

Abstract
The E2 component of pyruvate dehydrogenase has been engineered to form a caged, hollow dodecahedral protein assembly, and we have examined the feasibility of this scaffold to be used as a drug delivery system by introducing cysteines to the internal cavity (D381C). Fluorescent dye Alexa Fluor 532 (AF532M) and the antitumor drug doxorubicin were coupled to this internal cavity through maleimides on the guest molecules. The virus-like particle’s structure and stability remained intact after binding of the molecules within the interior of the nanocapsule. The pH-dependent hydrolysis of a hydrazone linkage to doxorubicin allowed 90% drug release from the D381C scaffold within 72 hrs at pH 5.0. Fluorescence microscopy of MDA-MB-231 breast cancer cells indicated significant uptake of the D381C scaffold incorporating AF532M and doxorubicin and suggested internalization of the nanoparticles through endocytosis. We observed that the protein scaffold does not induce cell death, but doxorubicin encapsulated in D381C is indeed cytotoxic, yielding an IC50 of 1.3 ± 0.3 μM. While the majority of particulate-based drug delivery strategies encapsulates drugs within polymeric nanoparticles, our results show the potential of using macromolecular protein assemblies. This approach yields a promising new opportunity for designing highly-defined nanomaterials for therapeutic delivery.
Keywords: Drug delivery, 6-maleimidocaproyl hydrazone derivative of doxorubicin, virus-like particle, pyruvate dehydrogenase, pH-dependent hydrolysis
1. Introduction
Recent advances have applied nanotechnology and nanoparticles as diagnostic and therapeutic agents toward the treatment of cancer.[1,2] As a better understanding of cellular processes at the molecular level is gained, novel nanoscale materials can be designed that exploit molecular and physiological characteristics of tumor cells or tissues to deliver therapeutic compounds more effectively. Traditional particulate drug delivery systems have improved the efficacy of drugs and have included nanoparticle platforms such as polymeric hydrogels, micelles, liposomes, dendrimers, and polymersomes.[3,4]
Ideal nanoparticle dimensions for targeted intracellular delivery have been experimentally determined to be within the range of approximately 25–75 nm.[5,6] Once inside the cell, precise delivery into organelles additionally depends on overcoming other different obstacles, and delivery vehicles with multiple functionalities are needed to overcome individual internal barriers.[7–9] Other functionalities which can be important in nanodelivery include cell-specific targeting,[10] pH-responsive behavior to release the drug and promote endosomal escape,[11] and the ability to carry multiple molecules, such as both therapeutic and imaging agents for simultaneous treatment and visualization of tumors.[12] However, limitations in synthesizing such small nanoparticles often preclude the ability to rationally incorporate multiple functionalities or obtain a uniform size distribution, which in turn restricts available delivery strategies.
In contrast, nature has already created functional macromolecular assemblies and structures that can be manipulated for nanobiotechnology applications. Protein engineering coupled with synthetic chemical strategies can be used to redesign structure and function, yielding particles with very narrow size distributions and multiple functionalities. Virus-like particles and caged protein structures, which are self-assembled from a defined number of stable subunits, have been investigated as nanocarriers for introducing non-native functionalities.[13,14] For example, the interiors of these structures can encapsulate inorganic and macromolecular cargo, such as metals, polymers, and horseradish peroxidase.[15–18] Various moieties can be incorporated on the exterior surface of viruses to display folic acid for specific targeting[19] or different antigenic peptides and epitopes for vaccines.[20,21] Virus-like particles have also been manipulated to incorporate an external peptide or polymer while simultaneously encapsulating guest molecules.[22–24] Although numerous examples of virus-like proteins functionalized with non-native properties have been reported, we are not aware of studies that examine their efficacy in controlled drug release within cancer cells.
We have investigated a virus-like protein scaffold based on the dihydrolipolyl acyltransferase subunit (E2) from pyruvate dehydrogenase[25–29] for its versatility in biomolecular design. The dodecahedral E2 protein cage consists of 60 identical subunits, self-assembling to form a hollow nanoparticle that is 25 nm in diameter. Genetic modifications to the three different interfaces of the scaffold (exterior, subunit-subunit, interior) have been evaluated, demonstrating that these changes can still enable the assembly of the scaffold. Domingo et al. reported the display of antigentic epitopes on the exterior of the E2 scaffold which could induce specific antibodies and T-cell responses.[30,31] By redesigning the interactions of the subunit-subunit interface, we showed the protein assembly can be engineered to remain stable at pH 7.4 but dissociate at acidic pH, and the pH-transition point of this change can be modulated.[26,27] With twelve 5-nm openings per scaffold, guest molecules can diffuse into the internal cavity. Introduction of cysteines to this cavity has enabled covalent coupling and encapsulation of fluorescent dye molecules while still retaining the scaffold’s dodecahedral structure.[25]
In this work, we demonstrate that conjugation of the anti-tumor drug doxorubicin with the E2 nanoparticle can be achieved, and we evaluate the efficacy of this system for drug delivery applications. Drug release from the protein is pH-controlled to coincide with the physiological pH decrease when taken up by cells through the endocytic pathway. This strategy has been reported in many studies,[32–39] most of which are polymer-based carriers. However, the advantages of protein-based macromolecular systems over traditional polymeric ones include their biodegradation into non-toxic amino acid subunits, ability to obtain very narrow size distributions, and degree of architectural and multifunctional control afforded by recombinant synthesis strategies. While the release of doxorubicin from a heat shock protein has been shown in solution,[32] its therapeutic efficacy in cells has not been reported. Here we have engineered the E2 protein scaffold of pyruvate dehydrogenase to encapsulate drug molecules in the internal cavity, and we assess, for the first time, its effect on mammalian cells. The cellular uptake and efficacy of inducing cell death in cancer cells was examined, and our results demonstrate the potential of the E2 nanoparticle platform in drug delivery.
2. Materials and Methods
2.1 Materials
Alexa Fluor 532 C5 maleimide (AF532M), Dulbecco’s phosphate-buffered saline (PBS), 3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), and Hoechst 33342, trihydrochloride trihydrate were purchased from Invitrogen. The (6-maleinimidocaproyl) hydrazone derivative of doxorubicin (DOXO-EMCH, Figure 1A) was synthesized and characterized as previously described,[35,40] and doxorubicin hydrochloride was obtained from Yic-Vic (Hong Kong). Sodium phosphate dibasic, sodium phosphate monobasic, sodium chloride (NaCl), sodium dodecyl sulfate (SDS), N,N-dimethylformamide (DMF), and 1 N hydrochloric acid (HCl) were purchased from EMD. Dimethyl sulfoxide (DMSO) was obtained from Thermo Scientific. Tris(2-carboxyethyl) phosphine hydrochloride (TCEP) was from Pierce. Dulbecco’s Modified Eagle’s Medium (DMEM) was purchased from Sigma. Fetal bovine serum (FBS) and L-glutamine were obtained from Mediatech. Acetic acid, acetonitrile, formic acid, potassium phosphate monobasic, and potassium phosphate dibasic were purchased from Fisher Scientific. Water was purified by a Milli-Q system (Millipore) to a final resistivity of 18.2 MΩ-cm.
