

Abstract
Inactivation of manganese superoxide dismutase (MnSOD), a mitochondrial antioxidant, has been associated with renal disorders and often results in detrimental downstream events that are mechanistically not clear. Development of an animal model that exhibits kidney-specific deficiency of MnSOD would be extremely beneficial in exploring the downstream events that occur following MnSOD inactivation. Using Cre-Lox recombination technology, kidney-specific MnSOD deficient mice (both 100% and 50%) were generated that exhibited low expression of MnSOD in discrete renal cell types and reduced enzymatic activity within the kidney. These kidney-specific 100% KO mice possessed a normal life-span, although it was interesting that the mice were smaller. Consistent with the important role in scavenging superoxide radicals, the kidney-specific KO mice showed a significant increase in oxidative stress (tyrosine nitration) in a gene-dose dependent manner. In addition, loss of MnSOD resulted in mild renal damage (tubular dilation and cell swelling). Hence, this novel mouse model will aid in determining the specific role (local and/or systemic) governed by MnSOD within certain kidney cells. Moreover, these mice will serve as a powerful tool to explore molecular mechanisms that occur downstream of MnSOD inactivation in renal disorders or possibly in other pathologies that rely on normal renal function.
Keywords: Cre-Lox technology, Kidney, MnSOD, Cre recombinase, Superoxide, Nitrotyrosine
Introduction
Manganese superoxide dismutase (MnSOD), also known as SOD2, is the major mitochondrial antioxidant responsible for scavenging superoxide radicals generated by the respiratory chain activity or via mitochondrial stressors [1,2]. This enzyme is encoded by a single copy nuclear gene that consists of five exons and four introns [3,4], and upon translation MnSOD is transported to mitochondria via an amino-terminal targeting sequence [5]. Studies using global MnSOD knockout (KO) mice have shown that complete loss of MnSOD can result in massive oxidative stress and neonatal death caused by cardiomyopathy, neurodegeneration, and metabolic acidosis [6,7]. Thus, it is clear that MnSOD provides an indispensable function within the mitochondria.
The balance of oxidants and antioxidants may play a primary role against the development of the cell and tissue injury. Damage caused by excess production of mitochondrial superoxide has been implicated in the pathogenesis of a number of disorders such as chronic inflammation, aging and cancer [8-12]. Reduced MnSOD enzymatic activity has been well-documented in several diseases [12-18] and can lead to significant oxidative stress within the mitochondria and/or cell. Inactivation of MnSOD has been frequently observed in renal disorders such as ischemia/reperfusion injury, transplant rejection as well as angiotensin-II induced hypertension [19-21]. Our laboratory has previously shown that MnSOD is susceptible to tyrosine nitration and oxidation which leads to inactivation of the enzyme, hence additional oxidant production [19,22]. These reports clearly demonstrated that loss of MnSOD protein did not account for loss of enzymatic activity during renal transplant injury; rather post-translational modifications of the enzyme were involved. In addition, these studies also showed that MnSOD inactivation preceded renal damage further suggesting that loss of MnSOD activity was a key event in renal damage following ischemia. However, the mechanistic pathways involved with the protection governed by MnSOD remain largely unknown. This has encouraged us to investigate the molecular events downstream to the reduced expression of MnSOD enzyme within the kidney using an in vivo model.
The development of transgenic and gene-KO mice in which the MnSOD gene is either over-expressed or knocked out, respectively, provides a powerful tool to study the consequence of reduced MnSOD in disease and/or to determine the enzyme's contributory role in normal physiology. However, as mentioned earlier, the complete MnSOD KO mice die [6,7], and the heterozygous/partial MnSOD KO mice exhibit decreased MnSOD activity in all tissues/organs [7,23] which makes interpretation of these data on specific organ function less clear. Thus, the goal of this study was to develop a novel mouse model that mimics a condition of renal inactivation of MnSOD in vivo. This paper describes the generation of a kidney-specific MnSOD KO mouse line using Cre mediated deletion of MnSOD allele (s). These renal-specific KO mice will serve as an invaluable tool to explore the molecular mechanisms that occur downstream of MnSOD inactivation in various renal disorders and could possibly be useful in other pathologies that rely on normal renal function.
Materials and Methods
Mice
Two transgenic mouse lines were used to develop kidney-specific MnSOD KO mice. The first transgenic mouse line (Ksp1.3/Cre) expressed Cre recombinase specifically in the kidney (distal tubules, collecting ducts, Loops of Henle, ureteric buds, and developing genitourinary tract) [24] and was a generous gift from Drs. Peter Igarashi and Paul Overbeek. This mouse line is on a C57BL/6 background. The second transgenic mouse line was the MnSOD floxed mouse (exon 3 of MnSOD allele was flanked with two LoxP sites in introns 2 and 3) on a C57BL6CrSlc background, which was a generous gift from Drs. Takuji Shirasawa and Ting-Ting Huang [25]. Mice were maintained according to the criteria outlined in the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH). All of the animal protocols were approved by the Institutional Animal Care and Use Committee at the University of Arkansas for Medical Sciences to perform as described in the paper.
Generation of mice with kidney-specific deletion of MnSOD
Heterozygous female MnSOD floxed mice (MnSODflox/wt) [25] were crossed with heterozygous male Kidney Cre mice that express Ksp1.3/Cre transgene specifically in the kidney (Kidney Cre/MnSODwt/wt) [24] as illustrated in Figure 1B. From the filial (F) 1 progeny, mice (male or female) with heterozygous deletion of MnSOD gene that harbor Ksp 1.3/Cre transgene (Kidney Cre/MnSODflox/wt or 50% KO) were selected. These 50% KO mice were further crossed with the opposite sex of MnSOD floxed mice (MnSODflox/wt) to obtain mice expressing complete deletion of MnSOD (Kidney Cre/MnSODflox/flox or 100% KO) in the F2 progeny. In addition, to increase the percentage of 100% KO mice in the F2 progeny crosses between MnSOD homozygous floxed mice (MnSODflox/flox) and 50% KO were also made.
Figure 1. Generation of novel kidney specific MnSOD knockout (KO) mice.
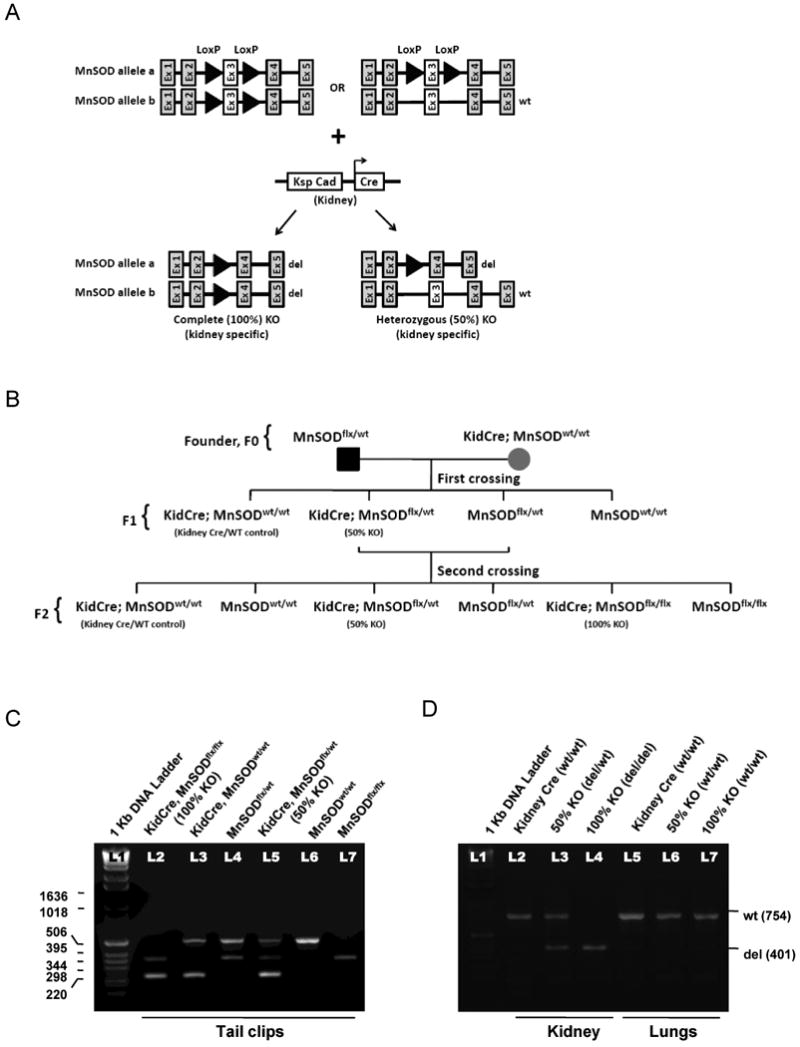
(A) Schematic diagram showing the LoxP sites flanking Exon 3 of MnSOD alleles that are targets for the Cre recombinase (CR) enzyme. As a result, Exon 3 will be deleted leaving a mutant form of the MnSOD allele (complete/100% KO and heterozygous/50% KO). (B) Breeding strategy using founders (F0) transgenic MnSOD floxed mice and Ksp/1.3 Cre transgenic mice to obtain kidney specific 50% and 100% MnSOD KO mice. (C) Multiplex PCR was used to determine Cre and MnSOD gene expression of all six genotypes using tail clip DNA. The yield of PCR products were as follows: 500 bp - MnSODwt, 358 bp - MnSODflox, and 235 bp - Cre. (D) DNA material isolated from mouse kidney and lung tissues were PCR amplified using P1 and P3 primers. Kidney DNA of 50% and 100% KO mice yielded PCR product of 401 bp (MnSODdel) while an additional 754 bp fragment of MnSODwt was amplified from the kidney DNA of 50% KO mice and Kidney Cre mice. Amplified lung DNA displayed only the MnSODwt band in all three genotypes using P1 and P3 primers.
