

Abstract
Extracellular matrix (stroma) regulation of mucosal T-cell function is incompletely understood. Here we uncovered a role for intestinal stromal products in the innate regulation of effector T-cells. Stroma-conditioned media (S-CM) derived from normal human intestinal stroma (TGF-βhi/IL-6lo/IL-1βlo) significantly down-regulated T-cell proliferation and IFN-γ production compared to S-CM derived from inflamed Crohn’s mucosa (TGF-βhi/IL-6hi/IL-1βhi). Antibody neutralization studies showed that TGF-β in normal S-CM inhibited T-cell proliferation and IFN-γ production, whereas IL-6 plus IL-1β in Crohn’s S-CM promoted T-cell proliferation, and the IL-1β alone promoted IFN-γ and IL-17 release. Importantly, normal S-CM inhibited T-bet expression, whereas Crohn’s S-CM activated STAT3, suggesting that discordant T-cell responses are regulated at the transcription factor and signaling levels. These findings implicate stromal TGF-β in the down-regulation of T-cell responses in normal intestinal mucosa but stromal IL-6 and IL-1β in the promotion of Th1 and Th17 responses in inflamed Crohn’s mucosa, suggesting innate regulatory function for the intestinal extracellular matrix.
INTRODUCTION
Mucosal homeostasis - the finely tuned balance between tolerance to commensal bacteria and inflammatory responses to pathogens - is maintained through a network of complementary regulatory processes. To begin to dissect this network in human intestinal mucosa, we have shown that intestinal extracellular matrix (stroma)-associated TGF-β promotes the recruitment of pro-inflammatory monocytes and mediates monocyte differentiation into non-inflammatory intestinal macrophages, thereby contributing to the absence of mucosal inflammation in the normal human small intestine 1–4. T-cells in normal intestinal mucosa also are down-regulated, reflected in their reduced capacity to proliferate and produce IFN-γ compared to circulating blood T-cells 5–7, but the homeostatic mechanisms responsible for T-cell down-regulation in normal mucosa are not well understood. In Crohn’s disease mucosa, inappropriate pro-inflammatory Th1 and Th17 responses to commensal bacteria 8 appear to be due, in part, to reduced TGF-β signaling 9. Factors that contribute to this impaired TGF-β signaling may include IL-6 and IL-1β10, 11. These cytokines, abundantly present in inflamed Crohn’s disease mucosa but not normal mucosa 8, promote Th1 responses 12, 13 and together with TGF-β promote Th17 responses 14–16. Although the source of these cytokines is presently unclear, the above findings suggest that the mucosal microenvironment in Crohn’s disease is involved in the pro-inflammatory responses of local effector T-cells.
Here we investigated whether factors associated with the stroma in human intestinal mucosa regulate T-cells using a novel system that recapitulates the in vivo exposure of newly recruited blood T-cells to the lamina propria stroma. We have defined stroma as the lattice of collagen, fibronectin and laminin, plus the cells responsible for their production, that mediate cytokine and cell adhesion and transmit information in a bi-directional manner to local immune cells 1, 2. We report that stroma-associated cytokines, especially TGF-β, from normal intestinal mucosa down-regulate effector T-cell responses, but stroma-associated pro-inflammatory cytokines IL-6 and IL-1β, together with TGF-β, from inflamed Crohn’s mucosa potentiate pro-inflammatory effector T-cell responses. These findings identify a previously under-appreciated contribution of the local extracellular matrix to the innate regulation of mucosal T-cells in normal and inflamed human intestinal mucosa.
MATERIALS AND METHODS
Intestinal tissue and blood lymphocytes
Tissue was obtained with IRB approval from normal jejunum from subjects undergoing elective gastric bypass, normal ileum from patients undergoing colectomy for adenocarcinoma, and inflamed ileum from patients undergoing resection for Crohn’s disease. Crohn’s disease was confirmed histologically, and all donors had not received immunosuppressive therapy for 4 weeks prior to surgery. Blood lymphocytes were isolated from healthy donors by gradient sedimentation and purified by magnetic cell sorting (MACS) using CD4+ beads (Miltiney, Auburn, CA). Mucosal T-cell isolation.
Stroma-conditioned media (S-CM)
Intestinal mucosa was dissected from the submucosa and digested to remove epithelial cells and mononuclear leukocytes as previously described 17. Culture supernatant from cell-depleted lamina propria stroma (1 g wet weight/mL) cultured in RPMI overnight to generate S-CM 2 was sterile filtered (0.2-µm Syringe Filter; Corning, Corning, NY), tested for endotoxin and protein by ELISA (Pierce, Rockford, IL; Bio-rad, Hercules, CA) and stored at −70°C. Only S-CM containing less than 1.5 endotoxin U/mL was used. Total protein concentration was determined using a bicinchoninic acid (BCA) protein assay kit (Pierce). In all experiments, S-CM was used at a normalized total protein concentration of 250 µg/mL, unless otherwise indicated. S-CM derived from normal intestinal mucosa is hereafter referred to as normal S-CM. S-CM derived from inflamed intestinal mucosa is hereafter referred to as Crohn’s S-CM. Importantly, S-CM derived from normal ileum and normal jejunum had similar effects on T-cell responses (data not shown).
T-cell function assays
CD4+ blood T-cells (106/mL) from normal donors were stained with carboxyflurescein succinimidyl ester (CFSE) (Invitrogen, Carlsbad, CA) and incubated in RPMI with 10% HuAB serum plus IL-2 (25 U/mL; R&D Systems, Minneapolis, MN) for 1 hr with (a) media; (b) S-CM; (c) S-CM pre-incubated for 30 min with or without the indicated neutralizing antibodies; (d) S-CM plus recombinant cytokines; or (e) media with recombinant cytokines, neutralizing antibody alone, or irrelevant control antibody alone, then stimulated with CD3/CD28 dynabeads (105/mL, Invitrogen) or PHA (5 µg/mL, Sigma Aldrich). On day 4, supernatants were harvested for IFN-γ or IL-17 determination by ELISA (R&D Systems), and cells were stained with CD3 PEcy7, CD4 APC (BD Biosciences, San Jose, CA) or irrelevant isotype control antibody, as previously described 2 and analyzed for proliferation using an LSRII with FACS Diva software. In additional experiments, T-cells were pre-activated for 3 days with CD3/CD28 or PHA and then cultured for an additional 4 days with or without normal or Crohn’s S-CM in the presence or absence of a second stimulation (CD3/CD28).