Figure 1.
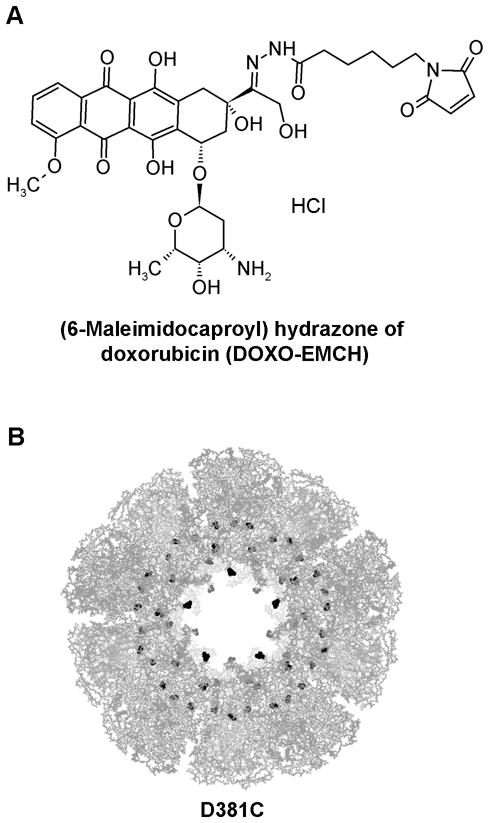
A. Chemical structure of DOXO-EMCH. B. Quaternary protein structure of the E2-D381C nanoparticle, comprising 60 subunits, viewed at the five-fold axis of symmetry. The Protein Data Bank crystallographic file (1b5s) is displayed using PyMOL (DeLano, 2002), and the doxorubicin conjugation sites (amino acid 381) are highlighted in black.
2.2 Expression and purification of protein scaffolds E2-WT and D381C
Introduction of thiols into the interior cavity of the E2 scaffold enables loading of foreign small molecules.[25] The initial E2 scaffold with native wild-type sequence (E2-WT) has only one native cysteine in each subunit (60 subunits per scaffold), but this cysteine is surface-inaccessible due to its buried location. To encapsulate foreign guest molecules within the scaffold cavity, we mutated the aspartic acid at position 381 of E2 scaffold to cysteine, resulting in 60 thiols within the interior cavity (Figure 1B). This mutant scaffold is designated “D381C”.
Experimental details for mutagenesis, expression, and purification of the wild-type control (E2-WT) and the D381C protein scaffolds were performed as described in Dalmau et al.[25] In summary, the gene for the D381C nanoparticle was created using site-directed mutagenesis. This cysteine exists in the internal cavity of the nanoparticle and allows guest molecules to be encapsulated within the protein scaffold. The plasmid containing the E2-WT or D381C gene was transformed into E .coli strain BL21(DE3) (Stratagene), the protein expression was induced, and the cells were harvested and lysed. The protein nanocapsules were purified on a fast protein liquid chromatography (FPLC) system (ÄKTA, Amersham Biosciences), using a Q Sepharose column followed by a Superose 6 PG column in buffer containing 50 mM potassium phosphate (pH 7.4), 100 mM NaCl, 0.02% sodium azide, and 5 mM EDTA, and proteins were concentrated and stored in this solution. Proteins were exchanged into “phosphate solution” in subsequent experiments, which is defined in this protocol as 50 mM potassium phosphate (pH 7.4) with 100 mM NaCl.
2.3 Conjugation of AF532M and DOXO-EMCH to internal cysteines of D381C protein scaffold
We performed conjugation of guest molecules to the D381C protein scaffold using the previously reported protocol as a departure point.[25] For stock solutions, AF532M was dissolved in DMSO, and doxorubicin and DOXO-EMCH were dissolved in 10 mM sodium phosphate (pH 5.8) and used within 30 minutes. To enable conjugation to the cysteines within the internal cavity, thiols in D381C were reduced with 15 molar equivalents of TCEP at room temperature for 1 hr. The protein was mixed with AF532M or DOXO-EMCH for 2 hrs at room temperature at a ratio of one D381C protein subunit to three guest molecules, followed by 4°C incubation overnight. The unreacted AF532M or DOXO-EMCH was removed by dialysis, either using drop dialysis (MF-type cellulose ester membranes, 50 nm pore size, Millipore) for 4 hours, or cellulose ester dialysis membrane (MWCO 100 kDa, Spectra/Por Biotech) in phosphate solution (as defined above) for 48 hrs, with at least three buffer changes.
We quantified the coupling ratio of AF532M and DOXO-EMCH to D381C (abbreviated D381C-AF532M and D381C-DOX, respectively). The protein concentration was measured by a Micro BCA Protein Assay Kit (Pierce) using bovine serum albumin as a standard. The concentrations of conjugated AF532M and DOXO-EMCH were determined by measuring absorbance at 526 nm and 495 nm, respectively (SpectraMax M2, Molecular Devices) and calibrated to standard curves.
2.4 Characterization of D381C-AF532M and D381C-DOX particles
We measured hydrodynamic particle size of D381C, D381C-AF532M, and D381C-DOX using dynamic light scattering (DLS) with a Zetasizer Nano ZS (Malvern Instruments). Measurements were performed on 1 mg/mL protein samples in phosphate solution.
We confirmed the molecular weights of the unconjugated and conjugated protein nanoparticles with electrospray mass spectrometry. To remove salt, 10 μL of 1 mg/mL protein was dialyzed with water for 2 hrs by drop dialysis (MF-type cellulose ester membranes, 50 nm pore size, Millipore). The proteins were mixed with 100 μL of a H2O/acetonitrile (v/v 1/1) solution containing 0.1% formic acid and injected into a Micromass LCT Premier Mass Spectrometer (Waters). The molecular weight was deconvoluted using the “Max Ent” program within MassLynx v. 4.0.
