

Abstract
Structure-based drug design relies on static protein structures despite significant evidence for the need to include protein dynamics as a serious consideration. In practice, dynamic motions are neglected because they are not understood well enough to model – a situation resulting from a lack of explicit experimental examples of dynamic receptor-ligand complexes. Here, we report high-resolution details of pronounced ~1 ms timescale motions of a receptor-small molecule complex using a combination of NMR and X-ray crystallography. Large conformational dynamics in Escherichia coli dihydrofolate reductase are driven by internal switching motions of the drug-like, nanomolar-affinity inhibitor. Carr-Purcell-Meiboom-Gill relaxation dispersion experiments and NOEs revealed the crystal structure to contain critical elements of the high energy protein-ligand conformation. The availability of accurate, structurally resolved dynamics in a protein-ligand complex should serve as a valuable benchmark for modeling dynamics in other receptor-ligand complexes and prediction of binding affinities.
INTRODUCTION
High-resolution crystal structures have classically provided the information that drives structure-based drug design. However, such structures are static models and are not representative of the dynamic nature of proteins under physiological conditions in vitro or in vivo. Proteins undergo constant motions in solution (dynamics), and they can also flex their structures such that the time-averaged, ‘static’ coordinates change significantly (flexibility). Both complicate the process of structure-based drug design1,2 and hence are often ignored in the design of small molecule inhibitors.3 This is one of the main reasons why prediction of binding affinities (and efficacies) is fraught with inaccuracies and drug design is dominated by an empirical approach. Although computational methods are being developed to account for molecular dynamics in free energy calculations, dynamics can exist over a wide range of timescales, some of which are still inaccessible to those methods.4 We propose here that experimental determination of the dynamic properties of protein-small molecule complexes will speed the development of reliable methods to more accurately predict ligand binding affinities.
There are several ways in which knowledge of protein flexibility and/or dynamics can aid structure-based drug design, according to different views. Flexibility is most commonly acknowledged from multiple crystal structures of the same protein bound to different ligands, in which the protein adopts different conformations (‘induced fit’). This is now often viewed as reflecting the inherent flexibility in the absence of ligand (‘selected fit’). A priori knowledge of flexible residues (e.g., from crystal structures) can be used to model active site conformational changes that might occur, even in a homologous protein, on binding a given small molecule.1 Induced fit behavior is also seen from the ligand side: minor changes to ligand structure can drastically affect its mode of binding, resulting in different orientations in the binding site.2,4,5 The second view, orthogonal to induced and selected fit, recognizes that binding free energy is not restricted to arise only from non-covalent bonding within the binding site. For example, changes in the nature of the conformational ensemble can influence the overall entropy.6 Thus, the dynamics of the whole system, both the free and bound states (of protein and ligand), become important. Third, as there is often a relationship between dynamics and function, drugs may be developed to inhibit (or activate) functional dynamics, as opposed to acting directly on the binding site.7 This strategy figures prominently in the development of allosteric drugs.8,9 Finally, it has been proposed that dynamics play an important role in mediating drug resistance, as demonstrated in a recent study on the Bcr-Abl fusion kinase.10 In principle, accounting for dynamics should improve prediction accuracy of binding affinities. This is underscored by the recent finding that 85% of the proteins with deposited structures have 1-3 “flexible” residues within their ligand binding pockets,11 and that most ligand receptors show an increase of atomic mobility for some ligand binding site atoms.12
Given the large number of examples from crystallographic studies implicating conformational heterogeneity as an important consideration for small molecule design, it is surprising that relatively few studies have reported more direct characterizations of dynamics in complexes of small, drug-like molecules with their targets. It stands to reason that accurate information on target and small molecule flexibility in solution should be gained to lay a foundation for developing more sophisticated methods that incorporate dynamics into drug design. Here, we have identified a small molecule-target enzyme interaction that is inherently dynamic. The target, E. coli dihydrofolate reductase (DHFR), is a popular target for drug design against microbial infections, and the human enzyme is the target for cancer chemotherapy agent methotrexate.13 The bacterial enzyme bound to a quinazoline derivative is shown here to exhibit conformational dynamics, both in the enzyme and the small molecule. From NMR spectroscopy and X-ray crystallography, the compound was found to bind in an unorthodox orientation but switch internally to drive a dynamic conformational loop change in the protein. The two methods used jointly are highly complementary, and both are necessary to develop a full, accurate picture of this small molecule complex.
RESULTS
Compound 1 is a high-affinity, competitive inhibitor of DHFR
In studying a larger panel of ~10 DHFR inhibitors, 5-(4-chlorophenylthio)-quinazoline-2,4-diamine (compound 1, Figure 1) was identified as exhibiting interesting NMR line-broadening properties when bound in a ternary complex with DHFR and NADPH (referred to as E:NADPH:1).
Figure 1.
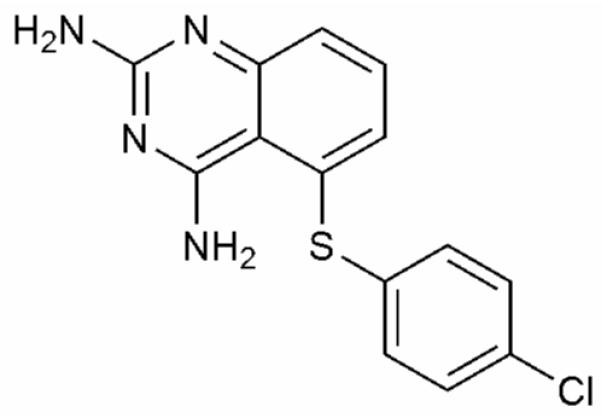
Structure of Compound 1 (5-(4-chlorophenylthio)-quinazoline-2,4-diamine).
