut Ph. Development of ELISAs for detecting domoic acid, okadaic acid, and saxitoxin and their applicability for the detection of marine toxins in samples collected in Belgium. Food Addit Contamin. 2010;27(6):859–868. doi: 10.1080/19440041003662881. [DOI] [PubMed] [Google Scholar]
- Etheridge SM. Paralytic shellfish poisoning: Seafood safety and human health perspectives. Toxicon. 2010;56:108–122. doi: 10.1016/j.toxicon.2009.12.013. [DOI] [PubMed] [Google Scholar]
- European Commission. EC Regulation No. 853/2004 of the European Parliament and of the Council of April 29, 2004 laying down specific hygiene rules for food of animal origin. Off J Eur Comm. 2004;139:55–205. [Google Scholar]
- European Food Safety Authority (EFSA) Scientific opinion of the Panel on Contaminants in the Food Chain on a request from the European Commission on marine biotoxins in shellfish-saxitoxin group. EFSA J. 2009;1019:1–76. [Google Scholar]
- Fonfria ES, Vilarino N, Campbell K, Elliott C, Haughey SA, Ben-Gigirey B, Vieites JM, Kawatsu K, Botana LM. Paralytic shellfish poisoning detection by surface plasmon resonance-based biosensors in shellfish matrices. Anal Chem. 2007;79:6303–6311. doi: 10.1021/ac070362q. [DOI] [PubMed] [Google Scholar]
- Garthwaite I, Ross KM, Miles CO, Briggs LR, Towers NR, Borrell T, Busby P. Integrated enzyme-linked immunosorbent assay screening system for amnesic, neurotoxic, diarrhetic, and paralytic shellfish poisoning toxins found in New Zealand. J AOAC Int. 2001;84(5):1643–1648. [PubMed] [Google Scholar]
- Gawley RE, Mao H, Haque M, Thorne JB, Pharr JS. Visible fluorescence chemosensor for saxitoxin. J Org Chem. 2007;72(6):2187–2191. doi: 10.1021/jo062506r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gawley RE, Pinet S, Cardona CM, Datta PK, Ren T, Guida WC, Nydick J, Leblanc RM. Chemosensors for the marine toxin saxitoxin. J Am Chem Soc. 2002;124(45):13448–13453. doi: 10.1021/ja027507p. [DOI] [PubMed] [Google Scholar]
- Genenah AA, Shimizu Y. Specific toxicity of paralytic shellfish poisons. J Agric Food Chem. 1981;29:1289–1291. doi: 10.1021/jf00108a047. [DOI] [PubMed] [Google Scholar]
- Gestwicki JE, Hsieh HV, Pitner JB. Using receptor conformational change to detect low molecular weight analytes by surface plasmon resonance. Anal Chem. 2001;73:5732–5737. doi: 10.1021/ac0105888. [DOI] [PubMed] [Google Scholar]
- Hall AJ, Emgenbroich M, Sellergren B. Imprinted polymers. Top Curr Chem. 2005;249:317–349. [Google Scholar]
- Henry OYF, Cullen DC, Piletsky SA. Optical interrogation of molecularly imprinted polymers and development of MIP sensors: a review. Anal Bioanal Chem. 2005;382(4):947–956. doi: 10.1007/s00216-005-3255-8. [DOI] [PubMed] [Google Scholar]
- Huang X, Hsu KH, Chu FS. Direct competitive enzyme linked immunosorbent assay for saxitoxin and neosaxitoxin. J Agri Food Chem. 1996;44:1029–1035. [Google Scholar]
- Jellett JF, Doucette LI, Belland ER. The MIST (TM) shipable cell bioassay kits for PSP: an alternative to the mouse bioassay. J Shellfish Res. 1998;17(5):1653–1655. [Google Scholar]
- Jenison RD, Gill SC, Pardi A, Polisky B. High resolution molecular discrimination by RNA. Science. 1994;263:1425–1434. doi: 10.1126/science.7510417. [DOI] [PubMed] [Google Scholar]
- Johnston HM, Frey PA, Angelotti R, Campbell JE, Lewis KH. Haptenic properties of paralytic shellfish poison conjugated to proteins by formaldehyde treatment. Proc Soc Exp Biol Med. 1964;117:425–430. doi: 10.3181/00379727-117-29599. [DOI] [PubMed] [Google Scholar]
- Karlsson R. Real-time competitive kinetic analysis of interactions between low molecular weight ligands in solution and surface immobilised receptors. Anal Biochem. 1994;221:142–151. doi: 10.1006/abio.1994.1390. [DOI] [PubMed] [Google Scholar]
- Karlsson R. SPR for molecular interaction analysis: a review of emerging application areas. J Mol Recognit. 2004;17(3):151–161. doi: 10.1002/jmr.660. [DOI] [PubMed] [Google Scholar]
- Kawatsu K, Hamano Y, Sugiyama A, Hashizume K, NoGuchi T. Development and application of an enzyme immunoassay based on a monoclonal antibody against gonyautoxin components of paralytic shellfish poisoning toxins. J Food Protect. 2002;65:1304–1308. doi: 10.4315/0362-028x-65.8.1304. [DOI] [PubMed] [Google Scholar]