Far-UV circular dichroism (CD) was used to monitor the folding and secondary structure of the protein scaffolds. The measurements were performed on a Jasco 810 spectropolarimeter equipped with a Jasco Peltier temperature controller (Jasco). Proteins were diluted to 0.06 mg/mL in phosphate solution and scanned from 190 nm to 260 nm at a rate of 10 nm/min in 0.1 cm path length quartz cells at 25 °C. To determine thermal stability, the ellipticity at 222 nm was measured as temperature was increased from 40 °C to 95 °C using a scanning rate of 1 °C/min. The apparent midpoint of unfolding transition temperature, Tm, was determined as previously described[25] using an in-house MATLAB program. CD measurements were performed three times and results were averaged.
We used transmission electron microscopy (TEM) to confirm the intact structure of the protein nanoparticles before and after conjugation with AF532M and DOXO-EMCH. Ten μL of proteins (0.03 mg/mL) were deposited on a carbon-Formvar TEM grid (Ted Pella) and negatively-stained for 2 min with 2% uranyl acetate. Images were acquired with a Philips CM-20 transmission electron microscope operating at 80 kV.
2.5 In vitro release of doxorubicin from D381C-DOX at pH 7.4 and pH 5.0
To evaluate the pH-dependent release of doxorubicin from the protein nanoparticles, we dialyzed D381C-DOX at pH 7.4 and pH 5.0. D381C-DOX (0.5 mL, 1.5 mg/mL protein in phosphate solution, 70 μM doxorubicin) was placed in a Slide-A-Lyzer Dialysis Cassette G2 (0.5 mL, MWCO 20 kDa, Thermo Scientific) in 50 mL phosphate solution (50 mM potassium phosphate, 100 mM NaCl) at pH 7.4 or pH 5.0. Experiments were carried out at 4 °C and at constant stirring rate. At different time points, 500 μL of the dialysate was withdrawn and the amount of doxorubicin released was determined by fluorescence spectroscopy. Fluorescence measurements of 100 μL dialysate were performed in triplicate, using excitation and emission wavelengths of 478 nm and 594 nm, respectively, and compared with doxorubicin standard curves. The measured solution containing doxorubicin was returned to the dialysate to bring the volume back up to 50 mL. In vitro release experiments were terminated after 5 days dialysis because no further increase of doxorubicin in the dialysate was detected at pH 5.0 and mass spectrometry of the retentate showed no remaining peak corresponding to doxorubicin-conjugated nanoparticle (D381C-DOX).
Data were plotted and analyzed using KaleidaGraph (Version 4.1, Synergy Software). Free doxorubicin gave a higher fluorescence reading than nanoparticle-encapsulated doxorubicin, an observation which has also been reported by others;[41] thus, 100% release was normalized to the fluorescence of the maximum equilibrium value.
2.6 Imaging of D381C-AF532M, D381C-DOX, and doxorubicin uptake into human breast cancer cells
Cellular assays were performed with the human breast carcinoma cell line MDA-MB-231 (ATCC). For our cellular studies, the growth medium is defined as DMEM supplemented with 10% fetal bovine serum and 1% L-glutamine. We seeded 104 cells per well in 96-well tissue-culture treated plates (BD Falcon) and incubated them at 37°C in 5% CO2 atmosphere. After 16–20 hrs, the growth medium was replaced with 160 μL of fresh growth medium containing D381C-AF532M (at final concentrations of 0.075 mg/mL protein and 5 μM AF532M), and cells were incubated for an additional 0, 12, 24, 48, and 72 hrs. For controls containing no protein nanoparticles, cells were incubated with free AF532M and with growth medium alone.
A similar procedure was used to investigate the uptake of D381C-DOX. Cells were incubated with 160 μL of growth medium containing D381C-DOX at doxorubicin-equivalent concentrations of 0.5, 1.5, and 30 μM for 72 hrs. For controls with no protein nanoparticles, cells were incubated with the equivalent concentrations of free doxorubicin and with growth medium alone.
To image uptake of AF532M and doxorubicin, cells were washed twice with ice-cold PBS and covered with 160 μL of PBS. Fluorescence images were obtained with an Olympus IX51 microscope equipped with a QICAM FAST camera. FITC and TRITC filters were used for D381C-AF532M and D381C-DOX, respectively.
We investigated the intracellular distribution of doxorubicin and D381C-DOX with confocal laser scanning microscopy (CLSM). Cells (104 per well) were seeded in an 8-well Lab-Tek chambered coverglass (Thermo Scientific Nunc) and incubated at 37°C in 5% CO2 for 16–20 hrs. The growth medium was replaced with 160 μL of fresh medium containing doxorubicin (3 μM) or D381C-DOX (at final concentration of 0.065 mg/mL protein and 3 μM equivalent doxorubicin) and incubated for 52 hr. Cells were then incubated with fresh medium (160 μL) containing 0.2 μg/mL Hoechst 33342 for 30 min to stain the nucleus, washed twice with ice-cold PBS, and covered with 160 μL of PBS. CLSM images were obtained using an Olympus Fluoview FV1000, at excitation/emission filter wavelengths of 488 nm/505–605 nm for doxorubicin and 405 nm/430–470 nm for Hoechst 33342.
2.7 Cytotoxicity of D381C-AF532 and D381C-DOX to human breast cancer cells
To evaluate cytotoxicity of D381C-AF532M, we used an MTT assay based on previously described protocols.[42] Cells were seeded at 104 cells per well, grown overnight, and incubated with growth medium containing D381C-AF532M at a final protein concentration of 0.075 mg/ml. In control samples containing no nanoparticles, cells were incubated with the equivalent concentration of free AF532M and with medium alone. After incubating for 0, 24, 48, and 72 hrs, cells were washed twice with medium and covered with 160 μL medium. Twenty-five μL of 5 mg/mL MTT in PBS was added into each well and incubated at 37°C for 2 hrs. The reaction was terminated by adding 70 μL of lysing buffer (20% w/v sodium dodecyl sulfate in 50:50 dimethylformamide/water, with 2.5% acetic acid and 2.5% 1 N HCl). The plates were incubated at 37 °C overnight and the formazan content was measured by absorbance spectroscopy at 570 nm. Percent cell viability was calculated relative to cells grown in media alone, and a minimum of 4 replicates per sample was averaged.
