

Abstract
The discovery of the multidrug transporter P-glycoprotein (Pgp) over 35 years ago in drug resistant cells prompted several decades of work attempting to overcome drug resistance by inhibition of drug efflux. Despite convincing laboratory data showing that drug transport can be inhibited in vitro, efforts to translate this discovery to the clinic have not succeeded. Since overexpression of Pgp and related transporters including ABCG2 and members of the ABCC family have been linked with poor outcome, it remains a reasonable hypothesis that this poor outcome is linked to reduction of drug exposure by efflux, and thus to drug resistance. In this review, we will discuss the question of whether ABC transporters mediate drug resistance in cancer through a reduction in drug accumulation in tumors, and whether the “Pgp inhibition hypothesis” might be wrong. The hypothesis, which holds that increased chemotherapy effectiveness can be achieved by inhibiting Pgp-mediated drug efflux has only been validated in model systems. Possible explanations for the failure to validate this clinically include the existence of other modulators of drug accumulation and uptake in tumors. Despite these difficulties, a potential role has emerged for drug transporters as therapeutic targets in the central nervous system (CNS). Both lines of investigation point to the need for imaging agents to facilitate the study of drug accumulation in human cancer. This is a critical need for targeted therapies where an important dose-response relationship is likely to exist, and where drug resistance renders many of the novel targeted agents ineffective in a subset of patients.
Keywords: ABC transporters, drug resistance, P-glycoprotein
Introduction
Of the 48 human ATP-binding cassette transporters that have been described, three have predominantly been linked to a potential role in drug resistance. These include P-glycoprotein (Pgp, encoded by the MDR-1/ABCB1 gene), the multidrug resistance-associated protein 1 (MRP1, encoded by the MRP1/ABCC1 gene) and the breast cancer resistance protein (BCRP or ABCG2, encoded by the ABCG2 gene). Pgp was the first ABC transporter described and is by far the best characterized. Its substrates are numerous and include chemotherapeutic agents including anthracyclines, vinca alkaloids, taxanes, and etoposide; tyrosine kinase inhibitors such as imatinib, nilotinib and dasatinib; HIV protease inhibitors and HMG-CoA inhibitors [1, 2]. High endogenous levels of Pgp are found in the placenta, kidney, liver, brain microvasculature and gastrointestinal tract [1, 2]. Pgp has been shown to form part of the blood-brain barrier as well as limit oral drug bioavailability [1, 3].
MRP1 was the second ABC transporter discovered and was found to confer resistance to a narrower range of chemotherapeutics including anthracyclines, vinca alkaloids, and etoposide as well as organic anions and glutathione and glucuronide conjugates [1]. Expression of MRP1 is ubiquitous, but high levels of expression are found in the vessel endothelium of the brain suggesting a protective role for MRP1 at the blood-brain barrier [1, 3]. ABCG2 is the third major transporter studied and has been shown to transport a wide range of substrates including chemotherapy drugs such as mitoxantrone, topotecan, irinotecan; tyrosine kinase inhibitors including imatinib and gefitinib; as well as a range of non-chemotherapy substrates such as antibiotics and HMG-CoA inhibitors [4]. ABCG2 is expressed in the placenta, liver, adrenal glands, lung, prostate, and gastrointestinal tract [1, 4]. In addition, ABCG2 has been shown to form part of the maternal-fetal, blood-brain and blood-testis barriers and has also been shown to modulate oral drug absorption [4].
ABC transporters were discovered and inhibitors taken to the clinic before the terms molecular target and targeted therapy became ingrained in the oncologist’s vocabulary. Nonetheless, they can be considered as potential therapeutic targets. As such, the presence of the therapeutic target in the tumor would be the first requirement before introducing a potential therapy. Unlike molecular targets such as HER2 or EGFR or Bcr-Abl, targeting the ABC transporters can only modify the effectiveness of another “active” therapy. If the active therapy were only partially effective, then decreasing drug accumulation could be an important component in rendering the tumor drug resistant. But the converse is not true. Increasing drug accumulation may have no benefit if other mechanisms of drug resistance are equally important. Thus, there is a critical need to demonstrate the predominance of ABC transporters in determining drug accumulation and drug sensitivity before considering them therapeutic targets.
Determining relevance of the target by clinical outcome
Two strategies were concurrently undertaken by the cancer therapeutics community to evaluate the relevance of ABC transporters to drug resistance. The first has been the study of expression and correlation with outcome. These data have been extensively reviewed earlier [1, 5, 6], and, in sum, the most consistent and convincing data have been obtained in acute myelogenous leukemia. In this disease, two decades of data have repeatedly shown that leukemic cells overexpress Pgp in about 45% of patients with newly diagnosed AML, while leukemic cells in about 65%; of patients with relapsed or refractory disease overexpress this protein. Numerous studies have demonstrated this, with differences in the fraction of patients reported, based on the sensitivity of the assays used [7]. Assays have included immunostaining and detection by flow cytometry, quantitative RNA analysis, cDNA array, and drug efflux assays. Other ABC transporters were evaluated in leukemia after also in vitro documentation of their ability to confer resistance to chemotherapeutics, including MRP1, MRP3 and ABCG2 [6]. For several of them, correlation with poor outcome was demonstrated in one or more studies [6, 8]. It is possible that the prevalence of ABC transporters in leukemia relates to their high expression in hematopoietic stem cells, and that expression is dysregulated in leukemia. There also may be some element of publication bias in the reports that individually examine only one or two transporters.
To overcome potential publication bias in answering this question, systematic studies are needed that assay all candidate transporters simultaneously. An unsupervised clustering of cDNA array data obtained from 178 older acute myeloid leukemia (AML) patients revealed that a subset of samples with the worst overall survival and highest rate of resistant disease had high expression of the multidrug transporters ABCB1 and ABCG2 [9]. These results again support the theory that ABC transporters are targets in leukemia.
In solid tumors, the relationship of ABC transporter overexpression and outcome has been more complex. Clearly there are tumor types with high levels of expression as part of the inherent tumor phenotype. These include renal cancer, adrenocortical cancer, and pancreatic and colorectal cancer [1]. These tumors are characterized by general resistance to cytotoxic compounds and insensitivity to both substrate and non-substrate drugs. Consequently, it is unlikely that a transporter phenotype would be the dominant mechanism of resistance to cytotoxic therapies in these tumors. Thus, a more interesting question is whether tumor types that have inherent sensitivity to chemotherapy develop resistance due to ABC transporter overexpression. There are multiple studies showing expression in tumors such as breast, ovary, and lung cancer, and increased expression following treatment with cancer chemotherapeutics (reviewed in [5, 10, 11]). No studies have appeared examining expression following treatment with molecularly targeted agents including lapatinib in breast cancer, erlotinib or gefitinib in lung cancer, or the PARP inhibitor olaparib (AZD2281) in breast or ovarian cancer. Since these have only recently been shown to be transporter’s substrates, it would be of interest to have resistant tumors examined in the context of assays that evaluate multiple mechanisms of resistance.