Genotype analysis
Genomic DNA was extracted either using the HotSHOT method [26], from tail clips of 4 weeks old pups or using a commercialized kit (DNeasy® Tissue Kit, Qiagen Inc, Valencia, CA) from kidney and liver tissues after sacrificing the mice at 8-10 wks of age (this was performed to detect the deleted MnSOD allele). Five different published PCR primer pairs (P1: 5′CGA GGG GCA TCT AGT GGA GAA G; P2: 5′TTA GGG CTC AGG TTT GTC CAT AA; P4: 5′ AGC TTG GCT GGA GGT AA; Cre1: 5′ AGG TTC GTT CAC TCA TGG A and Cre2: 5′ TCG ACC AGT TTA GTT ACC C) were routinely used to detect the MnSODwt and MnSODflox alleles and the inserted Cre gene by multiplex PCR analysis [24,25]. The multiplex PCR conditions were as follows: 95°C for 15 min, then 32 cycles of 94°C for 35 sec, 58°C for 35 sec, 72°C for 35 sec, and finally 72°C for 10 min. The MnSODwt allele was detected by using primer pairs P1 and P2, which amplified a 500-bp fragment; whereas the MnSODflox allele was detected by using primer pairs P1 and P4, which gave a 358-bp fragment. The Ksp1.3/Cre transgene was detected by using the primer pairs Cre1 and Cre2, which amplified a 235-bp fragment. An additional primer P3 (5′ CTA GTG AGA TGG CTC AGC-3′) [27] was used to identify the deleted MnSOD allele (MnSODdel). Using primer pairs P1 and P3, a 401-bp product of MnSODdel was detected in the complete (100%) KO mice, whereas the heterozygous deletion (50% KO) gave an additional 754-bp wild type (WT) product. The PCR conditions to amplify deleted MnSOD allele were slightly different: 95°C for 15 min, then 30 cycles of 94°C for 45 sec, 62°C for 45 sec, 72°C for 1 min, and finally 72°C for 10 min.
Organ isolation
Mice were anesthetized with Isofluorane, which was delivered as 5% for induction and 2% for maintenance anesthesia using an “ISOTEC” vaporizer. An incision was made superior to the symphysis pubis up to the tip of the xyphoid process. Bilateral nephrectomy was performed immediately after clamping of renal vessels. Both kidneys were weighed and processed as follows: half of the right kidney was fixed in neutral buffered formalin (NBF), the other half and whole left kidneys were saved for biochemical assays (e.g. MnSOD activity assay). The blood was collected from the inferior vena cava. Liver, heart, and lungs were isolated, weighed and saved for histology.
Histological evaluation
Two cross-sections of 4 to 5-μm thickness from each paraffin block were mounted on a glass slide (Fisherbrand Superfrost Plus, Fisher Scientific, Pittsburgh, PA, USA) and deparaffinized through xylene and a series of graded ethanol washes. The sections were further processed as described in separate sections below. Counterstaining was performed with Mayer's Hematoxylin (Electron Microscopy Science, Hatfield, PA, USA) and bluing was carried out by dipping in 0.125 % ammonia blue solution. Finally, the slides were dehydrated and covered with Cytoseal-60 (Electron Microscopy Science, Hatfield, PA, USA) and mounted with a cover slip. All images were taken using Nikon Eclipse E800 microscope (Q Capture imaging and Nikons Elements software).
Periodic Acid-Schiff reaction
Renal sections were assessed for tissue injury using the Periodic Acid-Schiff (PAS) reaction using standard procedures. For each kidney, cross sections containing the cortex and medulla were measured objectively by a pathologist for the severity of cellular damage. The PAS stained sections of KO (50% and 100%) mice were compared to Kidney Cre mice. Evaluation was conducted based on the following criteria: tubular dilation, cast in lumen, and cell swelling/enlargement. All parameters were graded on a scale of 0 - no change, 1 - minimal change, 2 - mild change, and, 3 - prominent change.
Immunohistochemistry
For immunohistochemical analysis, antigens were retrieved by heating sections in 10 mM sodium citrate buffer (pH 6.0) for 20 min. Endogenous peroxidase was quenched by incubating the sections with Peroxidase Suppressor (Thermo Scientific, Rockford, IL, USA) for 15 min at RT. The slides were blocked with Non Serum Protein Block (Dako, Carpinteria, CA, USA) for 20 min at RT. Primary antibodies were prepared in antibody diluent solution (0.5% non fat dry milk and 1% BSA in TBS) and incubated overnight at 4 °C, except for Cre-recombinase (1h at RT). The concentration of primary antibody and dilution were as follows: Anti-MnSOD, 1:250 (Millipore, Temecula, CA, USA); Anti-Cre recombinase, 1:1000 (Covance, Emeriville, CA, USA), Anti-Nitrotyrosine, 1:6000 (Millipore, Temecula, CA, USA). The specificity of nitrotyrosine antibody binding in the renal tissue was confirmed by blocking the antibody with 3-nitrotyrosine (10 mM). Immunoreactivity was detected by Dako Envision+ System-HRP (Dako, Carpinteria, CA, USA). Semi quantitative evaluation of nitrotyrosine staining was performed based on the percentage of positive tubules in 10 high power fields (200×) from cortex and medulla using following scores: 0 - null/negative; 1 - less than 10% positivity; 2 - 10% to 50% positivity; 3 - greater than 50% positivity.
Serum creatinine assay
Serum creatinine was determined using a modified Jaffe's method (Pointe Scientific, Canton, MI, USA) in a Cobas Mira clinical analyzer (Roche Diagnostics, Indianapolis, IN, USA). The values were expressed as mg/dl.
Blood glucose determination
An ACCU-CHEK® Compact Plus meter (Cat. # 3149137) was used to measure the fasting blood glucose levels.
Systolic blood pressure measurement
Systolic blood pressure was recorded in conscious mice using the tail-cuff method [28].
MnSOD activity
Enzymatic activity of MnSOD was determined in renal extracts by the Cytochrome c reduction method in the presence of 1 mM KCN to inhibit Cu, Zn SOD activity as previously described [29].
Statistical analysis
Results are presented as mean ± standard error of the mean (SEM). One way analysis of variance (ANOVA) was used to compare the mean values among the different groups, followed by Tukey's test to compare differences in mean between two groups at 95% level of confidence using the Origin 6.0 statistical software. Differences with a P value less than 0.05 were considered statistically significant.
Results
Generation of kidney specific MnSOD deficient mice
Using Cre-Lox recombination technology, novel kidney specific MnSOD KO mice (complete or 100% KO and heterozygous or 50% KO) were generated. Two different transgenic mouse lines were utilized for breeding: 1) floxed MnSOD mice [25], and 2) Ksp1.3/Cre transgenic (Kidney Cre) mice [24]. The LoxP sites that flank exon 3 of the mouse MnSOD gene are targets for Cre recombinase (CR) that is expressed in the kidney of the same mouse (Fig 1A), thus, exon 3 is deleted leaving the other four exons present in the genome. All six different genotypes were obtained in the second or F2 crossing (Fig 1B). DNAs from tail clips from all mice were PCR amplified using multiplex PCR primers. As shown in Fig 1C, mice with complete deletion of MnSOD allele within the kidney (100% KO) expressed a 358-bp fragment for MnSODflox and a 235-bp fragment for Ksp1.3/Cre transgene (Lane 2). An additional 500-bp fragment for MnSODwt allele was detected in the kidney specific 50% KO mice (Lane 5). Similarly, a single band of 358-bp for mice homozygous for floxed MnSOD (Lane 7) and a single band of 500-bp for mice homozygous for the WT MnSOD allele (Lane 6) were observed. Kidney Cre mice (Lane 3) and heterozygous MnSOD floxed mice (Lane 4) expressed an additional 500-bp wild type MnSOD allele.
