

Abstract
Genome-wide association studies have identified prostate cancer susceptibility alleles on chromosome 11q13. As part of the Cancer Genetic Markers of Susceptibility (CGEMS) Initiative, the region flanking the most significant marker, rs10896449, was fine mapped in 10 272 cases and 9123 controls of European origin (10 studies) using 120 common single nucleotide polymorphisms (SNPs) selected by a two-staged tagging strategy using HapMap SNPs. Single-locus analysis identified 18 SNPs below genome-wide significance (P< 10−8) with rs10896449 the most significant (P= 7.94 × 10−19). Multi-locus models that included significant SNPs sequentially identified a second association at rs12793759 [odds ratio (OR) = 1.14, P= 4.76 × 10−5, adjusted P= 0.004] that is independent of rs10896449 and remained significant after adjustment for multiple testing within the region. rs10896438, a proxy of previously reported rs12418451 (r2= 0.96), independent of both rs10896449 and rs12793759 was detected (OR = 1.07, P= 5.92 × 10−3, adjusted P= 0.054). Our observation of a recombination hotspot that separates rs10896438 from rs10896449 and rs12793759, and low linkage disequilibrium (rs10896449–rs12793759, r2= 0.17; rs10896449–rs10896438, r2= 0.10; rs12793759–rs10896438, r2= 0.12) corroborate our finding of three independent signals. By analysis of tagged SNPs across ∼123 kb using next generation sequencing of 63 controls of European origin, 1000 Genome and HapMap data, we observed multiple surrogates for the three independent signals marked by rs10896449 (n= 31), rs10896438 (n= 24) and rs12793759 (n= 8). Our results indicate that a complex architecture underlying the common variants contributing to prostate cancer risk at 11q13. We estimate that at least 63 common variants should be considered in future studies designed to investigate the biological basis of the multiple association signals.
INTRODUCTION
Prostate cancer is the most common non-cutaneous cancer in developed countries (1,2). Established risk factors for this malignancy are increased incidence with age, ethnic background and familial history of prostate cancer (3). Genetic risk factors have been estimated to account for nearly 40% of the risk (4). Genome-wide association studies (GWAS) have identified at least 35 loci across the genome associated with prostate cancer risk, primarily discovered in men of European background (5–16), but recently new loci have been discovered in men of Japanese background (17). Current estimates suggest that there are at least an equal number more of common variants associated with the risk for prostate cancer (18). To date, GWAS have successfully identified new regions associated with the overall prostate cancer risk but have not conclusively identified novel regions associated with advanced prostate cancer disease. Similarly, none of the loci identified by GWAS have been firmly associated with clinical prognosis. In three of the regions associated with the overall prostate cancer risk, additional independent common variants have been detected neighboring the initial associations detected by GWAS. For prostate cancer risk, there are at least four independent loci in 8q24, two in HNF1B on chromosome 17q12 and recently two loci in 11q13 (5,7,8,10,11,15,16,19,20).
Two GWAS have identified a pair of highly correlated single nucleotide polymorphisms (SNPs) on chromosome 11q13, rs7931342 and rs10896449 (r2= 0.97, HapMap phase II CEU) associated with prostate cancer risk (10,15). A second independent locus (rs12418451, chr11:68 691 995, r2= 0.06 with both rs7931342 and rs10896449) was reported in association with prostate cancer (20). Both loci map to a 203.5 kb intergenic region flanked by TPCN2 at its centromeric end and by MYEOV at its telomeric end. TPCN2 encodes two-pore segment channel 2, which was recently found to contain two coding SNPs, associated with blond versus brown hair color (21). MYEOV is frequently overexpressed in multiple myeloma, breast cancer and oral and esophageal squamous cell carcinomas (22,23). The markers previously identified by GWAS are not correlated with markers in the flanking genes. Recently, GWAS have identified two independent loci telomeric to the prostate cancer region on 11q13 associated with the risk for kidney and breast cancer, respectively (24,25); the strongest markers, rs7105934 and rs614367 are 245 and 334 kb telomeric of rs10896449, respectively; Supplementary Material, Figure S1 depicts the positions of the notable SNPs relative to the local candidate genes of interest in 11q13.
We report the results of fine-mapping the region flanking the most notable SNP, rs10896449, initially associated with prostate cancer risk in 11q13. One hundred and twenty common SNPs chosen in a two-stage tagging approach were genotyped in 10 272 cases and 9123 controls of men of European background. A tagged SNP approach with re-sequence data across a ∼123 kb region of 11q13 together with 1000 Genome and HapMap data was used to estimate a comprehensive set of surrogate variants worthy of consideration for follow-up functional studies.
RESULTS
Single-SNP analysis adjusted for age, study, center and principal components of population stratification confirmed the association for rs10896449 [P= 7.94 × 10−19, trend test, heterozygote odds ratio (OR): 0.83, 95% confidence interval (CI): 0.80–0.87; homozygote OR: 0.69, 95% CI: 0.64–0.75]. Seventeen additional SNPs were significant below the threshold for genome-wide significance (P-values of <10−8; rs10896437, rs10896438, rs2924540, rs2924538, rs11228551, rs11228553, rs4255548, rs12793759, rs4495900, rs12281017, rs4620729, rs7931342, rs9787877, rs7950547, rs7939250, rs10896450 and rs11228583; Table 1, Fig. 1, Supplementary Material, Table S1). To determine the degree of linkage disequilibrium (LD) among the genotyped 18 SNPs, we conducted a tagging analysis that placed highly correlated SNPs into ‘bins' [r2 ≥ 0.8, minor allele frequency (MAF) ≥ 5%]. Fourteen of 18 SNPs segregated into three bins; bin1 (average MAF = 0.257): rs10896438, rs2924538, rs11228551 and rs11228553; bin2 (average MAF = 0.380): rs4255548, rs4495900 and rs7950547; bin3 (average MAF = 0.495): rs4620729, rs7931342, rs10896449, rs9787877, rs7939250, rs10896450 and rs11228583 (Fig. 2C). Although 4 SNPs (rs10896437, rs2924540, rs12793759 and rs12281017) were not highly correlated (r2 < 0.8 with any of the other 14 SNPs), it is notable that rs10896437 and rs2924540 were in LD with SNPs in bin1 (average pair-wise r2= 0.73 and 0.70, respectively) whereas LD was observed between rs12793759 and rs12281017 (r2= 0.68).