Cytokine determination
Among 16 normal and 15 Crohn’s S-CMs provided by the Human Tissue Core of the UAB Mucosal HIV and Immunobiology Center, 7 Crohn’s and 4 normal S-CMs were analyzed for immunomodulating cytokines by Luminex assay (Invitrogen) (kindly performed by SM Wahl, NIH), according to the manufacturer’s instructions. Samples for the luminex assay were randomly selected and normalized to a total protein concentration of 1.8 mg/mL, which was the lowest protein concentration in the assayed S-CMs. Based on these results, which identified significantly elevated levels of IL-6 and IL-1β in Crohn’s S-CM compared to normal S-CM, and our previous identification of stromal TGF-β as an important modulator 2, 4, TGF-β, IL-6 and IL-1β levels were determined by ELISA (R&D systems) for each S-CM used in this study. Luminex and ELISA values for these cytokines were similar. IFN-γ and IL-17 in T-cell supernatants were analyzed by ELISA.
Western blot
Whole cell preparations made from CD4+ T-cells (5×106) using standard techniques 4 were analyzed by western blot with antibodies to T-bet (1:750; Santa Cruz Biotechnologies), STAT3 or p-STAT3 (1:1000; Cell Signaling Technologies, Boston, MA), as we have previously described 4.
Statistical Analysis
Student’s t-test was used to determine statistical significance. A paired t-test was used to compare experimental results to control results, and an unpaired t-test was used to compare results of T-cells cultured in normal S-CM to T-cells cultured in Crohn’s S-CM.
RESULTS
Stromal factors derived from normal intestinal mucosa down-regulate T-cell responses more effectively than stromal factors from Crohn’s mucosa
To recapitulate in vitro the exposure of newly recruited blood T-cells to the intestinal lamina propria and determine whether stroma-associated molecules in the lamina propria regulate T-cells, we cultured blood T-cells in intestinal stroma-conditioned media (S-CM) and measured T-cell proliferation and IFN-γ production. Blood CD4+ T-cells from normal subjects pre-incubated with increasing concentrations of normal or Crohn’s S-CM showed differential dose-dependent reductions in CD3/CD28- (Figure 1A) and PHA- (Supplemental Figure 1A) stimulated proliferation. Inhibition of T-cell proliferation was significantly greater for T-cells cultured in normal S-CM than T-cells cultured in Crohn’s S-CM at both 250 µg/mL (p < 0.05) and 500 µg/mL (p < 0.05) (Figure 1B and Supplemental Figure 1B). Moreover, stromal inhibition of T-cell proliferation by normal S-CM was evident not only in a reduced percentage of proliferating T-cells, but also in a reduced number of generations of daughter cells.
Figure 1. S-CM inhibition of CD4+ T-cell responses.
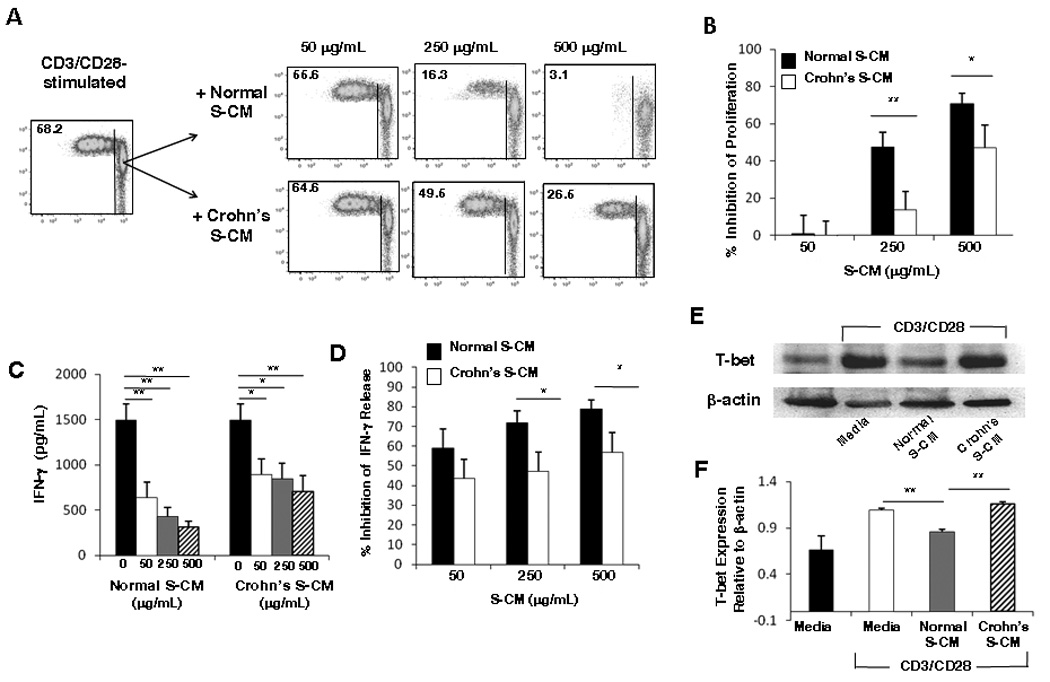
(A) CD3/CD28-stimulated T-cells cultured in media alone or the presence of a representative normal or Crohn’s S-CM were analyzed on day 4 for proliferation by flow cytometry for CFSE dilution. The percent of cells that proliferated is indicated in the top left corner of the plots. (B) Percent inhibition of proliferation for CD3/CD28-stimulated T-cells cultured in normal S-CM (n=7) or Crohn’s S-CM (n=6). (C) IFN-γ release by CD3/CD28-stimulated T-cells cultured for 4 days in the presence of media, normal S-CM (n=5) or Crohn’s S-CM (n=6). (D) Percent inhibition of IFN-γ release by CD3/CD28-stimulated T-cells cultured for 4 days in the presence of normal S-CM (n=5) or Crohn’s S-CM (n=6) compared to stimulated T-cells cultured in media alone. (E) Western blot analysis of T-bet protein expression in CD3/CD28-stimulated T-cells cultured 24 hr in the presence of a representative normal S-CM or Crohn’s S-CM (n=3 each). (F) Densitometric analysis of T-bet relative to β-actin (n=3). For B–D and F, error bars indicate the standard error of the mean. * p ≤ 0.05, ** p ≤ 0.01.
Inducible T-cell production of IFN-γ, the prototypic Th1 cytokine, also was reduced in the presence of normal and Crohn’s S-CM (p < 0.05 at each concentration) (Figure 1C and Supplemental Figure 1C), and, similar to the inhibition of proliferation, normal S-CM more potently down-regulated IFN-γ production than Crohn’s S-CM, reaching significance at S-CM concentrations of 250 µg/mL (p < 0.05) and 500 µg/mL (p < 0.04) (Figure 1D). Importantly, the percent of T-cells producing inducible IFN-γ was 5% in normal S-CM and 25% in Crohn’s S-CM (data not shown), indicating that Crohn’s but not normal S-CM permits selective Th1 cell expansion. Moreover, T-cell expression of the Th1 transcription factor T-bet was more potently down-regulated by normal S-CM than Crohn’s S-CM (Figure 1E, F, and Supplemental Figure 1D), indicating that stromal modulation of T-cell function was likely regulated at the transcription factor level.
