

Viruses infect specific cells within host organisms, replicate, destroy the cells, and spread from cell to cell in infectious cycles, thus causing disease.[1] These viral properties have inspired synthetic designs of various delivery vehicles,[2–6] particularly for toxic anticancer agents that exhibit numerous side effects. Drug-delivery vehicles often mimic viral aspects, such as size and surface properties, to improve cell entry and residence within the body before being cleared.[2–6] Recent efforts in biomimetic drug-carrier design have aimed to endow advanced functionality.[3] Herein, we describe a synthetic nanosized polymer vehicle that mimics viral properties more significantly than any known delivery systems so far reported. This virus-mimetic nanogel (VM-nanogel) should prove valuable for treating several major disease classes, such as tumors, with greater efficacy.
The VM-nanogel we have developed consists of a hydrophobic polymer core and two layers of hydrophilic shell (Figure 1). A particle core made of poly(L-histidine-co-phenylalanine) (poly(His32-co-Phe6),[4] where the numerals indicate the numbers of His and Phe units in the block) is loaded with a model anticancer drug, doxorubicin (DOX). Polyethylene glycol (PEG; number-average molecular weight 2000 Da) forms the inner shell. One PEG end is linked to the core polymer and another to bovine serum albumin (BSA), which forms a capsid-like outer shell.[5] The core and inner shell were constructed by an oil-in-water emulsion method.[2,6] A single BSA molecule could be linked to multiple PEG ends, as depicted in Figure 1. The BSA and PEG components on the surface may help to avoid potential immune responses.[7]
Figure 1.
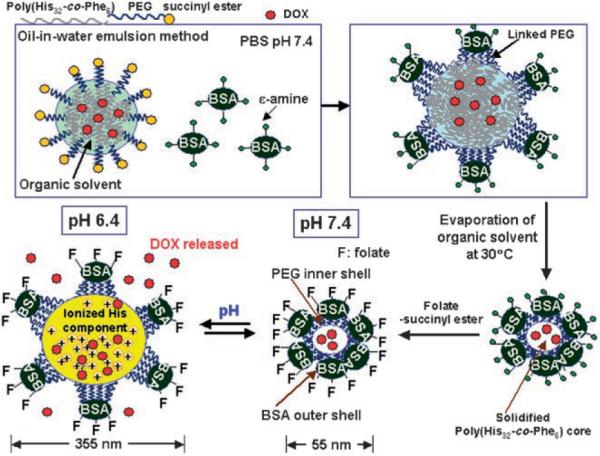
Preparation and structure of the VM-nanogel. See text for details; PBS = phosphate-buffered saline.
Extensive study of the biocompatibility and toxicity of PEG-b-poly(His-co-Phe) is under way in our laboratory, and no apparent cytotoxicity or systemic toxicity have been found in mouse models for PEG-b-polyHis. The BSA shell is subsequently conjugated with folate ligands for specific recognition of the folate receptor (FR), which is overexpressed on many tumors.[8] Folate conjugation will contribute to the enhancement of the antitumor activity of VM-nanogels by providing an active entry mechanism.[9]
The sensitivity of VM-nanogel to the pH of the solution was assessed by the optical transmittance of an aqueous VM-nanogel solution (Figure 2a). The percentage transmittance (% T) dropped sharply over the narrow pH range of 6.6–6.4 upon reduction of the pH from 7.4 to 5.5. When the pH was increased again, % T recovered with a small hysteresis. The observed change in % T is attributed to the dynamic dimensional changes of the VM-nanogel with pH. The pKb of L-histidine in the core poly(His32-co-Phe6) is near 6.4,[4] as measured by an acid–base titration method. At high pH, the VM-nanogel core is rigid. However, the core swells spontaneously when the polymer His residues are protonated at lower pH values[4] (Figure 1).
Figure 2.

a) Relative transmittance of the VM-nanogel solution measured at selected pH values with respect to transmittance at pH 7.4. The solution pH is gradually changed from 7.4 to 5.5 (●) and from pH 5.5 to 7.4 (◯). b) Size changes of VM-nanogel (determined by dynamic light scattering) with pH value. The solution pH is successively adjusted in the order A (pH 7.4)→B (pH 6.8)→A→B (upper row) and A (ph 7.4)→C (pH 6.4)→A→C (lower row), with equilibration for 1 h at each pH. The parcticle size distribution at each pH was obtained from the particle scattering intensity. c) Confocal slice images of the VM-nanogels in a living cell. Ovarian A2780 cells were incubated with FITC-labeled VM-nanogels (10 μgmL−1, pH 7.4) and confocal slice images were obtained at 60, 80, and 90 min.