The dose-response and cytotoxicity of D381C-DOX was determined in a similar manner. After seeding, MDA-MB-231 cells were incubated for 48 and 72 hrs with 160 μL of growth medium containing D381C-DOX at final doxorubicin concentrations of 0.005, 0.01, 0.05, 0.15, 0.5, 1.5, 10, 20 and 30 μM. As controls, cells were treated with the same equivalent concentrations of doxorubicin (drug-alone in phosphate solution, no nanoparticles), the same equivalent protein scaffold concentrations (E2-WT protein nanoparticles alone in phosphate solution, no doxorubicin), and the same dilution volumes of the phosphate solution alone. The MTT assay was performed as described above. To calculate the IC50 value, the percent cell viability versus concentration of drug was plotted. This dose-response curve was fitted to a 4-parameter Hill model.
2.8 Statistical analysis
For cytotoxicity studies, samples were performed in replicates of n = 4 or 6, and data were reported as the average ± standard deviation. The differences in cell viabilities between AF532M and D381C-AF532M treated cells across all time points were determined with Student’s t-test. A “P” value below 0.05 is considered statistically different between the groups.
3. Results and Discussion
3.1 D381C protein nanoparticles encapsulating AF532M and doxorubicin remain intact and stable
The D381C protein nanoparticles produced for this investigation were characterized, and results were consistent with those previously published for this variant of the scaffold.[25] In summary, mass spectrometry yielded an average molecular weight of 28,105 Daltons, corresponding to the theoretical value (28,105 Da). CD analysis demonstrated that D381C is folded with comparable secondary structure and high thermal stability as the baseline wild-type scaffold.[25] Correct 60-mer assembly was also confirmed, with an average hydrodynamic diameter of 25.4 ± 0.4 nm (Supporting Information, Figure SI-1) and TEM images showing distinct nanoparticles.
The 25-nm D381C nanoparticle scaffold has 12 openings which are 5 nm in diameter, allowing for small molecules (e.g., dye, drug) to diffuse into the internal cavity. The maleimide groups of AF532M and DOXO-EMCH are expected to form thioether linkages through a Michael addition with the surface-accessible thiols at site cysteine-381, which are located in this cavity. In this set of experiments, we determined the subunit:AF532M ratio for D381C-AF532M to be 1:1.9 to 1:2, corresponding to approximately 120 AF532M molecules per scaffold. For D381C-DOX, the final subunit:DOXO-EMCH ratio ranged from 1:1.4 to 1:1.9, corresponding to 84 to 114 doxorubicin molecules per scaffold. The covalent coupling of guest molecules to the protein scaffold is confirmed by electrospray mass spectrometry, with molecular weights of 28,898 Da and 28,856 Da for D381C-AF532M and D381C-DOX, respectively. (Theoretical values are 28,895 and 28,856 Da.) Because the experimental mass spectrometry conditions will denature the protein structure, we expect only chemically-attached molecules to be detected in the protein subunit’s molecular weight, and our results are consistent with this.
Theoretically, a maximum of 60 guest molecules can chemically attach to one nanoparticle scaffold, giving a maximum ratio of 1:1 (protein subunit:molecule). Our higher ratios, measured by absorbance spectroscopy, indicate that additional molecules are most likely bound due to nonspecific interactions. This is supported by experiments in which the wild-type scaffold (E2-WT) was mixed with free doxorubicin. Even with no maleimides present for chemical conjugation, we observed an average of approximately 1:0.4 (protein subunit:doxorubicin) ratio, showing a nonspecific affinity of doxorubicin with the protein scaffold itself of approximately 24 molecules per nanoparticle. Such nonspecific doxorubicin binding to proteins such as albumin have been reported and attributed to its preference for hydrophobic environments.[43,44]
Our results reveal that conjugation of AF532M and doxorubicin to D381C yields intact and stable protein nanoparticles. The characteristics of D381C-AF532M have already been reported,[25] and our data are consistent with these results (Supporting Information, Figure SI-1). For D381C-DOX, the average hydrodynamic diameter was 33.8 ± 4.1 nm, with the maximum distribution peak at 28.2 nm. The CD analysis of the drug-conjugated scaffold yielded the characteristic profile for a correctly-folded D381C that is still rich in α-helical content, with the midpoint of unfolding temperature (Tm) at approximately 90.5°C (Figure 2A), comparable to the Tm for unconjugated D381C.[25] TEM confirmed the intact dodecahedral structure of D381C-DOX (Figure 2B). These results show that conjugation of doxorubicin to the protein scaffold does not alter the protein assembly’s structure or stability.
Figure 2.

The mostability and structural characterization of protein scaffolds encapsulating doxorubicin (D381C-DOX). A. Far-UV circular dichroism thermostability scan at 222 nm revealed Tm to be 90.5°C. B. Transmission electron micrograph of D381C-DOX. Samples were stained with 2% uranyl acetate. Scale bar is 50 nm.
3.2 Doxorubicin is released from D381C-DOX at pH 5
We investigated the in vitro release of doxorubicin from D381C-DOX through the acid-dependent hydrolysis of the hydrazone linker.[35,39] Similar to the wild-type E2 scaffold,[25] D381C is also intact and stable at both pH 7.4 and pH 5.0 (Supporting Information, Figure SI-2). We expected, therefore, that under acidic conditions the protein scaffold in D381C-DOX would remain stable while doxorubicin is released.
As shown in Figure 3, the release rate of doxorubicin at pH 5.0 is significantly faster than at pH 7.4. At pH 5.0, approximately 50%, 85%, and 90% of doxorubicin was released within 24, 48, and 72 hrs, respectively. In contrast, at pH 7.4, only approximately 7% and 15% of doxorubicin release was observed over 48 and 120 hrs respectively. Previous studies investigating the stability of hydrazone linkage reported the half-life of doxorubicin release was 2 to 30 hrs (from doxorubicin-PEG conjugates)[39] and 25 min (from doxorubicin-albumin conjugate) at pH 5.0,[35] and <10% doxorubicin was released after 48 hrs at pH 7.4. Our pH 7.4 data is consistent with these prior values, while our results at pH 5.0 yielded slower release relative to previous rates at pH 5.0. The differences in the hydrolysis assays explain our slower rate; our assay uses a lower temperature and additionally requires the extra barrier of molecular diffusion through dialysis membrane.
Figure 3.

In vitro release of doxorubicin from D381C-DOX at pH 7.4 and pH 5.0.