We next determined whether stromal factors can similarly regulate T-cell proliferation and cytokine release in previously activated (dividing) T-cells, thereby mimicking the arrival of activated T-cells into the mucosa. Proliferation and IFN-γ release by T-cells pre-activated for 3 days with CD3/CD28 or PHA and then cultured for a further 4 days in the presence or absence of a second stimulation (CD3/CD28) was also profoundly inhibited by S-CM from normal mucosa (Supplemental Figures 2 and 3). S-CM derived from Crohn’s mucosa was less efficient at inhibiting these T-cell functions than normal S-CM, consistent with our earlier findings (Figure 1). These findings suggest that stromal products from normal intestinal mucosa powerfully inhibit T-cell function of both resting and stimulated T-cells, and are significantly more effective at down-regulating T-cell function than products from Crohn’s mucosa, indicating that reduced innate stromal regulation likely contributes to the inflammatory T-cell response in Crohn’s disease.
Crohn’s disease intestinal stroma contains higher levels of pro-inflammatory cytokines than normal intestinal stroma
Since cytokines play a critical role in regulating T-cell function, we analyzed S-CM generated from normal mucosa and Crohn’s mucosa for eight key regulatory cytokines. Levels of TGF-β and IL-10 in normal S-CM and Crohn’s S-CM were similar, with the level of TGF-β markedly higher than that of IL-10 (Table 1). In contrast, levels of the pro-inflammatory cytokines IL-6 and IL-1β were more than 10-fold higher in Crohn’s S-CM compared to normal S-CM (p = 0.03). Although the level of IFN-γ was higher in Crohn’s S-CM than normal S-CM, overall the level was very low (< 2.3 pg/mL). TNF-α also was present at barely detectable levels in S-CM derived from normal and Crohn’s mucosa. The markedly low levels of IFN-γ and TNF-α (as well as both IL-2 and IL-10) in S-CM is consistent with the absence of effector T-cells and macrophages in the hematopoetic cell-depleted stroma used to generate S-CM, underscoring the relevance of S-CM as a model for the extracellular matrix. These data suggest that the loss of homeostatic down-regulation in Crohn’s disease mucosa is not due to lower levels of regulatory cytokines (TGF-β and IL-10) and raise the possibility that stroma-associated IL-6 and IL-1β shift the balance from tolerance to inflammation in Crohn’s mucosa.
Table I.
Levels of immune-modulating cytokines in stroma-conditioned media derived from normal mucosa and inflamed Crohn’s mucosa
| Normal mucosa (n=4) |
Crohn’s mucosa (n=7) |
||||
|---|---|---|---|---|---|
| Mean (pg/mL) |
SEM |
Mean (pg/mL) |
SEM |
p-value |
|
| TGF-β | 210.00 | 75 | 275.00 | 37.00 | 0.40 |
| IL-10 | 3.06 | 0.10 | 3.89 | 0.76 | 0.44 |
| IL-6 | 6.37 | 1.73 | 255.40 | 70.39 | 0.03 |
| IL-1β | 7.14 | 0.37 | 71.40 | 29.00 | 0.03 |
| IFN-γ | 1.59 | 0.05 | 2.29 | 0.20 | 0.03 |
| IL-17 | 10 | 0.7 | 16 | 2 | <0.5 |
| IL-2 | 2.28 | 0.36 | 4.21 | 1.15 | 0.25 |
| TNF-α | 1.18 | 0.36 | 2.88 | 0.65 | 0.04 |
Cytokine concentrations are standardized to 1.8 mg/mL total protein
Stromal TGF-β promotes down-regulation of T-cell responses
We have shown that stromal TGF-β mediates the differentiation of pro-inflammatory monocytes into cells with the phenotype and functional profile of non-inflammatory intestinal macrophages 2–4. Others have shown that blockade of TGF-β in mucosal biopsies leads to increased mucosal Th1 responses 6. Since TGF-β associates with the extracellular matrix 18 and is released into S-CM 2, 4, we assessed the contribution of stromal TGF-β to the down-regulation of T-cell function mediated by normal S-CM. Neutralization of TGF-β in normal S-CM significantly inhibited S-CM down-regulation of inducible T-cell proliferation (Figure 2A, B and Supplemental Figure 4A) and IFN-γ production (Figure 2C and Supplemental Figure 4B) in a dose-dependent manner, implicating a critical role for TGF-β in normal S-CM down-regulation of T-cell function. In parallel experiments, equivalent amounts of irrelevant isotype control antibody did not affect T-cell proliferation or cytokine release (Supplemental Figure 5A–C), and neutralization of TGF-β in complete media with human serum containing low levels of TGF-β also had no effect on T-cell proliferation and IFN-γ release. We next evaluated whether the level of TGF-β in S-CM was sufficient to down-regulate T-cell proliferation and cytokine production. rhTGF-β alone at 50–500 pg/mL induced dose-dependent down-regulation of T-cell proliferation (Figure 3A and Supplemental Figure 4C). However, rhTGF-β alone at 50 pg/mL and 500 pg/mL was less effective at down-regulating T-cell proliferation than normal S-CM (250 µg/mL) (p < 0.01 for each concentration) (Figure 3B). (Note: 50 pg/mL rhTGF-β is equivalent to the level of TGF-β in normal S-CM at a concentration of 250 µg protein/mL (Supplemental Table 1)). rhTGF-β at 50 – 500 pg/mL also inhibited T-cell IFN-γ release (p < 0.05 – 0.01) (Figure 3C and Supplemental Figure 4D). These findings suggest that TGF-β is necessary for T-cell down-regulation in the mucosa, although alone it is not sufficient to achieve the full down-regulation of T-cell responses induced by normal S-CM.
Figure 2. Neutralization of TGF-β reverses normal S-CM-mediated down-regulation of CD4+ T-cell responses.