The second strategy taken to evaluate ABC transporters as a target was to inhibit function and thereby reverse resistance in the clinic. The outcome of clinical trials aimed at overcoming resistance mediated by Pgp has been the subject of intense scrutiny and disappointment. Clinical trials aimed at overcoming Pgp were undertaken almost as soon as verapamil was discovered as an inhibitor [12]. Early inhibitors lacked potency, which led to the development of compounds that were more potent but also exhibited drug-drug interactions, requiring reduction of anticancer drug doses in most studies [13–15]. Numerous trials were performed with these inhibitors and often more toxicity was observed in the experimental arm. Subsequent inhibitors with less drug-drug interaction also did not fare well in the clinic, although by then the enthusiasm for a drug resistance reversal strategy involving cytotoxic compounds had waned and been replaced by enthusiasm for truly targeted therapy such as that seen with imatinib. Nonetheless, two large randomized trials testing tariquidar in lung cancer closed early for toxicity in the experimental arm, despite the lack of observed drug-drug interactions [16]. This, plus the negative trials with valspodar, resulted in almost total abandonment of the hypothesis that drug resistance could be overcome by the addition of an efflux inhibitor. Only scattered clinical trials remain with CBT-1, tariquidar, and zosuquidar. Notably, a recent trial testing zosuquidar in AML failed to demonstrate an improvement in overall survival when added to standard therapy [17]. The trial design may have been limited by the short infusion time of zosuquidar, but its recent publication adds to the list yet another example of a clinical trial that failed to show benefit of the addition of a Pgp inhibitor to standard therapy (Table 1). Structures for some of the inhibitors are provided in figure 1.
Table 1.
Phase III trials of P-glycoprotein inhibitors
| Inhibitor | Disease | Outcome | Reference |
|---|---|---|---|
| Valspodar (PSC 833) | Ovarian Cancer | No benefit | [66] |
| Valspodar (PSC 833) | Myeloma | No benefit | [67] |
| Valspodar (PSC 833) | Leukemia | No benefit | [68] |
| Valspodar (PSC 833) | MDS | No benefit | [69] |
| Dofequidar (MS-209) | Breast Cancer | Improved outcome | [70] |
| Tariquidar (XR9576) | Lung Cancer | Closed due to toxicity | [16] |
| Zosuquidar (LY335979) | AML | No benefit | [17] |
| CBT-1 | Lung Cancer | Trial ongoing |
Figure 1.

Chemical structures of Pgp inhibitors
Several points must be made concerning the trials. Typically, Pgp was not assayed in tumors and there was no selection of patients whose tumors displayed overexpression of Pgp. Many trials enrolled patients following multiple lines of therapy, so that drug resistance was likely to be multifactorial. Many involved combination chemotherapy in which the dominant drug was not an ABC transporter substrate. The need to reduce the dose of the chemotherapeutic was a major flaw that, while perhaps equalizing the area under the curve because of delayed clearance in the presence of the inhibitor, almost certainly resulted in decreased time above a concentration threshold. Finally, and perhaps most importantly, these inhibitor trials did not and could not control for the presence of other drug efflux mechanisms, or lack of drug uptake mechanisms. Taken together, there are enough problems with the trials to conclude that the Pgp inhibition hypothesis – defined as the idea that drug resistance could be overcome by inhibiting drug efflux – was never adequately tested. It is also possible that the hypothesis was wrong.
Could the Pgp inhibition hypothesis have been wrong?
Patel and Tannock evaluated this question in xenograft models, examining the effect of Pgp inhibitors on drug accumulation [18]. It has been recognized for some time that a steep gradient exists for doxorubicin and other cancer chemotherapeutics as distance increases from blood vessels [19]. Patel and Tannock examined this gradient in the presence of the Pgp inhibitor valspodar (Table 2). They found a definite increase in drug accumulation in the cells adjacent to blood vessels, but a steeper gradient so that at 100 microns from the vessel, levels of uptake were comparable to those in doxorubicin-only xenografts [18]. Indeed, the impact of Pgp alone, without addition of inhibitor, was attenuated with distance away from the vessels, so that a 50%; reduction in drug accumulation in the cell layers adjacent became a 30%; reduction in accumulation at 60 microns [18]. Thus, the xenograft studies of Patel and Tannock suggest that another explanation for the failure of Pgp-mediated resistance reversal is that the impact of Pgp in vivo is limited – other determinants of drug diffusion dominate.
Table 2.
Doxorubicin intensity in distance from blood vessel using Image J software quantification of tissue sections obtained from tumors in mice 10 minutes after administration of 25 mg/kg doxorubicin. Data compiled from [18]
| ≤ 20 mm | ≤ 60 mm | ≤ 100 mm | |
|---|---|---|---|
| Control EMT6 | 29.4 ± 6.1 | 15.6 ± 1.9 | 11.3 ± 2.4 |
| Pgp-expressing AR1 cells | 17.1 ± 2.9 | 10.5 ± 2.8 | 7.4 ± 1.6 |
| Difference (% reduction) | 12.3 (42%) | 5.1 (33%) | 3.9 (35%) |
| Control MCF-7 | 23.8 ± 4.2 | 13.7 ± 2.5 | 10.4 ± 2.3 |
| Pgp-expressing BC19 cells | 13.0 ± 4.3 | 9.6 ± 2.8 | 8.7 ± 3.5 |
| Difference (% reduction) | 10.8 (45%) | 4.1 (30%) | 1.7 (16%) |
Opposing data, however, can be found in the genetically engineered breast cancer model described by Rottenberg et al [20]. In this murine model, derived from a p53 and BRCA1-deleted spontaneous breast cancer that is orthotopically transplanted, resistance that develops during repeated therapy with doxorubicin [21] or the PARP inhibitor olaparib (AZD2281) [20] is largely Pgp-mediated and tumors can be sensitized by the addition of the Pgp inhibitor tariquidar (14, 15) (Figure 2, 3). Similarly, resistance to topotecan is largely ABCG2-mediated [22]. The model system was not chosen with a goal of generating drug transporter mediated resistance; this was the outcome after administration of known transporter substrate drugs. Interestingly, transporters did not emerge as a resistance mechanism to cisplatin. Emergence of resistance to doxorubicin (Figure 2A) and to olaparib (AZD2281, Figure 2B) was delayed in the majority of tumors by the addition of the Pgp inhibitor tariquidar [20]. Similarly, when Abcg2 was deleted in the implanted tumor cells, overall survival increased in topotecan-treated animals, although this did not result in complete tumor eradication, shown in Figure 3 [22].
Figure 2.