Table 1.
Results of the pooled dichotomous association analysis of 18 SNPs with genome-wide significance (P < 10−8)
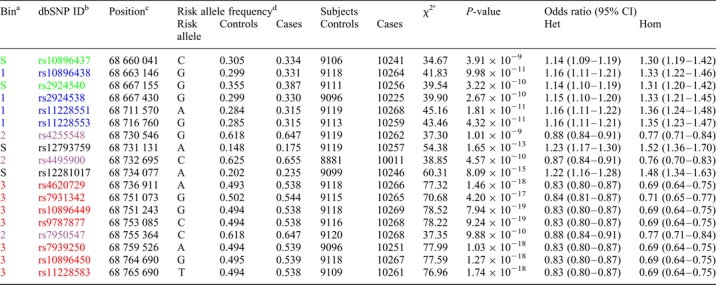 |
The results of the dichotomous logistic regression of the pooled genotypes generated from the ten studies in a total of 10 272 prostate cancer cases and 9123 controls—adjusted for age, study, center and four eigenvectors to control population stratification—are shown for the 18 SNPs with P-values of <10−8.
OR, odds ratio; Het, heterozygous; Hom, homozygous for minor allele; CI, 95% confidence interval.
aCorrelation bin with r2 > 0.8; S, Singleton bin with no proxy under an r2 > 0.8 threshold.
bNCBI dbSNP identifier. SNPs were color coded to show correlation bins (r2 > 0.8)—Green and black, singletons with no proxy at r2 > 0.8; light blue, bin1; purple, bin2; red, bin3.
cChromosomal position based on NCBI Human genome Build 36.
dSNP allele that confers susceptibility to prostate cancer and its frequency in controls and cases.
e1-d.f. score test.
Figure 1.

Association analysis result, recombination hotspots and LD of 11q13 region. The upper panel shows P-values for association testing from stages 1–3 combined CGEMS prostate cancer scan across a region of 11q13 bounded by rs930782 and rs4584599 (Chr11:68 628 370–68 870 174). Shaded regions ‘Centro’ and ‘Telo’ correspond to the centromeric region and the telomeric region, respectively. The line graph shows likelihood ratio statistics for recombination hotspot by SequenceLDhot software and five different colors represent 5 tests of 900 combined controls without resampling. The upper horizontal line indicates a genome-wide significance level (P-value of <10−8) and the lower horizontal line indicates a likelihood ratio statistic cutoff to predict the presence of a hotspot with a false-positive rate of 1 in 3700 independent tests (26). The lower panel shows an enlarged view of the region bounded by rs1123608 and rs4131929 (Chr11:68 659 896–68 768 714). The pair-wise r2 correlation coefficient for SNPs in the region was estimated using TagZilla and plotted using SnpPlotter (41). The 18 SNPs with genome-wide significance were color coded. Light-blue represents correlation bin1 SNPs (rs10896438, rs2924538, rs11228551 and rs11228553), purple represents bin2 SNPs (rs4255548, rs4495900 and rs7950547) and red represents bin3 SNPs (rs4620729, rs7931342, rs10896449, rs9787877, rs7939250, rs10896450 and rs11228583). Green and black represent singleton SNPs with no proxy (r2 > 0.8), but colored to show separation by a recombination hotspot (green, rs10896437 and rs2924540; black, rs12793759 and rs12281017). A black arrow indicates the recombination hotspot that separates the region into centromeric and telomeric regions.
Figure 2.

Sequential multi-locus model of SNPs in the 11q13 region. The colored vertical boxed area represents the region of the observed recombination hotspot. Eighteen SNPs that showed genome-wide significance (P< 10−8) in the single-SNP analysis (Table 1) were color coded comparably with Figure 1. SNPs in gray were observed to be not genome-wide significant in the single-SNP model but of interest in the multi-locus modeling. (A) Shows the two SNP multi-locus analysis conditioned on rs10896449 (a red diamond on the x-axis). Twenty eight SNPs remained significant at an alpha of 0.05 (horizontal line). rs12793759 is the most significant SNP (per allele OR = 1.14, 95% CI: 1.07–1.21, P = 4.76 × 10−5, adjusted P = 0.004). (B) Shows the three-SNP multi-locus analysis conditioned on rs12793759 and rs10896449 (black diamond and red diamonds on x-axis, respectively). Thirteen SNPs showed significance at an alpha level of 0.05 (horizontal line) with rs10896438 being the most significant SNP (per allele OR = 1.07, 95% CI: 1.02–1.12, P = 5.92 × 10−3, adjusted P = 0.054). (C) Depicts the correlation patterns of the 18 genome-wide significant SNPs with color coding as per Figure 1. Correlation bins were defined with an r2 > 0.8 threshold and based on the analysis of all controls of European background in this study. Four SNPs that had no proxy with the threshold were denoted as ‘singleton'. The pair-wise (singleton versus singleton) or average (singleton versus bin, SNPs within a bin) correlation values are expressed by r2 (D′).
We analyzed the region to search for statistically independent signals by sequential multi-locus models (Table 2, Fig. 2A and B, Supplementary Material, Table S2). When conditioned on the original marker rs10896449, 28 SNPs remained significant at an alpha error of 0.05 and the most significant SNP rs12793759 (per allele OR = 1.14, 95% CI: 1.07–1.21, P = 4.76 × 10−5) achieved region-wide significance after adjustment for multiple testing (adjusted P = 0.004; Table 2, Fig. 2A). When we investigated the multi-locus 3-SNP models conditioned on both rs10896449 and rs12793759, 13 SNPs remained significant. In the multi-locus model, rs10896438 showed the strongest association (per allele OR = 1.07, 95% CI: 1.02–1.12, P = 5.92 × 10−3) and after adjustment for multiple tests, the significance remained noteworthy (adjusted P = 0.054; Table 2, Fig. 2B). After excluding the CAPS and JHH samples used in the previous report (20), rs12793759 remained the most significant (per allele OR = 1.13, 95% CI: 1.05–1.21, P = 1.48 × 10−3, adjusted P = 0.0027) conditioning on the original marker rs10896449, and in the 3-SNP model conditioned on both rs10896449 and rs12793759, the signal due to rs10896438 is still noteworthy (per allele OR = 1.09, 95% CI: 1.03–1.53, P = 1.61 × 10−3, adjusted P = 0.0945; Supplementary Material, Table S3). The low correlation between rs10896449 and rs12793759 (r2= 0.17; rs10896449 and rs10896438, r2= 0.10; rs12793759 and rs10896438, r2= 0.12) corroborates our test results.