To determine whether a CR mediated complete ablation of MnSOD allele occurred specifically in the kidney, genomic DNA extracted from kidney and lungs were PCR amplified using P1 and P3 primers. The deleted MnSOD allele (MnSODdel) was detected as a single 401-bp fragment from the kidney of 100% KO mice, whereas the 50% KO mice gave an additional 754-bp product, which corresponded to WT MnSOD (Fig 1D). Amplification of lung DNA resulted in a single WT MnSOD band (754-bp), with no evidence of the deleted allele, for all genotypes, which confirms that this breeding strategy results in generation of kidney specific MnSOD KO mice (50% and 100%). Additional studies revealed no differences between WT or Kidney Cre mice in any of the parameters tested (data not shown); therefore, Kidney Cre results are shown as WT control throughout this study.
Histochemical evidence of Cre mediated MnSOD deletion in the kidney
MnSOD immunohistochemistry (IHC) was used to examine the extent and localization of MnSOD knockdown in both KO mice (50% and 100%). Kidney sections from KO mice revealed a gene-dose dependent decline of MnSOD protein expression when compared to the Kidney Cre mice. A predominant loss of MnSOD was observed within the medullary region (both inner, IM; and outer, OM) of KO mice (Fig 2A, a-c). Whereas, MnSOD protein expression in proximal tubules (pt) and glomeruli (gl) of the cortical area remained unchanged, the cortical distal tubules (dt) showed modest and substantial reduction in MnSOD protein expression in 50% and100% KO mice respectively (Fig 2A, d-f). Discrete MnSOD knockdown was observed in the outer stripe of the outer medullary region, where thick ascending limb (TAL) of Loops of Henle (indicated by arrow head) and the collecting ducts (indicated by arrow) showed a gene-dose dependent reduction in MnSOD protein expression with the greatest reduction observed in the 100% KO mice (Fig 2A, g-i). A dramatic decline of MnSOD protein expression was observed in the collecting ducts (arrow) and thin limb of Loops of Henle (arrow head) of the inner medullary region of 100% KO mice, while 50% KO mice exhibited only modest reduction of MnSOD protein in these tubules (Fig 2A, j-l).
Figure 2. MnSOD protein expression and activity is reduced in kidney-specific MnSOD KO mice.
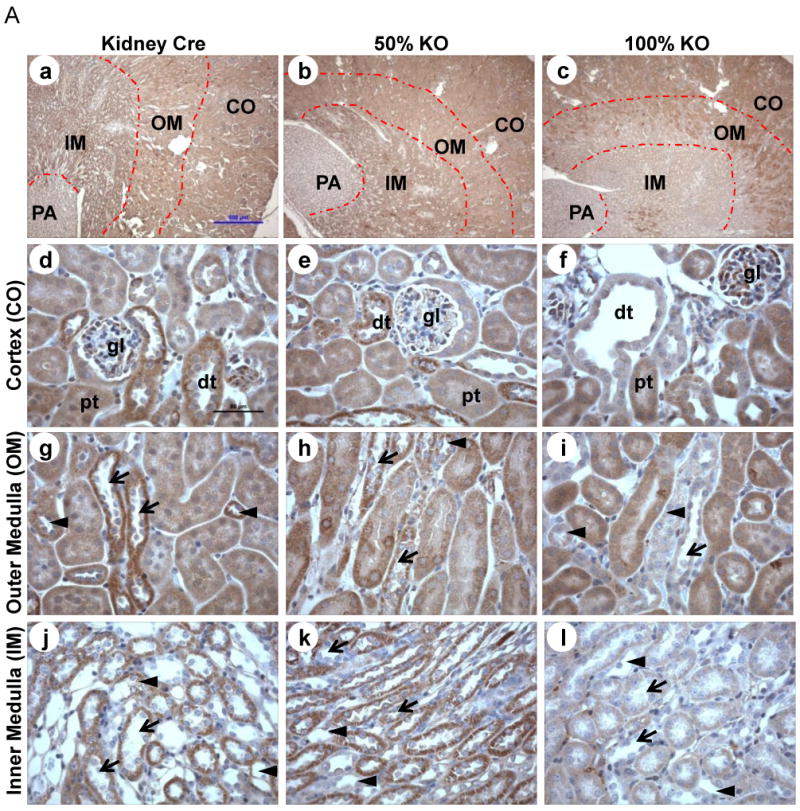
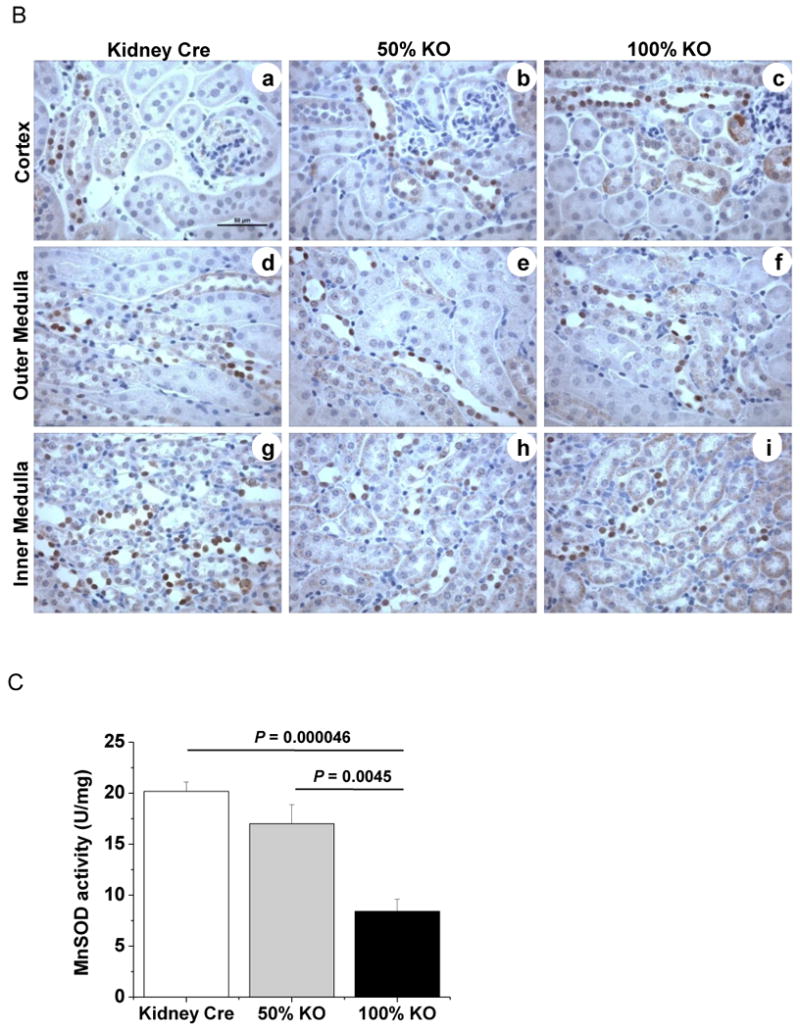
MnSOD and Cre-recombinase immunohistochemistry was performed using 4 – 5 μm kidney sections (n = 6). (A) MnSOD protein expression: representative 40× cross-sectional images for Kidney Cre, 50% KO, and 100% KO (a-c respectively). MnSOD protein was expressed in the cortical (CO), outer medullary (OM), inner medullary (IM), and papillary (PA) regions. 400× images of cortex (d-f); outer medulla (g-i) and inner medulla (j-l) from the respective 40× images are shown (dt: distal tubules; pt: proximal tubules; arrow head indicates Loops of Henle; arrow indicates collecting ducts). (B) Cre recombinase protein expression in kidney sections from Kidney Cre, 50% KO, and 100% KO mice within the cortex, outer and inner medulla (a-i). Representative bars indicate 500 μm for 40× images and 50 μm for 400× images. (C) MnSOD activity was determined in total renal homogenates using the cytochrome c reduction method. Activity decreased ∼2 to 3 –fold in 100% KO mice when compared to 50% KO (P =0.0045) and Kidney Cre (P =0.000046) mice respectively. Error bar represents Mean ± SEM (n = 5).