(A) From Pajic M et al. Cancer Res 2009;69:6396–6404: Doxorubicin-resistant Brca1−/−; p53−/− mammary tumors were generated in K14cre;Brca1F/F;p53F/F mice, and then orthotopically transplanted into syngeneic mice. The figure shows results following 10 mg tariquidar/kg i.v. (turquoise), 5 mg doxorubicin/kg i.v. (pink), or 10 mg tariquidar/kg i.v. followed 15 min later by 5 mg doxorubicin/kg i.v. (in three separate animals, shown in green, orange, and brown). Tumors were treated when the size reached ~200 mm3. (B) From Rottenberg S et al. PNAS 2008;105:17079–17084: Orthotopically transplanted mice were treated with 50 mg olaparib (AZD2281) per kg for 28 days. When tumors relapsed to 100%; of their original volume, they were retreated by i.p. injection of 2 mg of tariquidar per kg every other day (light blue line) or 50 mg of AZD2281 per kg daily (red line) or both (dark blue line). Relative tumor volume (RTV, ratio of tumor volume to initial size at start of treatment) is shown as a function of time.
Figure 3. From Zander S A et al. Cancer Res 2010;70:1700–1710.
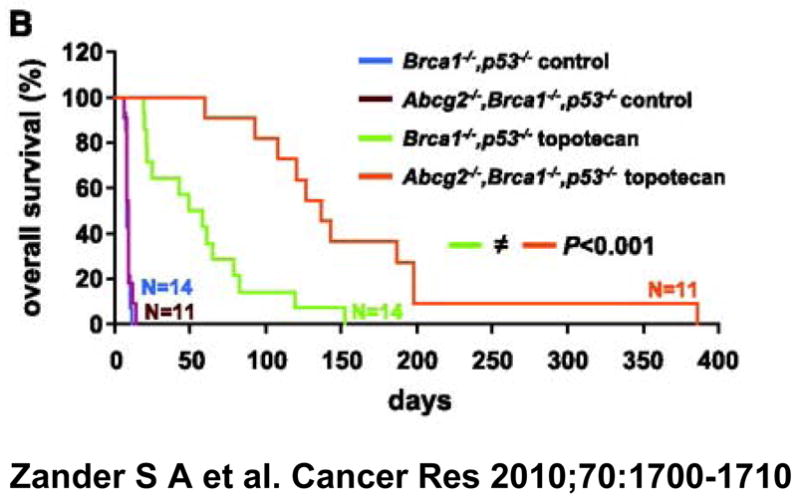
Overall survival (%;) over time of wild-type animals carrying orthotopically transplanted Abcg2-proficient or ABCG2-deficient Brca1−/−, p53−/− mammary tumors. Topotecan was administered at 4 mg/kg topotecan i.p. on days 0 to 4 and 14 to 18. When tumors relapsed or showed progression (tumor size, ≥50%), treatment was resumed.
Several important observations were made in the studies of Rottenberg and Borst. One is that they observed significant tumor heterogeneity in terms of sensitivity to the anticancer agent used. In some animals resistance emerged early and in others very late; resistance to topotecan could emerge as early as 20 days or as late as 150 days after drug exposure. There was heterogeneity of transporter expression, ranging from 2.5- to 90-fold increase in MDR1 mRNA [23]. Low levels of MDR1 mRNA expression were able to confer resistance and the resistance could be inhibited by tariquidar. Pgp or ABCG2 is the dominant mechanism of drug resistance in many of the tumors, but not in all. In other words, these studies suggest that Pgp or ABCG2 may emerge, depending upon the challenging agent, and they suggest that, at least in some of the tumor clones, other mechanisms of resistance are more important than Pgp overexpression. These studies resonate with the clinical experience in which heterogeneity of tumor response to therapy is commonly observed among patients. Nonetheless, these studies offer proof-of-principle that Pgp can emerge as a mechanism of resistance and that Pgp inhibitors can be used to reverse Pgp-mediated resistance.
Other transporter mechanisms have been cited as explanations for reduced drug accumulation. ABCG2 and MRP have been studied as biomarkers of drug resistance in human tumor tissue, but most of the other 45 ABC transporters have not been evaluated in detail. Other ABC transporters such as ABCA2 and ABCC5 have also been shown to transport drug substrates, albeit with a narrower substrate profile than the three most intensively studied [24, 25]. In addition, there is a very large family of solute carriers (SLC) – over 300 arranged in 47 families – including organic anion and cation transporters able to regulate uptake and efflux of a number of anticancer substrates [26]. The number of SLC proteins is so large that expression studies need to include unbiased evaluation of the transporters rather than more traditional candidate gene approaches.
Tumor microenvironment may also play a role in drug uptake. While investigators assume homogeneous drug delivery, Tannock et al discuss a number of factors contributing to heterogeneity in drug distribution [18]. Blood flow is impaired in tumors; vasculature is irregular, shunted and disordered, leading to reduced pressure gradients and increased viscosity and geometric resistance. Marginalized lymphatics fail to drain tissue, further increasing interstitial fluid pressure. Poor nutrient delivery, hypoxia and acidity are also hallmarks of tissue not proximal to vasculature. These characteristics may select for a slow-growing, drug resistant cellular phenotype - drugs targeting proliferative cells will be less effective and drugs optimized in alkaline conditions will be compromised [18]. Furthermore, Tannock discusses the possibility of sequestration of drug in perivascular cells, thus compromising tissue at the distal end of the distance gradient in a tumor microenvironment [18].
Thus, the Pgp inhibition hypothesis – that inhibition of Pgp can reduce drug efflux and thereby reduce drug resistance can be viewed in a more complex light today than previously. We need to know whether Pgp or other ABC transporter is dominant in controlling drug accumulation. And, whether there are coexisting ABC transporters providing redundancy, so that blocking Pgp will not be sufficient to reverse resistance. We need to know whether uptake transporters impact drug accumulation, and about the contribution of tumor microenvironment. These factors contribute individually to drug accumulation and thereby drug resistance, and each have differing degrees of importance for any given tumor. Recently, a renewed interest in Darwin’s theory of evolution has allowed scientists to consider how a cancer can evolve, collecting additional mutations and epigenetic changes over time [27]. These changes allow the fittest cells to survive in the metastatic niche, and survive during exposure to anticancer agents. Whether any one mutation, epigenetic change, or gene expressed dominates and will confer resistance to the next selected therapy will determine its suitability as an anticancer drug target. The schematic in figure 3 illustrates the point that the ABC transporters must be dominant if inhibition is to succeed in reversal of resistance. While it is true that any increase in drug accumulation may also overcome weak cellular resistance mechanisms, it is also true that a 50%; increase in drug accumulation will not matter if there is a mutation that confers a very high level of resistance. Considering the complexity of drug resistance and drug accumulation illustrated in the figure, it is remarkable, and yet frequently observed, that overexpression of Pgp or any other ABC transporter is able to independently confer poor outcome.
Determining relevance of the target by surrogate assay
Taken together, these data suggest that we still do not know whether the ABC transporters in tumors serve to reduce drug accumulation to a magnitude that they become the dominant mechanism controlling intracellular concentration and drug sensitivity. We also do not know whether or not transporter inhibitors are able to increase drug accumulation in human tumors. We have scant data available to answer this question. The best direct data available are studies of imaging agents, including surrogates, or radiolabeled cancer chemotherapeutics.