Table 2.
Sequential multi-locus regression analysis identified three independent, prostate cancer susceptibility SNPs in 11q13
| SNP | Per allele odds ratio (95% CI) | P-value |
|---|---|---|
| rs10896449 | 1.14 (1.09–1.20) | 8.69 × 10−9 |
| rs12793759 | 1.11 (1.04–1.18) | 1.41 × 10−3 |
| rs10896438 | 1.07 (1.02–1.12) | 5.92 × 10−3 |
A three-SNP model conditioned on the original CGEMS hit rs10896449 and the novel second signal rs12793759 was used to assess whether a third independent signal was among the remaining SNPs. The most significant SNP in that analysis was rs10896438, which is in strong LD with a marker previously reported (20); it achieved borderline significance after permutation-based adjustment for multiple testing (adjusted P = 0.054). Reported P-values are unadjusted.
On the basis of the analysis of a 241 805 bp region of chromosome 11q13 (68 628 370–68 870 174) using SequenceLDhot (26), we observed a recombination hotspot that separates the centromeric and telomeric regions, dividing the 18 genome-wide significant SNPs into two genetically independent clusters (Fig. 1). The location of the recombination hotspot corresponds to that reported for HapMap (Release 21). Specifically, we detected strong evidence for a recombination hotspot between rs11228553 and rs4255548 (P = 9.15 × 10−8 to 4.03 × 10−11 with five non-overlapping pooled control sets of 900 individuals each; Fig. 1). A copy number variant (CNV) has been reported overlapping the observed recombination hotspot (68 706 481–68 723 072, an insertion of ∼16.6 kb segment, Database of Genomic Variants); it is defined by six contiguous SNPs (rs7128814, rs11228551, rs4495899, rs11228553, rs11228554 and rs11602052). Although this CNV was not systematically investigated in our study, it notably includes two SNPs from bin1, rs11228551 and rs11228553, which displayed genome-wide significance. The observation is consistent with the hotspot inference given the frequent occurrence of CNVs due to recombination events.
On the basis of the position of the recombination hotspot, the original GWAS signal, rs10896449 resides on the telomeric side along with two bins (bin2 and bin3) and two singletons, rs12793759 and rs12281017. The centromeric region includes bin1 (rs10896438, rs2924538, rs11228551 and rs11228553), as well as two additional SNPs with moderate LD, rs10896437 and rs2924540 (Fig. 2C). On the basis of HapMap data, SNPs in bin1 are in high LD with rs12418451, which we did not test (rs10896438, rs2924538, rs11228551 and rs11228553, with r2 = 0.96, 0.96, 0.83 and 0.79, respectively, in HapMap CEU), but was recently reported as a second locus by two of the groups participating in this larger study (20).
The six SNPs in the centromeric region were not statistically independent of one another (Fig. 2, Supplementary Material, Table S2), which is not surprising because of the high LD (rs10896437, rs2924540 and bin1 SNPs, average r2= 0.73 and 0.70, respectively, between rs10896437 and rs2924540, r2= 0.59). However, we have established that the centromeric region contains an independent association signal because it remained significant after adjusting for the telomeric region in the 2-SNP and 3-SNP models (Fig. 2, Supplementary Material, Table S2).
For the telomeric region, SNPs in bin2 (rs4255548, rs4495900 and rs7950547) are highly correlated with SNPs in bin3, which includes rs10896449 (average pair-wise r2= 0.59). None of the bin2 SNPs remained significant in conditional logistic regression analysis after adjusting for rs10896449 or SNPs in bin3 (Fig. 2, Supplementary Material, Table S2). Singletons rs12793759 and rs12281017 (r2= 0.68) segregate within the telomeric region with bin2 SNPs and bin3 SNPs. When conditioned on the initial hit rs10896449, rs12793759 ranked highest and was highly significant even after adjustment for multiple testing within the region (adjusted P = 0.004).
To explore the complex genetic architecture, we constructed haplotypes and tested for haplotype-specific effect for the 18 genome-wide significant SNPs, covering 105.650 kb (Table 1, Supplementary Material, Table S4). Thirteen haplotypes with population frequency >1% (constituting 91.4% of all observed haplotypes) were inferred and tested. P-values obtained from the haplotype-based logistic regression analysis for global haplotypic effect showed significance (P= 9.61 × 10−14). The results for the global haplotype test performed separately for the centromeric and telomeric regions (Fig. 1) (centromeric, rs10896437–rs11228553; telomeric, rs4255548–rs11228583) demonstrated significant global haplotypic effects (P = 1.78 × 10−8 for the centromeric and P = 4.92 × 10−17 for the telomeric regions).
We performed a series of conditional haplotype analyses to determine if additional signals not captured by rs10896449, rs12793759 and rs10896438 could be identified (Supplementary Material, Table S5). When conditioned on both the telomeric SNPs rs10896449 and rs12793759, the global haplotype effect was borderline significant (P = 0.065), indicating that the highly significant global haplotype effect that we observed for the centromeric region could be partially ascribed to the moderate LD between the two regions (Fig. 1). When conditioned on rs10896438, rs12793759 and rs10896449, the global haplotype effect for the whole region was insignificant (P = 0.166), indicating that no additional signals that contribute to the global haplotype effect were among the 18 genome-wide significant SNPs.