Since the extent of MnSOD knockdown was present in discrete renal cells it was equally important to determine the localization of CR expression. In agreement with previous findings using Kidney Cre (Ksp 1.3/Cre) transgenic mice [24], our bi-transgenic MnSOD KO mice also exhibited intra-nuclear CR protein within the distal tubules, collecting ducts, and Loops of Henle (Fig 2B). CR-positive cells were rarely detected in the proximal tubules. Taken together, these results suggest that not all renal cells were the target for the Cre mediated MnSOD deletion, which explains the discrete nature of MnSOD knockdown within the kidney of these newly developed KO mice.
To determine whether knockdown of MnSOD protein also reduced enzymatic activity, renal tissue from Kidney Cre and KO (50% and 100%) mice were homogenized and utilized in the MnSOD activity assay. Consistent with the extent of protein reduction observed with IHC, MnSOD activity was reduced in a gene-dose dependent manner. Results obtained from five different mice within each genotype revealed that MnSOD activity was impaired by ∼16% and ∼60% in the 50% and 100% KO mice respectively (Fig 2C). The overall reduction in MnSOD activity correlated well with the extent of protein knockdown within the kidney in 50% and 100% KO mice, especially since the activity measurements included all regions (cortex and medulla) of the kidneys.
Cre-Lox mediated MnSOD knockdown was specific to kidney
The bi-transgenic Cre-Lox mice have global expression of MnSODflox and kidney specific Ksp 1.3/Cre transgene expression. In order to confirm that the CR protein expression is confined only in the kidney and not in other organs it was necessary to examine MnSOD protein expression outside of the kidney. Paraffin embedded sections from liver and heart of Kidney Cre, 50% and 100% KO mice were utilized for IHC of CR and MnSOD proteins. As expected, the Kidney Cre and KO (50% and 100%) mice did not show CR protein expression in heart (Fig 3, a-c) or liver (Fig 3, d-f). Similarly, no change in protein levels of MnSOD was observed in the examined organs – heart (Fig 3, g-i) and liver (Fig 3, j-l), confirming that the Cre mediated MnSOD knockdown is kidney specific in this newly developed MnSOD KO mice.
Figure 3. Tissue specific nature of Cre recombinase expression and lack of effect on Cu, Zn SOD protein expression.
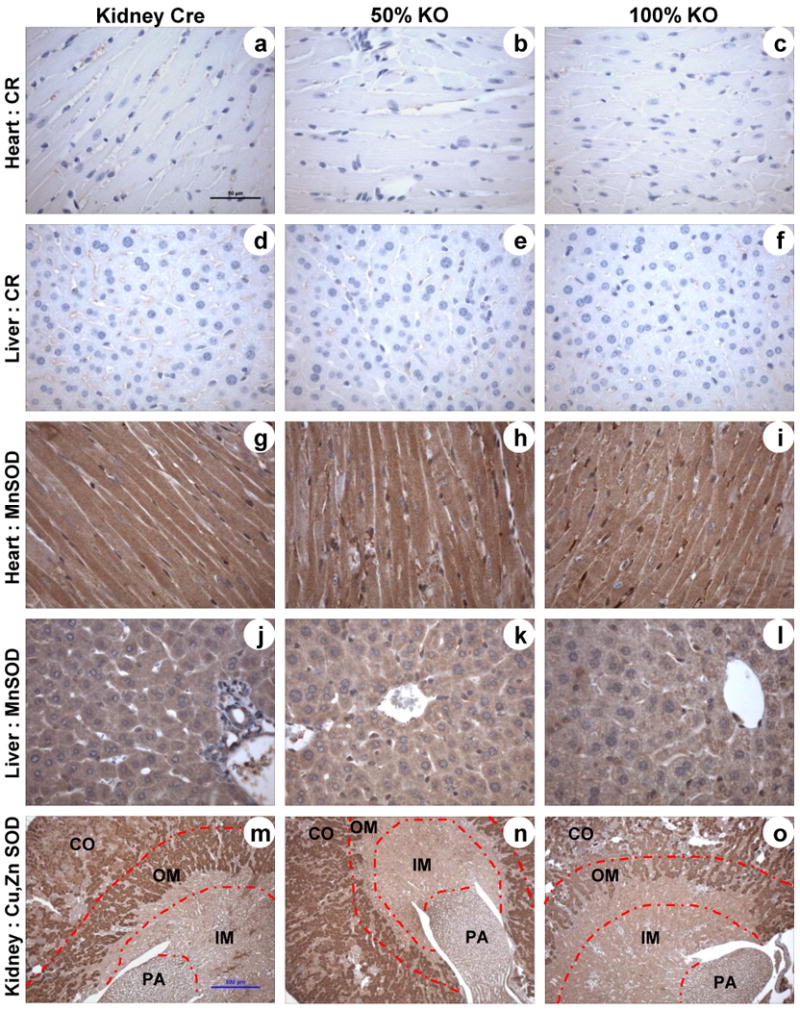
Representative immunohistochemistry micrographs of liver, heart, and kidney sections from Kidney Cre, 50% KO and 100% KO mice (n = 6). Sections were evaluated for Cre recombinase (a-f; heart and liver), MnSOD (g-l; heart and liver), and Cu, Zn SOD (m-o; kidney) protein expression. Representative bars indicate 50 μm for 400× images (a-l) and 500 μm for 40× images (m-o).
MnSOD knockdown did not alter Cu, Zn SOD protein levels
Superoxide dismutases (SOD) are a distinct group of antioxidant enzymes that catalyze the dismutation of superoxide into molecular oxygen and hydrogen peroxide. Among three isoforms of SOD, MnSOD and Cu, Zn SOD are localized intracellularly. Therefore, Cu, Zn SOD IHC was performed to examine whether low levels of MnSOD protein expression altered expression of the Cu, Zn SOD protein. These data showed that the Kidney Cre and 100% KO mice exhibited similar levels of Cu, Zn SOD protein in the kidney (Fig 3, m-o). Thus, the reduction in expression and activity of MnSOD did not alter the expression of Cu, Zn SOD suggesting that there is independent regulation of these two intracellular forms of SOD.
Kidney specific knockout mice showed no overt survival difference and body physiology change but were smaller in size
Mice with globally mutated MnSOD gene die within 21 days of birth [6,7]. Therefore, it was of interest to determine whether the deletion of MnSOD within the kidney had any altered effect on survivability and growth pattern. The kidney specific 100% KO mice showed no overt survival difference when monitored out to ∼22 months of age when compared to Kidney Cre mice (Table 1). Interestingly, kidney specific 100% KO mice exhibited a significant smaller body weight when compared to their Kidney Cre littermates (Fig 4 A, B). Further evaluation revealed no significant difference in the weight of vital organs (kidney, liver, heart and lungs) among the genotypes (Table 1). In addition, these KO mice had no gross abnormality in other basic physiological parameters including blood glucose and systolic blood pressure (Table 1).
Table 1. Survival and physiologic (8 - 10 wks) data in KO mice compared to Kidney Cre mice.
| Parameters | Kidney Cre | 50% KO | 100% KO |
|---|---|---|---|
| Survivability (months) | > 22 months | not studied | > 22 months |
| Serum Creatinine (mg/dl) | 0.22 ± 0.04 | 0.32 ± 0.04 | 0.25 ± 0.04 |
| Blood Glucose (mg/dl) | 169.00 ± 4.83 | 163.17 ± 7.52 | 162.50 ± 12.50 |
| Systolic Blood Pressure (mmHg) | 94.48 ± 9.98 | 95.49 ± 5.91 | 100.47 ± 4.48 |
| Weight of vital organs(g) | |||
| Kidney | 0.151 ± 0.010 | 0.169 ± 0.009 | 0.160 ± 0.008 |
| Liver | 1.065 ± 0.084 | 0.972 ± 0.086 | 0.937 ± 0.087 |
| Heart | 0.124 ± 0.006 | 0.147 ± 0.030 | 0.129 ± 0.007 |
| Lungs | 0.144 ± 0.008 | 0.170 ± 0.036 | 0.124 ± 0.007 |
Figure 4. Kidney specific 100% MnSOD KO mice are smaller in size.