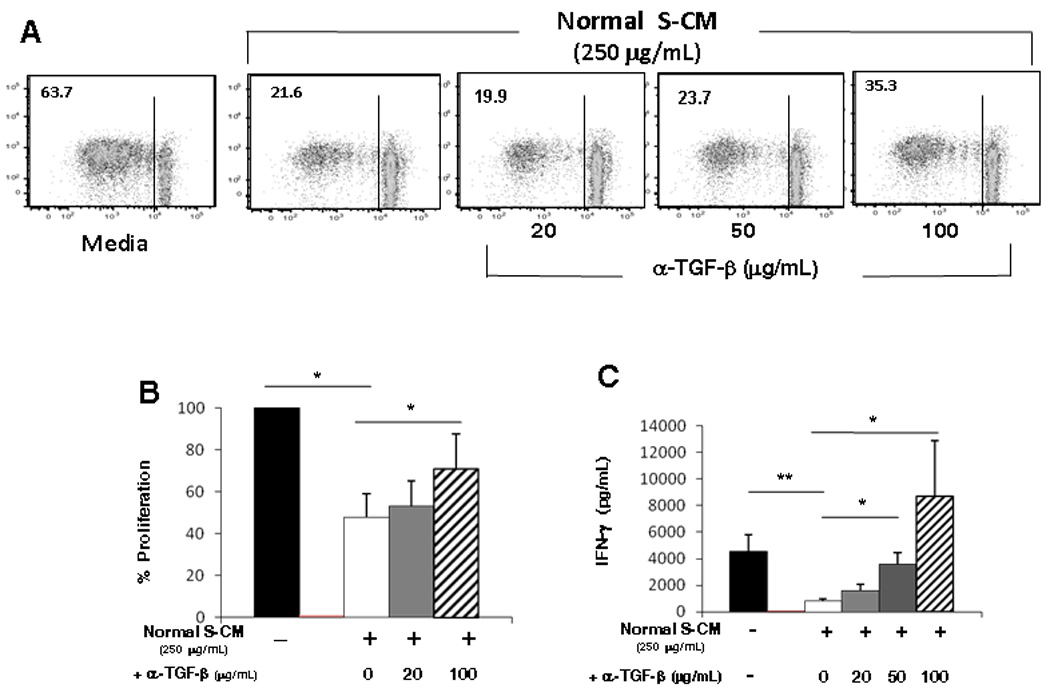
(A) Proliferation of CD3/CD28-stimulated T-cells cultured for 4 days in the presence of media or a representative normal S-CM (250 µg/mL) ± anti-TGF-β neutralizing antibody. (B) Percent proliferation of stimulated T-cells cultured in the presence of normal S-CM ± anti-TGF-β antibody compared to stimulated T-cells cultured in media alone (n=4). (C) IFN-γ release by CD3/CD28-stimulated T-cells cultured for 4 days in the presence of media or normal S-CM ± anti-TGF-β antibody (n=3). For B and C error bars indicate the standard error of the mean. * p ≤ 0.05 and ** p ≤ 0.01.
Figure 3. TGF-β promotes down-regulation of CD4+ T-cell responses.
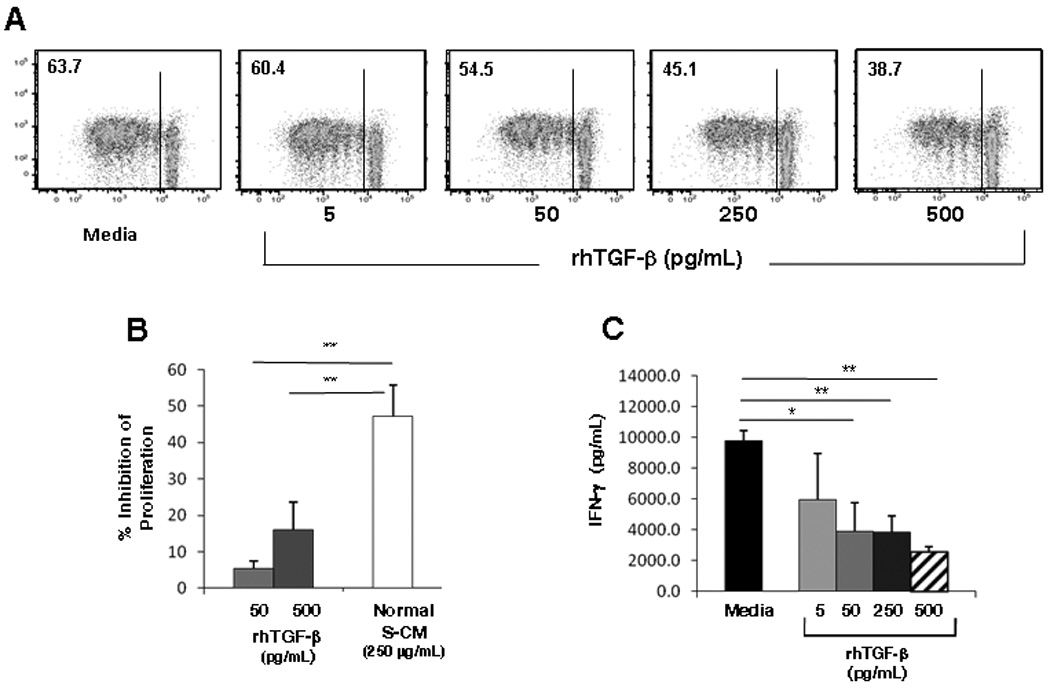
(A) Proliferation of CD3/CD28-stimulated T-cells cultured in the presence of rhTGF-β alone for 4 days in a representative experiment (p ≤ 0.05 at 50, 250 and 500 pg/mL for n=4). (B) Percent inhibition of CD3/CD28-stimulated T-cell proliferation by rhTGF-β alone (n=4) at 50 pg/mL or 500 pg/mL compared to normal S-CM (250 µg/mL) (n=7). (C) CD3/CD28-stimulated IFN-γ release by T-cells cultured in the presence rhTGF-β (n=3). For B and C, error bars indicate the standard error of the mean. * p ≤ 0.05 and ** p ≤ 0.01.
Stromal IL-6 and IL-1β derived from Crohn’s mucosa promote pro-inflammatory T-cell responses
TGF-β and IL-10 were present at similar levels in normal and Crohn’s S-CM, but the levels of IL-6 and IL-1β, which also associate with extracellular matrix 19, 20, were increased in Crohn’s S-CM (Table 1). Therefore, we investigated whether stromal IL-6 and IL-1β contribute to the reduced ability of Crohn’s S-CM to inhibit T-cell responses. Pre-incubation of Crohn’s S-CM with an optimal concentration of neutralizing antibody to IL-6, but not IL-1β, enhanced Crohn’s S-CM down-regulation of CD3/CD28-stimulated T-cell proliferation (Figure 4A). Moreover, simultaneous blockade of IL-6 and IL-1β further enhanced Crohn’s S-CM down-regulation of T-cell proliferation (n=4). Furthermore, neutralization of IL-1β alone or IL-1β plus IL-6 in Crohn’s S-CM significantly reduced inducible T-cell IFN-γ production (p < 0.05) (Figure 4B). These findings implicate discordant roles for IL-6 and IL-1β in the modulation of effector T-cell function in Crohn’s mucosa.
