

Abstract
Background
Previous studies suggest that the transient receptor potential vanilloid-1 (TRPV1) has a role in sepsis but it is unclear whether its effect on survival and immune response is beneficial or harmful.
Methods
We studied the effects of genetic (Trpv1-knock out vs. wild-type mice) and pharmacologic disruption of TRPV1 with resiniferatoxin (an agonist) or capsazepine (an antagonist) on mortality, bacterial clearance, and cytokine expression during lipopolysaccharide or cecal ligation and puncture (CLP)-induced sepsis.
Results
After CLP, genetic disruption of TRPV1 in Trpv1-knock out mice was associated with increased mortality risk [2.17 (1.23 to 3.81) hazard ratio (95% confidence interval), p=0.01] compared with wild-type. Further, pharmacologic disruption of TRPV1 with intrathecal resiniferatoxin, compared with vehicle, increased mortality risk [1.80 (1.05 to 3.2) hazard-ratio (95% confidence interval), p=0.03] in wild-type animals, but not in Trpv1-knock out mice. After lipopolysaccharide, neither genetic (Trpv1-knock out) nor pharmacologic disruption of TRPV1 with resiniferatoxin had significant effect on survival compared with respective controls. In contrast, after lipopolysaccharide, pharmacologic disruption of TRPV1 with capsazepine, compared with vehicle, increased mortality risk [1.92 (1.02 to 3.61) hazard-ratio (95% confidence interval), p=0.04] in wild-type animals. Further, after CLP, increased mortality in resiniferatoxin-treated-wild-type animals was associated with higher blood bacterial count (p=0.0004) and nitrate/nitrite levels and downregulation of tumor necrosis factor α expression (p=0.004) compared with controls.
Conclusions
Genetic or pharmacologic disruption of TRPV1 can affect mortality, blood bacteria clearance, and cytokine response in sepsis in patterns that may vary according to the sepsis-inducing event and the method of TRPV1 disruption.
Introduction
A growing body of literature supports the notion that sensory neurons have a role in modulating inflammatory response1–2. In fact, since the initial description of neurogenic inflammation - vasodilation, plasma extravasation, and pain – that results from stimulation of sensory neurons several studies have shed light on the mechanisms underlying the phenomenon3. Researchers have shown that when sensory neurons expressing the transient receptor potential vanilloid 1 (TRPV1) are activated by protons, lipoxygenase products, noxious heat, nitric oxide or vanilloid compounds, neuropeptides known to generate neurogenic inflammation [substance P and calcitonin gene-related peptide] are released4–5. The findings that animals genetically lacking the TRPV1 receptor or undergoing ablation of TRPV1 sensory neurons are unable to mount neurogenic proinflammatory responses to noxious stimuli or vanilloid agonists further support the notion that TRPV1 plays a role in inflammation4–6.
With regards to sepsis, some studies implicate TRPV1, calcitonin gene-related peptide, and substance P in its pathophysiology in humans and animals. For example, in septic patients, higher levels of calcitonin gene-related peptide and substance P correlate with levels of nitrate/nitrites, hypotension, and lethality7–9. In rats, lipopolysaccharide-induced toxicity is associated with augmented expression of TRPV1 receptors and increased density of calcitonin gene-related peptide positive mesenteric neurons10. In mice, genetic disruption of TRPV1 in Trpv1-knockout (Trpv1-KO) mice is associated with worse hypotension, increased cytokine levels in the peritoneum and worse liver injury during lipopolysaccharide-induced toxicity11. In rats, the TRPV1 competitive antagonist, capsazepine, decreased survival and worsened hypotension in lipopolysaccharide-induced toxicity12. Thus, taken together these studies suggest that TRPV1 has a protective role during sepsis.
However, contrary to reports suggesting a protective role of TRPV1, a recent study in mice showed that pharmacologic disruption of TRPV1 with capsazepine has beneficial effects during peritonitis and sepsis13. That study showed that in a model of polymicrobial sepsis, one dose of capsazepine administered 30 min before cecal ligation and puncture (CLP), increased survival and decreased liver and lung damages13. While one must recognize that capsazepine can affect receptors other than TRPV1 including nicotinic acetylcholine receptors14 and other voltage-activated calcium channels15, that study in capsazepine-treated mice suggests that TRPV1 has a potentially harmful effect during polymicrobial sepsis and its pharmacologic disruption with capsazepine is protective. Therefore, while animal studies have yielded conflicting results as to what role TRPV1 has in survival and multiple organ damage during sepsis, there is ample evidence that TRPV1 plays an incompletely understood role in the pathophysiology of sepsis.
The purpose of the present study was to investigate the role of TRPV1 in mortality, bacterial clearance, and cytokine expression in murine sepsis induced by bacteria during CLP or lipopolysaccharide challenge. These investigations are relevant because agents such as resiniferatoxin, a potent capsaicin analog and a TRPV1 agonist known to ablate TRPV1-expressing neurons by excessive calcium influx16–17, are currently being investigated as potential therapies for various pain syndromes and inflammatory states in animals and humans4,18–20.