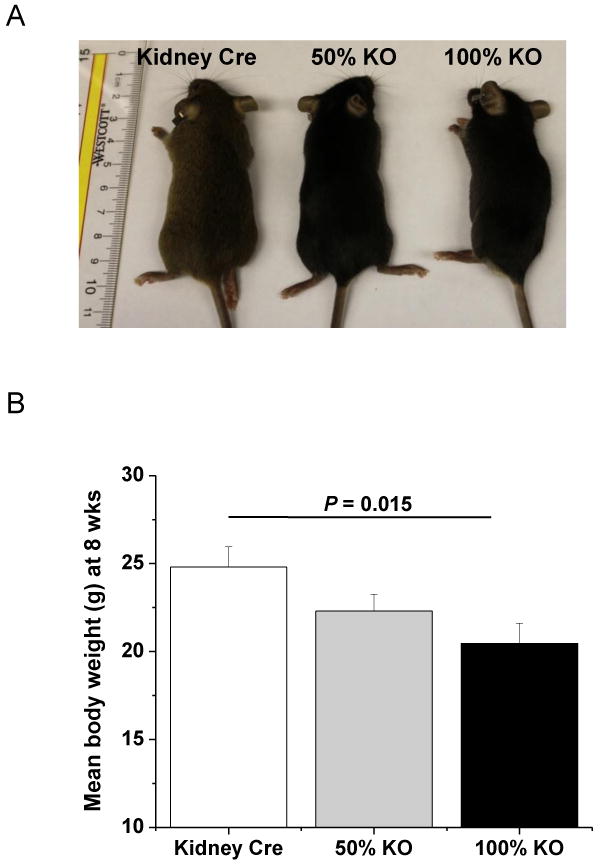
(A) Photograph showing phenotypic appearance of genetically modified mice at 8 weeks old. 100% KO mice (right) were smaller in size when compared to Kidney Cre (left) and 50% KO (middle) mice littermates. (B) Mean body weight (g) was determined at 8 weeks. 100% KO mice were significantly smaller in weight (g) than the Kidney Cre control mice (P = 0.015). Error bar represents Mean ± SEM (n = 10).
Kidney-specific MnSOD knockout mice exhibited altered kidney morphology with no overt damage in renal function
Periodic Acid-Schiff (PAS) staining was performed to examine histopathological changes in kidneys of the MnSOD KO mice. Interestingly, the 100% KO mice exhibited dilated distal tubules, within the cortex region (Fig 5, c and l). Semi quantitative evaluation based on the pathological scores (see methods) showed a significant tubular dilation in 100% KO mice when compared to Kidney Cre mice (Fig 5; P = 0.016). These dilated tubules of 100% KO mice exhibited a significant increase in proteinacious casts within the lumen compared to the Kidney Cre mice (Fig 5; P = 0.012). In addition, loss of MnSOD protein was associated with prominent epithelial cell swelling in the dilated distal tubules (Fig 5 l). This tubular cell swelling was significant both in the 50% and 100% KO mice (Fig 5; P = 0.0009, Kidney Cre vs. 100% KO, and P = 0.0003, 50% vs. 100% KO). These results indicate that the loss of MnSOD within the distal tubules appears to induce a stress mediated tubular dilation and cellular swelling.
Figure 5. Renal morphology is altered in kidney-specific MnSOD KO mice.
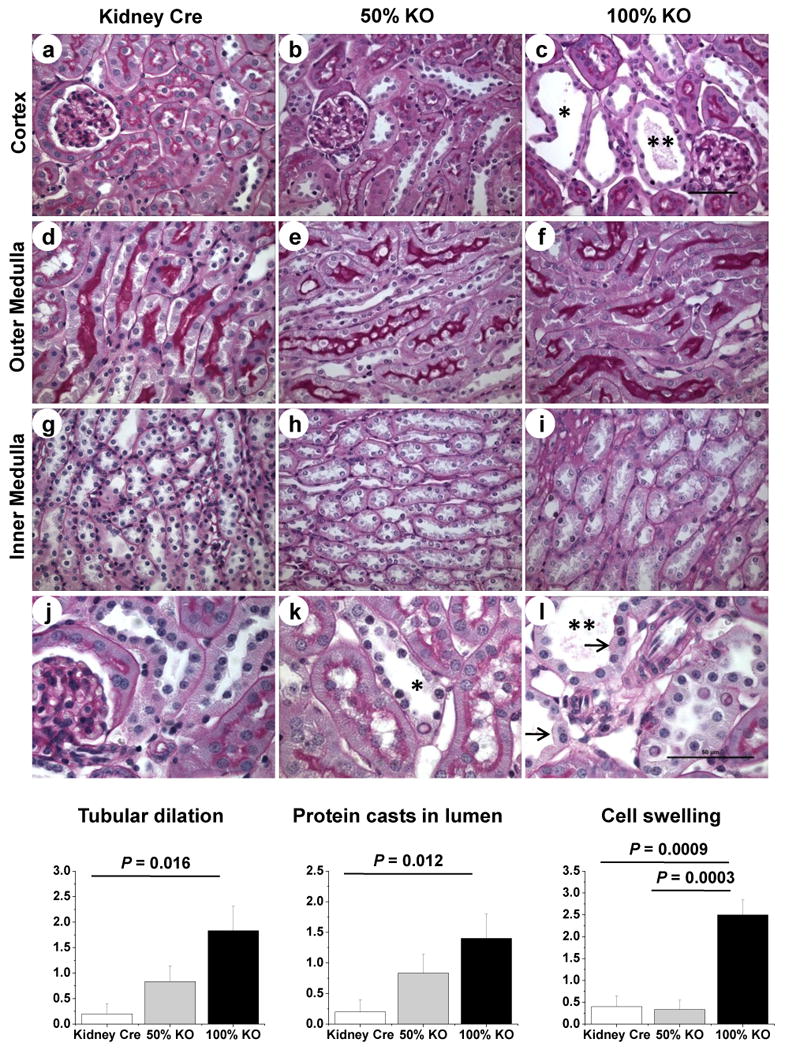
Representative 400× micrographs of PAS staining in renal cortex (a-c); outer medulla (d-f) and inner medulla (g-i) of Kidney Cre, 50% and 100% KO mice. Representative cortical sections (400×) were enlarged in panels j-l to show the dilation of distal tubules (asterisk), protein casts formation (double asterisks), and epithelial cell swelling (arrow) in tubules from MnSOD KO mice. Representative bars indicate 50 μm for 400 × images. The graphs show the pathological scoring for tubular dilation, protein casts in lumen, and cell swelling of Kidney Cre, 50% KO and 100% KO mice. Error bar indicates Mean ± SEM (n = 5 - 6).
Serum creatinine is a common marker of overt renal function. Significant changes in serum creatinine normally occur only after the kidney has sustained a marked injury. Using serum samples from the MnSOD KO mice, no significant difference in serum creatinine levels were detected (Table 1), indicating that these KO mice do not undergo severe renal dysfunction.
MnSOD knockdown augments oxidant production within the kidney
Previous reports from our laboratory, and others, have shown that MnSOD inactivation leads to increased nitrotyrosine levels [19,21,22,30]. Tyrosine nitration is considered a good marker of oxidant production [31,32]. Thus, it was of interest to evaluate the accumulation of nitrated proteins within the kidney as a consequence of MnSOD knockdown. Nitrotyrosine IHC data revealed a gene-dose dependent increase in tyrosine nitration in KO mice when compared to the basal level of expression in Kidney Cre mice (Fig 6A). The specificity of nitrotyrosine staining was also confirmed using nitrotyrosine antibody preabsorbed with excess 3-nitrotyrosine (Fig 6A, dhl). Similar to the discrete pattern of MnSOD protein expression within specific renal compartments (Fig 2A); tyrosine nitration staining also appeared to be localized. Specifically, high levels of tyrosine nitration were localized to cortical distal tubules in a gene-dose dependent manner (Fig 6A a-c). Medullary regions (both outer and inner) also showed gene-dose dependent localization of tyrosine nitration within the collecting ducts and Loops of Henle (TALs and thin segments) in both KO mice (Fig 6A e-g, i-k). Interestingly, acellular casts within distal tubules, collecting ducts, and Loops of Henle of KO mice showed positive staining for tyrosine nitration (Fig 6A c, j and k). Semi quantitative data based on the percentage of positive tubules (see methods) showed a significant increase in tyrosine nitration levels in the kidney sections of both KO mice (Fig 6B; P = 0.0008, Kidney Cre vs. 100% KO; and P = 0.0012, Kidney Cre vs. 50% KO). These results indicate that loss of MnSOD leads to increased oxidant production, tubular dilation, cell swelling, and cast formation.
Figure 6. Oxidative stress is induced in kidney-specific MnSOD KO mice.