99mTc-sestamibi and 99mTc-tetrofosmin are cardiac imaging agents that are Food and Drug Administration (FDA) approved to test myocardial function. Both have been used, mostly in small, single-institution studies, to image tumors. Several such studies found that sestamibi or tetrafosmin uptake in lung cancer was highly associated with chemotherapy response [28–34]. Given that sestamibi and tetrofosmin are substrates for both Pgp and MRP1 [35], co-expression in lung cancer could effect a dramatic reduction in accumulation. We recently confirmed a marked variability in sestamibi uptake in lung tumors. There was both interpatient and intrapatient variability – large tumors that were clearly visible on CT scan were not visualized by 99mTc-sestamibi [36]. The addition of tariquidar was previously shown to increase sestamibi retention in normal liver and kidney, presumably by inhibiting Pgp [37]. In addition, tariquidar was able to increase sestamibi accumulation in tumors of 8 patients by 36 to 263%; with the most pronounced effects in patients with adrenocortical cancers or renal cancers [37]. Disappointingly, only a 12–24%; increase in sestamibi uptake and accumulation in 8 of 10 visible lung tumors was observed with tariquidar [36]. If sestamibi is an adequate surrogate (and that is not a certainty) for drug accumulation in cancer, the lung tumor studies suggest that tariquidar is insufficient to increase drug accumulation in that disease. Since erlotinib and gefitinib have been experimentally radiolabeled for PET imaging [38, 39], clinical studies should be performed with these agents. A PET-labeled paclitaxel has been synthesized [40] and a clinical trial is open and accruing (clinicaltrials.gov). Uptake and accumulation studies in lung and breast tumors would provide valuable information on baseline variability and, confirming such, would allow testing of approaches to increase drug uptake in tumors. These approaches are needed regardless of whether or not ABC transporters are implicated in clinical drug resistance. Molecularly targeted therapies that do not reach their target represent a lost opportunity for improved patient outcome.
Could polymorphic variation in ABC transporters confound data?
One advance that has been made in the field since the termination of many of the randomized Phase III trials is the discovery of single nucleotide polymorphisms (SNPs) that affect the structure and function of several of the ABC transporters. For ABCB1, some commonly occurring SNPs have been linked to changes in protein function. The 3435C>T polymorphism has garnered intensive study recently. The polymorphism is synonymous and is often found associated with other variants in ABCB1. One of the most frequently found set of variants is the 1236C>T, 2677G/T, 3435C>T haplotype that is found in roughly 25–40%; of Caucasians and Asians but in only 5–8%; of Africans [41]. The 1236C>T mutation is synonymous, while the 2677G/T mutation is non-synonymous. Interestingly, the wild-type protein and the haplotype variant are expressed at similar levels at the cell membrane, but the presence of the 3435C>T polymorphism in the haplotype changes the folding and function of the protein, reducing the efficiency of transport of known substrates, but interestingly also reducing the efficacy of inhibitors such as cyclosporine A or verapamil [42]. This raises the possibility that the action of other inhibitors might be altered by the haplotype. Despite their potential impact on inhibitor efficacy, this idea is relatively new, and these haplotypes have been rarely examined in clinical studies with Pgp inhibitors.
On the other hand, numerous pharmacokinetic studies have evaluated the impact of the ABCB1 variants on area under the concentration curve (AUC) or half-life or other pharmacokinetic parameter [2, 43]. Although the findings have been controversial and conflicting, there is generally a consensus that patients bearing the variant alleles are likely to have reduced clearance and potentially increased toxicity if there is a narrow therapeutic window. For ABCG2, several SNPs have been described and associated with reduced clearance of substrates, and/or increased toxicity. These include topotecan, diflomotecan, and gefitinib [44–47].
Thus, the polymorphic variants of Pgp could have confounded results of inhibitor trials and genotyping for these variants has the potential to further our understanding of those trials. Additionally, pharmacokinetic studies of the variants may give us insight into the potential for Pgp or other transporter to affect drug response – will patients who have reduced clearance and higher drug concentrations have improved anticancer efficacy?
An alternate therapeutic target: ABC transporters in the CNS
While imaging studies that would determine the contribution of drug uptake and accumulation to resistance are developing, another potential therapeutic target among the ABC transporters has emerged. The blood-brain barrier (BBB) is a protective mechanism designed to prevent toxins, both endogenous and exogenous, from damaging the brain. In protecting the brain, however, the BBB also serves to prevent therapeutic drugs from entering, thus hampering efficacy. High levels of expression of Pgp, MRP1 and ABCG2 at the brain capillary endothelium suggest they form a major role in preventing drug uptake in the brain [3]. Studies in mice deficient in Abcb1/2, Abcc1 or Abcg2 have provided in vivo proof for the significance of these transporters at the mouse BBB [48–50].
What is striking about the mouse studies is the apparent cooperativity between Pgp and ABCG2 at the murine BBB. Interestingly, in mouse models, maximum brain penetration of tyrosine kinase inhibitors as well as topotecan and flavopiridol was observed when the Abcb1/2 and Abcg2 genes are knocked out [51–55]. Figure 5 shows results compiled from various studies in knockout mice. Similar results have also been obtained with erlotinib and sunitinib [56, 57]. These results raise concern that long-term control of systemic disease in breast, lung, and renal cancer obtained with targeted agents lapatinib, erlotinib, and sunitinib, respectively, could be accompanied by emergence of CNS metastases. Thus, it may be worthwhile to explore inhibition of both Pgp and ABCG2. Elacridar (GF120918) [58], tariquidar (XR9576) [59] and biricodar (VX-710) [60] have all been shown to inhibit both Pgp and ABCG2 in in vitro models and elacridar has been used extensively in mouse models as a dual Pgp and ABCG2 inhibitor [53, 54, 61]. It is possible that these agents could find utility in increasing substrate uptake in the CNS. Consideration has been given to this more in neurology and psychiatry than in oncology, to increase CNS uptake of anti-seizure and psychiatric medications [62, 63].
Figure 5.

Effects of ABC transporters on brain penetration of substrate compounds. The fold-increase in brain penetration of flavopiridol, imatinib, topotecan, dasatinib, lapatinib, and sorafenib was determined in mice lacking Abcb1a/b (Abcb1a/b −/−), Abcg2 (Abcg2 −/−) or both transporters (Abcb1a/b −/− Abcg2 −/−) compared to wild-type mice (WT) which were assigned a value of 1. Data compiled from [52–55].
Before undertaking clinical trials with these dual inhibitors attempting to increase accumulation of any agent, it is critical that a validated probe of ABCG2 (or Pgp) function in the human blood-brain barrier be developed. Without such a probe to prove inhibition of transporters at the blood-brain barrier, results of clinical trials combining a tyrosine kinase inhibitor with an ABC transporter inhibitor would be difficult to interpret. However, several imaging agents are being developed for this purpose, including N-desmethyl-loperamide for imaging Pgp in patients [64, 65]. Also, animal studies suggest that radiolabeled gefitinib may also be useful in this regard [39].