Materials and Methods
Animals
After approval from the National Institutes of Health Clinical Center Animal Care and Use Committee (Bethesda, MD, USA), we studied female C57Bl/6 (wild-type, WT) and B6.129X1-Trpv1tm1Jul/J (Trpv1-KO) mice (six to eight weeks-old and 15–20 g, Jackson Laboratories, Bar Harbor, ME). Animals were acclimated to their cages and housed in a temperature controlled facility with free access to food and water.
Experimental Protocol
Survival
In order to investigate the role of TRPV1 in sepsis and lipopolysaccharide-induced toxicity, we examined the role of pharmacologic disruption of the receptor with resiniferatoxin (a TRPV1 agonist) and capsazepine (a TRPV1 antagonist), as well as genetic disruption in Trpv1-KO in two murine models: CLP- induced polymicrobial sepsis and lipopolysaccharide-induced toxicity. Table 1 lists the number of WT and Trpv1-KO animals enrolled in each of the 12 study groups that included combinations of surgery (CLP), lipopolysaccharide administration, genotype (WT or Trpv1-KO), and pharmacological treatment (resiniferatoxin, a potent TRPV1 agonist, and capsazepine, a competitive TRPV1 antagonist or respective vehicle control). Surgery (CLP) or lipopolysaccharide-challenge experiments were conducted weekly and during each week animals from treatment and respective control groups (vehicle or no treatment) were studied simultaneously. In animals undergoing CLP, after isoflurane anesthesia was induced, a small midline abdominal incision was made, the cecum was located and exteriorized, then after a 4-0 silk ligature was placed at 1 cm from its tip the cecum was perforated once with a 16 G needle at the anti-mesenteric surface. A small amount of fecal material was then expressed through the puncture wound, the cecum was placed back into the abdomen and the incision was closed with metal clips21. One investigator, unaware of genotype or treatment groups, performed all CLP procedures. Saline was administered at surgery completion (50ml/kg) and with each antibiotic dose [Primaxin® (imipenem/cilastatin sodium), 25 mg/kg of imipenem, Merck, Whitehouse Station, NJ] starting 2 hours after surgery and every 12 h for 72 hrs (50 ml/kg of saline subcutaneously, amounting to 150ml/kg in the first 24h and 100ml/kg/24h for two more days). After surgery, animals were observed every 2h for the first 36, every 4h from 36 to 48, and once daily from 48 to 168h.
Table 1.
Experimental groups in the survival study and respective mortality rates*
| Genotype | Treatment | Number of Animals Enrolled | Number of non-survivors (Mortality Rate, %) |
|---|---|---|---|
| Cecal Ligation and Puncture | |||
|
| |||
| Wild-type | None | 20 | 8 (40) |
| Trpv1-KO | None | 20 | 9 (45) |
| Wild-type | Vehicle | 35 | 12 (34) |
| Wild-type | Resiniferatoxin | 42 | 25 (60) |
| Trpv1-KO | Vehicle | 30 | 18 (60) |
| Trpv1-KO | Resiniferatoxin | 30 | 19 (63) |
| Lipopolysaccharide | |||
|
| |||
| Wild-type | None | 21 | 13 (62) |
| Trpv1-KO | None | 42 | 26 (62) |
| Wild-type | Vehicle | 27 | 18 (67) |
| Wild-type | Resiniferatoxin | 57 | 29 (51) |
| Wild-type | Vehicle | 25 | 15 (60) |
| Wild-type | Capsazepine | 33 | 29 (88) |
Trpv1-KO represents animals lacking the transient receptor potential vanilloid 1 gene.
After CLP (at 6 and 24 h) and lipopolysaccharide-challenge (at 3, 6, and 24 h) randomly selected animals were anesthetized (isoflurane) for terminal blood draws for blood bacterial culture, serum cytokines and nitrate/nitrite measurements or spleens procurement for RNA isolation or peritoneal lavage for flow cytometry (lipopolysaccharide-challenged animals only).
Pharmacologic disruption of TRPV1 with resiniferatoxin or capsazepine
Resiniferatoxin was administered intrathecally (L5–L6 intervertebral space) 72 h prior to CLP or lipopolysaccharide-challenge to animals assigned to resiniferatoxin-groups under light isoflurane anesthesia as previously described22. The resiniferatoxin dose (25 ng suspended in 0.25% Tween-80/preservative free saline in a volume of 10 ul) was chosen after pilot studies determined that such dose elicited no morbidity and increased nocifensive behavior latency time on the hotplate (data not shown) indicating successful intrathecal resiniferatoxin injection. Controls received an intrathecal injection of the resiniferatoxin vehicle.
