

Abstract
Mutations in the gene encoding the cystic fibrosis transmembrane conductance regulator (CFTR) anion channel cause the autosomal recessive disease, cystic fibrosis (CF). The most common mutation is ΔF508, which deletes phenylalanine508. In vitro studies indicate that CFTR-ΔF508 is misprocessed, though in vivo consequences of the mutation are uncertain. To better understand effects of the ΔF508 mutation, we produced CFTRΔF508/ΔF508 pigs. Our biochemical, immunocytochemical and electrophysiological data on CFTR-ΔF508 in newborn pigs paralleled in vitro results. They also indicated that CFTRΔF508/ΔF508 airway epithelia retain a small residual CFTR conductance; maximal stimulation produced ~6% of wild-type function. Interestingly, cAMP agonists were less potent at stimulating current in CFTRΔF508/ΔF508 epithelia, suggesting that quantitative tests of maximal anion current may overestimate transport under physiological conditions. Despite residual CFTR function, four older CFTRΔF508/ΔF508 pigs developed lung disease strikingly similar to human CF. These results suggest that this limited CFTR activity is insufficient to prevent lung or gastrointestinal disease in CF pigs. These data also suggest that studies of recombinant CFTR-ΔF508 misprocessing predict in vivo behavior, which validates its use in biochemical and drug discovery experiments. These findings help elucidate the molecular pathogenesis of the common CF mutation and will guide strategies for developing new therapeutics.
INTRODUCTION
Cystic fibrosis (CF) is a common life-shortening, autosomal recessive disease caused by mutations in the gene encoding the CFTR anion channel (1). CFTR is expressed in epithelia of multiple organs and its loss causes airway, pancreatic, intestinal, liver, and vas deferens disease. The ΔF508 mutation (also called F508del), is the most common CF-causing mutation, accounting for ~70% of CF alleles; most patients carry at least one ΔF508 allele.
Numerous studies have expressed human CFTR-ΔF508 in vitro and found that its biosynthetic processing is disrupted; the mutant protein is retained in the endoplasmic reticulum (ER) and rapidly degraded (2–4). As a result, CFTR-ΔF508 fails to reach the apical membrane. CFTR-ΔF508 can be induced to traffic to the cell surface by reducing the incubation temperature or adding chemicals that facilitate folding, and once at the membrane, it retains channel function, although its lifetime and open state probability are reduced (5–9). These discoveries sparked an effort by academia and industry to therapeutically correct the CFTR-ΔF508 defects (10,11).
However, the conclusion that CFTR-ΔF508 biosynthesis is disrupted has relied largely on studies of recombinant protein (2–4). It has been much more difficult to study the endogenous protein because of limited human tissue availability, the small amount of CFTR in affected epithelia, and changes caused by inflammation and tissue remodeling of advanced CF. Studies of endogenous CFTR have sometimes reached conclusions that contrast strikingly with data from recombinant systems. For example, some reports indicated that CFTR-ΔF508 was processed and localized like wild-type CFTR (12,13). Other reports suggested that CFTR-ΔF508 reached the apical membrane, but in reduced amounts(14,15). Still other reports indicated that CFTR-ΔF508 was either not detectable or did not reach the apical membrane (16–18). In addition, although most studies did not detect Cl− channel function in freshly excised ΔF508/ΔF508 airway epithelial cells, others have identified residual Cl− transport (19,20). The reasons for these varying conclusions are uncertain, but obtaining the cells and tissues from airways with infection, inflammation and remodeling might have affected results (14).
Efforts to understand abnormalities produced by the ΔF508 mutation have been hindered by lack of an animal model that expresses CFTR-ΔF508 and manifests a typical CF phenotype. This limitation has also hindered attempts to target CFTR-ΔF508 therapeutically, and it has impeded efforts to understand pathogenesis. Unfortunately, mice with CFTR gene mutations, including ΔF508, do not develop airway disease typical of human CF (21). The newly developed CFTR−/− pig and ferret models may offer an opportunity to better understand disease pathogenesis (22,23). At birth, CFTR−/− pigs exhibit a phenotype like that in patients with CF, including pancreatic destruction, meconium ileus, early focal biliary cirrhosis, and microgallbladder (22,24). Like lungs from newborn humans with CF, lungs from newborn CFTR−/− pigs show no evidence of inflammation, but with time they spontaneously develop lung disease with the characteristic features of CF including inflammation, infection, mucus accumulation, tissue remodeling, and airway obstruction (25).
Encouraged by the phenotype of CFTR−/− pigs, we set out to generate CFTRΔF508/ΔF508 pigs. Our initial goal was to answer three key questions. First, would CFTRΔF508/ΔF508 pigs have the same or a different clinical phenotype as pigs with a complete lack of CFTR? Residual CFTR activity might ameliorate disease severity compared to CFTR−/− pigs, or alternatively, the presence of the mutant protein might worsen disease manifestations. Second, would porcine CFTR-ΔF508 be misprocessed in vivo? In earlier work, we found that although porcine CFTR-ΔF508 was largely misprocessed in vitro, a small fraction was correctly processed and delivered to the epithelial apical membrane where it generated some Cl− conductance (26). We wondered if this would also occur in vivo. The results would have important implications for studies of mechanism and for strategies to therapeutically target CFTR-ΔF508. Third, if CFTR-ΔF508 was partially processed in CFTRΔF508/ΔF508 pigs and retained some anion transport function, it would provide us with an opportunity to begin to answer an important question for therapeutic strategies, i.e., how much airway epithelial anion channel activity is sufficient to prevent CF lung disease?
RESULTS
We generated CFTRΔF508/ΔF508 pigs
We previously generated male CFTRΔF508/+ pigs using somatic cell nuclear transfer and embryo transfer (27). The nucleotide sequence …ATC-TTT-GGT…, which encodes …I507-F508-G509…, is identical in exon 10 of porcine and human CFTR. To reproduce the human mutation, we deleted C-TT (i.e., theΔF508 allele) to generate …ATT-GGT.., which encodes …I507-G509…. In addition, the intron downstream of exon 10 contains a NeoR cassette.
We crossed CFTRΔF508/+ males, which had a normal clinical phenotype, to wild-type females to generate CFTRΔF508/+ female pigs. We then crossed CFTRΔF508/+ males and females to generate CFTRΔF508/ΔF508 pigs. The ratio of CFTRΔF508/ΔF508:CFTRΔF508/+:CFTR+/+ pigs, 82:129:63, did not differ statistically (chi-squared test) from the predicted Mendelian ratio of 1:2:1.
Newborn CFTRΔF508/ΔF508 pigs have pathology like that of CFTR−/− pigs
Like CFTR−/− and CFTRΔF508/− piglets (24,25), CFTRΔF508/ΔF508 pigs had meconium ileus with 100% penetrance. The site of obstruction varied, but was generally near the ileocecal junction (Fig. 1A). Distal to the obstruction, the intestine was of small caliber and variably filled with mucocellular debris (Fig. 1B). We did not discern differences between the intestinal pathology or meconium ileus of CFTRΔF508/ΔF508 and CFTR−/− pigs.
Figure 1. Pathology of newborn CFTRΔF508/ΔF508 pigs.

A. Location (in cm) of meconium ileus obstruction in CFTRΔF508/ΔF508 (n=10) and CFTR−/− (n=9) pigs.
B. CFTRΔF508/ΔF508ileum distal to the obstruction had a small caliber and was heterogeneously filled with mucocellular debris (arrows). Ileal Peyer’s patches (asterisks) appeared similar in CFTR+/+ and CFTRΔF508/ΔF508 pigs. Bars=725 top and 145 μm bottom.