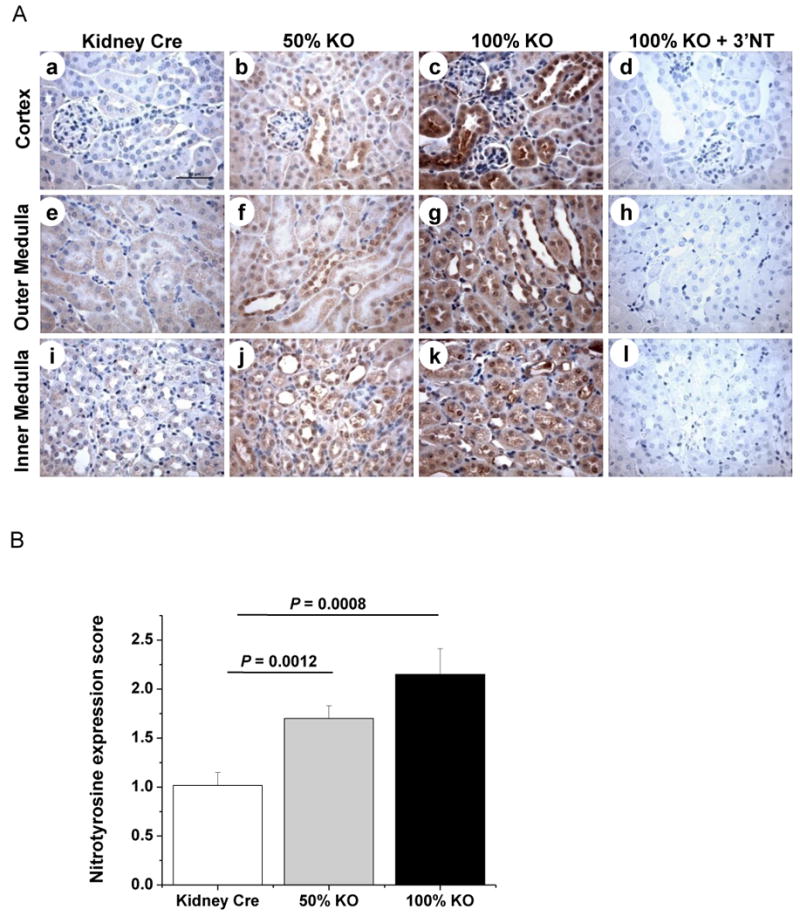
Increased nitrotyrosine protein expression was detected in the tubules of 100% KO (c, g, k) and 50% KO (b, f, j) mice when compared to Kidney Cre mice (a, e, i). The specificity of nitrotyrosine antibody binding in the renal tissue was confirmed by blocking the antibody with 3′nitrotyrosine (10 mM) using 100% KO mice kidney sections (d, h, l). Representative micrographs of 400× magnification are shown. Bar indicates 50 μm. (B) Expression level of nitrotyrosine was evaluated and scored (Kidney Cre vs. 100% KO, P = 0.0008 and Kidney Cre vs. 50 % KO, P = 0.0012). Error bar indicates Mean ± SEM (n = 6).
Discussion
There is growing evidence, from experimental and clinical studies, that oxidative stress may be implicated in the pathogenesis of renal dysfunction [33]. Low expression or decreased enzymatic activity of MnSOD can result in excessive generation of superoxide anions and more toxic downstream oxidants. Previous studies reported that the down-regulation of MnSOD protein [18] and reduced enzymatic activity were prevalent during renal failure [22]. However, the precise molecular events that lead to renal injury subsequent to MnSOD inactivation are not clear.
Current animal models that modulate the expression of MnSOD have been developed and have greatly contributed to scientific advancements. Global deletion (heterozygous) of MnSOD resulted in similar levels (∼50%) of enzyme dysfunction in all tissues/organs [7,23], limiting the use of this MnSOD KO mouse model for evaluation of the kidney-specific effects related to MnSOD inactivation. Therefore, it was imperative to design an in vivo model that would allow us to explore the resultant effect of kidney-specific MnSOD protein ablation. The transgenic mouse line carrying a floxed MnSOD gene allows for deletion of the MnSOD gene in cells that express the CR enzyme. This MnSOD floxed transgenic mouse line has been used in several other animal models to selectively delete MnSOD from liver [25], heart [34], brain [35], and muscle [27,36]. Another transgenic mouse line used in this study was the Ksp1.3/Cre transgenic mouse that specifically expresses Cre-recombinase in collecting ducts and loops of Henle (highest), distal tubules (substantial) and proximal tubules (occasional), but not in glomeruli, blood vessels, or renal interstitial cells [24]. Exploiting Cre/Lox recombination technology and these two mouse lines for breeding, we were able to generate kidney-specific MnSOD KO mice (50% and 100%) in which a Cre mediated deletion of exon 3 left a mutated version of MnSOD allele (s) specifically in the kidney (Fig 1). As a result, gene-dose dependent MnSOD protein knockdown was observed exclusively in the cells of distal tubules, collecting ducts, and Loops of Henle in these 50% and 100% KO mice. Reduction of MnSOD protein was dramatic in the inner medullary region of the 100% KO mice (Fig 2A c, l). Furthermore, this ablation of MnSOD protein resulted in ∼60% reduction in enzymatic activity within the kidney (Fig 2C). These findings suggest that this mouse model may be suitable for studying a consequent effect of discrete renal inactivation of MnSOD in vivo.
It has been shown that over-expression or deletion of Cu, Zn SOD does not regulate the expression of MnSOD protein and it appears that these two enzymes are regulated differently in vivo [37]. In line with this observation [6], we were able to show an independent regulation of MnSOD and Cu, Zn SOD enzyme expression in the kidney of our novel KO mouse models (Fig 3), which further makes these KO mice an excellent model for kidney-specific MnSOD KO in vivo.
Characterization of these novel KO mice showed that the kidney-restricted 100% KO mice resulted in a smaller body size with no developmental abnormalities (Fig 4) or change in survivability (Table 1). However, the smaller body size observed had no effect on the weight of other vital organs such as heart, lungs and liver (Table 1). These results raise an intriguing question as to whether renal knockdown of MnSOD (hence elevated mitochondrial superoxide) has an effect on the musculo-skeletal system. Future studies will address the link between decreased MnSOD within specific renal cells and the change in phenotype of these MnSOD KO mice. One possibility is that MnSOD KO may impact mineral metabolism critical to normal bone formation.
Surprisingly, the MnSOD KO mice exhibited normal kidney function (Table 1), although MnSOD knockdown did result in modest renal damage including tubular dilation, epithelial cell enlargement, and casts formation within the tubular lumen (Fig 5). The renal damage was localized to the distal part of the nephron, which was consistent with the localization of Cre recombinase and areas showing repressed MnSOD expression (distal tubules, Loops of Henle and collecting ducts). No evidence of glomerular injury was observed, which was comparable to unaltered MnSOD protein expression in the glomeruli. Since the primary function of distal tubular cells is to maintain ion homeostasis, impairment in these cell types (as evident by cell swelling) might not be sufficient to affect the overall glomerular function. This may explain why we did not see an overt decrease in renal function (as measured by serum creatinine level) in the MnSOD KO mice.
Distal tubules and collecting ducts are the primary sites for casts formation and these casts are normally excreted in the urine, which has been shown to be an early marker of renal injury [38]. Proteinacious/acellular casts were abundantly present in the dilated distal tubules and was associated with enlarged tubular cells (Fig 5 c, k, l). The mechanism leading to dilation of distal tubules in the KO mice is unknown. However, it is possible that injury to distal tubules increased casts formation which might lead to obstruction and dilation of the distal tubules in the KO mice. Furthermore, it is possible that a specialized group of cells in the Loops of Henle - thick ascending limbs (TALs), might also contribute to this dilation via synthesis of the glycoprotein (Tamm-horsfall protein or THP), which is normally secreted in the urine and can facilitate casts formation [39,40]. A transient increase of THP has been observed in the urine during oxygen free radical mediated damage in dog kidneys [41]. Moreover, it has been shown that THP can be tyrosine nitrated which leads to aggregation of calcium oxalate crystals [42,43]. Although, our studies did not address the role of THP, it is possible that it could contribute to tubular dilation observed in the current study.
The macula densa cells are specialized cells of distal tubules, and play an important role in blood pressure regulation via induction of renin production [44,45]. As there was evidence of morphological alteration of distal tubular cells following MnSOD knockdown, we hypothesized that this might result in altered blood pressure. However, the KO mice did not show a significant change in blood pressure (Table 1). Given the localized nature of MnSOD expression within renal cells, it is possible that this effect was not adequate to affect the BP in the 100% KO mice.