Conclusions
ABC transporters play an important role in normal pharmacology and in normal tissue protection. It is not clear what their role is in mediating drug resistance in tumors of patients – clinical trials testing inhibitors of Pgp failed to confirm the hypothesis that inhibition of Pgp could increase drug accumulation and thereby reduce resistance. Continued investigation is warranted to understand how, in the clinic, over-expression confers poor outcome, while concurrent addition of compounds that could reduce efflux though Pgp inhibition have failed. Uptake transporters may be equally important and have not been adequately studied in cancer. Absence of an uptake transporter or presence of deleterious elements of tumor microenvironment could render overexpression of an ABC transporter a mute point. Inadequate surrogates exist to answer the question of whether reduced drug accumulation (even on a simple pharmacologic basis) is responsible for drug resistance in patient tumors. Development of imaging agents will be a keystone of truly personalized therapy for patients, so that we can confirm that adequate levels of a molecularly targeted therapy have reached the tumor. Finally, these same considerations exist for both uptake and efflux transporters guarding the blood brain barrier. Substrate drugs prevented from CNS accumulation are candidates for future studies with modulators of drug uptake and may represent a therapeutic niche for the reservoir of learning that has been gathered in the field over the last 30 years.
Figure 4.

Schematic illustrating the proportions of drug resistance factors needed to successfully exploit ABC transporters as therapeutic targets. In the schematic in panel A, reduced drug accumulation must be the most important component of drug resistance. Decreasing drug accumulation confers increasing levels of resistance while other mechanisms occupy a relatively minor role. In turn, in panel B, ABC efflux transporters are the major contributor of reduced drug accumulation. These assumptions, although never proven, underlay the hypothesis that inhibition of drug efflux could reverse drug resistance and improve drug efficacy.
References
- 1.Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nature Rev Cancer. 2002;2(1):48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 2.Cascorbi I. Role of pharmacogenetics of ATP-binding cassette transporters in the pharmacokinetics of drugs. Pharmacol Ther. 2006;112(2):457–73. doi: 10.1016/j.pharmthera.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 3.Deeken JF, Loscher W. The blood-brain barrier and cancer: transporters, treatment, and Trojan horses. Clin Cancer Res. 2007;13(6):1663–1674. doi: 10.1158/1078-0432.CCR-06-2854. [DOI] [PubMed] [Google Scholar]
- 4.Robey RW, Polgar O, Deeken J, To KW, Bates SE. ABCG2: determining its relevance in clinical drug resistance. Cancer Metastasis Rev. 2007;26(1):39–57. doi: 10.1007/s10555-007-9042-6. [DOI] [PubMed] [Google Scholar]
- 5.Leonard GD, Fojo T, Bates SE. The role of ABC transporters in clinical practice. Oncologist. 2003;8(5):411–424. doi: 10.1634/theoncologist.8-5-411. [DOI] [PubMed] [Google Scholar]
- 6.van der Kolk DM, de Vries EG, Müller M, Vellenga E. The role of drug efflux pumps in acute myeloid leukemia. Leuk Lymphoma. 2002;43(4):685–701. doi: 10.1080/10428190290016773. [DOI] [PubMed] [Google Scholar]
- 7.Sonneveld P. Multidrug resistance in haematological malignancies. J Intern Med. 2000;247(5):521–534. [PubMed] [Google Scholar]
- 8.Steinbach D, Legrand O. ABC transporters and drug resistance in leukemia: was P-gp nothing but the first head of the Hydra? Leukemia. 2007;21(6):1172–1176. doi: 10.1038/sj.leu.2404692. [DOI] [PubMed] [Google Scholar]
- 9.Wilson CS, Davidson GS, Martin SB, Andries E, Potter J, Harvey R, Ar K, Xu Y, Kopecky KJ, Ankerst DP, Gundacker H, Slovak ML, Mosquera-Caro M, Chen IM, Stirewalt DL, Murphy M, Schultz FA, Kang H, Wang X, Radich JP, Appelbaum FR, Atlas SR, Godwin J, Willman CL. Gene expression profiling of adult acute myeloid leukemia identifies novel biologic clusters for risk classification and outcome prediction. Blood. 2006;108(2):685–696. doi: 10.1182/blood-2004-12-4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leonessa F, Clarke R. ATP binding cassette transporters and drug resistance in breast cancer. Endocr Relat Cancer. 2003;10(1):43–73. doi: 10.1677/erc.0.0100043. [DOI] [PubMed] [Google Scholar]
- 11.Takara K, Sakaeda T, Okumura K. An update on overcoming MDR1-mediated multidrug resistance in cancer chemotherapy. Curr Pharm Des. 2006;12(3):273–286. doi: 10.2174/138161206775201965. [DOI] [PubMed] [Google Scholar]
- 12.Ozols RF, Cunnion RE, Klecker RW, Hamilton TC, Ostchega Y, Parrillo JE, Young RC. Verapamil and adriamycin in the treatment of drug-resistant ovarian cancer patients. J Clin Oncol. 1987;5:641–647. doi: 10.1200/JCO.1987.5.4.641. [DOI] [PubMed] [Google Scholar]
- 13.Seiden MV, Swenerton KD, Matulonis U, Campos S, Rose P, Batist G, Ette E, Garg V, Fuller A, Harding MW, Charpentier D. A phase II study of the MDR inhibitor biricodar (INCEL, VX-710) and paclitaxel in women with advanced ovarian cancer refractory to paclitaxel therapy. Gynecol Oncol. 2002;86(3):302–310. doi: 10.1006/gyno.2002.6762. [DOI] [PubMed] [Google Scholar]
- 14.Fracasso PM, Brady MF, Moore DH, Walker JL, Rose PG, Letvak L, Grogan TM, McGuire WP. Phase II study of paclitaxel and valspodar (PSC 833) in refractory ovarian carcinoma: a gynecologic oncology group study. J Clin Oncol. 2001;19(12):2975–2982. doi: 10.1200/JCO.2001.19.12.2975. [DOI] [PubMed] [Google Scholar]
- 15.Chico I, Kang MH, Bergan R, Abraham J, Bakke S, Meadows B, Rutt A, Robey R, Choyke P, Merino M, Goldspiel B, Smith T, Steinberg S, Figg WD, Fojo T, Bates S. Phase I study of infusional paclitaxel in combination with the P-glycoprotein antagonist PSC 833. J Clin Oncol. 2001;19(3):832–842. doi: 10.1200/JCO.2001.19.3.832. [DOI] [PubMed] [Google Scholar]