- Lawrence JF, Niedzwiadek B. Quantitative determination of paralytic shellfish poisoning toxins in shellfish using prechromatographic oxidation and liquid chromatography with fluorescence detection. J AOAC Int. 2001;84:1099–1108. [PubMed] [Google Scholar]
- Lawrence JF, Niedzwiadek B, Menard C. Quantitative determination of paralytic shellfish poisoning toxins in shellfish using prechromatographic oxidation and liquid chromatography with fluorescence detection: interlaboratory study. J AOAC Int. 2004;87:83–100. [PubMed] [Google Scholar]
- Lawrence JF, Niedzwiadek B, Menard C. Quantitative determination of paralytic shellfish poisoning toxins in shellfish using prechromatographic oxidation and liquid chromatography with fluorescence detection: collaborative study. J AOAC Int. 2005;88:1714–1732. [PubMed] [Google Scholar]
- Laycock MV, Donovan MA, Easy DJ. Sensitivity of lateral flow tests to mixtures of saxitoxins and applications to shellfish and phytoplankton monitoring. Toxicon. 2010;55:597–605. doi: 10.1016/j.toxicon.2009.10.014. [DOI] [PubMed] [Google Scholar]
- Llewellyn LE. Saxitoxin, a toxic marine natural product that targets a multitude of receptors. Nat Prod Rep. 2006a;23:200–222. doi: 10.1039/b501296c. [DOI] [PubMed] [Google Scholar]
- Llewellyn LE. The behaviour of mixtures of paralytic shellfish toxins in competitive binding assays. Chem Res Toxicol. 2006b;19:661–667. doi: 10.1021/tx050277i. [DOI] [PubMed] [Google Scholar]
- Llewellyn LE, Doyle J. Microtitre plate assay for paralytic shellfish toxins using saxiphilin: gauging the effects of shellfish extract matrices, salts and pH upon assay performance. Toxicon. 2001;39:217–224. doi: 10.1016/s0041-0101(00)00118-5. [DOI] [PubMed] [Google Scholar]
- Lotierzo M, Henry OYF, Piletsky S, Tothill I, Cullen D, Kania M, Hock B, Turner APF. Surface plasmon resonance sensor for domoic acid based on grafted imprinted polymer. Biosens Bioelectron. 2004;20(2):145–152. doi: 10.1016/j.bios.2004.01.032. [DOI] [PubMed] [Google Scholar]
- Mahony JO, Nolan K, Smyth MR, Mizaikoff B. Molecularly imprinted polymers – potential and challenges in analytical chemistry. Anal Chim Acta. 2005;534:31–39. [Google Scholar]
- Marchesini GR, Hooijerink H, Haasnoot W, Nielen MWF, Buijs J, Campbell K, Elliott CT, Nielen MWF. Towards surface plasmon resonance biosensing combined with bioaffinity-assisted nano HILIC liquid chromatography/time-of-flight mass spectrometry identification of paralytic shellfish poisons. Trends in Anal Chem. 2009;28(6):792–803. [Google Scholar]
- Michalski M. Paralytic shellfish poisoning as a health risk. Medycyna Weterynaryjna. 2007;63:1530–1533. [Google Scholar]
- Micheli L, Di Stefano S, Moscone D, Palleschi G, Marini S, Coletta M, Draisci R, Quadri FD. Production of antibodies and development of highly sensitive formats of enzyme immunoassay for saxitoxin analysis. Anal Bioanal Chem. 2002;373:678–684. doi: 10.1007/s00216-002-1399-3. [DOI] [PubMed] [Google Scholar]
- Minkin VI, Dubonosov AD, Bren VA, Tsukanov AV. Chemosensors with crown ether-based receptors. ARKIVOC. 2008;4:90–102. [Google Scholar]
- Mok W, Li Y. Recent progress in nucleic acid based biosensors and bioassays. Sensors. 2008;8:7050–7084. doi: 10.3390/s8117050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngundi MM, Kulagina NV, Anderson GP, Taitt CR. Nonantibody-based recognition: alternative molecules for detection of pathogens. Expert Rev Proteomics. 2006;3(5):511–524. doi: 10.1586/14789450.3.5.511. [DOI] [PubMed] [Google Scholar]
- Oshima Y. Post column derivatization liquid chromatography method for paralytic shellfish toxins. J AOAC. Int. 1995;78:528–532. [Google Scholar]
- Powell CL, Doucette GJ. A receptor binding assay for paralytic shellfish poisoning toxins: recent advances and applications. Natural Toxins. 1999;7:393–400. doi: 10.1002/1522-7189(199911/12)7:6<393::aid-nt82>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Ramstrom O, Skudar K, Haines J, Patel P, Bruggemann O. Food analyses using molecularly imprinted polymers. J Agric Food Chem. 2001;49:2105–2114. doi: 10.1021/jf001444h. [DOI] [PubMed] [Google Scholar]