We performed a tag analysis across each data set, our re-sequence data (447 variants, 63 samples), 1000 Genome data (Nov 2010 release, 430 variants, 60 samples) and HapMap CEU (release 28, 114 variants, 60 samples) (Supplementary Material, Table S6). For our analysis, indels were included from both the re-sequence analysis and the 1000 Genome data set, in which we observed minor differences between the two. Not all variants were detected by re-sequence analysis, the 1000 Genome and HapMap projects, but when we combined the three sets, we observed at least 63 surrogates for the three-SNPs identified using a standard threshold of r2> 0.8 (Table 3). On the basis of our analysis we identified sets of surrogate variants for each of the three markers for the independent signals, rs10896449, rs12793759 and rs10896438 of at least 31, 24 and 8 common markers. Of note, the HapMap CEU data lacked a suitable surrogate for rs12793759, while re-sequence data and 1000 Genome data identified eight surrogates in total, four of which were binned separately in HapMap.
Table 3.
Number of surrogates by r2 > 0.8 in three data sets
| Re-sequence | 1000 Genomea | Hapmapb | Total | |
|---|---|---|---|---|
| rs10896449 | 25 | 29 | 17 | 31 |
| rs10896438 | 21 | 16 | 9 | 24 |
| rs12793759 | 6 | 8 | 0 | 8 |
| 52 | 53 | 26 | 63 |
Tag analyses were performed separately in the re-sequence data (447 variants, 63 samples), 1000 Genome data (Nov 2010 release, 430 variants, 60 samples) and HapMap CEU (release 28, 114 variants, 60 samples) in the re-sequenced region 11:68,642,75568,765,690 (UCSC genome build hg18) and number of surrogates for rs10896449, rs10896438 and rs12793759 by r2 > 0.8 was counted.
a1000 Genome CEU (Nov 2010 release, low-coverage data).
bHapMap III (release 2) CEU 60 founders.
DISCUSSION
In our large-scale fine-mapping analysis of the region of 11q13 associated with prostate cancer in men of European background, we confirmed a set of SNPs highly correlated with the initial signal, rs10896449. On the basis of our large sample size in subjects of similar continental origin (men of European background), we observed two more independent signals in the region, each of which is notable for unique sets of highly correlated surrogates. The evidence is based on a sequential multi-locus model that retained region-specific significance. This observation is similar to a recent study of the regulation of fetal hemoglobin by the HBS1L-MYB intergenic interval in which the authors demonstrated a more complex architecture, namely several independent loci that explain the effect of a set of variants on hemoglobin F levels (27). Our study is also notable because we observed a recombination hotspot located between two of the three independent markers. Moreover, haplotype analyses did not detect additional signals in the region, suggesting that there are at most three loci contributing to common variants on 11q13 associated with the risk of prostate cancer.
We estimate a comprehensive set of common genetic markers associated with prostate cancer risk to be pursued in follow-up functional analyses using data drawn from a re-sequence analysis of individuals without evidence of cancer in the Prostate, Lung, Colon and Ovarian (PLCO) cohort, together with publicly available data from the 1000 Genome Project and the International HapMap. For the SNP initially discovered by GWAS, rs10896449, a recent meta-analysis of prostate cancer risk in men of African-American background confirmed the association of rs7931342 (28), which is correlated in CEU (r2 = 0.966) and YRI (r2 = 0.456) in HapMap. These results suggest that one or more functional variants probably reside in this bin of surrogate SNPs. Further mapping studies in other populations, such as African-Americans and Japanese, in which the signal has been seen, could be useful to decrease the number of variants in each of the three loci for functional analyses based on differences in observed LD patterns. In light of the large number of surrogates identified across the three loci, the functional variants responsible for the direct association will have to be pursued based on a prioritization of variants using in silico analysis together with interest in SNPs that could influence the function or regulation of flanking genes, TPCN2 and MYEOV. High-priority variants include those observed in genomic analyses in publically available resources reporting massively parallel sequencing of chromatin immunoprecipitation (ChIP-seq) or whole transcriptomes. Similarly, SNPs residing in regions notable for a high degree of sequence conservation over multiple vertebrates (UCSC genome browser regulation tracks) could be prioritized on the assumption that they could indicate possible regulatory regions.
A limitation of the study is that the choice of tagging SNPs was determined based on the set of SNPs available in HapMap in 2008 and before the completion of Phase 1 of the 1000 Genome Project as well as our next generation sequence analysis (29). Consequently, our hypothesis interrogated common variants, namely those with MAF > 5% and thus could not investigate the possibility of the recently described ‘synthetic association’ predicated on a set of rare variants that can explain part or all of the common variant signals discovered in our GWAS and subsequent mapping reported herein (30).
Two groups have explored this region of chromosome 11q13, including one that has participated in CGEMS (20). Both groups have shown evidence for a second locus, but neither demonstrated three independent signals simultaneously as we report herein. Gudmundsson et al. explored the region with six SNPs, five of which we directly tested (rs7128814, rs11228563, rs11603288, rs7950547 and rs3884627), and the one not tested in CGEMS, rs11228565, was monitored with a proxy rs12281017 (r2 = 0.861 and D′ = 0.950 in HapMap CEU) (7). In our data, rs12281017 is one of the 18 SNPs that achieved genome-wide significance (P = 8.09 × 10−15) (Table 1) and demonstrated high correlation with rs12793759 (r2 = 0.68, D′ = 1.00, CGEMS control samples) that we report to be the most significant association in this region after adjustment for the initial signal rs10896449 (Figs 1 and 2). Because of the high LD, it is not surprising that in the multivariate conditional analysis, no significant association was observed for either SNP when adjusted for the other (rs12793759, adjusted P = 0.9890 conditioned on rs12281017; rs12281017, adjusted P = 0.1046 conditioned on rs12793759) (Supplementary Material, Fig. S2 and Table S2). rs12793759 is also strongly correlated with rs11228565 (r2 = 0.554, D′ = 0.826 in HapMap CEU), further suggesting that rs12793759, rs12281017 and rs11228565 point to a single locus. Zheng et al. reported on the centromeric locus, which we now confirm with six SNPs that reached genome-wide significance. In our study, rs10896438, which is a proxy for the previously reported (but not tested here) rs12418451 (20), showed the most promising association in the centromeric region after adjustment for the telomeric SNPs (Fig. 2, Table 2, Supplementary Material, Table S3).