The number and identity of sites experiencing line broadening differed greatly from that observed in the absence of 1 (E:NADPH).14 Based on this, we decided to carry out a full structural and dynamic characterization of this complex. Compound 1 was previously identified as a competitive inhibitor of E. coli DHFR from a high-throughput screen of 50,000 small molecules.15 Using a competition assay, 1 was confirmed to competitively inhibit DHFR with a Ki of 120 ± 9 nM.
Structural evidence of multiple conformations in E:NADPH:1
DHFR is one of the most thoroughly studied enzymes from both a structural and dynamic point of view.13,16-18 From these studies, it is known that the loops subdomain (Figure 2) is highly dynamic.
Figure 2.
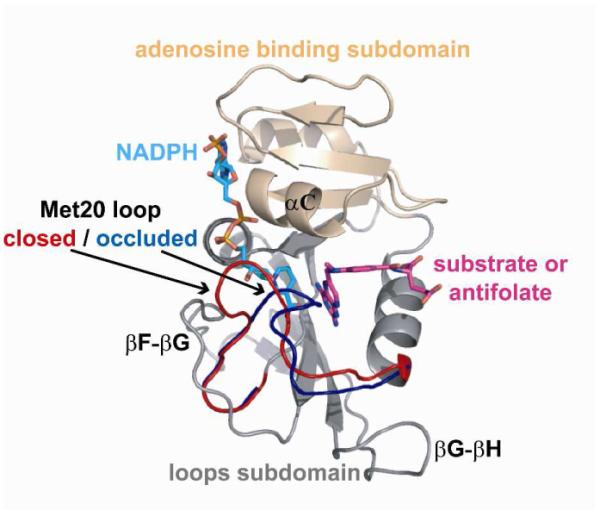
Structure of E. coli DHFR. Important subdomains, loops, and ligand binding sites are highlighted on a ternary complex of DHFR (PDB IDs 1RX3 and 1RX6 rendered using PyMOL.)
As DHFR progresses through its catalytic cycle, the enzyme undergoes a functionally important conformational change in its Met20 loop (residues 9-24) from the ‘closed’ state prior to hydride transfer, to the ‘occluded’ state following hydride transfer and leading up to product release.13,17 Stabilizing hydrogen bonds between the Met20 and F-G (residues 116-132) loops within the closed state are broken as the Met20 loop transitions to form new hydrogen bonds with the G-H (residues 142-150) loop in the occluded state. In the occluded conformation, the side chains of M16 and E17 occupy the active site, forcing the nicotinamide of NADPH out into solvent.
The structure of E:NADPH:1 in the P212121 space group was determined to a resolution of 2.1 Å (Supporting Information, Table S1A). This structure is isomorphous to those determined previously,17 thus minimizing structural differences due to crystal packing artifacts and allowing for direct comparisons to be made. Overall, it is very similar to the methotrexate (MTX) ternary complex, PDB ID 1RX3 (backbone rmsd = 0.33 Å). However, some notable differences are observed relative to other ternary or closed complexes.17 While the Met20 loop is found primarily in the closed conformation (Figure 3A), electron density for some regions of the loop is quite poor, suggestive of mobility.
Figure 3.
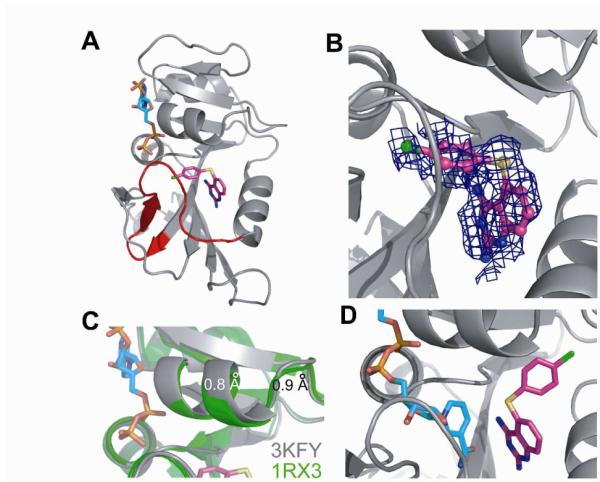
The Crystal Structure of E:NADPH:1. (A) The 2.1 Å resolution structure demonstrates that the Met20 loop is primarily in the closed conformation. NADPH is shown in cyan and 1 in magenta. (PDB ID 3KFY rendered using PyMOL.) (B) The 2Fo-Fc electron density for 1 is shown with a cover radius of 3 Å to remove extraneous electron density that complicates this view (pose B). Electron density on the 4-chlorophenyl group is a convolution of density of 1 and weak electron density of nicotinamide. (C) α-Helix C above the inhibitor binding site shifts away from the drug by approximately 1 Å in E:NADPH:1 (grey) relative to 1RX3 (green). (D) NOE and chemical shift data suggest an alternative, ground state binding pose (pose A) for 1 in solution.