C. Pancreas from CFTRΔF508/ΔF508 pigs had increased connective tissue (asterisks) and destruction compared to CFTR+/+. Histopathological changes in CFTRΔF508/ΔF508 pancreas were slightly less severe than in CFTR−/−. HE stain. Bar=457 μm.
D. Lobular parenchyma in CFTRΔF508/ΔF508 pigs (n=17) was reduced compared to CFTR+/+ (n=9, * P<0.001) and greater than in CFTR−/− (n=19, # P<0.05, Dunn’s post-test). Data from CFTR+/+ and CFTR−/− pigs were previously published (24).
E. Liver from newborn CFTRΔF508/ΔF508and CFTR−/− pigs showed portal areas that were focally expanded (arrows) by inflammation, duct proliferation and connective tissue. HE stain. Bar=46 μm.
F. CFTRΔF508/ΔF508 pigs had microgallbladder variably filled by mucus and bile. HE stain, bars=928 μm (+/+) and 463 μm (ΔF508/ΔF508).
G. Lung from newborn CFTRΔF508/ΔF508 pigs lacked mucus accumulation or inflammatory changes. HE stain. Bar=93 μm.
H–J. Bronchoalveolar lavage liquid analyses from newborn pigs, including total cell counts (H), neutrophil percentages (I), and IL-8 concentrations (J) revealed no statistically significant differences between genotypes. CFTR+/+ (n=5) combined with CFTR+/ΔF508 (n=4); CFTRΔF508/ΔF508 (n=11).
In the pancreas, lobular parenchyma was decreased in CFTRΔF508/ΔF508 pigs (Fig. 1C). Pancreatic acini and ducts were often dilated by zymogen concretions with scattered neutrophils, macrophages and mucus, like that found in newborn CFTR−/− pigs (24). CFTRΔF508/ΔF508 pancreata had reduced parenchyma compared to CFTR+/+, but the destruction was slightly less severe than in CFTR−/− (Fig. 1D).
The CFTRΔF508/ΔF508 liver had focal portal areas expanded by bile duct proliferation, inflammation, and/or increased connective tissue, changes characteristic of early focal biliary cirrhosis (Fig. 1E). The frequency and severity of changes were similar to those we observed in CFTR−/− pigs (24). Likewise, the microgallbladder and mucinous changes in gallbladder epithelia observed in CFTR−/− pigs were ubiquitous in CFTRΔF508/ΔF508 animals (Fig. 1F).
Airway epithelia of newborn CFTRΔF508/ΔF508 pigs were normal in appearance and lacked evidence of mucus accumulation. Like CFTR−/− pigs, on histopathological examination CFTRΔF508/ΔF508 airways lacked inflammatory cells (Fig. 1G) (25). The alveolar and airway epithelia were indistinguishable in CFTR+/+, CFTRΔF508/ΔF508, and CFTR−/− pigs. Analysis of bronchoalveolar lavage of newborn pigs revealed no statistically significant differences in total cell counts, differential cell counts, or IL-8 concentrations between CFTR+/+ and CFTRΔF508/ΔF508pigs (Fig. 1H–J).
Tracheal abnormalities occur in humans with CF and CFTR−/− pigs and mice (28,29). Compared to wild-type trachea, CFTRΔF508/ΔF508 trachea had an altered lumen area, circumference, submucosal gland area, and smooth muscle area (Fig. 2A–2E). However, changes in smooth muscle area were not as severe as those in CFTR−/− pigs.
Figure 2. Morphometry of newborn CFTRΔF508/ΔF508 trachea.

A. Cross section of trachea. MT stain. Bars=1 mm. Images from CFTR+/+ and CFTR−/− are from reference (29).
B–E. Tracheal morphometry in CFTR+/+ (n=20), CFTRΔF508/ΔF508 (n=19), and CFTR−/− (n=18) newborn pigs. * indicates different from CFTR+/+ and # indicates different from CFTR−/− (* P<0.05 vs. CFTR+/+, and # P<0.05 vs. CFTR−/−, 1-way ANOVA with Bonferroni’s post test).
B. Tracheal lumen cross-sectional area.
C. Tracheal circumference.
D. Submucosal gland area normalized to tracheal lumen circumference.
E. Smooth muscle area normalized to tracheal lumen area.
Thus, newborn CFTRΔF508/ΔF508 pigs are remarkably similar to their CFTR−/− counterparts with the exception of slightly less severe abnormalities in pancreas and tracheal smooth muscle.
CFTRΔF508/ΔF508 lungs develop disease with time
Meconium ileus would prevent survival of all CFTRΔF508/ΔF508 pigs. Therefore, to learn whether disease in CFTRΔF508/ΔF508 pigs would progress after birth, surgical intervention was necessary to bypass the intestinal obstruction. Therefore, we placed an ileostomy or cecostomy in the pigs within 15 hr after birth. The procedures and treatments were the same as we previously described for CFTR−/−pigs (25).
We examined the histopathology of four CFTRΔF508/ΔF508 pigs ranging in age from 13 to 87 days at time of euthanasia (Table 1). Over time, CFTRΔF508/ΔF508 pigs lost pancreatic parenchyma, which was replaced with fatty and fibrous tissue (Fig. 3A). In the liver, changes varied from minimal to diffuse steatosis (Fig. 3B,C). One animal (Case 2) had portal areas with focal to bridging fibrosis, duct proliferation and inflammation, the changes typical of progressive focal biliary cirrhosis (24) (Fig. 3D).
TABLE 1.
CFTRΔF508/ΔF508 pigs two or more weeks old.
| Case | Sex | Genotype | Type of Surgery | Age | Reason for Euthanasia |
|---|---|---|---|---|---|
| 1 | M | ΔF508/ΔF508 | loop ileostomy | 13 d | prolapsed ostomy |
| 2 | F | ΔF508/ΔF508 | cecostomy | 62 d | gastric ulcer* |
| 3 | F | ΔF508/ΔF508 | cecostomy | 77 d | poor oral intake, weight loss ** |
| 4 | F | ΔF508/ΔF508 | cecostomy | 87 d | prolapsed ostomy |
Gastric ulcer has previously been reported to occur in both non-CF and CF pigs (25).
Etiology of poor oral intake and weight loss was not identified. Pig was hypothermic prior to euthanasia and on post-mortem examination the stomach and proximal small intestine had a large quantity of luminal material suggestive of an ileus. Clinically, the animal appeared septic, but bacterial cultures were negative.
Figure 3. Disease progression in pigs ~2-weeks of age and older.

A. Pancreas from a 77-day old CFTRΔF508/ΔF508 pig and 69-day old CFTR+/+ pig for comparison. Islands of degenerative, fibrotic and inflamed CFTRΔF508/ΔF508 pancreas were surrounded by abundant adipose tissue (asterisk). HE stain. Bar=75 μm.
B–D. Porcine liver. MT stain. Bars=570 top and 57 μm bottom. B. Liver from a 136-day old CFTR+/+ pig. C. Diffuse zone 1 steatosis (black arrows) in a 77-day old CFTRΔF508/ΔF508 pig. D. A 62-day old CFTRΔF508/ΔF508 pig had focal to bridging expansion (black arrows) of triads by fibrosis, duct proliferation and inflammation.
E–M. Histopathological evaluation of CFTRΔF508/ΔF508 lungs. E–H are HE stain and I–M are PAS stain.
E. Lung from a 69-day old CFTR+/+ pig; changes like those in panels F–H were not observed in wild-type pigs.
F–H. Lung from 13-day old CFTRΔF508/ΔF508 pig.
F. Lungs showed mucopurulent airway obstruction (arrow) and adjacent atelectasis (asterisks). Bar=757 μm.
G. Affected airway lumens often contained a heterogeneous mixture of mucopurulent debris obstructing the airway (arrows) and adjacent atelectasis (asterisks). Bar = 378 μm.