- 16.Fox E, Bates SE. Tariquidar (XR9576): a P-glycoprotein drug efflux pump inhibitor. Expert Rev Anticancer Ther. 2007;7(4):447–459. doi: 10.1586/14737140.7.4.447. [DOI] [PubMed] [Google Scholar]
- 17.Cripe LD, Uno H, Paietta EM, Litzow MR, Ketterling RP, Bennett JM, Rowe JM, Lazarus HM, Luger S, Tallman MS. Zosuquidar, a novel modulator of P-glycoprotein, does not improve the outcome of older patients with newly diagnosed acute myeloid leukemia: a randomized, placebo-controlled trial of the Eastern Cooperative Oncology Group 3999. Blood. 2010;116(20):4077–4085. doi: 10.1182/blood-2010-04-277269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Patel KJ, Tannock IF. The influence of P-glycoprotein expression and its inhibitors on the distribution of doxorubicin in breast tumors. BMC Cancer. 2009;9:356. doi: 10.1186/1471-2407-9-356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lankelma J, Dekker H, Luque FR, Luykx S, Hoekman K, van der Valk P, van Diest PJ, Pinedo HM. Doxorubicin gradients in human breast cancer. Clin Cancer Res. 1999;5(7):1703–1707. [PubMed] [Google Scholar]
- 20.Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA, Derksen PW, de Bruin M, Zevenhoven J, Lau A, Boulter R, Cranston A, O’Connor MJ, Martin NM, Borst P, Jonkers J. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci U S A. 2008;105(44):17079–17084. doi: 10.1073/pnas.0806092105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pajic M, Iyer JK, Kersbergen A, van der Burg E, Nygren AO, Jonkers J, Borst P, Rottenberg S. Moderate increase in Mdr1a/1b expression causes in vivo resistance to doxorubicin in a mouse model for hereditary breast cancer. Cancer Res. 2009;69(16):6396–6404. doi: 10.1158/0008-5472.CAN-09-0041. [DOI] [PubMed] [Google Scholar]
- 22.Zander SA, Kersbergen A, van der Burg E, de Water N, van Tellingen O, Gunnarsdottir S, Jaspers JE, Pajic M, Nygren AO, Jonkers J, Borst P, Rottenberg S. Sensitivity and acquired resistance of BRCA1; p53-deficient mouse mammary tumors to the topoisomerase I inhibitor topotecan. Cancer Res. 2010;70(4):1700–1710. doi: 10.1158/0008-5472.CAN-09-3367. [DOI] [PubMed] [Google Scholar]
- 23.Rottenberg S, Nygren AO, Pajic M, van Leeuwen FW, van der Heijden I, van de Wetering K, Liu X, de Visser KE, Gilhuijs KG, van Tellingen O, Schouten JP, Jonkers J, Borst P. Selective induction of chemotherapy resistance of mammary tumors in a conditional mouse model for hereditary breast cancer. Proc Natl Acad Sci U S A. 2007;104(29):12117–12122. doi: 10.1073/pnas.0702955104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boonstra R, Timmer-Bosscha H, van Echten-Arends J, van der Kolk DM, van den Berg A, de Jong B, Tew KD, Poppema S, de Vries EG. Mitoxantrone resistance in a small cell lung cancer cell line is associated with ABCA2 upregulation. Br J Cancer. 2004;90(12):2411–2417. doi: 10.1038/sj.bjc.6601863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ritter CA, Jedlitschky G, Meyer zu Schwabedissen H, Grube M, Köck K, Kroemer HK. Cellular export of drugs and signaling molecules by the ATP-binding cassette transporters MRP4 (ABCC4) and MRP5 (ABCC5) Drug Metab Rev. 2005;37(1):253–278. doi: 10.1081/dmr-200047984. [DOI] [PubMed] [Google Scholar]
- 26.Franke RM, Gardner ER, Sparreboom A. Pharmacogenetics of drug transporters. Curr Pharm Des. 2010;16(2):220–230. doi: 10.2174/138161210790112683. [DOI] [PubMed] [Google Scholar]
- 27.Gerlinger M, Swanton C. How Darwinian models inform therapeutic failure initiated by clonal heterogeneity in cancer medicine. Br J Cancer. 2010;103(8):1139–1143. doi: 10.1038/sj.bjc.6605912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ceriani L, Giovanella L, Bandera M, Beghe B, Ortelli M, Roncari G. Semi-quantitative assessment of 99Tcm-sestamibi uptake in lung cancer: relationship with clinical response to chemotherapy. Nucl Med Commun. 1997;18(11):1087–1097. doi: 10.1097/00006231-199711000-00013. [DOI] [PubMed] [Google Scholar]
- 29.Nishiyama Y, Yamamoto Y, Satoh K, Ohkawa M, Kameyama K, Hayashi E, Fujita J, Tanabe M. Comparative study of Tc-99m MIBI and TI-201 SPECT in predicting chemotherapeutic response in non-small-cell lung cancer. Clin Nucl Med. 2000;25(5):364–369. doi: 10.1097/00003072-200005000-00010. [DOI] [PubMed] [Google Scholar]
- 30.Yüksel M, Cermik T, Doğanay L, Karlikaya C, Cakir E, Salan A, Berkarda S. 99mTc-MIBI SPET in non-small cell lung cancer in relationship with Pgp and prognosis. Eur J Nucl Med Mol Imaging. 2002;29(7):876–881. doi: 10.1007/s00259-002-0804-7. [DOI] [PubMed] [Google Scholar]
- 31.Komori T, Narabayashi I, Matsui R, Sueyoshi K, Aratani T, Utsunomiya K. Ann Nucl Med. 2000;14(6):415–420. doi: 10.1007/BF02988286. [DOI] [PubMed] [Google Scholar]
- 32.Fuster D, Vinolas N, Mallafre C, Pavia J, Martin F, Pons F. Tetrofosmin as predictors of tumour response. Q J Nucl Med. 2003;47(1):58–62. [PubMed] [Google Scholar]
- 33.Shih C, Shiau Y, Wang J, Ho S, Kao A. Using technetium-99m tetrofosmin chest imaging to predict taxol-based chemotherapy response in non-small cell lung cancer but not related to lung resistance protein expression. Lung. 2003;181(2):103–111. doi: 10.1007/s00408-003-1011-4. [DOI] [PubMed] [Google Scholar]
- 34.Mohan HK, Miles KA. Cost-effectiveness of 99mTc-sestamibi in predicting response to chemotherapy in patients with lung cancer: systematic review and meta-analysis. J Nucl Med. 2009;50(3):376–381. doi: 10.2967/jnumed.108.055988. [DOI] [PubMed] [Google Scholar]
- 35.Gomes CM, Abrunhosa AJ, Pauwels EK, Botelho MF. P-glycoprotein versus MRP1 on transport kinetics of cationic lipophilic substrates: a comparative study using [99mTc]sestamibi and [99mTc]tetrofosmin. Cancer Biother Radiopharm. 2009;24(2):215–227. doi: 10.1089/cbr.2008.0539. [DOI] [PubMed] [Google Scholar]