Hydrazone hydrolysis at acidic pH was confirmed by electrospray mass spectrometry. After five days dialysis at pH 5, the molecular weight of the remaining D381C-DOX scaffold corresponded to the expected molecular weight of a D381C subunit which has released its doxorubicin payload after hydrolysis but still contains the maleimide linker attached to cysteine (28,330 Da) (Supporting Information, Figure SI-3). No peak was observed that corresponded to the molecular weight of D381C-DOX with conjugated doxorubicin remaining, indicating complete release of doxorubicin. In contrast, for samples at pH 7.4, a significant amount of doxorubicin conjugated to D381C was still observed, in addition to the hydrolyzed doxorubicin-cleaved scaffold. The average diameters of the dialyzed D381C-DOX after release experiment were 49.1 ± 10.6 nm for pH 7.4 and 32.7 ± 0.9 nm for pH 5.0 (Supporting Information, Figure SI-1). Although these averages are a little higher than expected, distribution peaks were at 24.4 nm and 28.2 nm, respectively, indicating the vast majority of D381C remained stable, intact, and soluble after drug release.
Previous experiments revealed the uptake of nanoparticles through endocytosis to be strongly dependent on the particle size, with the optimal range to be on the order of 25–75 nm.[5,6] Therefore, our nanoparticles are likely to be taken up by endocytosis. Because internalization into the endocytic pathway results in an environmental pH change from pH 7.4 to pH 5, acidic pH-dependent release of drug from the nanoparticle would be desirable in controlled delivery. Since we observed complete pH-induced hydrolysis and release of doxorubicin in buffer, we investigated the efficacy and cytotoxicity of E2 and doxorubicin-coupled protein nanoparticles within human breast cancer cells.
3.3 Protein nanoparticles are internalized by human breast cancer cells and are not toxic
To investigate cellular uptake of the protein nanoparticles, MDA-MB-231 human breast cancer cells were incubated with D381C labeled with AF532M. Uptake of D381C-AF532M by cells was observed at the first time point investigated (12 hrs), and fluorescence intensity continued to increase over time (Figure 4). However, no observed intracellular fluorescence was apparent for the cells treated with free dye over all incubation times examined.
Figure 4.

MDA-MB-231 cells incubated with (top) free AF532M and (bottom) protein nanoparticle-conjugated AF532M (D381C-AF532M) at different times. Figures are overlays of phase contrast and fluorescence images. Scale bar is 50 μm.
The images indicate cellular uptake and an accumulation of D381C-AF532M within intracellular compartments, but minimal uptake of free AF532M. This difference between free and conjugated AF532M suggests divergent physical properties and uptake pathways. AF532M is a negatively-charged and hydrophilic fluorescent dye,[45] suggesting low permeability through cell membranes. In contrast, nanoparticles within our size range will be actively endocytosed.[5,6] Our results show that small molecules which do not naturally cross the cell membrane barrier can, in fact, be delivered into a cell by attachment to the E2 protein nanoparticle.
The lower level of uptake for the free dye relative to the nanoparticle-conjugated dye is not due to decreased cell viability, as MTT results for cells incubated with AF532M and D381C-AF532M are comparable (Supporting Information, Figure SI-4). Comparison of cell viability data between AF532M and D381C-AF532M treated cells across all time points (0 to 72 hrs) gave a P > 0.05, indicating no statistical difference in toxicity between free and nanoparticle-bound AF532M. Continued incubation and increased intracellular accumulation of free and nanoparticle-bound AF532M does not appear to increase toxicity.
To investigate the cytotoxicity of the protein nanoparticle scaffold alone, different concentrations of E2-WT (0.084 – 502 μg/ml protein, equivalent to the protein concentrations used in the D381C-DOX studies) were incubated with cells, and the equivalent buffer-alone volume (without protein) was used as a control. The resulting dose response curves for both samples are nearly coincident, with a gradual decrease in cell viability as volume of the sample added was increased (Supplemental Information, Figure SI-5). This shows that the protein nanoparticles are not toxic to the cells, but the observed cell death can be attributed to dilution with buffer. Since increasing the degree of phosphate buffer dilution decreases cell viability at the high dilutions, we attribute this cell death to the exhaustion of nutrients in the medium after such a long incubation time. The lack of toxicity of the E2 protein scaffold itself and the dye-encapsulating scaffold suggest that the E2 nanoparticle can be further pursued as a potential drug delivery and imaging platform, much like that reported for fluorescently-labeled cowpea mosaic virus.[46]
3.4 D381C-DOX protein nanoparticles are internalized by human breast cancer cells and exhibit cytotoxicity to cells
Fluorescence microscopy and confocal laser scanning microscopy (CLSM)
We visualized the uptake of doxorubicin and doxorubicin-coupled protein nanoparticles by fluorescence imaging. Figure 5 shows the cellular uptake of free doxorubicin and conjugated doxorubicin (D381C-DOX). As doxorubicin concentration increased, greater intracellular accumulation of the drug was observed. Furthermore, at the low drug concentration, fluorescence appeared to exist more diffusely for cells incubated with free doxorubicin (relative to D381C-DOX) and suggest its presence in the cytoplasm and nucleus.
Figure 5.

MDA-MB-231 cells incubated with (top) free doxorubicin and (bottom) protein nanoparticle-conjugated doxorubicin (D381C-DOX) at different doxorubicin concentrations for 72 hrs. Figures are overlays of phase contrast and fluorescence images. Scale bar is 50 μm.
To investigate the intracellular distribution of doxorubicin and D381C-DOX in the MDA-MB-231 tumor cells, we performed CLSM (Figure 6). After incubation for two days with free doxorubicin, the drug existed in both the nucleus and in the cytoplasm, with small regions of punctuated accumulation in cellular compartments. In contrast, doxorubicin conjugated to D381C nanoparticles were primarily localized in subcellular vesicles or organelles within the cytoplasm, with only small amounts in the nucleus, which supports endosomal or lysosomal accumulation. These results indicate that free and conjugated doxorubicin use different uptake mechanisms.
Figure 6.

Confocal laser scanning microscopy images of MDA-MB-231 cells treated with (top) free doxorubicin and (bottom) protein nanoparticle-conjugated doxorubicin (D381C-DOX) at 3 μM doxorubicin-equivalent concentration. (A) Doxorubicin is red, (B) cell nuclei are blue, and (C) overlay of the two images are presented. Scale bar is 50 μm.
Our data is consistent with prior microscopy studies of tumor cells. Other investigators have also reported free doxorubicin to be mainly concentrated in the nucleus while doxorubicin-conjugated serum proteins, polymers, or nanoparticles localize in the cytoplasm or subcellular organelles, such as lysosomes, mitochondria, and the Golgi apparatus.[39,47–52] Furthermore, our results are consistent with previous investigations of size-dependent nanoparticle endocytosis. In those studies, both nanoparticles that displayed surface ligands for specific cell receptors[5,53] and nanoparticles that did not incorporate ligands[6] were taken up by receptor-mediated endocytosis. For the latter group, it was hypothesized that serum proteins adsorbed to the nanoparticle surface, thereby inducing receptor-mediated uptake. Protein adsorption can occur to varying degrees; therefore it is possible our protein nanoparticles could also use this mechanism as well. However, detailed endocytosis studies would be required to elucidate internalization mechanism.