H. Airways sometimes showed nominal inflammatory changes in the wall (asterisks) adjacent to luminal neutrophils (arrows) suggesting the dispersion of the luminal mucocellular debris from more severely affected airways. Bar=38 μm.
I–J. Lung from 87-day old CFTRΔF508/ΔF508 pig.
I. Some airways showed focal airway mucus obstruction (arrow). Bar=162 μm.
J. The surface epithelium showed focal goblet cell hyperplasia (black arrows) and inflammation in the airway wall around submucosal glands (arrowheads). Bar=81 μm. Insets: magnified PAS-stained images of airway epithelia of 4.5-mo CFTR+/+ (top) and 87-day CFTRΔF508/ΔF508 (bottom) pigs.
K–L. Lung from 62-day old CFTRΔF508/ΔF508 pig.
K. Lungs showed mucopurulent inflammation associated with focal dilated submucosal glands and ducts (arrow). Bar=40 μm.
L. Airway lumens showed mucopurulent material in lumen (arrow) with epithelial proliferation and wall inflammation. Bar=40 μm.
M. Lung from 77-day old pig showed lesions included complete lobular atelectasis (arrows), although in this image airway obstruction was not present. Bar=378 μm.
Like CFTR−/− and CFTRΔF508/− piglets (25), all the CFTRΔF508/ΔF508 pigs showed changes consistent with CF lung disease. Disease severity varied from animal to animal, and changes within lungs of individual pigs were heterogeneous such that some areas of lung showed no abnormality. As early as two weeks of age (Case 1), CFTRΔF508/ΔF508 lung showed mucopurulent material obstructing some airways with areas of adjacent atelectasis (Fig. 3E–H). In cases 2 and 4 (62 and 87 days old), lung changes included scattered mucopurulent debris in airway lumens with chronic purulent to lymphoid airway wall inflammation (Fig. 3I–L). The surface epithelium showed areas of goblet cell hyperplasia, and mucocellular material was detected in some submucosal glands. In case 3 (77 days old), the lungs showed a range of severity from nominal mucinous changes to lobular atelectasis consistent with airway obstruction (Fig. 3M).
At the time of necropsy, lung samples were aseptically removed for bacterial culture from three of the four animals. Bacteria were present in the cultures, but in relatively low numbers ranging from 10–1650 cfu/g lung tissue (Table 2). As in CFTR−/− pigs, a variety of bacterial species were isolated. This result suggests a host-defense defect for many bacterial species and is consistent with data from humans with early CF lung disease (25). In contrast, no bacteria were isolated from lungs of three of the four control pigs, and in the fourth only 10 cfu/g were cultured. In addition, CFTRΔF508/ΔF508 pigs, but not CFTR+/+ pigs, received some systemic antibiotics (Materials and Methods), which may have suppressed bacterial recovery and minimized differences between the two groups.
TABLE 2.
Microbiology of CFTRΔF508/Δ508 lung.
| Case # | Lung bacteria
|
Cultured species | ||
|---|---|---|---|---|
| (avg CFU/g) | (range) | |||
| ΔF508/ΔF508 | 1 | - | - | * |
| 2 | 10 | 10** | Coagulase-negative Staphylococcus spp. | |
| 3 | 115 | 0–230 | Acinetobacter lwoffii, Escherichia coli, Leclercia adecarboxylata | |
| 4 | 743 | 230–1650 | Alpha-hemolytic Streptococcus spp. (3 morphologies), coagulase-negative Staphylococcus spp. (3 morph), E. coli (2 morphologies), Enterococcus spp., Haemophilus spp., Pasteurella aerogenes, Salmonella spp. | |
| +/+ | 1 | 10 | 10 | Alpha-hemolytic Streptococcus spp., Diphtheroids |
| 2 | 0 | 0 | ||
| 3 | 0 | 0 | ||
| 4 | 0 | 0 | ||
samples were not collected for microbiology culture.
n = 1
These results indicate that CFTRΔF508/ΔF508 pigs spontaneously develop lung disease that resembles that in CFTR−/− pigs and humans homozygous for the ΔF508 mutation.
CFTRΔF508/ΔF508 pigs produce CFTRΔF508 mRNA
Because the phenotype of CFTRΔF508/ΔF508 pigs was like that of CFTR−/− pigs, we asked whether newborn CFTRΔF508/ΔF508 pigs produced CFTR transcripts. We assessed expression from the ΔF508 allele using quantitative RT-PCR. Amounts of wild-type CFTR mRNA decreased from proximal to distal intestine and were lower in cultured airway epithelia than in intestine (Fig. 4A). In CFTRΔF508/ΔF508 pig intestine, CFTR transcripts followed a similar axial pattern and did not statistically differ from those in CFTR+/+ pigs. Northern blots of CFTRΔF508/ΔF508 duodenum were consistent with the RT-PCR data (Fig. 4B). In cultured airway epithelia, CFTR+/+ and CFTRΔF508/ΔF508had the same abundance of CFTR transcripts. These data suggest that the NeoR cassette in intron 10 has relatively minor effects on transcription from the CFTRΔF508 allele. These results are also consistent with our earlier estimate that CFTRΔF508 mRNA was present at ~70% of the wild-type amount (27).
Figure 4. mRNA and protein expression in intestine and airway.

A. Quantitative RT-PCR of CFTR mRNA in CFTRΔF508/ΔF508 and CFTR+/+ pigs. Data are from triplicate assays repeated on multiple days. For each tissue, amounts of CFTR mRNA were normalized to β-actin mRNA. These normalized values were then expressed relative to that in wild-type duodenum. Data are mean±SE from intestinal tissues from 6 CFTR+/+ and 6 CFTRΔF508/ΔF508piglets, and from cultured nasal epithelia from 1 CFTR+/+ piglet (n=3) and 1 CFTRΔF508/ΔF508piglet (n=4).
B. Northern blot analysis of duodenal CFTR mRNA, indicated by arrow.
C. Immunoprecipitated and in vitro phosphorylated CFTR isolated from intestine. “rec” (lanes 1,8,13) indicates recombinant protein. Lanes 2–7, proximal intestine. CFTR+/+ and CFTR−/− 500 μg and CFTRΔF508/ΔF508 750 μg. Lanes marked with * show enhanced exposure. Lanes 6 and 7 are same as lanes 4 and 5. Lanes 9–12, distal intestine. CFTR+/+ 200 μg and CFTRΔF508/ΔF508 1000 μg. Lanes 11 and 12 are same as 9 and 10.
D. Immunoprecipitated and in vitro phosphorylated CFTR isolated from airway epithelia. Recombinant protein, lanes 1,6,7. Lanes 2–5, trachea; CFTR+/+ 623 μg and CFTRΔF508/ΔF508 1208 μg. Lanes 4 and 5 are same as lanes 2 and 3. Lanes 8–10, cultured bronchial epithelia; each lane 750 μg.
The amount of CFTR-ΔF508 is reduced compared to wild-type CFTR
Processing of CFTR can be assessed by its migration on an SDS gel; immature CFTR (band B) has undergone core glycosylation in the endoplasmic reticulum (ER) and mature CFTR (band C) has been fully glycosylated in the Golgi complex (2,3). When expressed in vitro, most wild-type human CFTR migrates as band C, although a substantial amount of immature band B protein is also present. In human CFTR-ΔF508, band C is generally undetectable, and the predominance of band B indicates ER retention (2–4,26). In our earlier studies of recombinant porcine CFTR, wild-type protein behaved like wild-type human CFTR, whereas some of the mutant porcine protein processed to band C (26).