In fact, residues 16-20 fit poorly to the density observed (Figure S1A). Similarly, portions of NADPH and inhibitor have weak density, indicating that both cofactor and inhibitor sample multiple binding poses. Electron density for the 2,4-diaminoquinazoline moiety of 1 is well ordered, which overlays nicely with the corresponding moiety in MTX. However, the thiophenyl substituent is much less well defined (Figure 3B and S1B). Inspection of the density led to the only feasible conclusion, namely, that the thiophenyl group samples a previously unobserved pose for E. coli DHFR in which it is oriented towards the nicotinamide binding pocket of the active site (Figure 3A-B). Such a pose has been observed for an analogous inhibitor bound to C. albicans DHFR.19 The binding pose of 1 was studied further via induced fit docking20 against the E:NADPH:1 crystal structure, but with NADPH removed (see Supporting Information, Text S1). The lowest energy docking pose observed shows the thiophenyl bound within the nicotinamide binding site (Figure S2A). A second thiophenyl pose is not observable from the electron density within the active site region, suggesting a sampling of an unknown number of additional poses.
Consistent with 1 and cofactor sampling the same binding site, the nicotinamide-ribose moiety of NADPH samples multiple conformations. As mentioned above, poor electron density for nicotinamide-ribose is observed within the active site (Figure S1C). Surprisingly, electron density from both nicotinamide and thiophenyl groups overlay in this pocket, showing that the calculated density must result from the sum of different conformational poses within the crystal. Presumably, the nicotinamide-ribose group also samples a solvent exposed state, similar to that observed when the Met20 loop is occluded (e.g. bound to 5,10-dideazatetrahydrofolate),17 to make room for the binding of 1’s thiophenyl ring.
To add structural insight into the ambiguities within the crystal structure, NMR chemical shifts within the Met20 loop were analyzed. Nearly all residues within the Met20 loop are broadened, suggesting conformational exchange (Supporting Information, Table S1B); yet, the chemical shift values are indicative of a closed Met20 loop (Figure S3A). Chemical shift perturbations (CSPs) were calculated relative to model complexes with closed (E:NADP+:folate, access. no. 5470) or occluded (E:5,6-dihydroNADPH:folate, access. no. 5471) loops, using data deposited in the Biological Magnetic Resonance Bank (BMRB). Of the nearly twenty resonances with 1HN and/or 15N chemical shifts sensitive to the conformation of the Met20 loop (i.e. ‘markers’),21 only V13 possessed a shift more similar to an occluded loop conformation (Figure S3B-C). In other words, essentially all chemical shift markers indicate that the Met20 loop is primarily closed in E:NADPH:1. Furthermore, calculating CSPs for (E:NADPH:1 – E:NADPH) and (E:NADPH:trimethoprim (TMP) – E:NADPH) allowed for the identification of site-specific changes elicited by the two inhibitors (Figure 4A).
Figure 4.

CSPs of Inhibitor Binding. (A) CSPs of E:NADPH:1 and E:NADPH:TMP relative to E:NADPH. Outliers, shown in blue, were identified using a standard box plot function. (B) CSP outliers upon the binding of 1, highlighted in blue spheres, do not localize to the Met20, F-G, or G-H loops. Significant CSPs are noted in α-helix C and β-sheet B above this helix, suggesting that thiophenyl could bind in this region. (C) Outliers upon the binding of TMP do not localize to the Met20, F-G, or G-H loops, nor to α-helix C or β-sheet B.
The Met20 loop of the E:NADPH holoenzyme complex is known to be predominantly closed in solution.14,18 Relative to this closed complex, no significant changes in chemical shift were observed for any residues within the Met20 or F-G loops in the presence of either inhibitor (Figure 4B-C). The one outlier found in the G-H loop in binding both inhibitors is distal to the hydrogen bonds that form and break during Met20 loop switching motions. Since our previous analysis of the E:NADPH:TMP complex using residual dipolar couplings (RDCs) demonstrated that its Met20 loop is closed in solution, the current chemical shift comparisons indicate that the Met20 loop in E:NADPH:1 is predominantly closed.14 This is further supported by measurements of RDCs for E:NADPH:1 (Supporting Information, Table S1C and Text S1).
Interestingly, CSPs upon binding of 1 are seen above the inhibitor binding site, in the C-terminus of helix C and residues 40 and 41 of β-strand B (Figure 4B). Closer inspection of the crystal structure shows that, relative to E:NADPH:MTX, helix C is shifted about 1 Å away from the folate binding site (Figure 3C). In addition, significant CSPs upon binding 1 were not seen for the majority of residues lining the nicotinamide binding pocket. This raises the possibility of a preferred binding pose for the thiophenyl ring of 1 in solution, in which the substituent could be pointing toward α-helix C above the folate binding site (Figure 3D). Such a pose has been observed for an analog of 1 when bound to C. albicans DHFR (PDB ID 1IA2), in which the active site is several angstroms wider than in E. coli.22
Intermolecular NOEs reveal the bound inhibitor conformation
Given the suggestion from CSPs of a solution-preferred orientation of 1 different from the crystal structure, a 3D 13C-edited/filtered NOESY spectrum was collected on E:NADPH:1 to obtain intermolecular NOEs and determine the solution conformation of 1 within the active site. Five bound 1H chemical shifts of 1 (1-5 in Table 1) were observed to have NOEs to protein. 2D 15N,13C-filtered TOCSY showed that these five protons subdivided into two groups of J-coupled networks (Figure S4), corresponding to three signals for the quinazoline and two for the thiophenyl group (Table 1). Strong and medium intensity NOEs to the quinazoline moiety were consistent with the crystal structure, implicating 1H(3), 1H(4), and 1H(5) signals as arising from quinazoline (Table 1).
Table 1.