When the pH was cycled between 7.4 and 6.8 (the typical cytosolic pH range[10]), the VM-nanogels reversibly swelled and shrank. However, the magnitude of the reversible changes of the VM-nanogel between pH 7.4 and 6.4 (early endosomal pH[11]) was enormous (Figure 2b): 55 nm in diameter at pH 7.4 and 355 nm at pH 6.4. Apparently, the structure of the VM-nanogel is maintained during this size change by virtue of the capsid-like[5] BSA shell that holds multiple interior core block copolymers by PEG linkages, which prevents them from dissociation even after ionization of the core His component at lower pH values. Flexible tethering of the BSA corona to the pH-sensitive core by the PEG units accommodates a large amount of expansion without particle disassembly. Hydrophobic interaction among Phe components in the core[4] is also thought to help prevent dissociation.
VM-nanogels are designed to interact with the target cells through folate–FR coupling[8,9] and subsequent localization within early endosomes.[8] Figure 2c presents the coexistence of unswollen and swollen VM-nanogels in live human ovarian carcinoma A2780 cells, traced by a series of confocal slice images taken for a single cell. The red and white arrows indicate unswollen and swollen VM-nanogels, respectively. Swollen VM-nanogels appear after 60 min as a hollow sphere, as a result of surface-localized fluorescein isocyanate (FITC, green fluorescence) on the BSA shell. The dimensions of the VM-nanogels in both states coincide with the particle sizes measured by light scattering (Figure 2b).
This substantial volumetric expansion of VM-nanogels and the known proton buffering effect of polyHis or poly-(His32-co-Phe6)[4] are considered able to physically disrupt endosomal membranes (see the Supporting Information). This allows the VM-nanogels to transfer from the endosomes to the cytosol, where the VM-nanogels rapidly shrink back to their original size. Furthermore, the pH-induced reversible swelling/deswelling of the core is closely linked to the release rate of incorporated DOX[9] (see the Supporting Information). The VM-nanogels released a significant amount of DOX at endosomal pH (e.g., pH 6.4), while reducing the DOX release rate at cytosolic or extracellular pH (e.g., pH 6.8–7.4).
To verify VM-nanogel migration to neighboring cells (Figure 3), an A2780/AD[12] cell cluster seeded on a glass plate was pretreated with DOX-loaded VM-nanogels for primary infection for 4 h (see the Supporting Information). A neighboring glass plate with adherent untreated cells was used as a potential site for secondary VM-nanogel infection. The primary infected cell group (Figure 3, VM-nanogel, A) can be seen by confocal microscopy, where cells appear red because of the intrinsic DOX fluorescence within the VM-nanogel. DOX spreads evenly within the cells, even in the nucleus, while the untreated cell group appears dark (Figure 3, VM-nanogel, B-0). After co-incubation of the primary VM-nanogel “infected” cells and untreated cells for 20 h, the red DOX fluorescence appears in the untreated cell group, even in the cell nucleus (Figure 3, VM-nanogel, B-1). This may be because DOX action on the cells induces apoptosis and the VM-nanogels are released from the dead cells for subsequent infection in neighboring cells (see the Supporting Information).
Figure 3.
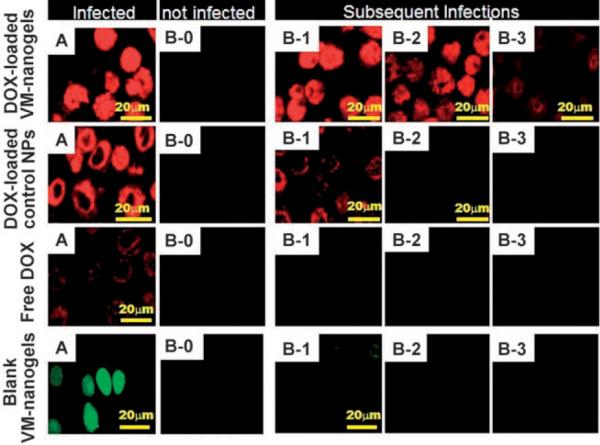
Migration of DOX-loaded VM-nanogels (equivalent DOX 10 μgmL−1) from infected A2780/AD cells to untreated cells. A2780/ AD cells grown on a glass slide fragment (dimensions 1 × 1 cm; A) were pretreated with DOX-loaded VM-nanogels for 4 h. After washing the treated cell plate with PBSof pH 7.4, it was co-cultured with fresh cells (B-0) seeded on another glass slide fragment for 20 h in fresh culture medium. The two plates were 1 mm apart in a culture dish, continuously sharing the culture medium. B-0 became B-1 after 20 h of co-culture with A. The procedure was repeated to yield B-2 and B-3 in subsequent cycles (that is, B-2 refers to the cells on a plate co-cultured with B-1 for 20 h). Three control experiments were conducted: DOX-loaded control NPs (equivalent DOX 10 μgmL−1), free DOX (10 μgmL−1), and FITC-labeled blank VM-nanogels (no DOX, VM-nanogel 50 μgmL−1). All confocal images were sliced.