In proximal small intestine from wild-type pigs, we detected band C and very little band B CFTR (Fig. 4C). This result suggests that most wild-type protein was processed to the mature form, consistent with maturation of endogenous wild-type human CFTR to band C (30). Thus, presence of band B in vitro may be due to overexpression of recombinant protein. The amount of CFTR recovered from proximal CFTRΔF508/ΔF508 intestine was markedly reduced compared to CFTR+/+ intestine, and we had to increase both the amount of protein studied and enhance the exposure to detect the mutant protein (Fig. 4C, lanes 2–7). Distal small intestine yielded similar results (Fig. 4C, lanes 9–12). In both cases, CFTR-ΔF508 was present in the mature band C and immature band B forms.
Because the intestine is affected by meconium ileus, we also assessed airway epithelia, which do not show secondary changes from the disease at birth. The data paralleled results from intestine. First, we detected little band B in either excised trachea (Fig. 4D, lanes 2–5) or differentiated cultures (lanes 8–10) of wild-type nasal epithelia; the preponderance of CFTR was in band C. Migration of band C protein was slightly slower than recombinant wild-type CFTR, suggesting some differences in glycosylation of CFTR in vivo compared to recombinant CFTR. Second, we detected both band B and band C forms of CFTR-ΔF508. Third, the amount of CFTR-ΔF508 protein was decreased compared to wild-type CFTR, although the reduction was less marked in cultured than excised epithelia.
These results agree with our earlier in vitro studies of recombinant porcine wild-type and ΔF508 CFTR (26). They suggest that porcine CFTR-ΔF508 has a biosynthetic defect. However, they also indicate that a fraction of the mutant protein is processed to the mature form.
Immunostaining reveals a reduced amount of CFTR-ΔF508
We used immunocytochemistry as an additional way to evaluate CFTR-ΔF508. In small intestine, we detected wild-type CFTR in the apical membrane of crypt, but not villus cells (Fig. 5A). In CFTRΔF508/ΔF508 intestine, we detected some immunostaining throughout the small intestine and the spiral colon. However, the signal was very weak and not uniformly detectable; the third panel of Fig. 5A shows an example in which we have electronically enhanced the CFTR (green) fluorescence so that the staining could be appreciated. CFTR−/− intestine had no immunostaining.
Figure 5.

Immunocytochemical localization of CFTR in intestinal and airway epithelia of newborn pigs. Data are stacks of confocal images, except as noted. Scale bars=10 μm.
A. Sections of intestine from newborn pigs. Third panel (asterisk) shows an electronically enhanced image of second panel. CFTR is green, ZO-1 is red, and nuclei are blue. Nonspecific staining was occasionally found in lumen of some CFTRΔF508/ΔF508and CFTR−/− crypts in areas of extensive mucus.
B. Sections of trachea.
C. Images of cultured airway epithelia. Top panels are enface images, and bottom panels are single vertical sections. Images from CFTRΔF508ΔF508 epithelia are electronically enhanced (*) to show CFTR. Cell size heterogeneity was observed with all genotypes.
In excised trachea and differentiated primary cultures of nasal epithelia, wild-type CFTR localized almost exclusively at the apical membrane (Fig. 5B,C). In CFTRΔF508/ΔF508 tracheal tissue and cultures, immunostaining was barely detectable. In Fig. 5B, we show a rare example from CFTRΔF508/ΔF508 trachea where we detected CFTR and found it localized similarly to that in CFTR+/+ trachea. For cultured CFTRΔF508/ΔF508 tracheal epithelia, we electronically amplified the signal post-collection to detect CFTR immunostaining that differed from that in wild-type cultured epithelia; staining extended from the apical membrane into the cytoplasm (Fig. 5C). We detected no CFTR immunostaining in excised or cultured CFTR−/− tracheal epithelia. The marked decrease in immunostaining in CFTRΔF508/ΔF508 intestinal crypts and airway epithelia is consistent with the greatly reduced amounts of CFTR recovered from these tissues. These data predicted that ion transport by CFTRΔF508/ΔF508 epithelia would be abnormal.
CFTRΔF508/ΔF508 airway epithelia show reduced but not absent CFTR Cl− transport
We designed electrolyte transport studies to answer two questions. First, does transepithelial ion transport in newborn CFTRΔF508/ΔF508 airway epithelia differ from that in wild-type epithelia? Second, is Cl− transport in CFTRΔF508/ΔF508 airway epithelia greater than that in CFTR−/− epithelia? We studied airway epithelia so that we could compare data to results from CFTR−/− pigs (31). We examined both nasal epithelia, which are often used to evaluate CF ion transport, and tracheal/bronchial epithelia because of their potential contribution to disease. We studied excised tissues as well as primary cultures of differentiated airway epithelia. We show data for excised tracheal epithelia in Fig. 6; the other data are in Fig. S1–S3.
Figure 6.
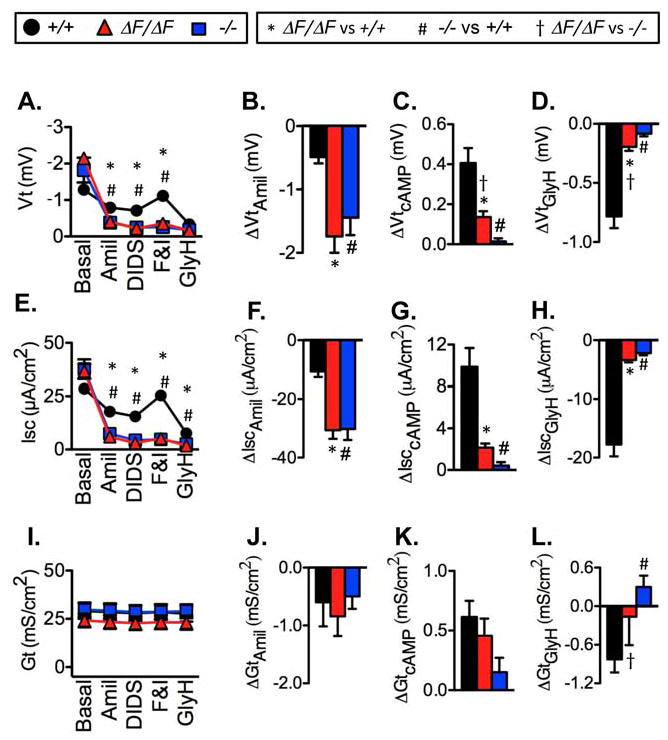
Electrophysiological properties of freshly excised porcine tracheal epithelia. Data are from CFTR+/+(23 tissues, 23 pigs), CFTRΔF508/ΔF508 (19 tissues, 17 pigs), and CFTR−/−(16 tissues, 14 pigs) epithelia. Data from CFTR−/− and most CFTR +/+ pigs were previously reported(31). * indicates CFTRΔF508/ΔF508 differs from CFTR+/+, # indicates CFTR−/− differs from CFTR +/+, and † indicates CFTRΔF508/ΔF508 differs from CFTR−/−, all at P<0.017 by unpaired t test with Welch’s correction.
A). Transepithelial voltage (Vt) and response to sequential apical addition of 100 μM amiloride, 100 μM DIDS, 10 μM forskolin and 100 μM IBMX, and 100 μM GlyH-101.
B). ΔVtamil indicates change in Vt with addition of amiloride.
C). ΔVtcAMP indicates change in Vt with addition of forskolin and IBMX.
D). ΔVtGlyH indicates change in Vt with addition of GlyH-101.
E–H). Short-circuit current (Isc) measurements corresponding to Vt measurements in panels A–D.
I–L). Transepithelial conductance (Gt) measurements corresponding to Vt measurements in panels A–D. Changes in Vt, Isc, and Gt with DIDS were small and did not differ by genotype.