Observed intermolecular NOEs for E:NADPH:1.
| 1H(1) | 1H(2) | 1H(3) | 1H(4) | 1H(5) | |
|---|---|---|---|---|---|
| DHFR 1H | |||||
| A7 – 1Hβ | - | - | - | vw | - |
| M20 – 1Hε | vw | w | m | s | s |
| D27 – 1Hβ | - | - | - | w | - |
| 1Hα | - | - | - | vw | - |
| L28 – 1Hδ1 | - | - | s | s | s |
| 1Hδ2 | m | - | s | s | s |
| F31 – 1Hβ | w | w | - | w | - |
| T35 – 1Hγ2 | w | w | - | w | - |
| M42 – 1Hε | m | w | - | - | - |
| I50 – 1Hδ1 | s | s | - | - | s |
| 1Hγ2 | s | m | - | - | - |
| L54 – 1Hδ1 | s | s | - | - | - |
| 1Hδ2 | s | m | - | - | - |
| I94 – 1Hδ1 | m | m | - | - | - |
| 1Hγ2 | w | w | - | - | - |
Abbreviations: very weak (vw), weak (w), medium (m), and strong (s). Gray shading indicates residues expected to have medium to strong NOEs to protons on the quinazoline moiety of 1. Bound chemical shifts of 1 (denoted 1-5) are 7.5, 7.1, 7.3, 6.93, and 6.88 ppm, respectively.
For the thiophenyl substituent, two binding orientations were considered: (A) bound above the substrate binding site, directed towards α-helix C, as suggested by CSPs perturbations (Figure 3D), or (B) bound within the nicotinamide site, as observed in the crystal structure (Figure 3B). Amino acids expected to be within 5-6 Å of 1 in these two conformations were identified for poses A and B (Supporting Information, Text S1). No pose B residues were observed to have NOEs to 1, except M20, whose side-chain is typically highly flexible.14 By contrast, five pose A residues showed mostly strong and medium NOEs to 1H(1) and 1H(2) (Table 1, non-shaded residues). This solidified the chemical shift assignments of 1 and strongly suggested that in solution the thiophenyl group exists primarily pointed in the direction of α-helix C (Figure 3D). Induced fit docking20 against the E:NADPH:1 crystal structure in the presence of NADPH (see Supporting Information, Text S1) shows binding pose A to be the lowest energy conformation for the thiophenyl ring (Figure S2B). The interproton distance patterns between receptor and the lowest energy docked conformation of 1 (Supporting Information, Table S1D) were found to agree well with most intermolecular NOEs in Table 1.
Extensive μs-ms motions in the E:NADPH:1 complex
What is the true nature of 1’s side-chain orientation if it appears well-positioned in solution and disordered in the crystal form? Proteins exist in multiple conformations and thus there may be no single “correct” conformation for 1. Protein motional dynamics occur over a broad range of timescales and include both small-scale bond rotations and large-scale conformational rearrangements.23,24 The latter often occur on the “slow”, or μs-ms timescale and have been implicated in the biological functions of proteins, including ligand binding and release, allosteric regulation, and catalysis-related events.18,25-27 Indeed, μs-ms dynamics are critical for movement of DHFR through its catalytic cycle.18 NMR relaxation studies of E:NADP+:folate, proposed as a surrogate for the reactive complex of DHFR, have shown that the Met20 loop closed-to-occluded switching event occurs in solution on the μs-ms timescale.16,27
For the E:NADPH:1 ternary complex, extensive μs-ms motion was detected by 15N Carr-Purcell-Meiboom-Gill (CPMG)-relaxation dispersion experiments. These experiments allow for decomposition of the transverse relaxation rate R2 into Rex, the relaxation rate component due to slow timescale conformational exchange, and R2o, the remaining contributions to transverse relaxation on a faster timescale.28 Assuming a two-state exchange process, R2 depends on the exchange rate constant (kex), the populations of ground state A and excited state B (pA and pB), and the difference in chemical shift between states A and B (Δω).28 Thus, kinetic, thermodynamic, and structural information, respectively, are potentially obtained to describe the dynamic sampling of two states.
Rex was identified at 55 residues in E:NADPH:1 (Figures 5A-B and S5).
Figure 5.
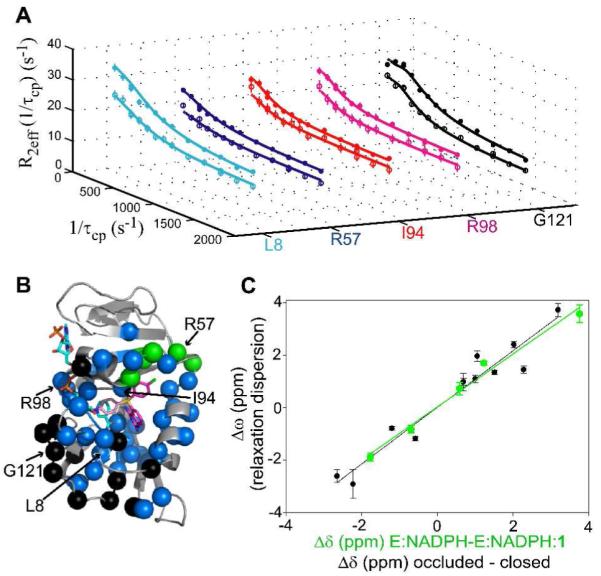
15N Relaxation Dispersion of E:NADPH:1. (A) Relaxation dispersion curves generated from 700 (closed circles) and 500 MHz (open circles) data are shown for several residues. Standard errors were determined by peak intensity analysis of duplicate experiments for specific 1/τcp values. (B) Residues that exhibit R2 dispersion are highlighted in colored spheres. NADPH and 1 are shown in cyan and magenta sticks, respectively. Thiophenyl poses A and B are shown as dark and faded sticks, respectively. (C) Sites surrounding the thiophenyl moiety of 1 show a linear correlation of Δω to Δδ for the loss of thiophenyl in the excited state, with a slope of 1.01 and R = 0.99 (green correlation and spheres in (B)). The comparison of Δω to Δδ for the sites participating in Met20 loop switching motion fit to a line with a slope of 1.08 and R = 0.97 (black correlation and spheres in (B)). The sign of Δω was determined from peak positions in HMQC and HSQC spectra.29 Errors in Δω were determined from Monte Carlo simulations in the global fitting procedure.