- 36.Kelly RJ, Draper D, Chen CC, Robey RW, Figg WD, Piekarz RL, Chen X, Gardner ER, Balis FM, Venkatesan AM, Steinberg SM, Fojo AT, Bates SE. A Pharmacodynamic Study of Docetaxel in Combination with the P-glycoprotein Antagonist Tariquidar (XR9576) in Patients with Lung Ovarian and Cervical Cancer. Clin Cancer Res. 2010 doi: 10.1158/1078-0432.CCR-10-1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Agrawal M, Abraham J, Balis FM, Edgerly M, Stein WD, Bates S, Fojo T, Chen CC. Increased 99mTc-sestamibi accumulation in normal liver and drug-resistant tumors after the administration of the glycoprotein inhibitor, XR9576. Clin Cancer Res. 2003;9(2):650–656. [PubMed] [Google Scholar]
- 38.Memon AA, Jakobsen S, Dagnaes-Hansen F, Sorensen BS, Keiding S, Nexo E. Positron emission tomography (PET) imaging with [11C]-labeled erlotinib: a micro-PET study on mice with lung tumor xenografts. Cancer Res. 2009;69(3):873–878. doi: 10.1158/0008-5472.CAN-08-3118. [DOI] [PubMed] [Google Scholar]
- 39.Kawamura K, Yamasaki T, Yui J, Hatori A, Konno F, Kumata K, Irie T, Fukumura T, Suzuki K, Kanno I, Zhang M. In vivo evaluation of P-glycoprotein and breast cancer resistance protein modulation in the brain using [(11)C]gefitinib. Nucl Med Biol. 2009;36(3):239–246. doi: 10.1016/j.nucmedbio.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 40.Kurdziel KA, Kalen JD, Hirsch JI, Wilson JD, Agarwal R, Barrett D, Bear HD, McCumiskey JF. Imaging multidrug resistance with 4-[18F]fluoropaclitaxel. Nucl Med Biol. 2007;34(7):823–831. doi: 10.1016/j.nucmedbio.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 41.Fung KL, Gottesman MM. A synonymous polymorphism in a common MDR1 (ABCB1) haplotype shapes protein function. Biochim Biophys Acta. 2009;1794(5):860–871. doi: 10.1016/j.bbapap.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kimchi-Sarfaty C, Oh JM, Kim IW, Sauna ZE, Calcagno AM, Ambudkar SV, Gottesman MM. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science. 2007;315(5811):525–528. doi: 10.1126/science.1135308. [DOI] [PubMed] [Google Scholar]
- 43.Ambudkar SV, Kimchi-Sarfaty C, Sauna ZE, Gottesman MM. P-glycoprotein: from genomics to mechanism. Oncogene. 2003;22(47):7468–7485. doi: 10.1038/sj.onc.1206948. [DOI] [PubMed] [Google Scholar]
- 44.Li J, Cusatis G, Brahmer J, Sparreboom A, Robey RW, Bates SE, Hidalgo M, Baker SD. Association of variant ABCG2 and the pharmacokinetics of epidermal growth factor receptor tyrosine kinase inhibitors in cancer patients. Cancer Biol Ther. 2007;6(3):432–438. doi: 10.4161/cbt.6.3.3763. [DOI] [PubMed] [Google Scholar]
- 45.Cusatis G, Gregorc V, Li J, Spreafico A, Ingersoll RG, Verweij J, Ludovini V, Villa E, Hidalgo M, Sparreboom A, Baker SD. Pharmacogenetics of ABCG2 and adverse reactions to gefitinib. J Natl Cancer Inst. 2006;98(23):1739–1742. doi: 10.1093/jnci/djj469. [DOI] [PubMed] [Google Scholar]
- 46.Sparreboom A, Loos WJ, Burger H, Sissung TM, Verweij J, Figg WD, Nooter K, Gelderblom H. Effect of ABCG2 Genotype on the Oral Bioavailability of Topotecan. Cancer Biol Ther. 2005;4(6):650–658. doi: 10.4161/cbt.4.6.1731. [DOI] [PubMed] [Google Scholar]
- 47.Sparreboom A, Gelderblom H, Marsh S, Ahluwalia R, Obach R, Principe P, Twelves C, Verweij J, McLeod HL. Diflomotecan pharmacokinetics in relation to ABCG2 421C>A genotype. Clin Pharmacol Ther. 2004;76(1):38–44. doi: 10.1016/j.clpt.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 48.Schinkel AH, Smit JJ, van Tellingen O, Beijnen JH, Wagenaar E, van Deemter L, Mol CA, van der Valk MA, Robanus-Maandag EC, te Riele HP, Berns AJ, Borst P. Disruption of mouse mdr-1a p-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell. 1994;77:491–502. doi: 10.1016/0092-8674(94)90212-7. [DOI] [PubMed] [Google Scholar]
- 49.Wijnholds J, deLange EC, Scheffer GL, van den Berg DJ, Mol CA, van der Valk M, Schinkel AH, Scheper RJ, Breimer DD, Borst P. Multidrug resistance protein 1 protects the choroid plexus epithelium and contributes to the blood- cerebrospinal fluid barrier. J Clin Invest. 2000;105(3):279–285. doi: 10.1172/JCI8267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Breedveld P, Pluim D, Cipriani G, Wielinga P, van Tellingen O, Schinkel AH, Schellens JH. The effect of Bcrp1 (Abcg2) on the in vivo pharmacokinetics and brain penetration of imatinib mesylate (Gleevec): implications for the use of breast cancer resistance protein and P-glycoprotein inhibitors to enable the brain penetration of imatinib in patients. Cancer Res. 2005;65(7):2577–2582. doi: 10.1158/0008-5472.CAN-04-2416. [DOI] [PubMed] [Google Scholar]
- 51.de Vries NA, Zhao J, Kroon E, Buckle T, Beijnen JH, van Tellingen O. P-glycoprotein and breast cancer resistance protein: two dominant transporters working together in limiting the brain penetration of topotecan. Clin Cancer Res. 2007;13(21):6440–6449. doi: 10.1158/1078-0432.CCR-07-1335. [DOI] [PubMed] [Google Scholar]
- 52.Polli J, Olson K, Chism J, John-Williams L, Yeager R, Woodard S, Otto V, Castellino S, Demby V. An unexpected synergist role of P-glycoprotein and breast cancer resistance protein on the central nervous system penetration of the tyrosine kinase inhibitor lapatinib (N-{3-chloro-4-[(3-fluorobenzyl)oxy]phenyl}-6-[5-({[2-(methylsulfonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine; GW572016) Drug Metab Dispos. 2009;37(2):439–442. doi: 10.1124/dmd.108.024646. [DOI] [PubMed] [Google Scholar]
- 53.Lagas JS, van Waterschoot RA, van Tilburg VA, Hillebrand MJ, Lankheet N, Rosing H, Beijnen JH, Schinkel AH. Brain accumulation of dasatinib is restricted by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) and can be enhanced by elacridar treatment. Clin Cancer Res. 2009;15(7):2344–2351. doi: 10.1158/1078-0432.CCR-08-2253. [DOI] [PubMed] [Google Scholar]