- Rawn DFK, Niedzwiadek B, Campbell K, Higgins HC, Elliott CT. Evaluation of surface plasmon resonance relative to high pressure liquid chromatography for the determination of paralytic shellfish toxins. J Agric Food Chem. 2009;57:10022–10031. doi: 10.1021/jf902176q. [DOI] [PubMed] [Google Scholar]
- Reis Costa P, Baugh KA, Wright B, RaLonde R, Nance SL, Tatarenkova N, Etheridge SM, Lefebvre KA. Comparative determination of paralytic shellfish toxins (PSTs) using five different toxin detection methods in shellfish species collected in the Aleutian Islands, Alaska. Toxicon. 2009;54:313–320. doi: 10.1016/j.toxicon.2009.04.023. [DOI] [PubMed] [Google Scholar]
- Ruberu SR, Liu YG, Wong CT, Perera SK, Langlois GW, Doucette GJ, Powell CL. Receptor binding assay for paralytic shellfish poisoning toxins: optimization and inter-laboratory comparison. J AOAC Int. 2003;86:737–745. [PubMed] [Google Scholar]
- Schneider E, Terplan G, Laycock MV. Two formats of enzyme immunoassay for the detection of saxitoxin and other paralytic shellfish poisoning toxins. Food Addit Contamin. 1995;12(3):405–413. doi: 10.1080/02652039509374322. [DOI] [PubMed] [Google Scholar]
- Stead SL, Ashwin H, Johnston B, Tarbin JA, Sharman M, Kay J, Keely BJ. An RNA-aptamer-based assay for the detection and analysis of malachite green and leucomalachite green residues in fish tissue. Anal Chem. 2010;82(7):2652–2660. doi: 10.1021/ac902226v. [DOI] [PubMed] [Google Scholar]
- Stoltenburg R, Reinemann C, Strehlitz B. SELEX — a revolutionary method to generate high affinity nucleic acid ligands. Biomol Eng. 2007;24:381–403. doi: 10.1016/j.bioeng.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Subrahmanyam S, Piletsky SA, Turner AP. Application of natural receptors in sensors and assays. Anal Chem. 2002;74:3942–3951. doi: 10.1021/ac025673+. [DOI] [PubMed] [Google Scholar]
- Tombelli S, Minunni M, Mascini M. Analytical applications of aptamers. Biosens Bioelectron. 2005;20:2424–2434. doi: 10.1016/j.bios.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Tombelli S, Minunni M, Mascini M. Aptamers-based assays for diagnostics, environmental and food analysis. Biomol Eng. 2007;24(2):191–200. doi: 10.1016/j.bioeng.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249(4968):505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- Usleber E, Dietrich R, Bürk C, Schneider E, Märtlbauer E. Immunoassay methods for paralytic shellfish poisoning toxins. J AOAC Int. 2001;84(5):1649–1656. [PubMed] [Google Scholar]
- Usleber E, Schneider E, Terplan G. Effect of heterologous paralytic shellfish poisoning toxin enzyme conjugates on the cross-reactivity of a saxitoxin enzyme immunoassay. Lett Appl Microbiol. 1991;13:275–277. [Google Scholar]
- van Dolah FM, Leighfield TA, Doucette GJ, Bean L, Niedzwiadek B, Rawn DF. Single-laboratory validation of the microplate receptor binding assay for paralytic shellfish toxins in shellfish. J AOAC Int. 2009;92:1705–1713. [PubMed] [Google Scholar]
- van Egmond HP, van Apeldoorn ME, Speijers GJA. Marine biotoxins. Geneva (Switzerland): Food and Agriculture Organization (FAO) of the United Nations. 2004. Food and Nutrition Paper No. 80.
- Vieites JM, Botana LM, Vieytes MR, Leira FJ. Canning process that diminishes paralytic shellfish poison in naturally contaminated mussels (Mytilus gallo-provincialis) J Food Protect. 1999;62:515–519. doi: 10.4315/0362-028x-62.5.515. [DOI] [PubMed] [Google Scholar]
- Wang W, Jia LY. Progress in aptamer screening methods. Chin J Analyt Chem. 2009;37(3):454–460. [Google Scholar]
- Wiegele JB, Barchi RL. Analysis of saxitoxin binding in isolated rat synaptosomes using a rapid filtration assay. Fed Eur Biochem Soc Lett. 1978;91:310–314. doi: 10.1016/0014-5793(78)81199-5. [DOI] [PubMed] [Google Scholar]
- Wilner I, Zayats M. Electronic aptasensors. Agnew Chem Int Ed. 2007;46:6408–6418. doi: 10.1002/anie.200604524. [DOI] [PubMed] [Google Scholar]