Capsazepine15, a TRPV1 antagonist, was suspended in 10% dimethyl sulfoxide/physiological saline and 50 μg in 0.1 mL was injected subcutaneously. Animals assigned to the capsazepine experiments received capsazepine or the same volume of vehicle subcutaneously (controls) twice daily for 72 hours before and 72 hours after lipopolysaccharide challenge.
Lipopolysaccharide (Escherichia coli 0111:B4, Sigma, St Louis, MO) was suspended in sterile lipopolysaccharide-free saline (Hospira Inc., Lake Forest, IL) and mice were injected with 25 mg/kg intraperitoneally
Quantitative real-time reverse transcriptase polymerase chain reaction
RNA was isolated using RNeasy Mini kit (Qiagen, Valencia, CA). RNA quality was evaluated by Agilent 2100 Bioanalyzer using RNA 6000 Nano kits (Agilent Technologies, Santa Clara, CA) and quantity was measured using NanoDrop (NanoDrop Technologies, Wilmington, DE). Tumor necrosis factor (TNF)α, macrophage inflammatory protein 1α (CCL3) and interleukin-10 (IL-10) gene expressions were quantified by real-time reverse transcriptase polymerase chain reaction using One-Step qRT-PCR Kit in the Abi Prism 7500 (both from Applied Biosystems, Foster City, CA). Fold changes in gene expression were calculated after normalization to endogenous glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and were expressed relative to that detected in spleens of WT or vehicle control injected animals (normalized to 1) using the comparative Ct method23.
Serum cytokine and chemokine levels
Serum TNFα, CCL3 and IL-10 levels were measured using a multiplex sandwich enzyme-linked immunosorbent assay (SearchLight Mouse Cytokine array, ThermoFisher Scientific, Woburn, MA).
Flow cytometry analysis of peritoneal cells
In lipopolysaccharide-challenge experiments, peritoneal cells were obtained from peritoneal lavage (5 ml of 1× phosphate buffered saline, Invitrogen, Carlsbad, CA). Lavage fluid was centrifuged, supernatant removed, and cell pellet resuspended in Dulbecco's phosphate buffered saline (Invitrogen) supplemented with 0.5% BSA. One million cells were stained with either CD19 (B-cell marker), F4/80 (macrophage marker), Ly6g (neutrophil marker), or their respective isotype controls (eBioscience, San Diego, CA) for 30 minutes on ice. After staining, cells were washed and resuspended in Cytofix (BD Biosciences, San Jose, CA) and analyzed on a FACSCalibur (BD Biosciences) within 48 hours.
Nitrate/nitrite measurements
Plasma was de-proteinized using 10K centrifugal filters (Millipore, Billerica, MA) and the flow-through was then mixed with vanadium chloride (5 ml of 0.4% in 1 N HCl) in a heated to 90°C purge vessel through which helium was continuously bubbled. The resulting nitric oxide from nitrate and nitrite in the samples was detected with a nitric oxide analyzer (Sievers 280, GE Analytical, Boulder, CO. Measurements were based on comparisons to control samples with known concentrations of nitrate.
Temperature measurements
In order to measure body temperature, an electronic thermosensor weighing 120 mg (Bio Medic Data Systems, Seaford, DE) was implanted subcutaneously between the mouse shoulder blades, with general anesthesia and using a sterile trocar (12-gauge needle). Body temperature was measured serially before and after lipopolysaccharide challenge.
In vitro effects of capsazepine in splenocytes treated with lipopolysaccharide
WT mice were euthanized under isoflurane anesthesia. Spleens were removed, mechanically disassociated and red blood cells were lysed using red cell lysing buffer (ACK). Three experiments were conducted and each experiment performed with cells pooled from spleens from two mice. Cells were resuspended in DMEM (Delbecco's Modified Essential Media) supplemented with fetal calf serum, Pen/Strep and GlutaMax (all solutions from Invitogen, Grand Island, NY), then plated at 10 × 106cells/ml in 6 well plates. Capsazepine (10μM) and lipopolysaccharide (10ng/ml) were added and cells were incubated for 16 hours at 37°C. Cell viability was determined prior to RNA isolation by trypan blue dye exclusion.
Statistical Analysis
The data were analyzed using SAS version 9.13 (SAS, Cary, NC). Survival analyses were conducted using Cox proportional hazard model to analyze the effects of Trpv1-KO or resiniferatoxin treatment vs. WT control in either CLP or lipopolysaccharide-challenge models, the effect of Trpv1-KO treated with resiniferatoxin vs. Trpv1-KO control in CLP or lipopolysaccharide model, and the effect of capsazepine vs. its vehicle in WT animals. As prospectively designed, WT or Trpv1-KO groups that received no treatment or vehicle in cohort experiments and did not differ in survival were combined as WT or Trpv1-KO controls respectively for survival analysis. Hazard ratio of death [95% confidence interval] was used to express the effect of each treatment on mortality compared with its own control. We also performed post hoc analyses to compare the effects of resiniferatoxin on survival between the two sepsis models, CLP vs. lipopolysaccharide and to compare the effects of pharmacologic disruption of TRPV1 with capsazepine vs. genetic and pharmacologic disruption with resiniferatoxin on survival of lipopolysaccharide-challenged animals.