Figure 4. IL-6 and IL-1β in Crohn’s S-CM promote pro-inflammatory CD4+ T-cell responses.
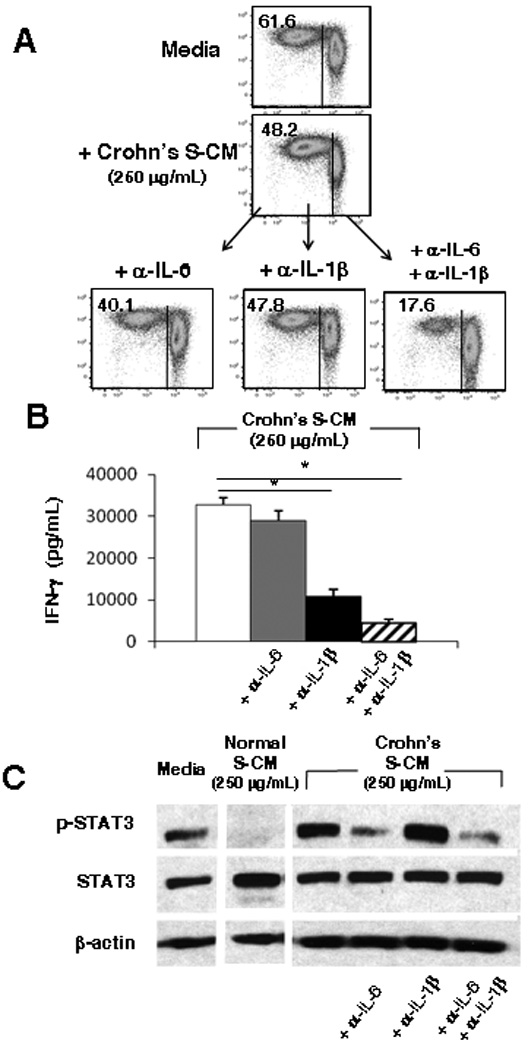
(A) Proliferation (n=3) and (B) IFN-γ release (n=2; each performed in triplicate) by CD3/CD28-stimulated T-cells cultured for 4 days in media or a representative Crohn’s S-CM ± anti-IL-6 and/or anti-IL-1β neutralizing antibodies (1 µg/mL). (C) Western blot analysis of STAT3 and p-STAT3 protein expression by T-cells exposed for 30 min to media, a representative normal S-CM or a representative Crohn’s S-CM ± anti-IL-6 and/or anti-IL-1β antibodies (1 µg/mL) (n=2). For B, error bars indicate the standard error of the mean. * p ≤ 0.05 .
Since IL-6 mediates its response through STAT3 signaling, we analyzed STAT3 phosphorylation in T-cells exposed to normal S-CM or Crohn’s S-CM. T-cells exposed to Crohn’s, but not normal, S-CM phosphorylated STAT3 (Figure 4C). Neutralization of IL-6, or IL-6 plus IL-1β, but not IL-1β alone, in Crohn’s S-CM substantially reduced STAT3 phosphorylation in T-cells exposed to Crohn’s S-CM, suggesting that the effect of IL-6 in Crohn’s S-CM on effector T-cell proliferation and cytokine release is mediated through STAT3 activation.
Stromal factors in Crohn’s disease promote Th17 responses
STAT3 activation plays a critical role in the development of IL-17-producing T-cells 16. Therefore, we determined whether the discordant regulation of T-cell function by normal versus Crohn’s stromal factors extended to IL-17 secretion. In the presence of normal S-CM, CD3/CD28-stimulated T-cell production of IL-17 was reduced, albeit not significantly, compared to control T-cells (Figure 5A). However, in the presence of increasing concentrations of Crohn’s S-CM, T-cells secreted markedly higher levels of IL-17 compared to T-cells cultured in media alone (p < 0.01), with similar amounts of IL-17 released at all concentrations of Crohn’s S-CM (Figure 5A and inset). Since the proportion of pro-inflammatory (IL-6 and IL-1β) to down-regulatory (TGF-β) cytokines was constant at each concentration of Crohn’s S-CM, these findings suggest that the relative proportion of the cytokines was more important than their absolute concentration in the regulation of IL-17 secretion in the presence of Crohn’s S-CM.
Figure 5. Crohn’s S-CM promotes IL-17 release by CD4 T-cells in an IL-1β -dependent manner.
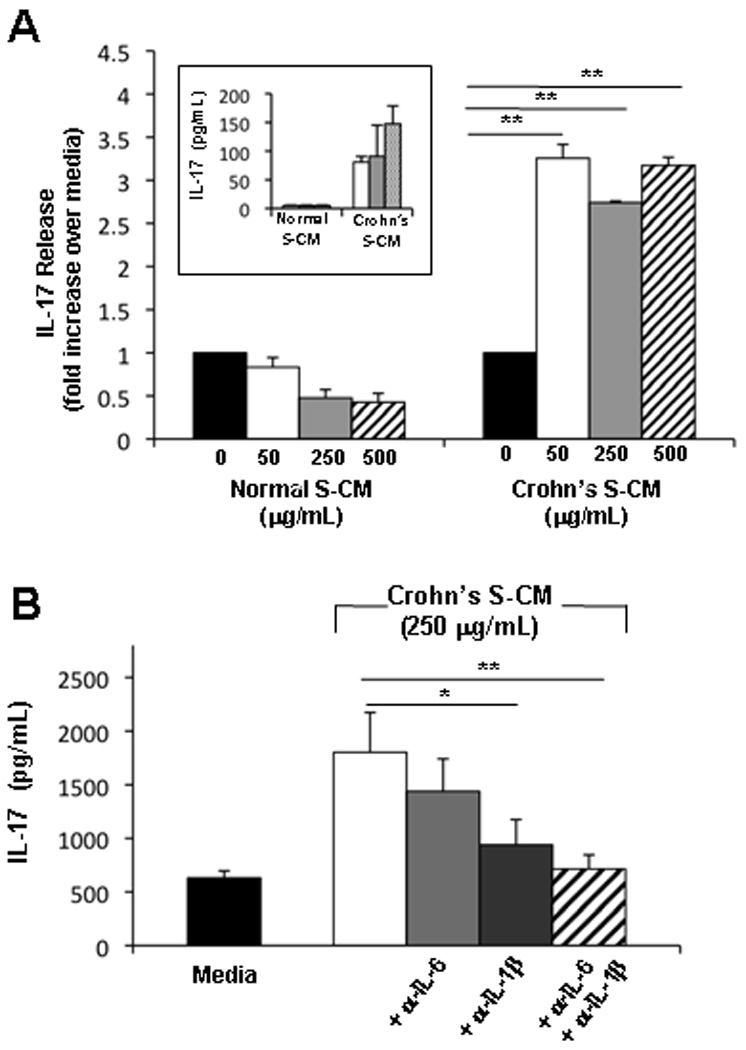
(A) IL-17 release by CD3/CD28-stimulated T-cells cultured 4 days in the presence of normal S-CM or Crohn’s S-CM compared to stimulated T-cells cultured in media alone (n=3). Inset shows results from a representative experiment (n=3) with IL-17 release (pg/mL) by CD3/CD28-stimulated blood T-cells cultured for 4 days in the presence of normal or Crohn’s S-CM. (B) IL-17 release by CD3/CD28-stimulated T-cells cultured for 4 days in Crohn’s S-CM ± anti-IL-6 antibody and/or anti-IL-1β antibodies (1 µg/mL) (n=2, each performed in triplicate). Error bars indicate the standard error of the mean. *p ≤ 0.05 and ** p ≤ 0.01.