- 54.Lagas J, van Waterschoot R, Sparidans R, Wagenaar E, Beijnen J, Schinkel A. Breast cancer resistance protein and P-glycoprotein limit sorafenib brain accumulation. Mol Cancer Ther. 2010;9(2):319–326. doi: 10.1158/1535-7163.MCT-09-0663. [DOI] [PubMed] [Google Scholar]
- 55.Zhou L, Schmidt K, Nelson FR, Zelesky V, Troutman MD, Feng B. The effect of breast cancer resistance protein and P-glycoprotein on the brain penetration of flavopiridol, imatinib mesylate (Gleevec), prazosin, and 2-methoxy-3-(4-(2-(5-methyl-2-phenyloxazol-4-yl)ethoxy)phenyl)propanoic acid (PF-407288) in mice. Drug Metab Dispos. 2009;37(5):946–955. doi: 10.1124/dmd.108.024489. [DOI] [PubMed] [Google Scholar]
- 56.Tang SC, Lagas JS, Lankheet N, Rosing H, Beijnen JH, Schinkel AH. P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) limit sunitinib brain accumulation which can be enhanced by oral elacridar treatment. Proc Amer Assoc Can Res. 2010;51 [Google Scholar]
- 57.Kodaira H, Kusuhara H, Ushiki J, Fuse E, Sugiyama Y. Kinetic analysis of the cooperation of P-glycoprotein (P-gp/Abcb1) and breast cancer resistance protein (Bcrp/Abcg2) in limiting the brain and testis penetration of erlotinib, flavopiridol, and mitoxantrone. J Pharmacol Exp Ther. 2010;333(3):788–796. doi: 10.1124/jpet.109.162321. [DOI] [PubMed] [Google Scholar]
- 58.de Bruin M, Miyake K, Litman T, Robey R, Bates SE. Reversal of resistance by GF120918 in cell lines expressing the ABC half-transporter, MXR. Cancer Lett. 1999;146(2):117–126. doi: 10.1016/s0304-3835(99)00182-2. [DOI] [PubMed] [Google Scholar]
- 59.Robey RW, Steadman K, Polgar O, Morisaki K, Blayney M, Mistry P, Bates SE. Pheophorbide a is a specific probe for ABCG2 function and inhibition. Cancer Res. 2004;64(4):1242–1246. doi: 10.1158/0008-5472.can-03-3298. [DOI] [PubMed] [Google Scholar]
- 60.Minderman H, O’Loughlin KL, Pendyala L, Baer MR. VX-710 (biricodar) increases drug retention and enhances chemosensitivity in resistant cells overexpressing P-glycoprotein, multidrug resistance protein, and breast cancer resistance protein. Clin Cancer Res. 2004;10(5):1826–1834. doi: 10.1158/1078-0432.ccr-0914-3. [DOI] [PubMed] [Google Scholar]
- 61.Bihorel S, Camenisch G, Lemaire M, Scherrmann JM. Modulation of the brain distribution of imatinib and its metabolites in mice by valspodar, zosuquidar and elacridar. Pharm Res. 2007;24(9):1720–1728. doi: 10.1007/s11095-007-9278-4. [DOI] [PubMed] [Google Scholar]
- 62.Loscher W, Potschka H. Blood-brain barrier active efflux transporters: ATP-binding cassette gene family. NeuroRx. 2005;2(1):86–98. doi: 10.1602/neurorx.2.1.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lazarowski A, Czornyj L, Lubienieki F, Girardi E, Vazquez S, D’Giano C. ABC transporters during epilepsy and mechanisms underlying multidrug resistance in refractory epilepsy. Epilepsia. 2007;48(Suppl 5):140–149. doi: 10.1111/j.1528-1167.2007.01302.x. [DOI] [PubMed] [Google Scholar]
- 64.Kannan P, John C, Zoghbi S, Halldin C, Gottesman M, Innis R, Hall M. Imaging the function of P-glycoprotein with radiotracers: pharmacokinetics and in vivo applications. Clin Pharmacol Ther. 2009;86(4):368–377. doi: 10.1038/clpt.2009.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kreisl WC, Liow JS, Kimura N, Seneca N, Zoghbi SS, Morse CL, Herscovitch P, Pike VW, Innis RB. P-glycoprotein function at the blood-brain barrier in humans can be quantified with the substrate radiotracer 11C-N-desmethyl-loperamide. J Nucl Med. 2010;51(4):559–566. doi: 10.2967/jnumed.109.070151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lhomme C, Joly F, Walker JL, Lissoni AA, Nicoletto MO, Manikhas GM, Baekelandt MM, Gordon AN, Fracasso PM, Mietlowski WL, Jones GJ, Dugan MH. Phase III study of valspodar (PSC 833) combined with paclitaxel and carboplatin compared with paclitaxel and carboplatin alone in patients with stage IV or suboptimally debulked stage III epithelial ovarian cancer or primary peritoneal cancer. J Clin Oncol. 2008;26(16):2674–2682. doi: 10.1200/JCO.2007.14.9807. [DOI] [PubMed] [Google Scholar]
- 67.Friedenberg WR, Rue M, Blood EA, Dalton WS, Shustik C, Larson RA, Sonneveld P, Greipp PR. Phase III study of PSC-833 (valspodar) in combination with vincristine, doxorubicin, and dexamethasone (valspodar/VAD) versus VAD alone in patients with recurring or refractory multiple myeloma (E1A95): a trial of the Eastern Cooperative Oncology Group. Cancer. 2006;106(4):830–838. doi: 10.1002/cncr.21666. [DOI] [PubMed] [Google Scholar]
- 68.Baer MR, George SL, Dodge RK, O’Loughlin KL, Minderman H, Caligiuri MA, Anastasi J, Powell BL, Kolitz JE, Schiffer CA, Bloomfield CD, Larson RA. Phase 3 study of the multidrug resistance modulator PSC-833 in previously untreated patients 60 years of age and older with acute myeloid leukemia: Cancer and Leukemia Group B Study 9720. Blood. 2002;100(4):1224–1232. [PubMed] [Google Scholar]
- 69.Greenberg PL, Lee SJ, Advani R, Tallman MS, Sikic BI, Letendre L, Dugan K, Lum B, Chin DL, Dewald G, Paietta E, Bennett JM, Rowe JM. Mitoxantrone, etoposide, and cytarabine with or without valspodar in patients with relapsed or refractory acute myeloid leukemia and high-risk myelodysplastic syndrome: a phase III trial (E2995) J Clin Oncol. 2004;22(6):1078–1086. doi: 10.1200/JCO.2004.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Saeki T, Nomizu T, Toi M, Ito Y, Noguchi S, Kobayashi T, Asaga T, Minami H, Yamamoto N, Aogi K, Ikeda T, Ohashi Y, Sato W, Tsuruo T. Dofequidar fumarate (MS-209) in combination with cyclophosphamide, doxorubicin, and fluorouracil for patients with advanced or recurrent breast cancer. J Clin Oncol. 2007;25(4):411–417. doi: 10.1200/JCO.2006.08.1646. [DOI] [PubMed] [Google Scholar]