This is more extensive than any other reported complexes of DHFR. Motions are observed only on the front face of the enzyme, seen throughout the active site (folate + nicotinamide binding site) and at many residues within the Met20 (8 sites), F-G (6 sites), and G-H loops (4 sites), including G121 which is an important marker of Met20 loop conformational switching.27 All 55 sites were grouped together for global fitting, yielding shared kex and pB values of 844 ± 59 s−1 and 2.6 ± 0.1 %, respectively (Supporting Information, Table S2).30 As will be described further below, the overall pattern of residues is consistent with two coupled motions: (1) switching of the thiophenyl group from preferred pose A above the substrate binding site (Figure 3D), as supported by NOEs, to alternative pose B observed in the crystal structure and (2) switching of the Met20 loop from closed to occluded, in order to accommodate the multiple poses of the inhibitor’s thiophenyl moiety. This model of structural dynamics reconciles the X-ray and NMR data, suggesting that the crystal structure captures a minor, transient state for the thiophenyl ring, whereas the NOEs and chemical shifts reflect the major state in solution.
Concerted small-molecule and receptor conformational switching
In contrast to our previous study of DHFR dynamics in the presence of MTX and TMP,14 a number of residues surrounding the solution-preferred pose of 1’s thiophenyl group exhibit Rex. These sites (residues 37, 40, 50, 52, and 57) were speculated to be undergoing exchange due to the switching of the thiophenyl from pose (A) above the substrate binding site to pose (B) within the nicotinamide binding site. Therefore, an analysis of chemical shift changes was undertaken. Dynamic chemical shift changes (Δω) from the relaxation dispersion analysis were plotted against changes in single-state chemical shifts (Δδ). Values of Δδ representative of loss of inhibitor (E:NADPH – E:NADPH:1) for all residues experiencing slow motions were calculated using assignments of E:NADPH:1 and E:NADPH. A correlation plot of Δω and Δδ for 5 sites surrounding the thiophenyl group (see Supporting Information, Text S1 for residue exclusions) (green correlation in Figure 5C) yields a Pearson coefficient of 0.99, indicating a two-state motion of the thiophenyl from pose (A) in the ground state to a different pose in the excited state, likely pose B as is discussed below. We interpret these to be motions occurring while 1 is bound (i.e., not from dissociation) based on thermodynamic and kinetic grounds (see Supporting Information, Text S1 and Table S3), and also because residues surrounding the anchored quinazoline moiety do not show this correlation.
A similar chemical shift analysis was undertaken for the residues known to be markers of the closed-to-occluded transition of the Met20 loop.18,21,27 Values of Δδ for all residues experiencing slow motions were calculated using the deposited resonance assignments mentioned previously (E:DHNADPH:folate – E:NADP+:folate).21 The correlation of Δω and Δδ for 13 sites (see Supporting Information, Text S1 for residue exclusions) (black correlation in Figure 5C) resulted in a Pearson coefficient of 0.97, indicating a concerted, two-state motion of the Met20 loop from closed to occluded in the E:NADPH:1 ternary complex. Using the shared kex and pB values from the global fit, the switching motion of the Met20 loop and the movement of the thiophenyl group away from pose (A) occurs at a forward rate (kf) of 21.9 ± 1.6 s−1. This rate translates into a ΔG†f of 15.6 kcal/mol, and an overall ΔG of 2.2 kcal/mol for the transition from ground to excited states, based on the populations. This value matches well with what has been observed previously by NMR for the transition, and also with what has been determined via simulation.31
Because the two motions are coupled, we hypothesize that the excited state pose of the thiophenyl group is one in which it occupies the nicotinamide binding site (pose B). This pose was (i) observed in the crystal structure and is further supported by (ii) the poor electron density for both the nicotinamide of NADPH and the Met20 loop, (iii) the relaxation dispersion results, (iv) induced fit docking of 1 to DHFR in the absence of NADPH (Figure S2A), and (v) induced fit docking of 1 to DHFR when the Met20 loop is in the occluded conformation (Figure S2C). The combination of these results strongly suggests that, in the excited state, the thiophenyl ring of 1 occupies the nicotinamide binding site. Regardless of the precise thiophenyl orientation in the excited state, it is clear that the binding of 1, unlike MTX and TMP,14 drives reversible Met20 loop switching from the closed to the occluded conformation. Despite the fact that 1 is an inhibitor, from a mechanistic point of view 1 can be considered a ‘dynamics agonist’. Upon binding (and thiophenyl insertion), 1 elicits a functional loop motion in a distal loop by competitively displacing nicotinamide, which allows adoption of the occluded conformation of the Met20 loop. Met20 loop motion was previously detected in E:NADP+:folate;27 however, motion of folate was not observed. Direct observation of movement of a non-biological inhibitor while bound to its target has implications for drug design.
In summary, the E:NADPH:1 complex is presented as a highly dynamic complex on the μs-ms timescale. Ligand, receptor, and cofactor are in a continuous state of shared conformational flux, with the ligand dynamics driving the cofactor and receptor dynamics. The thiophenyl group of 1 prefers to bind at the upper end of the active site, but it also samples a higher energy pose in the nicotinamide binding pocket, which expels cofactor nicotinamide. This, in turn, allows the Met20 loop to move between closed and occluded conformations.