All other data, i.e., bacterial count, serum cytokines, messenger RNA levels, were analyzed using 2-way analysis of variance taking into account time point and strain or challenge or treatment. Log transformation of the data was performed where applicable. Tukey test was conducted where applicable to compare the difference between groups at each time point. A two- tailed p value (uncorrected) less than or equal to 0.05 was considered as statistical significance. All data were expressed as mean±SEM.
Results
After CLP and lipopolysaccharide challenges, animals showed weakness, lethargy, and decreased mobility and over 168h, CLP and lipopolysaccharide challenges produced mortality in all groups.
Effect of pharmacologic disruption of TRPV1 with resiniferatoxin, a TRPV1 agonist
Effect of resiniferatoxin on survival after bacterial peritonitis and sepsis
Table 1 lists experimental groups and respective mortality rates and Figures 1 and 2 describe the effect of genetic and pharmacologic disruption of TRPV1 on survival. After CLP-induced polymicrobial sepsis, disruption of TRPV1-sensory neurons with intrathecal resiniferatoxin in resiniferatoxin-treated-WT animals was associated with increased risk of mortality [1.80 (1.05 to 3.2) hazard ratio (95% confidence interval), p=0.03, Figures 1 and 2] compared with WT control. In contrast to what was observed in WT animals, in Trpv1-KO mice, intrathecal resiniferatoxin injection, compared with vehicle, had no significant effect on mortality (p=0.47) after CLP (Figure 1B and Figure 2). This finding therefore suggests that the harmful effect of resiniferatoxin on mortality after CLP requires integrity of the TRPV1 gene.
Figure 1.

Effect of disruption of transient receptor potential vanilloid 1 (TRPV1) on survival after polymicrobial sepsis (cecal ligation and puncture) or lipopolysaccharide challenge. Panel A shows effects of TRPV1 disruption by genetic deletion (Trpv1-knock out, KO) or pharmacologic ablation with intrathecal resiniferatoxin (RTX) [WT (wild-type) control group includes 55 WT animals (20 receiving no treatment and 35 vehicle-treated WT combined); Trpv1-KO group includes 50 animals (20 Trpv1-KO mice receiving no treatment and 30 receiving vehicle)]. Panel B shows the effect of intrathecal resiniferatoxin in animals lacking the TRPV1 gene (Trpv1-KO) after cecal ligation and puncture. Panel C shows the effects of TRPV1 disruption by genetic deletion or pharmacologic ablation with intrathecal resiniferatoxin after lipopolysaccharide-induced toxicity [WT control group includes 48 WT animals (21 receiving no treatment and 27 vehicle-treated WT combined)]. Panel D shows effects of TRPV1 disruption with capsazepine (CPZ) after lipopolysaccharide challenge.
Figure 2.

Effects of disruption of the transient receptor potential vanilloid 1 (TRPV1) by pharmacologic disruption [with resiniferatoxin (RTX) or capsazepine (CPZ)] or genetic deletion (Trpv1-KO) on hazard ratio of death (95% confidential interval) based on the respective controls as indicated in Figure 1. The triangle, circle, and square represent the hazard ratio and the horizontal brackets, the 95% confidence interval. P values indicate the significance level of hazard ratio of death of each treatment in either cecal ligation and puncture (CLP) or lipopolysaccharide (LPS) compared to their respective controls.
Effect of resiniferatoxin on survival after lipopolysaccharide-induced toxicity
During lipopolysaccharide-induced toxicity, pharmacologic disruption of TRPV1 in resiniferatoxin-treated-WT animals was associated with no significant effect on mortality compared with vehicle-treated WT animals (Table 1, Figure 1, p=0.22).
On a post hoc analysis, the authors compared the pattern of effects of disruption of the TRPV1 gene (either with resiniferatoxin or gene knock out) in polymicrobial infection in CLP vs. lipopolysaccharide-induced toxicity and found that there were significantly different patterns of effect on risk of death (p=0.003) according to the model used. Specifically, with polymicrobial infection in CLP, disruption of TRPV1 was associated with a harmful effect whereas with lipopolysaccharide-induced toxicity, with no significant effect [1.98(1.2 to 3.32) vs. 0.81(0.58 to 1.18) hazard ratio (95% confidence interval) for CLP vs. lipopolysaccharide respectively, p=0.003] Figure 2.
Effect of resiniferatoxin on bacterial clearance during bacterial infection
Figure 3 shows blood bacterial counts after CLP. After CLP, resiniferatoxin-treated–WT mice had a significantly different pattern of blood bacterial counts at 6 and 24 h (Figure 3A, p=0.0005) compared with vehicle-treated WT. Specifically, while by 24 h, there were significantly greater increases (p=0.0004) in bacteria colony forming units in the blood in resiniferatoxin-treated compared with vehicle-treated WT animals, at 6 h there were no statistically significant differences (Figure 3A).