We next investigated whether blockade of IL-6, IL-1β, or IL-6 plus IL-1β in Crohn’s S-CM would reverse the enhanced IL-17 production by T-cells. Neutralization of IL-1β, but not IL-6, in Crohn’s S-CM significantly reduced IL-17 production (p = 0.04), reaffirming the critical role of IL-1 in promoting human Th17 responses (Fig 5B). Furthermore, neutralization of IL-1β plus IL-6 in Crohn’s S-CM reduced IL-17 production to levels similar to CD3/CD28-stimulated IL-17 released by T-cells in media alone. Given the low level of the IL-17 in Crohn’s S-CM (16 ± 2 pg/mL, n=7; Table 1), these results implicate T-cells as the source of the IL-17 and suggest that the balance between IL-1β (with IL-6) and TGF-β contributes to the pathogenic Th17 responses in Crohn’s disease.
DISCUSSION
The gastrointestinal mucosa is continuously exposed to a huge array of immunostimulatory antigens and microorganisms, necessitating the evolution of mechanisms to down-regulate potentially harmful inflammatory responses. The remarkable plasticity of the normal mucosal immune system of the small intestine is evident in the capacity of the system to respond to commensal bacteria with tolerance but to pathogens with an inflammatory response to contain the microorganism. However, this homeostatic regulation is disrupted in certain inflammatory processes such as Crohn’s disease, when T-cells respond inappropriately and exuberantly to commensal bacteria with increased proliferation and cytokine (IFN-γ and IL-17) production 8, 25–27.
To elucidate the mechanisms responsible for the hypo-responsiveness of T-cells in normal mucosa, but hyper-responsiveness in Crohn’s disease mucosa, we developed a novel system that recapitulates the in vivo exposure of newly recruited blood T-cells to lamina propria extracellular matrix products derived from normal or inflamed Crohn’s disease mucosa. Using this system, we uncovered a previously underappreciated role for extracellular matrix-associated cytokines in the regulation of T-cell responses in the intestinal mucosa. We show that the stroma from normal and Crohn’s disease mucosa released similar levels of the down-regulatory cytokine TGF-β and nearly undetectable levels of IL-10. However, the stroma from Crohn’s disease mucosa released elevated levels of the pro-inflammatory cytokines IL-6 and IL-1β compared to normal intestinal stroma. The TGF-β released by cultured normal stroma inhibited T-cell function, whereas the IL-6 plus IL-1β released by Crohn’s stroma promoted T-cell proliferation, and stromal IL-1β alone induced T-cells to release both IFN-γ and IL-17.
Stromal products derived from normal intestinal mucosa down-regulated T-bet expression and IFN-γ production. However, Crohn’s stromal products did not inhibit T-bet expression but did reduce IFN-γ production, suggesting involvement by additional regulatory mechanisms, such as impaired accessibility of T-bet to its DNA binding site, decreased mRNA stability, and improper protein folding and export 28. Among the products released by Crohn’s tissue stroma, IL-6 induced STAT3 phosphorylation. In this regard, STAT3 activation plays a critical role in Th17 differentiation 16, and in mouse models IL-6 in the presence of TGF-β promotes the differentiation of Th17 cells. The differentiation of Th17 cells in humans, is less well understood 22, 23. However, we show here that Crohn’s stromal products enhanced IL-17 release from CD3/CD28-stimulated T-cells in an IL-1β-dependent manner and others have reported that IL-1β enhanced secretion of IL-17 from human memory T-cells 23, 24. Taken together, our results suggest that stromal factors in Crohn’s disease mucosa do not differentiate T-cells into Th17 cells, but more likely promote IL-17 secretion from Th17 memory cells that have entered the mucosa.
Our findings implicate a role for stromal TGF-β in the down-regulation of T-cell responses in normal intestinal mucosa but stromal IL-6 and IL-1β in the promotion of Th1 and Th17 responses in inflamed Crohn’s mucosa, despite the continued presence of local TGF-β. Since apoptotic cells are also a potential source of TGF-β, it is important that S-CM derived from both normal and Crohn’s disease mucosa had a slightly protective effect on T-cell apoptosis (data not shown), confirming that the TGF-β responsible for regulating T-cell function was derived from the stroma. Moreover, that intestinal stromal factors inhibit T-cell proliferation and cytokine release in pre-activated T-cells suggests that the mucosal microenvironment also may inhibit mucosal inflammation enhanced by newly recruited, activated T-cells. Thus, these findings identify an innate regulatory function for the intestinal extracellular matrix, reflected in the ability of intestinal stroma to regulate T-cell responses. In order to dissect out the specific role of the stroma, we used normal blood T-cells as indicator cells. Whether T-cells from patients with Crohn’s disease are resistant to mucosal down-regulation by normal stromal factors is the subject of a separate study.
Investigative attention has recently focused on the conditioning of mucosal macrophages 1–4 and DCs 29–34. Regarding DCs, epithelial cells produce TGF-β and transcriptionally active retinoic acid that act as co-factors to drive the differentiation of CD103+ tolerogenic DCs, which in turn promote regulatory T-cell development 29, 34. Here we show that the stroma of the intestinal lamina propria also directly regulates T-cell responses, independent of mucosal DC regulation, indicating redundant control of T-cells to limit potentially harmful inflammation in normal intestinal mucosa. The findings presented here are consistent with the notion that disruption of extracellular matrix-mediated regulation of T-cells in the intestinal lamina propria permits the expansion of Th1 and Th17 cells, driving the inflammation that characterizes Crohn’s disease pathology.