DISCUSSION
Protein flexibility and dynamics represent a complication to drug design that has just begun to attract major efforts to tackle this problem. Although the problem is complex, one clear reason for this is that accurately characterized examples of receptor-ligand dynamics are needed from which to build upon, and such examples are essentially non-existent.2,4 Here, we demonstrate that the ternary complex of DHFR, NADPH, and the drug-like compound 1 exists in at least two conformational states that are dynamically interconverting on a timescale of ~1 ms. The structural, temporal, and population aspects of the dynamics were captured by use of crystallography and NMR. This complex could therefore serve as a useful benchmark for the refinement and future development of modeling methods that incorporate receptor and ligand dynamics. This should lead to improvements in predicting binding affinities and provide insight into targeting dynamics.7
The application of both NMR and crystallography was critical to reveal the true nature of this ligand-receptor complex. The resultant picture of this dynamic complex is that, in solution, the dominant state (~97%) has DHFR in the closed conformation, with cofactor fully bound and thiophenyl of 1 directed towards helix C. The minor state (~3%) has DHFR in the occluded conformation, nicotinamide-ribose of cofactor ejected into solvent, and thiophenyl inserted into the nicotinamide binding site. These states represent actual dynamics within the complex since dissociation is slow relative to these conformational changes (Supporting Information, Text S1). Ligand structural heterogeneity has been observed previously in E. coli DHFR complexes. A recent ternary crystal structure of DHFR complexed with a novel inhibitor (Ki = 11 nM) showed the inhibitor with diminished electron density for half of the molecule.32 A second structure with a shorter inhibitor corresponding to the anchored region of the first inhibitor also showed evidence for multiple conformations. The second inhibitor has substantially reduced affinity, showing that even flexible portions of ligands can make large contributions to binding affinity.32
Despite the motion of this small molecule while bound to DHFR, the binding affinity of 1 for holoenzyme is still high. Do the multiple binding poses of 1 limit its clinical potential? It may be possible for drug resistant mutations to limit one binding pose while not affecting the other. Thus, two dynamically sampled ligand binding poses for one drug could limit drug resistance if protein inhibition is preserved in either binding mode. This was specifically observed in crystal structures of inhibitor TMC278 (rilpiverine) in complex with HIV-1 reverse transcriptase mutants.33 In principle, 1 would be valuable as an inhibitor of trimethoprim (TMP) resistant strains of bacteria due to its sampling of a non-canonical binding pose within the active site. Known mutations that confer TMP resistance would not affect the binding of the thiophenyl substituent of 1 within the nicotinamide binding site, as many of these mutations are concentrated in the folate binding site.34
The findings reported here, along with innumerable crystallographic studies, suggest that multiple ligand poses may be sampled more often than expected.4,35 This may be especially true for small, lipophilic ligands encountered in drug discovery. In most instances of apparent single-mode binding, minor conformers that are actually sampled to a significant extent would not be expected to crystallize or would lie below the noise threshold for NOE detection; the only way to detect these conformers would be from NMR relaxation dispersion experiments (as reported here) or MD simulations.36,37 An important class of receptors for signal transduction and pharmaceuticals is that of the ligand activated G-protein coupled receptors (GPCRs). The degree of conformational flexibility and dynamics in these receptors is impressive38 and likely to be more extensive than in DHFR. Germane to the results here, biophysical studies on the β2-adrenergic receptor (β2AR) show that agonist binding (at saturating levels) produces structural heterogeneity,39 rather than locking the receptor into a single conformation. Thus, although it remains to be seen if single GPCR ligands adopt multiple bound configurations, dynamic receptor-ligand complexes are likely to be of broad relevance for understanding mechanisms of signal transduction and their perturbation by drugs.5
It is instructive to compare the dynamic characterization here to one of the only other target-drug systems characterized in detail by crystallography and NMR: the Bcr-Abl fusion kinase in complex with the kinase inhibitor dasatinib.40 Dramatic line-broadening was observed in the activation and P-loops of Bcr-Abl, suggesting allosteric loop switching motions. Even though inhibitors imatinib and nilotinib stabilize different loop conformations, they also show some line-broadening in a few loops residues, although significantly less than in the dasatinib complex.40 Further detail on the μs-ms timescale dynamics from relaxation dispersion experiments were not available. We also note that dynamics in a small molecule was previously shown to exist on multiple timescales when bound to matrix metalloproteinease-1 (MMP-1).41 Thus, dynamics in both ligands and receptors clearly exist across very different classes of drug targets.
It has recently been suggested that many underexploited protein target classes are avoided due to the flexibility inherent to their function, such as ion channels and nuclear hormone receptors.2 However, these more challenging targets are likely to become important in future drug design efforts, as we continue to exhaust the less complex targets. Identification of multiple ligand conformations and flexibility within the active site for the E:NADPH:1 complex is an example that stresses the importance of continuing efforts toward an understanding of protein dynamics and how they are modulated by small molecules. Given the scarcity of studies identifying specific ligand-induced protein flexibility, the results of this study may find use in the advancement of computational docking methods that include protein dynamics.3 The transient, excited states detected in this approach could also be targeted and stabilized by small molecules, leading to new high-affinity modulators of protein function for disease treatment.
METHODS
Synthesis of compound 1
Compound 1 was prepared in one step by the method patented previously by Singh and Gurney.42 Characterization information can be found in the Supporting Information (Text S2).