The infection procedure was repeated and another infection cycle occurred (Figure 3, VM-nanogel, B-2, B-3). In addition, the weakened “infection signal” (that is, fluorescence) over repeated cycles is most probably caused by the loss of VM-nanogel by medium replacement, DOX depletion from the VM-nanogel, and less release of DOX during each cell cycle, which eventually falls below the drug concentration required to induce cell death.
Three control experiments support the unique repeated infection capabilities of the VM-nanogels (Figure 3). Control nanoparticles (NPs; ≈70 nm in diameter),[13] which comprise a pH-insensitive central core (poly(L-lactic acid) and DOX) and a hydrophilic shell (PEG–folate), have no capability for endosomal escape, thus resulting in cytosolically trapped DOX[13] and limited cell death. The control NPs were only able to produce secondary infection very weakly in the first cycle after 20 h.[13] Experiments with free DOX produced limited fractions of DOX diffusing into cells in the primary infection cycle because of the drug-resistant, active P-glycoprotein DOX efflux pumps in A2780/AD cells.[12] A third control utilized VM-nanogels without DOX (Figure 3, blank VM-nanogels). After entry into the primary infected cells, the blank VM-nanogel with FITC (green color) distributed evenly within the cells.[13] No noticeable secondary infection was detected because of a lack of cell death[13] and lytic release of VM-nanogels.
Recently, Nagasaki et al. reported pH-sensitive PEGy-lated nanogels with a sharp volume transition at pH values around 7.0, as a result of the protonation of cross-linked poly[2-(N,N-(diethylamino)ethyl methacrylate)] in the core.[14,15] These nanogels facilitated triggered DOX release at endo/lysosomal pH for effective chemotherapy.[15] However, these nanogels did not show cell specificity and recycled infection.
Importantly, when a drug carrier completely releases its payload upon cell internalization to achieve high drug concentrations within the cells,[9,13] this often results in excessive drug over the threshold of its cytotoxicity. The excess drug could be ineffective against neighboring cells if these cells are drug resistant.[12] However, the VM-nanogel system may be able to maximize the effect of the drug by pulsatile drug release modulated by pH and repeated entry into cancerous cells. Once the VM-nanogel escapes the endosomes, the DOX release rate is minimized. This indicates that the amount of DOX released while the VM-nanogels are entrapped in the endosomes for approximately 1–1.5 h (Figure 2c) before endosomal disruption should be high enough to provide a drug concentration sufficient to kill cancer cells. The amount of DOX released for 1 h at pH 6.4 in each cycle is expressed in the Supporting Information.
Figure 4 a presents an impressive contrast for cell killing in the various cases. The viability of both drug-sensitive A2780 cells and drug-resistant analogues was below 20 % when treated at a DOX equivalent concentration of 5 μgmL−1 carried by the VM-nanogels. However, DOX carried by the control NPs was not effective in cell killing. Free DOX at the same concentration showed a viability of 30 % for sensitive cells but 80 % for resistant cells. Similar results with free DOX were obtained when the same experiments were conducted at pH 6.8, which is close to the tumor extracellular pH.[4,9,13] The results suggest that the VM-nanogels do not distinguish between sensitive and resistant cells in tumor-cell killing capability, by preserving a sufficient cytosolic dose of DOX.[9] More significantly, the VM-nanogels do not infect (see the Supporting Information) or kill A549 human nonsmall lung carcinoma cells[16] which do not express the FR (Figure 4 b).
Figure 4.