Cytotoxicity of doxorubicin and D381C-DOX
The qualitative results observed through fluorescence imaging are consistent with the cell viability data observed after treatment of MDA-MB-231 cells with free and nanoparticle-bound doxorubicin. Using dose-response curves for cells incubated with doxorubicin and D381C-DOX for 72 hours, we determined the IC50 to be 0.6 ± 0.01 μM and 1.3 ± 0.3 μM, respectively (Figure 7). Free doxorubicin is expected to directly diffuse through the lipid bilayers into the cell and nucleus.[54,55] In contrast, our slightly higher level of toxicity for free doxorubicin relative to conjugated doxorubicin can be explained by the additional steps required for D381C-DOX to be taken up by the cells through the endocytic pathway, with gradual pH change and corresponding drug release from the nanoparticle, and escape from the endosome/lysosome compartments into the cytosol and nucleus. Hence, it is likely that 72 hrs may be an insufficient period for the complete release of doxorubicin from the D381C scaffold.
Figure 7.

Dose response curves of MDA-MB-231 cells incubated with free doxorubicin and protein nanoparticle-conjugated doxorubicin (D381C-DOX) for 72 hrs. IC50 values are 0.6 ± 0.01 μM for doxorubicin and 1.3 ± 0.3 μM for D381C-DOX.
These IC50 values are within the range of typical values obtained in other types of nanoparticle-based delivery systems. Prior literature has reported IC50 of doxorubicin in MDA-MB-231 to range between 0.1 μM and 2 μM for cells incubated 48 to 72 hr.[56–58] Investigations for doxorubicin conjugated to various types of nanoparticles have reported both higher[36,38,49,50,58] and lower[51,59,60] IC50 values relative to free drug.
This apparent discrepancy among nanodelivery approaches can be attributed to multiple differences in the systems, including nanoparticle materials, particle sizes, surface properties, and cell lines, all of which can affect the mechanism of particle uptake. For example, doxorubicin has been incorporated into different polymers using various strategies such as covalent bonding, nanoprecipitation and emulsion encapsulation, with particle size ranging from tens to hundreds of nanometers. Many of the more effective doxorubicin-conjugated nanoparticles are also functionalized with targeting moieties on the surface, which increase efficacy by delivering doxorubicin to specific cells in vivo while decreasing the overall amount of systemic drug exposure to healthy cells.[19,51,60–62] Prior investigations have shown the feasibility of attaching peptides to the surface of the E2 scaffold.[30,31] Therefore, although this current investigation yielded slightly lower cytoxicity values for doxorubicin conjugated to E2, attaching targeting ligands on the scaffold surface is a potential means of significantly increasing efficacy.
In interpreting the implications of IC50 values, it is important to examine the in vitro-in vivo relationships of reported nanoparticle delivery systems. Many investigations have described doxorubicin-bound nanomaterials that yield less effective in vitro cytotoxicity (i.e., higher IC50) than free doxorubicin in tumor cells, but still gave substantially superior antitumor efficacy in in vivo animal models relative to free doxorubicin.[35,36,38,63] This shows that while IC50 values can be used as a comparative result between nanoparticle delivery systems, additional in vivo experiments using D381C-DOX are needed to fully assess therapeutic activity. Interestingly, these cited examples also yielded in vitro doxorubicin release kinetics (from cleavage of the acid-sensitive hydrazone group) that were similar to our release rates. Their corresponding in vivo results gave complete or nearly-complete tumor remission and demonstrate that the release rates were indeed sufficient to obtain desirable therapeutic effects. Overall, our data shows the feasibility of using the E2 scaffold for delivery of drugs into cells and demonstrates that this system is an attractive candidate for evaluating antitumor efficacy in vivo.
4. Conclusions
This investigation probed, for the first time, the effects of E2 protein nanoparticles on breast cancer cells and their potential in intracellular molecular delivery. We showed that the E2 scaffold containing accessible thiol groups is able to bind the maleimide-bearing fluorescent dye AF532M and the 6-maleimidecaproyl hydrazone derivative of doxorubicin within its hollow cavity. Fluorescence imaging showed AF532M and doxorubicin can be internalized by breast cancer cells when coupled to D381C. Free doxorubicin distributed mainly into the nucleus and cytoplasm of cells, while D381C-DOX was localized in subcellular organelles, suggesting the conjugated doxorubicin was taken up by endocytosis. Our results further reveal that doxorubicin bound through an acid-sensitive linker within the scaffold will indeed induce cell death. The in vitro release data showed that doxorubicin was released at a significantly faster rate at pH 5.0 than at pH 7.4, which is desirable for controlled intracellular release. Collectively, these properties demonstrate that virus-like protein nanoparticles represent a promising new opportunity for nanomaterials in therapeutic delivery and imaging applications.
Supplementary Material
Acknowledgments
We gratefully acknowledge assistance from Dr. John Greaves in the UCI Mass Spectrometry Facility, Dr. Michelle Digman in the UCI Optical Biology Core Facility, Prof. Andrew Putnam and Dr. Carlos Huang for initial cell culture training, Prof. Daniel Mumm for image processing discussions, and Dr. Matthew Shindel for assistance with TEM images and Tm calculations. TEM was performed at the UCI Materials Characterization Center, and DLS and CD were performed at the UCI Laser Spectroscopy Facility. This work was supported by NSF (CBET-0827852), NIH (5R21EB010161-02), and the Department of Defense Breast Cancer Research Program (BC085958).