The results of an analysis looking for non-multiplicative interaction between the initial signal, rs10896449, and additional SNPs on chromosome 11q13 were not statistically significant after multiple testing adjustments. However, the three top SNPs identified by this analysis, namely rs7118561, rs11228608 and rs7103126, were of interest because of their possible biological significance (P-values = 4.21 × 10−3, 4.65 × 10−3 and 6.42 × 10−3, respectively, not adjusted for multiple tests) (Supplementary Material, Fig. S3). Two of the three map to the MYEOV gene: rs11228608 is in the 5′ untranslated region (31) and rs7103126 is a non-synonymous coding SNP in exon 3 (Val>Ala). The biological role of MYEOV is not yet known, but it is noteworthy that the putative, functional SNPs in the gene showed interaction with the original risk marker, rs10896449. It is also notable that rs10896450 (r2 = 0.967 with rs10896449, 1000 genome CEU Nov 2010 release) is significantly associated with CCND1 mRNA expression (LOD = 9.004, P-value = 1.2 × 10−10) in lymphoblastoid cell lines derived from 400 children from families recruited through a proband with asthma (32). It is also notable that four additional SNPs showed significance with CCDN1 mRNA expression from a 20 kb region telomeric of the rs10896450, but these reside beyond a recombination hotspot that flanked the prostate cancer signal (Fig. 1). Although there are no known genes in the region harboring the three genome-wide signals associated with prostate cancer risk, there are three putative transcripts reported between rs9787877 and rs7939250 (Chr11:68 753 085–68 759 526, EST GenBank accession numbers: AA303209, BG946037 and DB036467). DB036467 is evident in testis and appears to be a spliced EST. AA303209 and BG946037 are reported in testis and bladder tumor libraries. Further work is needed to investigate both the eQTL finding and the spliced ESTs as possible underpinnings of the biological basis for the association signals, particularly in the telomeric region of the 11q13 locus.
Recently, GWAS in breast cancer and kidney cancer have identified loci, telomeric to the three prostate cancer loci in 11q13 (Supplementary Material, Fig. S1) (24,25). Interestingly, there is no significant correlation (LD) between the disease-specific markers nor are the markers strongly correlated with SNPs within the coding exons and introns of flanking genes, TPCN2 and MYEOV. However, the signals for breast and kidney cancer do not appear to harbor multiple independent neighboring loci as we report for prostate cancer. Still, it is notable that all of the SNPs associated with prostate, kidney and breast cancer in 11q13 map to non-genic regions. It is plausible that these variants influence a set of regulatory events, either locally or at a distance, perhaps comparable to those discovered in the region of 8q24 centromeric to MYC.
In summary, we have fine-mapped the 11q13 region surrounding the original signal—rs10896449—and showed that the region harbors a complex genomic architecture characterized by multiple independent signals contributing to prostate cancer risk. Of the 120 SNPs directly genotyped across a 241 kb region of 11q13, 18 SNPs showed genome-wide significance (P-values of <10−8) in association with prostate cancer risk. Across this region, we observed three correlation bins (defined by pair-wise r2 > 0.8). Conditional analyses using multi-locus models revealed three independent signals: the initial signal rs10896449 and two novel markers, rs12793759, and rs10896438, the latter of which is a proxy for the previously reported marker, rs12418451 (20). Our results underscore the value of investigating GWAS loci in large-scale follow-up genotyping studies. Together with an analysis of targeted re-sequence analysis with 1000 Genome and Hapmap data, we have identified a comprehensive catalog of common variants in 11q13 associated with the overall risk for prostate cancer. Future studies will need to investigate the biological basis of common variants in 11q13, either those directly tested or surrogates of the established markers, in order to elucidate the molecular basis of the direct association of this region with prostate cancer susceptibility.
MATERIALS AND METHODS
Study population
The initial scan was conducted in a nested case–control study of 1172 screened cases (484 non-aggressive prostate cancer, Gleason score <7 and disease stage <III; 688 aggressive prostate cancer, Gleason score ≥7 and/or disease stage ≥III) and 1157 PSA-screened controls in men of European ancestry from the PLCO Cancer Screening Trial. The second scan was conducted in four additional replication studies totaling 4020 cases and 4028 controls [American Cancer Society Cancer Prevention Study II (CPSII), 1790/1797; the Health Professionals Follow-up Study (HPFS), 619/620; CeRePP French Prostate Case–Control Study (FPCC), 671/671; and Alpha-Tocopherol, Beta-Carotene Cancer Prevention Study (ATBC), 940/940].
For Stage 3 of CGEMS, reported here, subjects of European origin were drawn from ten studies that participate in the CGEMS initiative. Overall, 10 272 cases and 9123 controls were available for analysis after quality control metrics were applied and reported elsewhere (8). Seven cohort studies were included: the American Cancer Society Cancer Prevention Study II (CPSII), 1634/1640 (cases/controls); the Health Professionals Follow-Up Study (HPFS), 595/589; the PLCO Cancer Screening Trial, 972/927; Alpha-Tocopherol, Beta-Carotene Cancer Prevention Study (ATBC), 906/868; the Multiethnic Cohort genetic study (MEC), 676/682; the European Prospective Investigation into Cancer and Nutrition (EPIC), 682/990; and the Cohort of Norway (CONOR), 606/662. Three case–control studies were included: the CeRePP FPCC, 998/952; Cancer of the Prostate in Sweden (CAPS), 2213/1362; and a hospital-based case–control from the Johns Hopkins Hospital (JHH), 990/451.
Genotyping and re-sequencing
SNPs chosen for fine-mapping analysis of common variants in the region of chromosome 11q13 marked by rs10896449 were genotyped as part of a custom Infinium chip (Illumina, San Diego, CA, USA) that investigated several hypotheses: follow-up of ∼150 regions that had at least one SNP with an observed P < 10−3 after Stage 2 of CGEMS (15), fine-mapping of significant SNPs in other regions of the genome and an analysis of ∼1500 SNPs with (r2 < 0.004) for investigation of population substructure. The details of the other hypotheses were reported elsewhere (8).
SNPs were selected from a region of chromosome 11q13 defined using the 0.2cM HapMap recombination data flanking the most significant SNP from the Cancer Genetic Markers of Susceptibility (CGEMS) second stage GWAS (rs10896449, P= 1.76 × 10−9) (15). This spans the variants between rs930782 to rs4584599, which corresponds to positions 68628370 to 68870174 (Build 36 NCBI) and covers 241 805 bp. A two-staged tagging strategy was used to select SNPs with an MAF greater than 0.05 from HapMap (Build 26). The entire region was tagged at a D′= 0.6 using HapMap CEU with obligate includes of all significant SNPs (P-values of <10−3) from the second stage of CGEMS (15). Final tags were chosen if they were observed to be correlated with an r2 ≥ 0.8 in HapMap CEU, YRI and JPT + CHB with the obligate includes.