Figure 3.

Effect of disruption of the transient receptor potential vanilloid-1 (TRPV1) gene by pharmacologic ablation with intrathecal resiniferatoxin, in wild-type (WT) animals (panels A and C) or genetic deletion (Trpv1-Knock Out, KO, panels B and D) on bacterial clearance from the blood (panels A and B) and nitrate/nitrite (NOx) serum levels at 6 and 24 h (panels C and D). Results are shown as means±SEM, number of animals sacrificed at respective time points are shown in parenthesis inside each bar, and * indicates P<0.05 comparing groups with respective controls.
Effect of resiniferatoxin on serum nitrate/nitrite levels and cytokine gene expression profile after CLP
Tables 2 and 3 show the effects of pharmacologic and genetic disruptions of TRPV1 on cytokine gene expression, survival, blood culture, and nitrate/nitrite levels after CLP and lipopolysaccharide. After CLP, resiniferatoxin-treated-WT mice had significantly higher serum nitrate/nitrite levels (Figure 3C, p=0.037) at 6, but not at 24 h compared with WT animals. With regards to gene expression of pro- and anti-inflammatory cytokines and chemokines in splenocytes, at 24 h after CLP (Table 2), resiniferatoxin-treated-WT mice had significantly lower TNF-α mRNA levels (p=0.004, Table 2) and at 6 and 24 h, similar CCL3 and IL-10 gene expressions compared with vehicle-treated animals (p≥0.06) Table 2.
Table 2.
Effect of disruption of the transient receptor potential vanilloid 1 on cytokine and chemokine relative gene expression after cecal ligation and puncture and lipopolysaccharide challenge*
| Genotype | Treatment | Time | TNFα | CCL3 | IL-10 |
|---|---|---|---|---|---|
| Cecal Ligation and Puncture | |||||
|
| |||||
| Wild-type | Resiniferatoxin | 6 | 1.71±1.2 | 1.34 ±1.5 | 1.15±1.3 |
| Wild-type | Resiniferatoxin | 24 | 0.55± 0.07† | 0.58 ±0.18 | 0.59 ±0.14 |
| Trpv1-KO | None | 6 | 0.62 ±0.1 | 0.48±0.4 | - |
| Trpv1-KO | None | 24 | 0.92 ±1.2 | 2.16 ±1.3‡ | 2.44 ±1.3‡ |
|
| |||||
| Lipopolysaccharide | |||||
|
| |||||
| Wild-type | Capsazepine | 3 | 1.76 ±1.1 | 0.99 ±0.14 | 0.85±0.06‡ |
| Wild-type | Capsazepine | 6 | 1.23±0.29 | 1.20 ±0.17 | 1.63±0.30† |
| Wild-type | Capsazepine | 24 | 2.47±0.51† | 0.95 ±0.05 | 0.90±0.15 |
CCL3 indicates macrophage inflammatory protein-1 α, IL-10 interleukin 10, TNFα tumor necrosis factor, and Trpv1-KO animals lacking the transient receptor potential vanilloid 1 gene. Comparisons were made between the treatment group with its respective control normalized to 1 (calibrator).
indicates p<0.01
p<0.05.
Effect of resiniferatoxin on serum nitrate/nitrite levels and cytokine gene expression profile after lipopolysaccharide
There was significant upregulation of cytokine gene expression in all lipopolysaccharide-treated animals compared to phosphate buffered saline (PBS) injected controls (data not shown). After lipopolysaccharide, there were no significant differences in nitrate/nitrite levels, gene expression or serum levels of cytokines, or percentage of B-cells, macrophages and neutrophils in peritoneal lavage comparing resiniferatoxin-treated-WT and vehicle-treated animals (data not shown).
Effect of pharmacologic disruption of TRPV1 with capsazepine, a TRPV1 antagonist
Effect of capsazepine on survival after lipopolysaccharide-induced toxicity
After lipopolysaccharide-induced toxicity, pharmacologic disruption of TRPV1 in capsazepine-treated-WT animals significantly increased mortality compared with vehicle-treated-WT [1.92(1.02–3.61) hazard ratio (95% confidence interval), p=0.04, Figures 1 and 2]. Further as suggested on post hoc analysis, the pattern of harmful effect of pharmacologic disruption of TRPV1 with capsazepine was significantly different than that of genetic deficiency and pharmacologic disruption with resiniferatoxin (Figure 2, p=0.03).
Effect of capsazepine on serum nitrate/nitrite levels, cytokine gene expression profile, and peritoneal cell trafficking after lipopolysaccharide
In capsazepine-treated-WT mice, the increase in mortality after lipopolysaccharide was associated with significantly higher serum nitrate/nitrite levels (at 3h) compared to WT animals (140 ±4 vs. 90±10 μM, mean±SEM, capsazepine-treated-WT vs. WT respectively, p= 0.01, Table 3).