Basal Vt and Isc did not differ between excised trachea from CFTRΔF508/ΔF508 and CFTR+/+ pigs (Fig. 6). An inhibitor of epithelial Na+ channels (100 μM apical amiloride) reduced Vt (ΔVtamiloride) and Isc (ΔIscamiloride) in CFTRΔF508/ΔF508 more than in wild-type epithelia. Our earlier work indicates that the greater ΔVtamiloride and ΔIscamiloride in CF epithelia is due to reduced CFTR anion conductance rather than greater Na+ channel activity, and these data are consistent with that earlier study (31). Values of transepithelial electrical conductance (Gt) were large, probably because of “edge damage” associated with clamping epithelia in Ussing chambers (32) (compare with cultured epithelia in Fig. S2–S3). Amiloride reduced Gt (ΔGtamiloride) to a similar extent in CFTRΔF508/ΔF508 and CFTR+/+ epithelia.
In nasal epithelia, compared to wild-type, CFTRΔF508/ΔF508 epithelia had a greater basal Vt and ΔVtamiloride in culture and a greater basal Isc and ΔIscamiloride in excised epithelia (Fig. S1 and S2). These differences between CF and non-CF epithelia at the two locations are the result of differences between basal CFTR Cl− channel activity and other epithelial properties rather than differences in rates of Na+ transport (31). ΔGtamiloride in CFTRΔF508/ΔF508 nasal epithelia was less than or the same as that in wild-type epithelia consistent with the conclusion that Na+ conductance is not greater in CF than non-CF epithelia (31). In excised and cultured nasal and tracheal/bronchial epithelia, these electrophysiological properties in CFTRΔF508/ΔF508 epithelia (Fig. 6, S1–S3) were approximately the same as those in CFTR−/− epithelia.
To assess CFTR function, we added 4,4′-diisothiocyanostilbene-2,2′-disulfonic acid (DIDS) to block non-CFTR Cl− channels, followed by forskolin and IBMX to increase cellular concentrations of cAMP and phosphorylate and activate CFTR (33). In both tracheal/bronchial and nasal epithelia and in both excised tissue and cultured epithelia, the forskolin and IBMX- induced changes in Vt (ΔVtcAMP), Isc (ΔIsccAMP), and Gt (ΔGtcAMP) were markedly reduced in CFTRΔF508/ΔF508 compared to CFTR+/+ epithelia (Fig. 6, S1–S3). Interestingly, for most of the electrophysiological measurements, there was either a statistically significant difference or a non-significant trend for CFTRΔF508/ΔF508 epithelia to show more cAMP-stimulated Cl− conductance and/or Cl− transport than CFTR−/− epithelia. As an additional way of assessing CFTR-mediated Cl− transport, after adding forskolin and IBMX, we applied GlyH-101, which inhibits CFTR Cl− channels (34). The results paralleled what we found with cAMP-dependent stimulation; the response was markedly attenuated in CFTRΔF508/ΔF508 compared to wild-type epithelia, but often greater than in CFTR−/− epithelia.
In addition to Cl−, CFTR also transports HCO3− (35,36), and it has been proposed that defective HCO3− transport may be critical for CF pathogenesis (37). Therefore, we also examined changes in Isc and Gt when tracheal epithelia were bathed in a Cl−-free HCO3− solution. Like the reduction in Cl− conductance, HCO3−conductance was markedly reduced in CFTRΔF508/ΔF508 trachea (Fig. 7A,B).
Figure 7. HCO3− transport, apical Cl− currents, and effect of increasing cAMP-dependent stimulation.

A–B. Changes in Isc and Gt in tracheal epithelia bathed in Cl−-free solution containing 25 mM HCO3−. Change in Isc (ΔIsc) (A) and Gt (ΔGt) (B). stimulated by forskolin (10 μM) and IBMX (100 μM) (F&I) and inhibited by GlyH-101 (100 μM, apical). * indicates P < 0.05, unpaired t- test. N=7 CFTR+/+and 7 CFTRΔF508/ΔF508.
C–E Changes in Cl− current after permeabilization of basolateral membrane. N=7 CFTR+/+and 7 CFTRΔF508/ΔF508.
C). Current traces in response to indicated agents in CFTR+/+, CFTRΔF508/ΔF508, and CFTR−/− epithelia. Concentrations are those indicated in Fig. 6 legend; nystatin was 0.36 mg/ml.
D). Change in current in response to nystatin plus forskolin and IBMX (ΔINystatin+F&I).
E). Change in current in response to GlyH-101 (ΔIGlyH).
F). Examples of Isc current traces following addition of increasing forskolin and IBMX concentrations. For concentrations, see panel G.
G). Changes in Isc with increasing forskolin and IBMX concentrations. N=7 CFTR+/+and 6 CFTRΔF508/ΔF508.
H). Examples of Isc current traces following addition of increasing 8-cpt-cAMP concentrations. For concentrations, see panel I.
I). Changes in Isc with increasing 8-cpt-cAMP concentrations. N=6 CFTR+/+and 7 CFTRΔF508/ΔF508.
Thus, the ΔF508 allele greatly decreased both Cl− and HCO3− conductances, consistent with a substantial loss of CFTR. However, compared to CFTR−/−, CFTRΔF508/ΔF508 epithelia retained some apical CFTR function.
CFTRΔF508/ΔF508 epithelia have residual CFTR function
The finding that CFTRΔF508ΔF508 pigs develop lung disease and yet have some CFTR anion conductance provided us with an opportunity to begin to address the question of how much CFTR function is sufficient to prevent lung disease. As one assessment of residual CFTR function, we compared the forskolin and IBMX-induced increases in Isc (ΔIsccAMP) and Gt (ΔGtcAMP) in the presence of amiloride and under short-circuit conditions (i.e., Vt clamped to zero and symmetrical solutions) (Fig. 6 and S1–3). In CFTRΔF508/ΔF508 excised and cultured nasal and tracheal/bronchial epithelia, the ΔIsccAMP was 9–15% of wild-type values (Table 3). Edge damage effects prevented accurate assessments of ΔGtcAMP in excised epithelia, but in cultured epithelia, the CFTRΔF508/ΔF508 ΔGtcAMP was 6–16% of CFTR+/+ values. Although GlyH-101 can have effects in addition to inhibiting CFTR (38,39), we also calculated GlyH-101-induced changes (ΔIscGlyH and ΔGtGlyH) and found that they varied from 3–32% in CFTRΔF508/ΔF508 epithelia compared to CFTR+/+ epithelia. The mean of all these ΔIsc and ΔGt measurements was ~12% of values in CFTR+/+ epithelia.
Table 3.