A total of 155 SNPs were selected for analysis and 27 were excluded due to design failure or provided a monoallelic signal. After applying quality control metrics, 120 SNPs were available for subsequent analysis; further exclusions were due to a genotype completion rate <97%.
A 123 kb region (11q13: 68 642 755–68 765 690, UCSC genome build hg18) was re-sequenced in 63 individuals of European ancestry, 61 individuals who were cancer-free drawn from the PLCO cohort, and 2 from a CEPH pedigree 1350, which was based on the observed LD pattern flanking rs10896449 using HapMap CEU data (release 22, phase II). Next generation sequence analysis was conducted with the 454 Genome Sequencer FLX system (http://www.454.com/products-solutions/product-list.asp) after a custom Nimblegen solution-based sequence capture method targeted the region of interest. Sequence reads that passed quality check using vendor-supplied software were aligned to the target genomic region using MOSAIK software. Variants were called based on a set of heuristic rules; then for quality assurance, NextGEN2 and Consed were used to resolve ambiguous cases. Genotype completion, concordance, MAF estimations, deviations from fitness for the Hardy–Weinberg proportion, pair-wise LD and tag SNP analysis were performed using the GLU software package (Genotype Library and Utilities; http://code.google.com/p/glu-genetics/).
Analysis
Single-SNP analyses were conducted using unconditional logistic regression, adjusted for age, study, center and population stratification based on four principal components' analyses using the set of ∼1400 SNPs chosen because of minimal correlation (r2< 0.004) (33). In addition, multi-locus models were used to explore interactions and independent signals within this chromosomal region. A two-SNP model conditioned on the original CGEMS hit, rs10896449, was used to assess whether a second independent signal was among the remaining SNPs. The significance of the top-ranked SNP in that analysis was assessed through a parametric permutation method to account for multiple testing in an efficient manner that can account for the LD between the SNPs. Because a significant result was observed for rs12793759 in the two-SNP model, a similar analysis was performed for a three-SNP model conditioned on both rs10896449 and rs12793759. Tests for multiplicative interactions between rs10896449 and other SNPs in the region were performed using logistic regression modeling. In all analyses, the count for the minor allele at each SNP locus was coded as a continuous variable and the corresponding association/interaction tests were performed using a 1 degree of freedom χ2 test.
We performed a tag analysis using the re-sequence data (447 variants, 63 samples), 1000 genome data (Nov 2010 release, 430 variants, 60 samples) (29) and HapMap CEU (release 28, 114 variants, 60 samples) (34,35) across the region 11: 68 642 755–68 765 690 (UCSC genome build hg18). The program tagzilla implemented in the GLU (Genotyping Library and Utilities), an open source suite of tools (http://code.google.com/p/glu-genetics/), was used to compare surrogates of rs10896438, rs12793759 and rs10896449 across a range of r2 thresholds (0.8–1.0). Indels identified after preliminary quality control assessment of re-sequence data were included in the analyses. Similarly, indels from the 1000 Genome data were included to maximize possible surrogates.
To identify recombination hotspots in the region, we used SequenceLDhot (26), a program that uses the approximate marginal likelihood method (36) and calculates likelihood ratio statistics at a set of possible hotspots. We sequentially tested control samples from each study, by pooling control groups of 90 samples from each study as well as by continental groups. For the latter, we categorized control samples into three continental groups, European; EPIC and FPCC; Scandinavian; ATBC, CONOR and CAPS; and USA: CPSII, HPFS, MEC, JHH and PLCO; then two sets of control samples for each group were used without resampling. For the overall pooled control samples, 90 samples from each study were sampled five times without re-sampling to create 5 sets of 900 pooled control samples. Haplotypes and background recombination rates were inferred using PHASE v2.1 (37,38) and used as direct input for the SequenceLDhot program. We performed a conditional haplotype analysis using the program WHAP (39) implemented in PLINK software package (40).
URLs
CGEMS portal: http://cgems.cancer.gov/
Database of Genomic Variants: http://projects.tcag.ca/variation/
GLU: http://code.google.com/p/glu-genetics/
Tagzilla: http://cgf.nci.nih.gov/glu/docs/1.0b2/modules/ld/tagzilla.html
PLINK: http://pngu.mgh.harvard.edu/~purcell/plink/
SnpPlotter:http://cbdb.nimh.nih.gov/~kristin/snp.plotter.html
STRUCTURE: http://pritch.bsd.uchicago.edu/structure.html
EIGENSTRAT: http://genepath.med.harvard.edu/~reich/EIGENSTRAT.htm
SUPPLEMENTARY MATERIAL
FUNDING
This study was supported by the Intramural Research Program of the Division of Cancer Epidemiology and Genetics, National Cancer Institute, National Institutes of Health (NIH). The project was partially funded in part using grant CA129684 to J.X. and Contract Number HHSN261200800001E.