Table 3.
Overall effect of pharmacologic or genetic disruptions of the transient receptor potential vanilloid 1 on mortality, cytokine gene expression and serum nitrate/nitrite levels after cecal ligation and puncture or lipopolysaccharide challenge*
| Cecal Ligation and Puncture | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Perturbation | Mortality | Blood Culture | Cytokine gene expression in splenocytes | Serum levels | |||||||
| (168 h) | Colony forming units/ml | TNFα Time (h) | CCL3 Time (h) | IL-10 Time (h) | Nitrate/Nitrite Time (h) | ||||||
| Pharmacologic | 6 | 24 | 6 | 24 | 6 | 24 | 6 | 24 | 6 | 24 | |
| WT+RTX | ↑ | → | ↑ | → | ↓ | → | → | → | → | ↑ | → |
| Genetic | |||||||||||
| Trpv1-KO | ↑ | → | → | → | → | → | ↑ | → | ↑ | → | → |
| Trpv1-KO+RTX | → | ||||||||||
| Lipopolysaccharide Challenge | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Perturbation | Mortality | Cytokine gene expression in splenocytes | Serum levels | ||||||||||
| (168 h) | TNFα Time (h) | CCL3 Time (h) | IL-10 Time (h) | Nitrate/Nitrite Time (h) | |||||||||
| Pharmacologic | 3 | 6 | 24 | 3 | 6 | 24 | 3 | 6 | 24 | 3 | 6 | 24 | |
| Capsazepine | ↑ | → | → | ↑ | → | → | → | ↓ | ↑ | → | ↑ | → | → |
| Resiniferatoxin | → | → | → | → | → | → | → | → | → | → | → | → | → |
| Genetic | |||||||||||||
| Trpv1-KO | → | → | → | → | → | → | → | → | → | ↑ | → | → | → |
Arrows pointing up or down indicate statistically significant effects (in the direction of the arrow) of given disruptions compared to respective controls and arrows pointing to the right indicate no significant effect. Cells were left empty when measurements were not obtained. CCL3 indicates macrophage inflammatory protein-1 α, IL-10 interleukin 10, RTX resiniferatoxin, TNFα tumor necrosis factor, Trpv1-KO animals lacking the transient receptor potential vanilloid 1gene, and WT wild-type.
After lipopolysaccharide challenge, capsazepine-treated-WT animals had significantly lower mRNA levels of IL-10 at 3 hours (p=0.01) and higher mRNA levels of TNF-α at 24 (p=0.002), of IL-10 at 6 h (p=0.002) compared with vehicle treated controls (Tables 2 and 3). These changes in gene expression, however, were not associated with significant differences in serum cytokine levels (data not shown). In addition, in flow cytometry analyses the percentage of B-cells, macrophages and neutrophils in peritoneal lavage was similar comparing capsazepine-treated-WT animals and controls (data not shown).
Effect of genetic disruption of TRPV1 in Trpv1-KO mice
Effect of genetic disruption of TRPV1 gene on survival after bacterial peritonitis and sepsis and lipopolysaccharide-induced toxicity
After CLP-induced polymicrobial sepsis, genetic disruption of TRPV1 in Trpv1-KO mice was associated with increased risk of mortality [2.17(1.23 to 3.81) hazard ratio (95% confidence interval), p=0.01, Figures 1 and 2] compared with WT controls. In contrast, this harmful effect was not seen during lipopolysaccharide-induced toxicity, as genetic TRPV1-disruption in Trpv1-KO was associated with no significant effect on survival compared with WT animals (p=0.58).
Effect of genetic disruption of TRPV1 gene on gene expression profile and serum nitrate/nitrite levels after CLP
After CLP, both Trpv1-KO and WT mice had similar mRNA levels of TNF- α at 6 and 24 and CCL3 at 6 h (Table 2 and 3) as well as similar serum nitrate/nitrite levels at 6 and 24 h (Figure 3). However, Trpv1-KO mice had significantly higher mRNA levels of CCL3 (p=0.038) and IL-10 (p=0.026) at 24h after CLP compared with WT animals.
Effect of genetic disruption of TRPV1 gene on gene expression profile, serum nitrate/nitrite levels, and peritoneal cell trafficking after lipopolysaccharide
At 24 h after lipopolysaccharide, Trpv1-KO animals had significantly higher serum levels of IL-10 compared with WT animals (625±485 vs. 22±41 pg/ml, Trpv1-KO vs. WT respectively, p=0.02). After lipopolysaccharide, there were no significant differences in gene expression or serum levels of other cytokines, serum nitrate/nitrite levels (Table 3), and percentage of B-cells, macrophages and neutrophils in peritoneal lavage comparing Trpv1-KO and WT (data not shown).