Decreased MnSOD protein resulted in significant oxidant production as revealed by histochemical assessment of nitrotyrosine protein accumulation in the KO mice. The pattern of tyrosine nitration was localized in similar cortical regions that displayed significant reductions in MnSOD (dilated distal tubules and collecting ducts) as well as medullary regions including the collecting ducts and Loops of Henle. Future studies will evaluate the effect that elevated mitochondrial oxidant production has on mitochondrial function in the MnSOD KO mice.
In summary, we have generated a novel mouse model in which Cre mediated deletion of MnSOD allele was kidney-specific. To our knowledge, this is the first mouse model for kidney-specific deletion of MnSOD and could be an ideal model to study the metabolism of mitochondrial superoxide in the kidney and/or the specific targets of injury following MnSOD inactivation within the kidney. Homozygous (100%) deletion of MnSOD in the kidney resulted in a distinct phenotype with smaller body size but displayed normal life-span and body physiology. One of the most interesting aspects of these KO mice was the localized deletion of MnSOD in particular cell types (distal tubules, collecting ducts and Loops of Henle). Consequently, this mouse model could serve as a powerful tool in dissecting out the pathways that occur downstream of MnSOD inactivation, as well as the role that normal MnSOD activity has in function of specific renal cell types.
Acknowledgments
We thank the National Institutes of Health for financial support (NIH Grant 1RO1DK0789361); Dr. Ting-Ting Huang for providing MnSOD floxed mice; Drs. Peter Igarashi and Paul Overbeek for providing Kidney Cre transgenic mice that were developed in University of Texas Southwestern O'Brien Kidney Research Core Center (NIH P30DK079328); Mr. Sidney “Clay” Williams for his assistance with the Q Capture imaging and Nikons Elements software; Mr. Sujay Kharade for his assistance in measuring BP; and Dr. Naeem Patil for critical reading of the manuscript. We also thank the Experimental Pathology Core Lab at UAMS for the excellent service in processing paraffin embedded tissue blocks.
List of Abbreviations
- Bp
base pair
- BP
Blood pressure
- CO
Cortex
- CR
Cre-recombinase
- Cu, Zn SOD
Copper, Zinc superoxide dismutase
- dt
distal tubule
- IHC
Immunohistochemistry
- IM
Inner Medulla
- KO
knockout
- MnSOD
Manganese superoxide dismutase
- OM
Outer medulla
- PAS
Periodic Acid Schiff
- PCR
polymerase chain reaction
- pt
proximal tubule
- TAL
Thick ascending limb
- WT
Wild type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Murphy MP. How mitochondria produce reactive oxygen species. Biochemical Journal. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Turrens JF. Superoxide production by the mitochondrial respiratory chain. Bioscience Reports. 1997;17:3–8. doi: 10.1023/a:1027374931887. [DOI] [PubMed] [Google Scholar]
- 3.Jones PL, Ping D, Boss JM. Cloning, Characterization, and Regulation of The, Murine Mnsod Gene. Faseb Journal. 1995;9:A488. [Google Scholar]
- 4.Disilvestre D, Kleeberger SR, Johns J, Levitt RC. Structure and Dna-Sequence of the Mouse Mnsod Gene. Mammalian Genome. 1995;6:281–284. doi: 10.1007/BF00352417. [DOI] [PubMed] [Google Scholar]
- 5.Wispe JR, Clark JC, Burhans MS, Kropp KE, Korfhagen TR, Whitsett JA. Synthesis and Processing of the Precursor for Human Mangano-Superoxide Dismutase. Biochimica et Biophysica Acta. 1989;994:30–36. doi: 10.1016/0167-4838(89)90058-7. [DOI] [PubMed] [Google Scholar]
- 6.Lebovitz RM, Zhang HJ, Vogel H, Cartwright J, Dionne L, Lu NF, Huang S, Matzuk MM. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:9782–9787. doi: 10.1073/pnas.93.18.9782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li YB, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson TL, Noble LJ, Yoshimura MP, Berger C, Chan PH, Wallace DC, Epstein CJ. Dilated Cardiomyopathy and Neonatal Lethality in Mutant Mice Lacking Manganese Superoxide-Dismutase. Nature Genetics. 1995;11:376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- 8.Wen JJ, Vyatkina G, Garg N. Oxidative damage during chagasic cardiomyopathy development: Role of mitochondrial oxidant release and inefficient antioxidant defense. Free Radical Biology and Medicine. 2004;37:1821–1833. doi: 10.1016/j.freeradbiomed.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 9.Wen JJ, Dhiman M, Whorton EB, Garg NJ. Tissue-specific oxidative imbalance and mitochondrial dysfunction during Trypanosoma cruzi infection in mice. Microbes and Infection. 2008;10:1201–1209. doi: 10.1016/j.micinf.2008.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miyazawa M, Ishii T, Yasuda K, Noda S, Onouchi H, Hartman PS, Ishii N. The Role of Mitochondrial Superoxide Anion(O-2(-)) on Physiological Aging in C57BL/6J Mice. Journal of Radiation Research. 2009;50:73–82. doi: 10.1269/jrr.08097. [DOI] [PubMed] [Google Scholar]
- 11.Massaad CA, Washington TM, Pautler RG, Klann E. Overexpression of SOD-2 reduces hippocampal superoxide and prevents memory deficits in a mouse model of Alzheimer's disease. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:13576–13581. doi: 10.1073/pnas.0902714106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bize IB, Oberley LW, Morris HP. Superoxide-Dismutase and Superoxide Radical in Morris Hepatomas. Cancer Research. 1980;40:3686–3693. [PubMed] [Google Scholar]
- 13.Archer SL, Marsboom G, Kim GH, Zhang HJ, Toth PT, Svensson EC, Dyck JRB, Gomberg-Maitland M, Thebaud B, Husain AN, Cipriani N, Rehman J. Epigenetic Attenuation of Mitochondrial Superoxide Dismutase 2 in Pulmonary Arterial Hypertension A Basis for Excessive Cell Proliferation and a New Therapeutic Target. Circulation. 2010;121:2661–U108. doi: 10.1161/CIRCULATIONAHA.109.916098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown KA, Didion SP, Andresen JJ, Faraci FM. Effect of aging, MnSOD deficiency, and genetic background on endothelial function evidence for MnSOD haploinsufficiency. Arteriosclerosis Thrombosis and Vascular Biology. 2007;27:1941–1946. doi: 10.1161/ATVBAHA.107.146852. [DOI] [PubMed] [Google Scholar]
- 15.Faraci FM, Modrick ML, Lynch CM, Didion LA, Fegan PE, Didion SP. Selective cerebral vascular dysfunction in Mn-SOD-deficient mice. Journal of Applied Physiology. 2006;100:2089–2093. doi: 10.1152/japplphysiol.00939.2005. [DOI] [PubMed] [Google Scholar]
- 16.MacMillan-Crow LA, Cruthirds DL. Invited review - Manganese superoxide dismutase in disease. Free Radical Research. 2001;34:325–336. doi: 10.1080/10715760100300281. [DOI] [PubMed] [Google Scholar]
- 17.Landriscina M, Remiddi F, Ria F, Palazzotti B, Deleo ME, Iacoangeli M, Rosselli R, Scerrati M, Galeotti T. The level of MnSOD is directly correlated with grade of brain tumours of neuroepithelial origin. British Journal of Cancer. 1996;74:1877–1885. doi: 10.1038/bjc.1996.648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vaziri ND, Dicus M, Ho ND, Boroujerdi-Rad L, Sindhu RK. Oxidative stress and dysregulation of superoxide dismutase and NADPH oxidase in renal insufficiency. Kidney International. 2003;63:179–185. doi: 10.1046/j.1523-1755.2003.00702.x. [DOI] [PubMed] [Google Scholar]
- 19.Cruthirds DL, Novak L, Akhi KM, Sanders PW, Thompson JA, MacMillan-Crow LA. Mitochondrial targets of oxidative stress during renal ischemia/reperfusion. Archives of Biochemistry and Biophysics. 2003;412:27–33. doi: 10.1016/s0003-9861(03)00039-0. [DOI] [PubMed] [Google Scholar]