Supplementary Material
ACKNOWLEDGEMENTS
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services nor does the mention of trade names, commercial products or organization indicate endorsement by the U.S. Government. The authors thank Drs Christine Berg and Philip Prorok, Division of Cancer Prevention, NCI, the screening center investigators and staff of the PLCO Cancer Screening Trial, Mr Thomas Riley and staff at Information Management Services, Inc., and Ms. Barbara O'Brien and staff at Westat, Inc. for their contributions to the PLCO Cancer Screening Trial. Finally, we acknowledge the study participants for donating their time and making this study possible.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Ferlay J., Autier P., Boniol M., Heanue M., Colombet M., Boyle P. Estimates of the cancer incidence and mortality in Europe in 2006. Ann. Oncol. 2007;18:581–592. doi: 10.1093/annonc/mdl498. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A., Siegel R., Ward E., Hao Y., Xu J., Thun M.J. Cancer statistics, 2009. CA. Cancer J. Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 3.Crawford E.D. Understanding the epidemiology, natural history, and key pathways involved in prostate cancer. Urology. 2009;73:S4–S10. doi: 10.1016/j.urology.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 4.Lichtenstein P., Holm N.V., Verkasalo P.K., Iliadou A., Kaprio J., Koskenvuo M., Pukkala E., Skytthe A., Hemminki K. Environmental and heritable factors in the causation of cancer—analyses of cohorts of twins from Sweden, Denmark, and Finland. N. Engl. J. Med. 2000;343:78–85. doi: 10.1056/NEJM200007133430201. [DOI] [PubMed] [Google Scholar]
- 5.Al Olama A.A., Kote-Jarai Z., Giles G.G., Guy M., Morrison J., Severi G., Leongamornlert D.A., Tymrakiewicz M., Jhavar S., Saunders E., et al. Multiple loci on 8q24 associated with prostate cancer susceptibility. Nat. Genet. 2009;41:1058–1060. doi: 10.1038/ng.452. [DOI] [PubMed] [Google Scholar]
- 6.Eeles R.A., Kote-Jarai Z., Al Olama A.A., Giles G.G., Guy M., Severi G., Muir K., Hopper J.L., Henderson B.E., Haiman C.A., et al. Identification of seven new prostate cancer susceptibility loci through a genome-wide association study. Nat. Genet. 2009;41:1116–1121. doi: 10.1038/ng.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gudmundsson J., Sulem P., Gudbjartsson D.F., Blondal T., Gylfason A., Agnarsson B.A., Benediktsdottir K.R., Magnusdottir D.N., Orlygsdottir G., Jakobsdottir M., et al. Genome-wide association and replication studies identify four variants associated with prostate cancer susceptibility. Nat. Genet. 2009;41:1122–1126. doi: 10.1038/ng.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yeager M., Chatterjee N., Ciampa J., Jacobs K.B., Gonzalez-Bosquet J., Hayes R.B., Kraft P., Wacholder S., Orr N., Berndt S., et al. Identification of a new prostate cancer susceptibility locus on chromosome 8q24. Nat. Genet. 2009;41:1055–1057. doi: 10.1038/ng.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amundadottir L.T., Sulem P., Gudmundsson J., Helgason A., Baker A., Agnarsson B.A., Sigurdsson A., Benediktsdottir K.R., Cazier J.B., Sainz J., et al. A common variant associated with prostate cancer in European and African populations. Nat. Genet. 2006;38:652–658. doi: 10.1038/ng1808. [DOI] [PubMed] [Google Scholar]
- 10.Eeles R.A., Kote-Jarai Z., Giles G.G., Olama A.A., Guy M., Jugurnauth S.K., Mulholland S., Leongamornlert D.A., Edwards S.M., Morrison J., et al. Multiple newly identified loci associated with prostate cancer susceptibility. Nat. Genet. 2008;40:316–321. doi: 10.1038/ng.90. [DOI] [PubMed] [Google Scholar]
- 11.Gudmundsson J., Sulem P., Manolescu A., Amundadottir L.T., Gudbjartsson D., Helgason A., Rafnar T., Bergthorsson J.T., Agnarsson B.A., Baker A., et al. Genome-wide association study identifies a second prostate cancer susceptibility variant at 8q24. Nat. Genet. 2007;39:631–637. doi: 10.1038/ng1999. [DOI] [PubMed] [Google Scholar]
- 12.Gudmundsson J., Sulem P., Steinthorsdottir V., Bergthorsson J.T., Thorleifsson G., Manolescu A., Rafnar T., Gudbjartsson D., Agnarsson B.A., Baker A., et al. Two variants on chromosome 17 confer prostate cancer risk, and the one in TCF2 protects against type 2 diabetes. Nat. Genet. 2007;39:977–983. doi: 10.1038/ng2062. [DOI] [PubMed] [Google Scholar]
- 13.Kraft P., Pharoah P., Chanock S.J., Albanes D., Kolonel L.N., Hayes R.B., Altshuler D., Andriole G., Berg C., Boeing H., et al. Genetic variation in the HSD17B1 gene and risk of prostate cancer. PLoS Genet. 2005;1:e68. doi: 10.1371/journal.pgen.0010068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lou H., Yeager M., Li H., Bosquet J.G., Hayes R.B., Orr N., Yu K., Hutchinson A., Jacobs K.B., Kraft P., et al. Fine mapping and functional analysis of a common variant in MSMB on chromosome 10q11.2 associated with prostate cancer susceptibility. Proc. Natl. Acad. Sci. USA. 2009;106:7933–7938. doi: 10.1073/pnas.0902104106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thomas G., Jacobs K.B., Yeager M., Kraft P., Wacholder S., Orr N., Yu K., Chatterjee N., Welch R., Hutchinson A., et al. Multiple loci identified in a genome-wide association study of prostate cancer. Nat. Genet. 2008;40:310–315. doi: 10.1038/ng.91. [DOI] [PubMed] [Google Scholar]
- 16.Yeager M., Orr N., Hayes R.B., Jacobs K.B., Kraft P., Wacholder S., Minichiello M.J., Fearnhead P., Yu K., Chatterjee N., et al. Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat. Genet. 2007;39:645–649. doi: 10.1038/ng2022. [DOI] [PubMed] [Google Scholar]
- 17.Takata R., Akamatsu S., Kubo M., Takahashi A., Hosono N., Kawaguchi T., Tsunoda T., Inazawa J., Kamatani N., Ogawa O., et al. Genome-wide association study identifies five new susceptibility loci for prostate cancer in the Japanese population. Nat. Genet. 2010;42:751–754. doi: 10.1038/ng.635. [DOI] [PubMed] [Google Scholar]