Changes in current and conductance in CFTRΔF508/ΔF508 epithelia as a percentage of changes in CFTR+/+ epithelia. Data are changes in Isc in response to forskolin and IBMX and GlyH-101 (ΔIsccAMP, ΔIscGlyH), the corresponding changes in Gt (ΔGtcAMP, ΔGtGlyH), changes in apical current induced by adding basolateral nystatin and apical forskolin and IBMX (ΔINyst+cAMP) in the presence of a Cl− concentration gradient, and changes induced by the subsequent addition of GlyH-101 (ΔIGlyH). To correct for any changes in the absence of CFTR, we subtracted values obtained in CFTR−/− epithelia, and data were calculated from mean values of individual measurements as (CFTRΔF508/ΔF508− CFTR−/−)/(CFTR+/+−CFTR −/−).
|
CFTRΔF508/ΔF508(% CFTR+/+) |
||||
|---|---|---|---|---|
| Excised nasal | Excised tracheal | Cultured nasal | Cultured tracheal | |
| ΔIsccAMP | 13.7 | 9.1 | 15.1 | 12.4 |
| ΔGtcAMP | 15.7 | 5.6 | ||
| ΔIscGlyH | 3.2 | 9.1 | 31.6 | 16.5 |
| ΔGtGlyH | 13.1 | 4.2 | ||
| ΔINyst+cAMP | 7.7 | 7.0 | ||
| ΔIGlyH | 8.3 | 2.0 | ||
In addition to apical CFTR conductance, Isc and Gt are affected by other apical ion channels and basolateral membrane transport, and CFTR may be partially active before cAMP elevation. In addition, the relationship between CFTR conductance and Isc is not linear, and the percentage increase in Isc overestimates the amount of CFTR function (40,41). Therefore, we imposed a transepithelial Cl− concentration gradient and measured the current response to basolateral membrane permeabilization with nystatin and addition of forskolin and IBMX (Fig. 7C,D). CFTRΔF508/ΔF508 nasal and tracheal epithelia generated 7–8% as much current as wild-type controls (Table 3). Subsequent addition of GlyH-101 produced a current change in CFTRΔF508/ΔF508 epithelia that was 2–8% of that in CFTR+/+ epithelia (Fig. 7C,E). The mean of the changes in current was ~6% of values in CFTR+/+ epithelia (Table 3).
These studies were done under conditions of maximal CFTR stimulation. Therefore, we also examined the response to increasing concentrations of forskolin and IBMX (at a fixed ratio of forskolin:IBMX of 1:10) (Fig. 7F,G). The EC50 for CFTR+/+ epithelia was 0.07±0.01 μM forskolin. In contrast, the EC50 for forskolin in CFTRΔF508/ΔF508 epithelia was 0.60±0.19 μM.
Because forskolin and IBMX might generate different cellular cAMP concentrations in CFTRΔF508/ΔF508 and CFTR+/+ epithelia, we repeated the experiments with 8-CPT-cAMP, a membrane-permeable cAMP analog (Fig. 7H,I). The results were similar in that the EC50 in wild-type epithelia was 8.0±1.3 μM and in CFTRΔF508/ΔF508 it was 65.2±17.3 μM.
These results suggest that CFTRΔF508/ΔF508 epithelia have a reduced sensitivity to cAMP-dependent stimulation of Cl− transport.
DISCUSSION
CFTRΔF508/ΔF508 pigs are born with gastrointestinal disease and spontaneously develop airway disease
Our earlier work showed that pigs lacking CFTR develop disease that mimics that in humans with CF (22,24,25,29). Here we found that pigs expressing the ΔF508 protein manifest a similar phenotype. Newborn CFTRΔF508/ΔF508 pigs exhibit CF gastrointestinal disease with meconium ileus, pancreatic destruction, early evidence of focal biliary cirrhosis, and microgallbladder. At birth, the lungs showed no evidence of inflammation. However, within weeks of birth, they developed lung disease like that in patients with CF.
In vitro studies of CFTR-ΔF508 predict in vivo function
Studies of human tissues expressing CFTR-ΔF508 have yielded varying and conflicting results about the behavior of CFTR-ΔF508 (12–20); those data have raised questions about whether the in vitro behavior of CFTR-ΔF508 predicts its behavior in vivo. Our earlier in vitro studies showed that although most porcine CFTR-ΔF508 is misprocessed and degraded, a fraction of the mutant protein matures and reaches the apical membrane where it generates a small Cl− conductance (26). Our data in CFTRΔF508/ΔF508 pigs are consistent with those earlier studies. The total amount of CFTR was markedly reduced in CFTRΔF508/ΔF508 epithelia, consistent with substantial degradation (2–4). However, similar to in vitro, some CFTR-ΔF508 was processed to the mature form and trafficked to the apical membrane. As a result, CFTRΔF508/ΔF508 epithelia retained a small Cl− conductance. These data indicate that in vitro studies of recombinant wild-type and ΔF508 porcine CFTR predict the behavior of the protein in vivo, a result important for biochemical experiments and drug discovery efforts.
Disease in CFTRΔF508/ΔF508 pigs is likely due to loss of CFTR function rather than misprocessing of the mutant protein
The autosomal recessive genetics of CF suggested that loss of CFTR function causes the disease. However, misfolded CFTR-ΔF508 has also been proposed to cause deleterious effects. Indeed, some in vitro studies showed that recombinant CFTR-ΔF508 could cause endoplasmic reticulum stress and an unfolded protein response, although these effects might have been due in part to over-expression (42,43).
Thus, we might have expected a more severe phenotype in CFTRΔF508/ΔF508 than CFTR−/− pigs if expression of CFTR-ΔF508 had an adverse effect. That was not the case. Although we did not test for endoplasmic reticulum stress, our data suggest that if it occurs in CFTRΔF508/ΔF508 pigs, it is not likely to markedly worsen the disease. However, we also note that our studies were done on newborn pigs, and it would be interesting to test for an unfolded protein response in older animals.
Residual CFTR function in CFTRΔF508/ΔF508 pigs attenuates pancreatic and tracheal abnormalities
Our biochemical, immunostaining, and electrophysiological studies of excised and cultured epithelia all indicate that a fraction of porcine CFTR-ΔF508 escapes misprocessing and retains some Cl− channel function. Although this residual CFTR activity was not sufficient to prevent disease, it did impact severity in the pancreas and trachea. Pancreas is an organ that shows a strong correlation between CFTR genotype and phenotype in humans (1,44), and interestingly, pancreatic destruction was slightly less severe in CFTRΔF508/ΔF508 than CFTR−/− pigs. In addition, the changes in tracheal smooth muscle were less marked in CFTRΔF508/ΔF508 than CFTR−/− pigs. In contrast, we did not discern a milder or more severe phenotype in the intestine, liver, gallbladder, or lung of the CFTRΔF508/ΔF508 compared to CFTR−/− animals. However, heterogeneity of disease within those individual organs, variations in severity between animals, and difficulty in quantitating severity could have obscured minor differences.
Residual CFTR function in CFTRΔF508/ΔF508 pigs is not sufficient to prevent lung disease
Discovery that CFTR-ΔF508 is partially processed and functional in CFTRΔF508/ΔF508 pigs allowed us to begin to address a persistent question; how much CFTR activity is required to maintain a normal lung? Our results point to two relevant factors.
First, measuring cAMP-stimulated elevations and GlyH-101-induced inhibitions of Gt and Isc indicated that CFTRΔF508/ΔF508 epithelia generated ~12% as much conductance and current as wild-type epithelia. Measurements of CFTR Cl− current after basolateral membrane permeabilization suggested that CFTRΔF508/ΔF508 epithelia had ~6% as much current as wild-type epithelia. These analyses have several advantages. We were able to compare electrophysiological data to those in CFTR−/− pigs, thereby identifying CFTR-specific properties. By studying newborns, we could eliminate confounding effects of infection, inflammation and remodeling. We also made measurements in both excised and cultured epithelia, and in both nasal and tracheal epithelia.
Second, our data revealed that cAMP-dependent stimulation was less potent at increasing Isc in CFTRΔF508/ΔF508 than CFTR+/+ epithelia. Thus, under physiological conditions, where stimuli are unlikely to be maximal, the relative activity of CFTR-ΔF508 may be substantially less than ~6% of apical CFTR conductance or ~12% of Isc and Gt that we calculated for maximal stimulation. Hence, estimates based solely on studies with maximal stimulation may overestimate actual activity under more physiological conditions. That said, it is currently not possible to know where “physiological” conditions lie on the dose-response curve. The mechanisms responsible for the altered relationship between stimulation and CFTR activity are also unknown. However in Xenopus oocytes, CFTR-ΔF508 was ~10-fold less potently stimulated by cAMP elevation than wild-type CFTR (45). In addition, studies of CFTR-ΔF508 in membrane patches have reported either a slower apparent PKA-dependent activation rate or an accelerated inactivation rate (46,47). Additional studies seem warranted to investigate these phenomena.