While no baseline levels were obtained, after intraperitoneal injection of phosphate buffered saline, there were no significant differences in splenocyte TNF- α, CCL3, and IL-10 mRNA levels comparing WT (N=6) and Trpv1-KO (N=6), data not shown.
Effect of genetic disruption of TRPV1 gene on temperature changes after lipopolysaccharide
In a cohort of mice not enrolled in the survival study, we measured body temperature before and after lipopolysaccharide challenge. At baseline, Trpv1-KO mice had significantly higher temperatures compared with WT animals. After lipopolysaccharide, both, Trpv1-KO and WT animals had significant decreases up to 15 hours and then increases in body temperature, p<0.0001, Figure 4. However, after lipopolysaccharide, Trpv1-KO animals had less of a decrease in body temperature compared with WT animals (p<0.0005), Figure 4.
Figure 4.
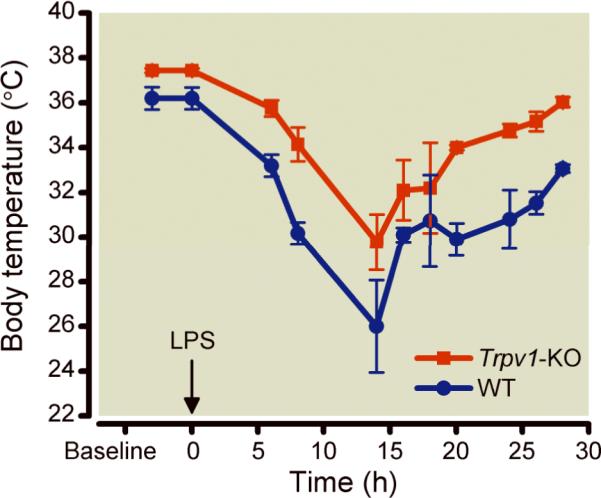
Serial mean±SEM body temperature measurements in Trpv1-knock out (Trpv1-KO) and wild-type (WT, N=4 per group) mice before and after lipopolysaccharide challenge.
In vitro effects of pharmacological inhibition of TRPV1 with capsazepine
In order to examine the effects of TRPV1 in lipopolysaccharide challenge in vitro, we measured inflammatory and anti-inflammatory cytokines mRNA levels using real-time quantitative reverse transcriptase-polymerase chain reaction in capsazepine-treated-WT and vehicle-treated-WT splenocytes challenged with 1 ug/ml lipopolysaccharide for 16 h (Figure 5). Cell viability was similar among all groups prior to RNA isolation as determined by trypan blue dye exclusion (data not show). In basal conditions (without lipopolysaccharide), capsazepine-treatment of WT splenocytes significantly decreased mRNA levels of IL-10 compared with vehicle-treatment (p=0.025). After lipopolysaccharide, capsazepine-treated-WT splenocytes had significantly less increases in mRNA levels of TNF-α (p=0.015), CCL3 (p=0.002), and IL-10 (p=0.003) compared with vehicle-treated- WT splenocytes, Figure5.
Figure 5.

In vitro effects of pharmacologic disruption with capsazepine (CPZ), an agonist of the transient receptor potential vanilloid-1 on wild-type splenocytes challenged with lipopolysaccharide for 16 h on tumor necrosis factor alpha (panel A), macrophage inflammatory protein (panel B), and interleukin-10 (panel C) gene expression. Relative expressions (y-axis) for each gene were normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and are relative to the gene expression in vehicle-treated WT splenocytes challenged with phosphate buffered saline (PBS). Results are shown as means±SEM of three separate experiments and each of these three experiments was performed by pooling the spleens of 2 animals in each of the groups studied. Log relative expressions and p values indicate comparison between groups indicated by brackets.
Discussion
We found that TRPV1 has an important role in survival, bacterial clearance from the blood, and cytokine expression profile during sepsis. During polymicrobial infection, such as in CLP, the TRPV1 receptor appears to be protective as its disruption either genetically or pharmacologically with intrathecal resiniferatoxin, which is know to result in ablation of TRPV1-expressing neurons17,24, increased mortality. Importantly, the detrimental effect of resiniferatoxin on mortality appears to be related to the presence of the TRPV1 gene as resiniferatoxin increased CLP mortality in WT but not in Trpv1-KO mice. In addition, these increases in mortality with intrathecal resiniferatoxin were associated with decreased bacterial clearance from the blood, downregulation of TNF gene expression, and increased serum levels of nitrate/nitrite early during the course of infection in WT animals. Therefore, our findings add to the existing body of literature indicating that the TRPV1 receptor and TRPV1-expressing sensory neurons have an important role in sepsis and inflammatory response.