- 18.Park J.H., Wacholder S., Gail M.H., Peters U., Jacobs K.B., Chanock S.J., Chatterjee N. Estimation of effect size distribution from genome-wide association studies and implications for future discoveries. Nat. Genet. 2010;42:570–575. doi: 10.1038/ng.610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun J., Zheng S.L., Wiklund F., Isaacs S.D., Purcell L.D., Gao Z., Hsu F.C., Kim S.T., Liu W., Zhu Y., et al. Evidence for two independent prostate cancer risk-associated loci in the HNF1B gene at 17q12. Nat. Genet. 2008;40:1153–1155. doi: 10.1038/ng.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zheng S.L., Stevens V.L., Wiklund F., Isaacs S.D., Sun J., Smith S., Pruett K., Wiley K.E., Kim S.T., Zhu Y., et al. Two independent prostate cancer risk-associated Loci at 11q13. Cancer Epidemiol. Biomarkers Prev. 2009;18:1815–1820. doi: 10.1158/1055-9965.EPI-08-0983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sulem P., Gudbjartsson D.F., Stacey S.N., Helgason A., Rafnar T., Jakobsdottir M., Steinberg S., Gudjonsson S.A., Palsson A., Thorleifsson G., et al. Two newly identified genetic determinants of pigmentation in Europeans. Nat. Genet. 2008;40:835–837. doi: 10.1038/ng.160. [DOI] [PubMed] [Google Scholar]
- 22.Janssen J.W., Cuny M., Orsetti B., Rodriguez C., Valles H., Bartram C.R., Schuuring E., Theillet C. MYEOV: a candidate gene for DNA amplification events occurring centromeric to CCND1 in breast cancer. Int. J. Cancer. 2002;102:608–614. doi: 10.1002/ijc.10765. [DOI] [PubMed] [Google Scholar]
- 23.Janssen J.W., Vaandrager J.W., Heuser T., Jauch A., Kluin P.M., Geelen E., Bergsagel P.L., Kuehl W.M., Drexler H.G., Otsuki T., et al. Concurrent activation of a novel putative transforming gene, myeov, and cyclin D1 in a subset of multiple myeloma cell lines with t(11;14)(q13;q32) Blood. 2000;95:2691–2698. [PubMed] [Google Scholar]
- 24.Purdue M.P., Johansson M., Zelenika D., Toro J.R., Scelo G., Moore L.E., Prokhortchouk E., Wu X., Kiemeney L.A., Gaborieau V., et al. Genome-wide association study of renal cell carcinoma identifies two susceptibility loci on 2p21 and 11q13.3. Nat. Genet. 2010;43:60–65. doi: 10.1038/ng.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Turnbull C., Ahmed S., Morrison J., Pernet D., Renwick A., Maranian M., Seal S., Ghoussaini M., Hines S., Healey C.S., et al. Genome-wide association study identifies five new breast cancer susceptibility loci. Nat. Genet. 2010;42:504–507. doi: 10.1038/ng.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fearnhead P. SequenceLDhot: detecting recombination hotspots. Bioinformatics. 2006;22:3061–3066. doi: 10.1093/bioinformatics/btl540. [DOI] [PubMed] [Google Scholar]
- 27.Galarneau G., Palmer C.D., Sankaran V.G., Orkin S.H., Hirschhorn J.N., Lettre G. Fine-mapping at three loci known to affect fetal hemoglobin levels explains additional genetic variation. Nat. Genet. 2010;42:1049–1051. doi: 10.1038/ng.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang B.L., Spangler E., Gallagher S., Haiman C.A., Henderson B.E., Isaacs W.B., Benford M.L., Kidd L.R., Cooney K., Strom S.S., et al. Validation of Genome-Wide Prostate Cancer Associations in Men of African Descent. Cancer Epidemiol. Biomarkers Prev. 2010;20:23–32. doi: 10.1158/1055-9965.EPI-10-0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Durbin R.M., Abecasis G.R., Altshuler D.L., Auton A., Brooks L.D., Gibbs R.A., Hurles M.E., McVean G.A. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dickson S.P., Wang K., Krantz I., Hakonarson H., Goldstein D.B. Rare variants create synthetic genome-wide associations. PLoS Biol. 2010;8:e1000294. doi: 10.1371/journal.pbio.1000294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Almeida R.A., Heuser T., Blaschke R., Bartram C.R., Janssen J.W. Control of MYEOV protein synthesis by upstream open reading frames. J. Biol. Chem. 2006;281:695–704. doi: 10.1074/jbc.M511467200. [DOI] [PubMed] [Google Scholar]
- 32.Dixon A.L., Liang L., Moffatt M.F., Chen W., Heath S., Wong K.C., Taylor J., Burnett E., Gut I., Farrall M., et al. A genome-wide association study of global gene expression. Nat. Genet. 2007;39:1202–1207. doi: 10.1038/ng2109. [DOI] [PubMed] [Google Scholar]
- 33.Yu K., Wang Z., Li Q., Wacholder S., Hunter D.J., Hoover R.N., Chanock S., Thomas G. Population substructure and control selection in genome-wide association studies. PLoS One. 2008;3:e2551. doi: 10.1371/journal.pone.0002551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Altshuler D.M., Gibbs R.A., Peltonen L., Dermitzakis E., Schaffner S.F., Yu F., Bonnen P.E., de Bakker P.I., Deloukas P., Gabriel S.B., et al. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–58. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frazer K.A., Ballinger D.G., Cox D.R., Hinds D.A., Stuve L.L., Gibbs R.A., Belmont J.W., Boudreau A., Hardenbol P., Leal S.M., et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fearnhead P., Donnelly P. Approximate likelihood methods for estimating local recombination rates. J. R. Stat. Soc. Ser. B-Stat. Methodol. 2002;64:657–680. [Google Scholar]
- 37.Crawford D.C., Bhangale T., Li N., Hellenthal G., Rieder M.J., Nickerson D.A., Stephens M. Evidence for substantial fine-scale variation in recombination rates across the human genome. Nat. Genet. 2004;36:700–706. doi: 10.1038/ng1376. [DOI] [PubMed] [Google Scholar]
- 38.Li N., Stephens M. Modeling linkage disequilibrium and identifying recombination hotspots using single-nucleotide polymorphism data. Genetics. 2003;165:2213–2233. doi: 10.1093/genetics/165.4.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Purcell S., Daly M.J., Sham P.C. WHAP: haplotype-based association analysis. Bioinformatics. 2007;23:255–256. doi: 10.1093/bioinformatics/btl580. [DOI] [PubMed] [Google Scholar]
- 40.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luna A., Nicodemus K.K. snp.plotter: an R-based SNP/haplotype association and linkage disequilibrium plotting package. Bioinformatics. 2007;23:774–776. doi: 10.1093/bioinformatics/btl657. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.