Protein expression and purification
Isotopically labeled wild-type Escherichia coli DHFR was over-expressed and purified as described previously.14 Purified apo-DHFR was frozen in a dry ice and ethanol bath, lyophilized, and stored in a desiccator at 4 °C until use.
Ki determination
Biochemical competition assays using a 96-well plate reader were used to determine the inhibition constant (Ki) for 1. 1 was added to a reaction of DHFR, NADPH, and dihydrofolate substrate, and depletion of NADPH was monitored by UV absorbance at 340 nm.15 The total reaction volume was 100 μL.
NMR Spectroscopy
NMR samples contained 1 mM isotopically labeled DHFR in NMR buffer (70 mM HEPES, 20 mM KCl, 1 mM EDTA, 1 mM DTT [pH 7.6]) along with 15 mM NADPH, 2.5 mM 1, 10 mM glucose-6-phosphate, 10 units of glucose-6-phosphate dehydrogenase, and 10% D2O. All samples were protected from light and air exposure by containment in amber NMR tubes flame-sealed under argon. Stock solutions of 1 were prepared in 10% D2O/90% H2O, and PULCON was used to determine the concentrations of stocks, relative to a tyrosine standard.43 All NMR experiments were conducted at 298 K on Varian spectrometers equipped with room temperature (500 MHz) or cryogenic (500 MHz and 700 MHz) probes. NMRPipe was used to process NMR data, and data visualization was accomplished with the combination of NMRDraw and NMRView.44,45 See Supporting Information (Text S1) for specific experimental details.
Protein crystallization, data collection and structure determination
Crystals of E:NADPH:1 were grown using similar conditions as described previously.17,32 See Supporting Information (Text S1) for details regarding crystallization and data collection and analysis.
Supplementary Material
Acknowledgements
The authors thank Randall Mauldin for methods contributions and Paul Sapienza for invaluable discussions. MJC gratefully acknowledges pre-doctoral fellowships from the ACS Division of Medicinal Chemistry (supported by Pfizer Global R&D) and the American Foundation for Pharmaceutical Education. This work was funded by NIH grant GM08359 (to ALL).
Footnotes
Supporting Information Available: Complete references for (10) and (36) can be found in the Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Bursavich MG, Rich DH. J Med Chem. 2002;45:541–58. doi: 10.1021/jm010425b. [DOI] [PubMed] [Google Scholar]
- (2).Teague SJ. Nat Rev Drug Discov. 2003;2:527–41. doi: 10.1038/nrd1129. [DOI] [PubMed] [Google Scholar]
- (3).B-Rao C, Subramanian J, Sharma SD. Drug Discov Today. 2009;14:394–400. doi: 10.1016/j.drudis.2009.01.003. [DOI] [PubMed] [Google Scholar]
- (4).Mobley DL, Dill KA. Structure. 2009;17:489–498. doi: 10.1016/j.str.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Bruning JB, Parent AA, Gil G, Zhao M, Nowak J, Pace MC, Smith CL, Afonine PV, Adams PD, Katzenellenbogen JA, Nettles KW. Nat Chem Biol. 6:837–43. doi: 10.1038/nchembio.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Frederick KK, Marlow MS, Valentine KG, Wand AJ. Nature. 2007;448:325–9. doi: 10.1038/nature05959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Peng JW. Structure. 2009;17:319–20. doi: 10.1016/j.str.2009.02.004. [DOI] [PubMed] [Google Scholar]
- (8).May LT, Leach K, Sexton PM, Christopoulos A. Annu Rev Pharmacol Toxicol. 2007;47:1–51. doi: 10.1146/annurev.pharmtox.47.120505.105159. [DOI] [PubMed] [Google Scholar]
- (9).Lee GM, Craik CS. Science. 2009;324:213–5. doi: 10.1126/science.1169378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Zhang J, et al. Nature. 2010;463:501–6. doi: 10.1038/nature08675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Najmanovich R, Kuttner J, Sobolev V, Edelman M. Proteins. 2000;39:261–8. doi: 10.1002/(sici)1097-0134(20000515)39:3<261::aid-prot90>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- (12).Yang CY, Wang R, Wang S. J Med Chem. 2005;48:5648–50. doi: 10.1021/jm050276n. [DOI] [PubMed] [Google Scholar]
- (13).Schnell JR, Dyson HJ, Wright PE. Annu Rev Biophys Biomol Struct. 2004;33:119–40. doi: 10.1146/annurev.biophys.33.110502.133613. [DOI] [PubMed] [Google Scholar]
- (14).Mauldin RV, Carroll MJ, Lee AL. Structure. 2009;17:386–394. doi: 10.1016/j.str.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Zolli-Juran M, Cechetto JD, Hartlen R, Daigle DM, Brown ED. Bioorg Med Chem Lett. 2003;13:2493–6. doi: 10.1016/s0960-894x(03)00480-3. [DOI] [PubMed] [Google Scholar]