The quantitative relationship between CFTR-mediated anion transport and lung disease has been difficult to elucidate without a model that develops typical pulmonary disease. The fact that humans who are heterozygotes for CFTR gene mutations do not have disease suggests that ~50% of CFTR mRNA is sufficient to prevent airway disease. A study using nested RT-PCR identified four individuals who did not have CF-like lung disease and had an estimated 8–27% of wild-type CFTR mRNA (48). In addition, it has been suggested that 20% of wild-type CFTR Cl− current rescues the intestinal phenotype of CFTR-null mice (49). Other studies found that ~20% of wild-type cells or CF epithelia containing ~10% of CFTR overexpressing cells generated 70–80% of the Cl− transport of wild-type airway epithelia, although the relationship to clinical disease could not be tested (40,41). Our studies set a value above which anion transport will be required to rescue the pulmonary phenotype in CF pigs; how much higher that value is, we do not know. However, some caveats are worth considering. These estimates are derived from CFTRΔF508/ΔF508 pigs and not humans with a ΔF508 allele; lung disease might be more or less severe in pigs than humans. In addition, modifier genes and Ca2+-activated Cl− channels might differ between pigs and humans. Environmental factors will also differ between the species; humans are exposed to respiratory viral infections, whereas such exposures are minimal for pigs in our animal care unit. Nevertheless, the striking similar lung disease between the two species suggests that similar pathogenic mechanisms are responsible.
In conclusion, our data indicate that CFTR-ΔF508 is misprocessed in pigs, and this produces a clinical phenotype that is strikingly similar to that in CFTR−/− pigs and humans with CF. These animals might be beneficial for investigating mechanisms of CFTR-ΔF508 biosynthesis in vivo, for understanding pathogenesis, and for assessing therapeutics to prevent lung disease. Finally, our results suggest strategies to further determine the amount of CFTR function required to prevent disease.
Supplementary Material
1). Materials and Methods.
2). Figure S1. Electrophysiological properties of freshly excised porcine nasal epithelia.
3). Figure S2. Electrophysiological properties of differentiated primary cultures of porcine nasal epithelia.
4). Figure S3. Electrophysiological properties of differentiated primary cultures of porcine tracheal epithelia.
Acknowledgments
We thank Pamela Hughes, Ping Tan, Theresa Mayhew, and Paul Naumann for excellent assistance. We thank Dr. Kathryn Chaloner for statistical advice. GlyH-101 was a generous gift from the Cystic Fibrosis Foundation Therapeutics and R. Bridges. This work was supported by the NIH (HL51670, HL091842, DK54759) and Cystic Fibrosis Foundation. DAS was supported by AI076671. MJW is an Investigator of the HHMI. MJW was a co-founder of Exemplar Genetics, a company licensing materials and technology related to this work.
References
- 1.Welsh MJ, Ramsey BW, Accurso F, Cutting GR. Cystic Fibrosis. In: Scriver CR, et al., editors. The Metabolic and Molecular Basis of Inherited Disease. McGraw-Hill; New York: 2001. pp. 5121–5189. [Google Scholar]
- 2.Cheng SH, et al. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell. 1990;63:827–834. doi: 10.1016/0092-8674(90)90148-8. [DOI] [PubMed] [Google Scholar]
- 3.Ward CL, Kopito RR. Intracellular turnover of cystic fibrosis transmembrane conductance regulator. Inefficient processing and rapid degradation of wild-type and mutant proteins. J Biol Chem. 1994;269(41):25710–25718. [PubMed] [Google Scholar]
- 4.Younger JM, et al. Sequential quality-control checkpoints triage misfolded cystic fibrosis transmembrane conductance regulator. Cell. 2006;126(3):571–582. doi: 10.1016/j.cell.2006.06.041. [DOI] [PubMed] [Google Scholar]
- 5.Denning GM, et al. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature. 1992;358:761–764. doi: 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]
- 6.Haws CM, et al. ΔF508-CFTR channels: kinetics, activation by forskolin, and potention by xanthines. Am J Physiol. 1996;270:C1544–C1555. doi: 10.1152/ajpcell.1996.270.5.C1544. [DOI] [PubMed] [Google Scholar]
- 7.Cholon DM, O’Neal WK, Randell SH, Riordan JR, Gentzsch M. Modulation of endocytic trafficking and apical stability of CFTR in primary human airway epithelial cultures. Am J Physiol Lung Cell Mol Physiol. 2010;298(3):L304–314. doi: 10.1152/ajplung.00016.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jurkuvenaite A, et al. Functional stability of rescued delta F508 cystic fibrosis transmembrane conductance regulator in airway epithelial cells. Am J Respir Cell Mol Biol. 2010;42(3):363–372. doi: 10.1165/rcmb.2008-0434OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Okiyoneda T, et al. Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science. 2010;329(5993):805–810. doi: 10.1126/science.1191542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pedemonte N, et al. Small-molecule correctors of defective DeltaF508-CFTR cellular processing identified by high-throughput screening. J Clin Invest. 2005;115(9):2564–2571. doi: 10.1172/JCI24898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Goor F, et al. Rescue of ΔF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am J Physiol Lung Cell Mol Physiol. 2006;290(6):L1117–11130. doi: 10.1152/ajplung.00169.2005. [DOI] [PubMed] [Google Scholar]
- 12.Kalin N, Claaß A, Sommer M, Puchelle E, Tummler B. ΔF508 CFTR protein expression in tissues from patients with cystic fibrosis. J Clin Invest. 1999;103(10):1379–1389. doi: 10.1172/JCI5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Barneveld A, et al. Functional analysis of F508del CFTR in native human colon. Biochim Biophys Acta. 2010;1802:1062–1069. doi: 10.1016/j.bbadis.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 14.Dupuit F, et al. CFTR and differentiation markers expression in non-CF and ΔF508 homozygous CF nasal epithelium. J Clin Invest. 1995;96:1601–1611. doi: 10.1172/JCI118199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Penque D, et al. Cystic fibrosis F508del patients have apically localized CFTR in a reduced number of airway cells. Lab Invest. 2000;80(6):857–868. doi: 10.1038/labinvest.3780090. [DOI] [PubMed] [Google Scholar]
- 16.Kartner N, Augustinas O, Jensen TJ, Naismith AL, Riordan JR. Mislocalization of ΔF508 CFTR in cystic fibrosis sweat gland. Nat Genet. 1992;1:321–327. doi: 10.1038/ng0892-321. [DOI] [PubMed] [Google Scholar]
- 17.Engelhardt JF, et al. Submucosal glands are the predominant site of CFTR expression in the human bronchus. Nat Genet. 1992;2:240–248. doi: 10.1038/ng1192-240. [DOI] [PubMed] [Google Scholar]
- 18.Kreda SM, et al. Characterization of wild-type and deltaF508 cystic fibrosis transmembrane regulator in human respiratory epithelia. Mol Biol Cell. 2005;16(5):2154–2167. doi: 10.1091/mbc.E04-11-1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sermet-Gaudelus I, et al. Normal function of the cystic fibrosis conductance regulator protein can be associated with homozygous (Delta)F508 mutation. Pediatr Res. 2002;52(5):628–635. doi: 10.1203/00006450-200211000-00005. [DOI] [PubMed] [Google Scholar]