References
- 1.Nie SM, Xing Y, Kim GJ, Simons JW. Annual Review of Biomedical Engineering. 2007;9:257. doi: 10.1146/annurev.bioeng.9.060906.152025. [DOI] [PubMed] [Google Scholar]
- 2.Ferrari M. Nature Reviews Cancer. 2005;5:161. doi: 10.1038/nrc1566. [DOI] [PubMed] [Google Scholar]
- 3.Doshi N, Mitragotri S. Advanced Functional Materials. 2009;19:3843. [Google Scholar]
- 4.Singh R, Lillard JW. Experimental and Molecular Pathology. 2009;86:215. doi: 10.1016/j.yexmp.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jiang W, Kim BYS, Rutka JT, Chan WCW. Nature Nanotechnology. 2008;3:145. doi: 10.1038/nnano.2008.30. [DOI] [PubMed] [Google Scholar]
- 6.Chithrani BD, Ghazani AA, Chan WCW. Nano Letters. 2006;6:662. doi: 10.1021/nl052396o. [DOI] [PubMed] [Google Scholar]
- 7.Belting M, Sandgren S, Wittrup A. Advanced Drug Delivery Reviews. 2005;57:505. doi: 10.1016/j.addr.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 8.Christie RJ, Grainger DW. Advanced Drug Delivery Reviews. 2003;55:421. doi: 10.1016/s0169-409x(02)00229-6. [DOI] [PubMed] [Google Scholar]
- 9.Rajendran L, Knolker HJ, Simons K. Nature Reviews Drug Discovery. 2010;9:29. doi: 10.1038/nrd2897. [DOI] [PubMed] [Google Scholar]
- 10.Byrne JD, Betancourt T, Brannon-Peppas L. Advanced Drug Delivery Reviews. 2008;60:1615. doi: 10.1016/j.addr.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 11.Kratz F, Beyer U, Schutte MT. Critical Reviews in Therapeutic Drug Carrier Systems. 1999;16:245. doi: 10.1615/critrevtherdrugcarriersyst.v16.i3.10. [DOI] [PubMed] [Google Scholar]
- 12.Nasongkla N, Bey E, Ren JM, Ai H, Khemtong C, Guthi JS, Chin SF, Sherry AD, Boothman DA, Gao JM. Nano Letters. 2006;6:2427. doi: 10.1021/nl061412u. [DOI] [PubMed] [Google Scholar]
- 13.Uchida M, Klem MT, Allen M, Suci P, Flenniken M, Gillitzer E, Varpness Z, Liepold LO, Young M, Douglas T. Advanced Materials. 2007;19:1025. [Google Scholar]
- 14.Manchester M, Singh P. Advanced Drug Delivery Reviews. 2006;58:1505. doi: 10.1016/j.addr.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 15.Douglas T, Young M. Nature. 1998;393:152. [Google Scholar]
- 16.Comellas-Aragones M, Engelkamp H, Claessen VI, Sommerdijk NAJM, Rowan AE, Christianen PCM, Maan JC, Verduin BJM, Cornelissen JJLM, Nolte RJM. Nature Nanotechnology. 2007;2:635. doi: 10.1038/nnano.2007.299. [DOI] [PubMed] [Google Scholar]
- 17.Meldrum FC, Wade VJ, Nimmo DL, Heywood BR, Mann S. Nature. 1991;349:684. [Google Scholar]
- 18.Kramer RM, Li C, Carter DC, Stone MO, Naik RR. Journal of the American Chemical Society. 2004;126:13282. doi: 10.1021/ja046735b. [DOI] [PubMed] [Google Scholar]
- 19.Destito G, Yeh R, Rae CS, Finn MG, Manchester M. Chemistry & Biology. 2007;14:1152. doi: 10.1016/j.chembiol.2007.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brennan FR, Jones TD, Hamilton WDO. Molecular Biotechnology. 2001;17:15. doi: 10.1385/MB:17:1:15. [DOI] [PubMed] [Google Scholar]
- 21.Chatterji A, Burns LL, Taylor SS, Lomonossoff GP, Johnson JE, Lin T, Porta C. Intervirology. 2002;45:362. doi: 10.1159/000067929. [DOI] [PubMed] [Google Scholar]
- 22.Uchida M, Flenniken ML, Allen M, Willits DA, Crowley BE, Brumfield S, Willis AF, Jackiw L, Jutila M, Young MJ, Douglas T. Journal of the American Chemical Society. 2006;128:16626. doi: 10.1021/ja0655690. [DOI] [PubMed] [Google Scholar]
- 23.Flenniken ML, Willits DA, Harmsen AL, Liepold LO, Harmsen AG, Young MJ, Douglas T. Chemistry & Biology. 2006;13:161. doi: 10.1016/j.chembiol.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 24.Kovacs EW, Hooker JM, Romanini DW, Holder PG, Berry KE, Francis MB. Bioconjugate Chemistry. 2007;18:1140. doi: 10.1021/bc070006e. [DOI] [PubMed] [Google Scholar]
- 25.Dalmau M, Lim S, Chen HC, Ruiz C, Wang SW. Biotechnology and Bioengineering. 2008;101:654. doi: 10.1002/bit.21988. [DOI] [PubMed] [Google Scholar]
- 26.Dalmau M, Lim S, Wang SW. Biomacromolecules. 2009;10:3199. doi: 10.1021/bm900674v. [DOI] [PubMed] [Google Scholar]
- 27.Dalmau M, Lim SR, Wang SW. Nano Letters. 2009;9:160. doi: 10.1021/nl8027069. [DOI] [PubMed] [Google Scholar]
- 28.Allen MD, Perham RN. FEBS Letters. 1997;413:339. doi: 10.1016/s0014-5793(97)00932-0. [DOI] [PubMed] [Google Scholar]
- 29.Izard T, AEA, Allen MD, Westphal AH, Perham RN, de Kok A, Hol WGJ. Proceedings of the National Academy of Sciences USA. 1999;96:1240. doi: 10.1073/pnas.96.4.1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Domingo GJ, Orru S, Perham RN. Journal of Molecular Biology. 2001;305:259. doi: 10.1006/jmbi.2000.4311. [DOI] [PubMed] [Google Scholar]
- 31.Domingo GJ, Caivano A, Sartorius R, Barba P, Backstrom M, Piatier-Tonneau D, Guardiola J, Berardinis PD, Perham RN. Vaccine. 2003;21:1502. doi: 10.1016/s0264-410x(02)00664-3. [DOI] [PubMed] [Google Scholar]
- 32.Flenniken ML, Liepold LO, Crowley BE, Willits DA, Young MJ, Douglas T. Chem Commun. 2005;447 doi: 10.1039/b413435d. [DOI] [PubMed] [Google Scholar]
- 33.Yoo HS, Lee EA, Park TG. Journal of Controlled Release. 2002;82:17. doi: 10.1016/s0168-3659(02)00088-3. [DOI] [PubMed] [Google Scholar]
- 34.Prabaharan M, Grailer JJ, Pilla S, Steeber DA, Gong SQ. Biomaterials. 2009;30:5757. doi: 10.1016/j.biomaterials.2009.07.020. [DOI] [PubMed] [Google Scholar]