Elucidating the pathophysiology of T-cell-mediated inflammation in Crohn’s disease mucosa and identification of the cytokines responsible for promoting these responses may facilitate the development of new therapeutic strategies that target these cytokines in the intestinal lamina propria of patients refractory to current therapies. Notably, our study reinforces the importance of IL-6 in promoting inflammation in Crohn’s disease and indicates an important role for IL-1β in promoting T-cell pro-inflammatory cytokine release in the presence of TGF-β. The recent administration of a fully humanized anti-IL-1β antibody (ACZ885) in patients with rheumatoid arthritis resulted in substantial clinical improvement 35, supporting the concept of IL-1β as a target for therapeutic intervention. Earlier studies in rabbits, mice and humans underscored the role of IL-1β in mediating colitis 36–39. These studies, coupled with the findings presented here, suggest that targeting of both local IL-1β and IL-6 may provide a more effective therapeutic strategy for patients with refractory Crohn’s disease.
Supplementary Material
ACKNOWLEDGMENTS
Grant support: NIH (DK-47322, DK-54495, AI-83027, AI-74438, AI-83539, DK-85898, DK-84063, Al-007051, GM-008361, RR-20136), the Mucosal HIV and Immunobiology Center (DK-64400), the Crohn’s and Colitis Foundation of America, and the Research Service of the Veterans Administration.
Footnotes
SUPPLEMENTARY MATERIAL FILES
Five supplemental figures and one supplemental table are included with this submission. The files are all in PDF format. The figures and the table are referenced in the text of the manuscript and legends are provided on the Legends page.
Supplementary Material is linked to the online version of the paper at http://www.nature.com/mi.
DISCLOSURE
The authors disclose that they have no conflict of interest.
REFERENCES
- 1.Smith PD, Smythies LE, Mosteller-Barnum M, Sibley DA, Russell MW, Merger M, et al. Intestinal macrophages lack CD14 and CD89 and consequently are down-regulated for LPS- and IgA-mediated activities. J Immunol. 2001;167(5):2651–2656. doi: 10.4049/jimmunol.167.5.2651. [DOI] [PubMed] [Google Scholar]
- 2.Smythies LE, Sellers M, Clements RH, Mosteller-Barnum M, Meng G, Benjamin WH, et al. Human intestinal macrophages display profound inflammatory anergy despite avid phagocytic and bacteriocidal activity. J Clin Invest. 2005;115(1):66–75. doi: 10.1172/JCI19229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smythies LE, Maheshwari A, Clements R, Eckhoff D, Novak L, Vu HL, et al. Mucosal IL-8 and TGF-beta recruit blood monocytes: evidence for cross-talk between the lamina propria stroma and myeloid cells. J Leukoc Biol. 2006;80(3):492–499. doi: 10.1189/jlb.1005566. [DOI] [PubMed] [Google Scholar]
- 4.Smythies LE, Shen R, Bimczok D, Novak L, Clements RH, Eckhoff DE, et al. Inflammation anergy in human intestinal macrophages is due to Smad-induced IκBα expression and NF-κB inactivation. J Biol Chem. 2010 doi: 10.1074/jbc.M109.069955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Targan SR, Deem RL, Liu M, Wang S, Nel A. Definition of a lamina propria T cell responsive state. Enhanced cytokine responsiveness of T cells stimulated through the CD2 pathway. J Immunol. 1995;154(2):664–675. [PubMed] [Google Scholar]
- 6.Di Sabatino A, Pickard KM, Rampton D, Kruidenier L, Rovedatti L, Leakey NA, et al. Blockade of transforming growth factor-β upregulates T-box transcription factor T-bet, and increases T helper cell type 1 cytokine and matrix metalloproteinase-3 production in the human gut mucosa. Gut. 2008;57(5):605–612. doi: 10.1136/gut.2007.130922. [DOI] [PubMed] [Google Scholar]
- 7.Zeitz M, Quinn TC, Graeff AS, James SP. Mucosal T cells provide helper function but do not proliferate when stimulated by specific antigen in lymphogranuloma venereum proctitis in nonhuman primates. Gastroenterology. 1988;94(2):353–366. doi: 10.1016/0016-5085(88)90422-2. [DOI] [PubMed] [Google Scholar]
- 8.Sartor RB. Mechanisms of disease: pathogenesis of Crohn's disease and ulcerative colitis. Nat Clin Pract Gastroenterol Hepatol. 2006;3(7):390–407. doi: 10.1038/ncpgasthep0528. [DOI] [PubMed] [Google Scholar]
- 9.Monteleone G, Kumberova A, Croft NM, McKenzie C, Steer HW, MacDonald TT. Blocking Smad7 restores TGF-beta1 signaling in chronic inflammatory bowel disease. J Clin Invest. 2001;108(4):601–609. doi: 10.1172/JCI12821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dominitzki S, Fantini MC, Neufert C, Nikolaev A, Galle PR, Scheller J, et al. Cutting edge: Trans-signaling via the soluble IL-6R abrogates the induction of FoxP3 in naive CD4+CD25− T cells. J Immunol. 2007;179(4):2041–2045. doi: 10.4049/jimmunol.179.4.2041. [DOI] [PubMed] [Google Scholar]
- 11.Bauge C, Legendre F, Leclercq S, Elissalde JM, Pujol JP, Galera P, et al. Interleukin-1beta impairment of transforming growth factor beta1 signaling by down-regulation of transforming growth factor beta receptor type II and up-regulation of Smad7 in human articular chondrocytes. Arthritis Rheum. 2007;56(9):3020–3032. doi: 10.1002/art.22840. [DOI] [PubMed] [Google Scholar]
- 12.Takeda K, Kaisho T, Yoshida N, Takeda J, Kishimoto T, Akira S. Stat3 activation is responsible for IL-6-dependent T cell proliferation through preventing apoptosis: generation and characterization of T cell-specific Stat3-deficient mice. J Immunol. 1998;161(9):4652–4660. [PubMed] [Google Scholar]
- 13.O'Sullivan BJ, Thomas HE, Pai S, Santamaria P, Iwakura Y, Steptoe RJ, et al. IL-1β breaks tolerance through expansion of CD25+ effector T cells. J Immunol. 2006;176(12):7278–7287. doi: 10.4049/jimmunol.176.12.7278. [DOI] [PubMed] [Google Scholar]