- 20.MacMillan-Crow LA, Cruthirds DL, Ahki KM, Sanders PW, Thompson JA. Mitochondrial tyrosine nitration precedes chronic allograft nephropathy. Free Radical Biology and Medicine. 2001;31:1603–1608. doi: 10.1016/s0891-5849(01)00750-x. [DOI] [PubMed] [Google Scholar]
- 21.Guo W, Adachi T, Matsui R, Xu SQ, Jiang BB, Zou MH, Kirber M, Lieberthal W, Cohen RA. Quantitative assessment of tyrosine nitration of manganese superoxide dismutase in angiotensin II-infused rat kidney. American Journal of Physiology-Heart and Circulatory Physiology. 2003;285:H1396–H1403. doi: 10.1152/ajpheart.00096.2003. [DOI] [PubMed] [Google Scholar]
- 22.MacMillanCrow LA, Crow JP, Kerby JD, Beckman JS, Thompson JA. Nitration and inactivation of manganese superoxide dismutase in chronic rejection of human renal allografts. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:11853–11858. doi: 10.1073/pnas.93.21.11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Remmen H, Salvador C, Yang H, Huang TT, Epstein CJ, Richardson A. Characterization of the antioxidant status of the heterozygous manganese superoxide dismutase knockout mouse. Archives of Biochemistry and Biophysics. 1999;363:91–97. doi: 10.1006/abbi.1998.1060. [DOI] [PubMed] [Google Scholar]
- 24.Shao XL, Somlo S, Igarashi P. Epithelial-specific Cre/lox recombination in the developing kidney and genitourinary tract. Journal of the American Society of Nephrology. 2002;13 doi: 10.1097/01.asn.0000016444.90348.50. [DOI] [PubMed] [Google Scholar]
- 25.Ikegami T, Suzuki Y, Shimizu T, Isono K, Koseki H, Shirasawa T. Model mice for tissue-specific deletion of the manganese superoxide dismutase (MnSOD) gene. Biochemical and Biophysical Research Communications. 2002;296:729–736. doi: 10.1016/s0006-291x(02)00933-6. [DOI] [PubMed] [Google Scholar]
- 26.Truett GE, Heeger P, Mynatt RL, Truett AA, Walker JA, Warman ML. Preparation of PCR-quality mouse genomic DNA with hot sodium hydroxide and tris (HotSHOT) Biotechniques. 2000;29:52–+. doi: 10.2144/00291bm09. [DOI] [PubMed] [Google Scholar]
- 27.Kuwahara H, Horie T, Ishikawa S, Tsuda C, Kawakami S, Noda Y, Kaneko T, Tahara S, Tachibana T, Okabe M, Melki J, Takano R, Toda T, Morikawa D, Nojiri H, Kurosawa H, Shirasawa T, Shimizu T. Oxidative stress in skeletal Muscle causes severe disturbance of exercise activity without muscle atrophy. Free Radical Biology and Medicine. 2010;48:1252–1262. doi: 10.1016/j.freeradbiomed.2010.02.011. [DOI] [PubMed] [Google Scholar]
- 28.Hayek T, Attias J, Coleman R, Brodsky S, Smith J, Breslow JL, Keidar S. The angiotensin-converting enzyme inhibitor, fosinopril, and the angiotensin II receptor antagonist, losartan, inhibit LDL oxidation and attenuate atherosclerosis independent of lowering blood pressure in apolipoprotein E deficient mice. Cardiovascular Research. 1999;44:579–587. doi: 10.1016/s0008-6363(99)00239-4. [DOI] [PubMed] [Google Scholar]
- 29.Mccord JM. FRIDOVIC.I Superoxide Dismutase An Enzymic Function for Erythrocuprein (Hemocuprein) Journal of Biological Chemistry. 1969;244:6049–&. [PubMed] [Google Scholar]
- 30.Mitchell T, Saba H, Laakman J, Parajuli N, MacMillan-Crow LA. Role of mitochondrial-derived oxidants in renal tubular cell cold-storage injury. Free Radical Biology and Medicine. 2010;49:1273–1282. doi: 10.1016/j.freeradbiomed.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ischiropoulos H, Almehdi AB. Peroxynitrite-Mediated Oxidative Protein Modifications. Febs Letters. 1995;364:279–282. doi: 10.1016/0014-5793(95)00307-u. [DOI] [PubMed] [Google Scholar]
- 32.Kooy NW, Royall JA, Ye YZ, Kelly DR, Beckman JS. Evidence for In-Vivo Peroxynitrite Production in Human Acute Lung Injury. American Journal of Respiratory and Critical Care Medicine. 1995;151:1250–1254. doi: 10.1164/ajrccm/151.4.1250. [DOI] [PubMed] [Google Scholar]
- 33.Zahmatkesh M, Kadkhodaee M, Mahdavi-Mazdeh M, Ghaznavi R, Hemati M, Seifi B, Golab F, Hasani K, Lessan-Pezeshki M, Einollahi B. Oxidative Stress Status in Renal Transplant Recipients. Experimental and Clinical Transplantation. 2010;8:38–44. [PubMed] [Google Scholar]
- 34.Nojiri H, Shimizu T, Funakoshi M, Yamaguchi O, Zhou H, Kawakami S, Ohta Y, Sami M, Tachibana T, Ishikawa H, Kurosawa H, Kahn RC, Otsu K, Shirasawa T. Oxidative stress causes heart failure with impaired mitochondrial respiration. Journal of Biological Chemistry. 2006;281:33789–33801. doi: 10.1074/jbc.M602118200. [DOI] [PubMed] [Google Scholar]
- 35.Misawa H, Nakata K, Matsuura J, Monwaki Y, Kawashima K, Shimizu T, Shirasawa T, Takahashi R. Conditional knockout of Mn superoxide dismutase in postnatal motor neurons reveals resistance to mitochondrial generated superoxide radicals. Neurobiology of Disease. 2006;23:169–177. doi: 10.1016/j.nbd.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 36.Lustgarten MS, Jang YC, Liu YH, Muller FL, Qi WB, Steinhelper M, Brooks SV, Larkin L, Shimizu T, Shirasawa T, McManus LM, Bhattacharya A, Richardson A, Van Remmen H. Conditional knockout of Mn-SOD targeted to type IIB skeletal muscle fibers increases oxidative stress and is sufficient to alter aerobic exercise capacity. American Journal of Physiology-Cell Physiology. 2009;297:C1520–C1532. doi: 10.1152/ajpcell.00372.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.White CW, Nguyen DDH, Suzuki K, Taniguchi N, Rusakow LS, Avraham KB, Groner Y. Expression of Manganese Superoxide-Dismutase Is Not Altered in Transgenic Mice with Elevated Level of Copper-Zinc Superoxide-Dismutase. Free Radical Biology and Medicine. 1993;15:629–636. doi: 10.1016/0891-5849(93)90166-r. [DOI] [PubMed] [Google Scholar]
- 38.Fogazzi GB, Garigali G. The clinical art and science of urine microscopy. Current Opinion in Nephrology and Hypertension. 2003;12:625–632. doi: 10.1097/00041552-200311000-00009. [DOI] [PubMed] [Google Scholar]
- 39.Sanders PW, Booker BB, Bishop JB, Cheung HC. Mechanisms of Intranephronal Proteinaceous Cast Formation by Low-Molecular-Weight Proteins. Journal of Clinical Investigation. 1990;85:570–576. doi: 10.1172/JCI114474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wangsiripaisan A, Gengaro PE, Edelstein CL, Schrier RW. Role of polymeric Tamm-Horsfall protein in cast formation: Oligosaccharide and tubular fluid ions. Kidney International. 2001;59:932–940. doi: 10.1046/j.1523-1755.2001.059003932.x. [DOI] [PubMed] [Google Scholar]
- 41.Bakris GL, Gaber AO, Jones JD. Oxygen Free-Radical Involvement in Urinary Tamm-Horsfall Protein Excretion After Intrarenal Injection of Contrast-Medium. Radiology. 1990;175:57–60. doi: 10.1148/radiology.175.1.2315505. [DOI] [PubMed] [Google Scholar]
- 42.Pragasam V, Kalaiselvi P, Sumitra K, Srinivasan S, Anandkumar P, Varalakshmi P. Immunological detection of nitrosative stress-mediated modified Tamm-Horsfall glycoprotein (THP) in calcium oxalate stone formers. Biomarkers. 2006;11:153–163. doi: 10.1080/13547500500421138. [DOI] [PubMed] [Google Scholar]
- 43.Pragasam V, Kalaiselvi P, Subashini B, Sumitra K, Varalakshmi P. Structural and functional modification of THP on nitration: Comparision with stone formers THP. Nephron Physiology. 2005:28–34. doi: 10.1159/000081800. [DOI] [PubMed] [Google Scholar]
- 44.Hackenthal E, Paul M, Ganten D, Taugner R. Morphology, Physiology, and Molecular-Biology of Renin Secretion. Physiological Reviews. 1990;70:1067–1116. doi: 10.1152/physrev.1990.70.4.1067. [DOI] [PubMed] [Google Scholar]
- 45.Hall JE. Historical perspective of the renin-angiotensin system. Molecular Biotechnology. 2003;24:27–39. doi: 10.1385/MB:24:1:27. [DOI] [PubMed] [Google Scholar]