- 35.Kratz F, Warnecke A, Scheuermann K, Stockmar C, Schwab J, Lazar P, Druckes P, Esser N, Drevs J, Rognan D, Bissantz C, Hinderling C, Folkers G, Fichtner I, Unger C. Journal of Medicinal Chemistry. 2002;45:5523. doi: 10.1021/jm020276c. [DOI] [PubMed] [Google Scholar]
- 36.MacKay JA, Chen MN, McDaniel JR, Liu WG, Simnick AJ, Chilkoti A. Nature Materials. 2009;8:993. doi: 10.1038/nmat2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hruby M, Konak C, Ulbrich K. Journal of Controlled Release. 2005;103:137. doi: 10.1016/j.jconrel.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 38.Lee CC, Gillies ER, Fox ME, Guillaudeu SJ, Frechet JMJ, Dy EE, Szoka FC. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:16649. doi: 10.1073/pnas.0607705103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rodrigues PCA, Beyer U, Schumacher P, Roth T, Fiebig HH, Unger C, Messori L, Orioli P, Paper DH, Mulhaupt R, Kratz F. Bioorganic & Medicinal Chemistry. 1999;7:2517. doi: 10.1016/s0968-0896(99)00209-6. [DOI] [PubMed] [Google Scholar]
- 40.Willner D, Trail PA, Hofstead SJ, King HD, Lasch SJ, Braslawsky GR, Greenfield RS, Kaneko T, Firestone RA. Bioconjugate Chemistry. 1993;4:521. doi: 10.1021/bc00024a015. [DOI] [PubMed] [Google Scholar]
- 41.Missirlis D, Kawamura R, Tirelli N, Hubbell JA. European Journal of Pharmaceutical Sciences. 2006;29:120. doi: 10.1016/j.ejps.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 42.Wang SW, Monagle J, McNulty C, Putnam D, Chen H. J Pharm Sci. 2004;93:2755. doi: 10.1002/jps.20183. [DOI] [PubMed] [Google Scholar]
- 43.Demant EJF, Friche E. Biochemical Pharmacology. 1998;56:1209. doi: 10.1016/s0006-2952(98)00255-x. [DOI] [PubMed] [Google Scholar]
- 44.Chassany O, Urien S, Claudepierre P, Bastian G, Tillement JP. Cancer Chemotherapy and Pharmacology. 1996;38:571. doi: 10.1007/s002800050529. [DOI] [PubMed] [Google Scholar]
- 45.Panchuk-Voloshina N, Haugland RP, Bishop-Stewart J, Bhalgat MK, Millard PJ, Mao F, Leung WY, Haugland RP. Journal of Histochemistry & Cytochemistry. 1999;47:1179. doi: 10.1177/002215549904700910. [DOI] [PubMed] [Google Scholar]
- 46.Lewis JD, Destito G, Zijlstra A, Gonzalez MJ, Quigley JP, Manchester M, Stuhlmann H. Nature Medicine. 2006;12:354. doi: 10.1038/nm1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dreher MR, Raucher D, Balu N, Colvin OM, Ludeman SM, Chilkoti A. Journal of Controlled Release. 2003;91:31. doi: 10.1016/s0168-3659(03)00216-5. [DOI] [PubMed] [Google Scholar]
- 48.Chavanpatil MD, Khdair A, Gerard B, Bachmeier C, Miller DW, Shekhar MPV, Panyam J. Molecular Pharmaceutics. 2007;4:730. doi: 10.1021/mp070024d. [DOI] [PubMed] [Google Scholar]
- 49.Shuai XT, Ai H, Nasongkla N, Kim S, Gao JM. Journal of Controlled Release. 2004;98:415. doi: 10.1016/j.jconrel.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 50.Liu SQ, Tong YW, Yang YY. Biomaterials. 2005;26:5064. doi: 10.1016/j.biomaterials.2005.01.030. [DOI] [PubMed] [Google Scholar]
- 51.Upadhyay KK, Bhatt AN, Mishra AK, Dwarakanath BS, Jain S, Schatz C, Le Meins JF, Farooque A, Chandraiah G, Jain AK, Misra A, Lecommandoux S. Biomaterials. 2010;31:2882. doi: 10.1016/j.biomaterials.2009.12.043. [DOI] [PubMed] [Google Scholar]
- 52.Beyer U, Rothen-Rutishauser B, Unger C, Wunderli-Allenspach H, Kratz F. Pharmaceutical Research. 2001;18:29. doi: 10.1023/a:1011018525121. [DOI] [PubMed] [Google Scholar]
- 53.Chithrani BD, Chan WCW. Nano Letters. 2007;7:1542. doi: 10.1021/nl070363y. [DOI] [PubMed] [Google Scholar]
- 54.Speelmans G, Staffhorst RWHM, Dekruijff B, Dewolf FA. Biochemistry. 1994;33:13761. [Google Scholar]
- 55.Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Pharmacological Reviews. 2004;56:185. doi: 10.1124/pr.56.2.6. [DOI] [PubMed] [Google Scholar]
- 56.Sevimli-Gur C, Akgun IH, Deliloglu-Gurhan I, Korkmaz KS, Bedir E. Journal of Natural Products. 2010;73:860. doi: 10.1021/np900778j. [DOI] [PubMed] [Google Scholar]
- 57.Pichot CS, Hartig SM, Xia L, Arvanitis C, Monisvais D, Lee FY, Frost JA, Corey SJ. British Journal of Cancer. 2009;101:38. doi: 10.1038/sj.bjc.6605101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gillies ER, Frechet JMJ. Bioconjugate Chemistry. 2005;16:361. doi: 10.1021/bc049851c. [DOI] [PubMed] [Google Scholar]
- 59.Yoo HS, Park TG. Journal of Controlled Release. 2001;70:63. doi: 10.1016/s0168-3659(00)00340-0. [DOI] [PubMed] [Google Scholar]
- 60.Soppimath KS, Liu LH, Seow WY, Liu SQ, Powell R, Chan P, Yang YY. Advanced Functional Materials. 2007;17:355. [Google Scholar]
- 61.Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Girard OM, Hanahan D, Mattrey RF, Ruoslahti E. Cancer Cell. 2009;16:510. doi: 10.1016/j.ccr.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.MacDiarmid JA, Amaro-Mugridge NB, Madrid-Weiss J, Sedliarou I, Wetzel S, Kochar K, Brahmbhatt VN, Phillips L, Pattison ST, Petti C, Stillman B, Graham RM, Brahmbhatt H. Nature Biotechnology. 2009;27:643. doi: 10.1038/nbt.1547. [DOI] [PubMed] [Google Scholar]
- 63.Kratz F. Journal of Controlled Release. 2008;132:171. doi: 10.1016/j.jconrel.2008.05.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.