- 14.Sutton C, Brereton C, Keogh B, Mills KH, Lavelle EC. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. The Journal of experimental medicine. 2006;203(7):1685–1691. doi: 10.1084/jem.20060285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFβ in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24(2):179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 16.Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441(7090):231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 17.Smith PD, Janoff EN, Mosteller-Barnum M, Merger M, Orenstein JM, Kearney JF, et al. Isolation and purification of CD14-negative mucosal macrophages from normal human small intestine. J Immunol Methods. 1997;202(1):1–11. doi: 10.1016/s0022-1759(96)00204-9. [DOI] [PubMed] [Google Scholar]
- 18.Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor β in human disease. N Engl J Med. 2000;342(18):1350–1358. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- 19.Mummery RS, Rider CC. Characterization of the heparin-binding properties of IL-6. J Immunol. 2000;165(10):5671–5679. doi: 10.4049/jimmunol.165.10.5671. [DOI] [PubMed] [Google Scholar]
- 20.Ramsden L, Rider CC. Selective and differential binding of interleukin (IL)-1α, IL-1β, IL-2 and IL-6 to glycosaminoglycans. Eur J Immunol. 1992;22(11):3027–3031. doi: 10.1002/eji.1830221139. [DOI] [PubMed] [Google Scholar]
- 21.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6(11):1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 22.Volpe E, Touzot M, Servant N, Marloie-Provost MA, Hupe P, Barillot E, et al. Multiparametric analysis of cytokine-driven human Th17 differentiation reveals a differential regulation of IL-17 and IL-22 production. Blood. 2009;114(17):3610–3614. doi: 10.1182/blood-2009-05-223768. [DOI] [PubMed] [Google Scholar]
- 23.Yang L, Anderson DE, Baecher-Allan C, Hastings WD, Bettelli E, Oukka M, et al. IL-21 and TGF-beta are required for differentiation of human T(H)17 cells. Nature. 2008;454(7202):350–352. doi: 10.1038/nature07021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu H, Rohowsky-Kochan C. Regulation of IL-17 in human CCR6+ effector memory T cells. J Immunol. 2008;180(12):7948–7957. doi: 10.4049/jimmunol.180.12.7948. [DOI] [PubMed] [Google Scholar]
- 25.Maloy KJ. The Interleukin-23 / Interleukin-17 axis in intestinal inflammation. J Intern Med. 2008;263(6):584–590. doi: 10.1111/j.1365-2796.2008.01950.x. [DOI] [PubMed] [Google Scholar]
- 26.Sellon RK, Tonkonogy S, Schultz M, Dieleman LA, Grenther W, Balish E, et al. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun. 1998;66(11):5224–5231. doi: 10.1128/iai.66.11.5224-5231.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Beelen AJ, Zelinkova Z, Taanman-Kueter EW, Muller FJ, Hommes DW, Zaat SA, et al. Stimulation of the intracellular bacterial sensor NOD2 programs dendritic cells to promote interleukin-17 production in human memory T cells. Immunity. 2007;27(4):660–669. doi: 10.1016/j.immuni.2007.08.013. [DOI] [PubMed] [Google Scholar]
- 28.Young HA, Bream JH. IFN-γ: recent advances in understanding regulation of expression, biological functions, and clinical applications. Curr Top Microbiol Immunol. 2007;316:97–117. doi: 10.1007/978-3-540-71329-6_6. [DOI] [PubMed] [Google Scholar]
- 29.Iliev ID, Mileti E, Matteoli G, Chieppa M, Rescigno M. Intestinal epithelial cells promote colitis-protective regulatory T-cell differentiation through dendritic cell conditioning. Mucosal Immunology. 2009;2(4):340–350. doi: 10.1038/mi.2009.13. [DOI] [PubMed] [Google Scholar]
- 30.Iliev ID, Spadoni I, Mileti E, Matteoli G, Sonzogni A, Sampietro GM, et al. Human intestinal epithelial cells promote the differentiation of tolerogenic dendritic cells. Gut. 2009;58(11):1481–1489. doi: 10.1136/gut.2008.175166. [DOI] [PubMed] [Google Scholar]
- 31.Jaensson E, Uronen-Hansson H, Pabst O, Eksteen B, Tian J, Coombes JL, et al. Small intestinal CD103+ dendritic cells display unique functional properties that are conserved between mice and humans. J Exp Med. 2008;205(9):2139–2149. doi: 10.1084/jem.20080414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Denning TL, Wang YC, Patel SR, Williams IR, Pulendran B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat Immunol. 2007;8(10):1086–1094. doi: 10.1038/ni1511. [DOI] [PubMed] [Google Scholar]
- 33.Suffia I, Reckling SK, Salay G, Belkaid Y. A role for CD103 in the retention of CD4+CD25+ Treg and control of Leishmania major infection. J Immunol. 2005;174(9):5444–5455. doi: 10.4049/jimmunol.174.9.5444. [DOI] [PubMed] [Google Scholar]
- 34.Coombes JL, Siddiqui KR, Arancibia-Carcamo CV, Hall J, Sun CM, Belkaid Y, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med. 2007;204(8):1757–1764. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alten R, Gram H, Joosten LA, van den Berg WB, Sieper J, Wassenberg S, et al. The human anti-IL-1β monoclonal antibody ACZ885 is effective in joint inflammation models in mice and in a proof-of-concept study in patients with rheumatoid arthritis. Arthritis Res Ther. 2008;10(3):R67. doi: 10.1186/ar2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Casini-Raggi V, Kam L, Chong YJ, Fiocchi C, Pizarro TT, Cominelli F. Mucosal imbalance of IL-1 and IL-1 receptor antagonist in inflammatory bowel disease. A novel mechanism of chronic intestinal inflammation. J Immunol. 1995;154(5):2434–2440. [PubMed] [Google Scholar]
- 37.Cominelli F, Nast CC, Duchini A, Lee M. Recombinant interleukin-1 receptor antagonist blocks the proinflammatory activity of endogenous interleukin-1 in rabbit immune colitis. Gastroenterology. 1992;103(1):65–71. doi: 10.1016/0016-5085(92)91096-m. [DOI] [PubMed] [Google Scholar]
- 38.Cominelli F, Nast CC, Clark BD, Schindler R, Lierena R, Eysselein VE, et al. Interleukin 1 (IL-1) gene expression, synthesis, and effect of specific IL-1 receptor blockade in rabbit immune complex colitis. J Clin Invest. 1990;86(3):972–980. doi: 10.1172/JCI114799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arai Y, Takanashi H, Kitagawa H, Okayasu I. Involvement of interleukin-1 in the development of ulcerative colitis induced by dextran sulfate sodium in mice. Cytokine. 1998;10(11):890–896. doi: 10.1006/cyto.1998.0355. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.