- (16).Bystroff C, Oatley SJ, Kraut J. Biochemistry. 1990;29:3263–77. doi: 10.1021/bi00465a018. [DOI] [PubMed] [Google Scholar]
- (17).Sawaya MR, Kraut J. Biochemistry. 1997;36:586–603. doi: 10.1021/bi962337c. [DOI] [PubMed] [Google Scholar]
- (18).Boehr DD, McElheny D, Dyson HJ, Wright PE. Science. 2006;313:1638–42. doi: 10.1126/science.1130258. [DOI] [PubMed] [Google Scholar]
- (19).Whitlow M, Howard AJ, Stewart D, Hardman KD, Chan JH, Baccanari DP, Tansik RL, Hong JS, Kuyper LF. J Med Chem. 2001;44:2928–32. doi: 10.1021/jm0101444. [DOI] [PubMed] [Google Scholar]
- (20).Sherman W, Day T, Jacobson MP, Friesner RA, Farid R. J Med Chem. 2006;49:534–53. doi: 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- (21).Osborne MJ, Venkitakrishnan RP, Dyson HJ, Wright PE. Protein Sci. 2003;12:2230–8. doi: 10.1110/ps.03219603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Whitlow M, Howard AJ, Stewart D, Hardman KD, Kuyper LF, Baccanari DP, Fling ME, Tansik RL. J Biol Chem. 1997;272:30289–98. doi: 10.1074/jbc.272.48.30289. [DOI] [PubMed] [Google Scholar]
- (23).Boehr DD, Dyson HJ, Wright PE. Chem Rev. 2006;106:3055–79. doi: 10.1021/cr050312q. [DOI] [PubMed] [Google Scholar]
- (24).Palmer AG., 3rd Chem Rev. 2004;104:3623–40. doi: 10.1021/cr030413t. [DOI] [PubMed] [Google Scholar]
- (25).Cole R, Loria JP. Biochemistry. 2002;41:6072–81. doi: 10.1021/bi025655m. [DOI] [PubMed] [Google Scholar]
- (26).Eisenmesser EZ, Bosco DA, Akke M, Kern D. Science. 2002;295:1520–3. doi: 10.1126/science.1066176. [DOI] [PubMed] [Google Scholar]
- (27).McElheny D, Schnell JR, Lansing JC, Dyson HJ, Wright PE. Proc Nat Acad Sci U S A. 2005;102:5032–5037. doi: 10.1073/pnas.0500699102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Palmer AG, 3rd, Kroenke CD, Loria JP. Methods Enzymol. 2001;339:204–38. doi: 10.1016/s0076-6879(01)39315-1. [DOI] [PubMed] [Google Scholar]
- (29).Skrynnikov NR, Dahlquist FW, Kay LE. J Am Chem Soc. 2002;124:12352–12360. doi: 10.1021/ja0207089. [DOI] [PubMed] [Google Scholar]
- (30).Mulder FA, Mittermaier A, Hon B, Dahlquist FW, Kay LE. Nat Struct Biol. 2001;8:932–5. doi: 10.1038/nsb1101-932. [DOI] [PubMed] [Google Scholar]
- (31).Arora K, Brooks CL., III Journal of the American Chemical Society. 2009;131:5642–5647. doi: 10.1021/ja9000135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Summerfield RL, Daigle DM, Mayer S, Mallik D, Hughes DW, Jackson SG, Sulek M, Organ MG, Brown ED, Junop MS. J Med Chem. 2006;49:6977–6986. doi: 10.1021/jm060570v. [DOI] [PubMed] [Google Scholar]
- (33).Das K, Bauman JD, Clark AD, Jr., Frenkel YV, Lewi PJ, Shatkin AJ, Hughes SH, Arnold E. Proc Natl Acad Sci U S A. 2008;105:1466–71. doi: 10.1073/pnas.0711209105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Watson M, Liu JW, Ollis D. FEBS J. 2007;274:2661–71. doi: 10.1111/j.1742-4658.2007.05801.x. [DOI] [PubMed] [Google Scholar]
- (35).Watkins RE, Wisely GB, Moore LB, Collins JL, Lambert MH, Williams SP, Willson TM, Kliewer SA, Redinbo MR. Science. 2001;292:2329–33. doi: 10.1126/science.1060762. [DOI] [PubMed] [Google Scholar]
- (36).Schames JR, Henchman RH, Siegel JS, Sotriffer CA, Ni H, McCammon JA. J Med Chem. 2004;47:1879–81. doi: 10.1021/jm0341913. [DOI] [PubMed] [Google Scholar]
- (37).Hazuda DJ, et al. Proc Natl Acad Sci U S A. 2004;101:11233–8. doi: 10.1073/pnas.0402357101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Kobilka BK, Deupi X. Trends Pharmacol Sci. 2007;28:397–406. doi: 10.1016/j.tips.2007.06.003. [DOI] [PubMed] [Google Scholar]
- (39).Ghanouni P, Gryczynski Z, Steenhuis JJ, Lee TW, Farrens DL, Lakowicz JR, Kobilka BK. J Biol Chem. 2001;276:24433–6. doi: 10.1074/jbc.C100162200. [DOI] [PubMed] [Google Scholar]
- (40).Vajpai N, Strauss A, Fendrich G, Cowan-Jacob SW, Manley PW, Grzesiek S, Jahnke W. J Biol Chem. 2008;283:18292–302. doi: 10.1074/jbc.M801337200. [DOI] [PubMed] [Google Scholar]
- (41).Moy FJ, Chanda PK, Chen J, Cosmi S, Edris W, Levin JI, Rush TS, Wilhelm J, Powers R. J Am Chem Soc. 2002;124:12658–9. doi: 10.1021/ja027391x. [DOI] [PubMed] [Google Scholar]
- (42).Singh J, Gurney ME. WO 2005-US19753 Preparation of 2,4-diamioquinazolines for treatment of spinal muscular atrophy. 2005 Jun 06;
- (43).Wider G, Dreier L. J Am Chem Soc. 2006;128:2571–2576. doi: 10.1021/ja055336t. [DOI] [PubMed] [Google Scholar]
- (44).Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- (45).Johnson BA, Blevins RA. J Biomol NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.