- 20.Ho LP, et al. Correlation between nasal potential difference measurements, genotype and clinical condition in patients with cystic fibrosis. Eur Respir J. 1997;10(9):2018–2022. doi: 10.1183/09031936.97.10092018. [DOI] [PubMed] [Google Scholar]
- 21.Grubb BR, Boucher RC. Pathophysiology of gene-targeted mouse models for cystic fibrosis. Physiol Rev. 1999;79(Suppl 1):S193–S214. doi: 10.1152/physrev.1999.79.1.S193. [DOI] [PubMed] [Google Scholar]
- 22.Rogers CS, et al. Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science. 2008;321(5897):1837–1841. doi: 10.1126/science.1163600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun X, et al. Disease phenotype of a ferret CFTR-knockout model of cystic fibrosis. J Clin Invest. 2010;120(9):3149–3160. doi: 10.1172/JCI43052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meyerholz DK, Stoltz DA, Pezzulo AA, Welsh MJ. Pathology of gastrointestinal organs in a porcine model of cystic fibrosis. Am J Pathol. 2010;176(3):1377–1389. doi: 10.2353/ajpath.2010.090849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stoltz DA, et al. Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Science Translational Medicine. 2010;2(29):29ra31 . doi: 10.1126/scitranslmed.3000928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ostedgaard LS, et al. Processing and function of CFTR-ΔF508 are species-dependent. Proc Natl Acad Sci U S A. 2007;104(39):15370–15375. doi: 10.1073/pnas.0706974104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rogers CS, et al. Production of CFTR null and ΔF508 heterozygous pigs by AAV-mediated gene targeting and somatic cell nuclear transfer. J Clin Invest. 2008;118(4):1571–1577. doi: 10.1172/JCI34773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bonvin E, et al. Congenital tracheal malformation in cystic fibrosis transmembrane conductance regulator-deficient mice. J Physiol. 2008;586(13):3231–3243. doi: 10.1113/jphysiol.2008.150763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meyerholz DK, et al. Loss of CFTR function produces abnormalities in tracheal development in neonatal pigs and young children. Am J Respir Crit Care Med. 2010;182:1251–1261. doi: 10.1164/rccm.201004-0643OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Varga K, et al. Efficient intracellular processing of the endogenous cystic fibrosis transmembrane conductance regulator in epithelial cell lines. J Biol Chem. 2004;279(21):22578–22584. doi: 10.1074/jbc.M401522200. [DOI] [PubMed] [Google Scholar]
- 31.Chen JH, et al. Loss of anion transport without increased sodium absorption characterize newborn porcine cystic fibrosis airway epithelia. Cell. 2010;143:911–923. doi: 10.1016/j.cell.2010.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Helman SI, Miller DA. Edge damage effect on electrical measurements of frog skin. Am J Physiol Cell Physiol. 1973;225(4):972–977. doi: 10.1152/ajplegacy.1973.225.4.972. [DOI] [PubMed] [Google Scholar]
- 33.Sheppard DN, Welsh MJ. Structure and function of the CFTR Cl− channel. Physiol Rev. 1999;79(1):S23–S45. doi: 10.1152/physrev.1999.79.1.S23. [DOI] [PubMed] [Google Scholar]
- 34.Muanprasat C, et al. Discovery of glycine hydrazide pore-occluding CFTR inhibitors: mechanism, structure-activity analysis, and in vivo efficacy. J Gen Physiol. 2004;124(2):125–137. doi: 10.1085/jgp.200409059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Poulsen JH, Fischer H, Illek B, Machen TE. Bicarbonate conductance and pH regulatory capability of cystic fibrosis transmembrane conductance regulator. Proc Natl Acad Sci U S A. 1994;91(12):5340–5344. doi: 10.1073/pnas.91.12.5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith JJ, Welsh MJ. cAMP stimulates bicarbonate secretion across normal, but not cystic fibrosis airway epithelia. J Clin Invest. 1992;89:1148–1153. doi: 10.1172/JCI115696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quinton PM. Cystic fibrosis: impaired bicarbonate secretion and mucoviscidosis. Lancet. 2008;372(9636):415–417. doi: 10.1016/S0140-6736(08)61162-9. [DOI] [PubMed] [Google Scholar]
- 38.Kelly M, et al. Cystic fibrosis transmembrane regulator inhibitors CFTRinh-172 and GlyH-101 target mitochondrial functions, independently of chloride channel inhibition. J Pharmacol Exp Ther. 2010;333(1):60–69. doi: 10.1124/jpet.109.162032. [DOI] [PubMed] [Google Scholar]
- 39.Caputo A, et al. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science. 2008;322(5901):590–594. doi: 10.1126/science.1163518. [DOI] [PubMed] [Google Scholar]
- 40.Farmen SL, et al. Gene transfer of CFTR to airway epithelia: low levels of expression are sufficient to correct Cl− transport and overexpression can generate basolateral CFTR. Am J Physiol Lung Cell Mol Physiol. 2005;280:L1123–1130. doi: 10.1152/ajplung.00049.2005. [DOI] [PubMed] [Google Scholar]
- 41.Johnson LG, et al. Efficiency of gene transfer for restoration of normal airway epithelial function in cystic fibrosis. Nat Genet. 1992;2:21–25. doi: 10.1038/ng0992-21. [DOI] [PubMed] [Google Scholar]
- 42.Bartoszewski R, et al. Activation of the unfolded protein response by deltaF508 CFTR. Am J Respir Cell Mol Biol. 2008;39(4):448–457. doi: 10.1165/rcmb.2008-0065OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hybiske K, et al. Effects of cystic fibrosis transmembrane conductance regulator and DeltaF508CFTR on inflammatory response, ER stress, and Ca2+ of airway epithelia. Am J Physiol Lung Cell Mol Physiol. 2007;293(5):L1250–1260. doi: 10.1152/ajplung.00231.2007. [DOI] [PubMed] [Google Scholar]
- 44.Wilschanski M, Durie PR. Pathology of pancreatic and intestinal disorders in cystic fibrosis. J R Soc Med. 1998;91(Suppl 34):40–49. doi: 10.1177/014107689809134s07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Drumm ML, et al. Chloride conductance expressed by ΔF508 and other mutant CFTRs in Xenopus oocytes. Science. 1991;254:1797–1799. doi: 10.1126/science.1722350. [DOI] [PubMed] [Google Scholar]
- 46.Wang F, Zeltwanger S, Hu S, Hwang TC. Deletion of phenylalanine 508 causes attenuated phosphorylation-dependent activation of CFTR chloride channels. J Physiol. 2000;524(Pt 3):637–648. doi: 10.1111/j.1469-7793.2000.00637.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schultz BD, Frizzell RA, Bridges RJ. Rescue of dysfunctional ΔF508-CFTR chloride channel activity by IBMX. J Membr Biol. 1999;170(1):51–66. doi: 10.1007/s002329900537. [DOI] [PubMed] [Google Scholar]
- 48.Chu CS, Trapnell BC, Curristin SM, Cutting GR, Crystal RG. Extensive posttranscriptional deletion of the coding sequences for part of nucleotide-binding fold 1 in respiratory epithelial mRNA transcripts of the cystic fibrosis transmembrane conductance regulator gene is not associated with the clinical manifestations of cystic fibrosis. J Clin Invest. 1992;90:785–790. doi: 10.1172/JCI115952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dorin JR, et al. A demonstration using mouse models that successful gene therapy for cystic fibrosis requires only partial gene correction. Gene Ther. 1996;3(9):797–801. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1). Materials and Methods.
2). Figure S1. Electrophysiological properties of freshly excised porcine nasal epithelia.
3). Figure S2. Electrophysiological properties of differentiated primary cultures of porcine nasal epithelia.
4). Figure S3. Electrophysiological properties of differentiated primary cultures of porcine tracheal epithelia.