Antitumor activity of DOX-loaded VM-nanogels (white), DOX-loaded control NPs (gray), and free DOX (black). The DOX equivalent content of each sample is 5 μgmL−1. a) A2780 or resistant A2780/AD tumor cells were treated with each sample for 48 h in culture media of pH 7.4 or 6.8. b) A549 cells were treated with each sample for 48 h in culture medium (pH 6.8). Each data point in (a) and (b) represents an average standard deviation (n = 9); ** indicates p<0.05 for free DOX.
In conclusion, the VM-nanogels infected cells in a receptor-dependent manner, effectively killed the tumor cells, and migrated to neighboring cells as a virus does. When coupled with other specific targeting moieties or gene vectors, this VM-nanogel nanotechnology could have great potential for treating solid tumors, inflamed tissues, and other diseases in an effective manner. To confirm and realize this potential, further in vitro evaluations and in vivo investigations are required.
Experimental Section
The synthesis of the VM-nanogels and other experimental procedures are available as Supporting Information.
Supplementary Material
Acknowledgments
We thank Dr. Grainger (Department of Pharmaceutics and Pharmaceutical Chemistry, University of Utah) for his kind suggestions and proofreading. This work was funded by NIH CA122356 and NIH CA101850.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- [1].De Vries P, Uytdehaag FG, Osterhaus AD. J. Gen. Virol. 1988;69:2071–2083. doi: 10.1099/0022-1317-69-8-2071. [DOI] [PubMed] [Google Scholar]
- [2].Vinogradov SV, Bronich TK, Kabanov AV. Adv. Drug Delivery Rev. 2002;54:135–147. doi: 10.1016/s0169-409x(01)00245-9. [DOI] [PubMed] [Google Scholar]
- [3].Zhu J, Xue J, Guo Z, Zhang L, Marchant RE. Bioconjugate Chem. 2007;18:1366–1369. doi: 10.1021/bc700212b. [DOI] [PubMed] [Google Scholar]
- [4].Kim GM, Bae YH, Jo WH. Macromol. Biosci. 2005;5:1118–1124. doi: 10.1002/mabi.200500121. [DOI] [PubMed] [Google Scholar]
- [5].Tkachenko A, Xie H, Franzen S, Feldheim D. Methods Mol. Biol. 2005;303:85–99. doi: 10.1385/1-59259-901-X:085. [DOI] [PubMed] [Google Scholar]
- [6].Peppas NA, Bures P, Leobandung W, Ichikawa H. Eur. J. Pharm. Biopharm. 2000;50:27–46. doi: 10.1016/s0939-6411(00)00090-4. [DOI] [PubMed] [Google Scholar]
- [7].Lu W, Zhang Y, Tan YZ, Hu KL, Jiang XG, Fu SK. J. Controlled Release. 2005;107:428–448. doi: 10.1016/j.jconrel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- [8].Sabharanjak S, Mayor S. Adv. Drug Delivery Rev. 2004;56:1099–1109. doi: 10.1016/j.addr.2004.01.010. [DOI] [PubMed] [Google Scholar]
- [9].Lee ES, Na K, Bae YH. J. Controlled Release. 2005;103:405–418. doi: 10.1016/j.jconrel.2004.12.018. [DOI] [PubMed] [Google Scholar]
- [10].Schwartz MA, Both G, Lechene C. Proc. Natl. Acad. Sci. USA. 1989;86:4525–4529. doi: 10.1073/pnas.86.12.4525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Schmaljohann D. Adv. Drug Delivery Rev. 2006;58:1655–1670. doi: 10.1016/j.addr.2006.09.020. [DOI] [PubMed] [Google Scholar]
- [12].Tijerina M, Fowers K, Kopeckova P, Kopecek J. Biomaterials. 2000;21:2203–2210. doi: 10.1016/s0142-9612(00)00161-7. [DOI] [PubMed] [Google Scholar]
- [13].Mohajer G, Lee ES, Bae YH. Pharm. Res. 2007;24:1618–1627. doi: 10.1007/s11095-007-9277-5. [DOI] [PubMed] [Google Scholar]
- [14].Hayashi H, Iijima M, Kataoka K, Nagasaki Y. Macromolecules. 2004;37:5389–5396. [Google Scholar]
- [15].Oishi M, Hayashi H, Iijima M, Nagasaki Y. J. Mater. Chem. 2007;17:3720–3725. [Google Scholar]
- [16].Oh IK, Mok H, Park TG. Bioconjugate Chem. 2006;17:721–727. doi: 10.1021/bc060030c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.