The findings that genetic disruption of TRPV1 and intrathecal administration of resiniferatoxin increases mortality and decreases bacterial clearance from the blood in a sepsis model are relevant because resiniferatoxin is currently being pursued as a potential therapy for various pain syndromes and inflammatory states in humans and animals1,19,25–26. How might disruption of TRPV1-expressing sensory neurons possibly impact on bacterial clearance from the blood in resiniferatoxin-treated animals? One possibility is that intrathecal resiniferatoxin, which has been shown to ablate 70% of TRPV1-expressing sensory neurons in mice24, could thereby ablate a peripheral neural network that is relevant for the inflammatory response. In support of this hypothesis are studies showing that, in addition to TRPV1, sensory neurons co-express toll like receptor 4 and CD14,27–28 the receptors that recognize bacterial wall products such as lipopolysaccharide and trigger the innate immune response to bacterial infections. Others have also shown that rats' dorsal root ganglia sensory neurons co-express calcitonin gene-related peptide, TRPV1, and TNF receptors29. Therefore, one could postulate that ablation of TRPV1-expressing sensory neurons by intrathecal administration of resiniferatoxin, possibly by altering expression of CD14, TNF receptors, and toll like receptor 4 (co-expressed in those sensory neurons), could thereby impair the host's ability to recognize bacterial wall products and mount the immune response to clear bacterial infection.
Our finding that pharmacologic disruption of TRPV1 with resiniferatoxin and genetic disruption of the receptor in Trpv1-KO mice might have different effects on mortality depending on whether inflammation is caused by polymicrobial sepsis or lipopolysaccharide-induced toxicity has been previously reported in other mouse models. For example, the C3H/HeJ mice that have a toll-like receptor 4 mutation are know to be resistant to lipopolysaccharide-induced shock and death and to be highly susceptible to gram-negative bacterial infection30–31. Researchers have shown that this susceptibility to gram-negative infection could be reversed by pretreatment of the C3H/HeJ mice with recombinant TNF and IL-1α32 thus suggesting that cytokine generation by the host has an important role in the response to bacterial infection. In the present investigation, we found that after CLP, resiniferatoxin induced decreases in bacterial clearance from the blood that was coupled with downregulation of TNF expression and increases in nitrate/nitrite levels. These findings then suggest that the resiniferatoxin-ablation of the TRPV1-sensory neurons was detrimental to survival possibly due to alterations on host response to bacterial infection.
Our findings that capsazepine administered prophylacticaly (72 hours before) and continued for 72 h after lipopolysaccharide challenge significantly increases mortality are in concert with results reported by others. Rats treated with capsazepine before intravenous administration of lipopolysaccharide had significantly decreased survival and worsened lipopolysaccharide-induced hypotension12. In that rat study of lipopolysaccharide-induced toxicity, capsazepine's deleterious effects on survival were associated with attenuation of sepsis-induced sympathetic response which likely explains worsening lipopolysaccharide-induced hypotension. Conversely, in a study of CLP-induced polymicrobial sepsis in mice, one dose of capsazepine administered 30 min before the onset of sepsis significantly increased survival and decreased sepsis-induced pulmonary and hepatic neutrophil infiltration13. While the discrepancy of these results could possibly be related to differences in species studied, dose, and timing of administration of capsazepine and various TRPV1 agonists and antagonists, these conflicting results further suggest that the overall effect of TRPV1 disruptions during sepsis depends on the sepsis-inducing event and the method of disruption of the receptor and the TRPV1-expressing neurons.
It is interesting that during lipopolysaccharide-induced toxicity, TRPV1 antagonism with capsazepine decreased survival in a pattern that appears to be different than that observed with genetic or pharmacologic disruption (with resiniferatoxin) of the receptor. These discrepant results raise the possibility that the detrimental effects of capsazepine on lipopolysaccharide-induced mortality could result from TRPV1-unrelated effects. In fact, some previous reports showing that capsazepine can affect channels other than TRPV1 support this possibility. For example, capsazepine has been shown to reversibly inhibit the effects of nicotine in sensory neurons14 and to block other voltage-activated calcium channels in dorsal root ganglia15. One could postulate that capsazepine's inhibitory effects on acetylcholine receptors14 might in part have contributed to the increased mortality in lipopolysaccharide-induced toxicity. Recent reports showing that activation of nicotinic acetylcholine receptors (by vagal stimulation) attenuate the inflammatory response to lipopolysaccharide in vitro and in vivo and increases survival in lipopolysaccharide-induced toxicity33 support that possibility. Further supporting the hypothesis that capsazepine worsens survival in sepsis independently of TRPV1 are findings that capsazepine inhibits the sympathetic response to sepsis and thereby worsens lipopolysaccharide-induced hypotension in septic rats12. Therefore, the findings that capsazepine but not resiniferatoxin-treatment or genetic deficiency of TRPV1 worsened survival after lipopolysaccharide, suggest that the detrimental effects of capsazepine during lipopolysaccharide-induced toxicity are possibly TRPV1-unrelated.
Our findings that TRPV1 disruption impacts on cytokine and chemokine expression during sepsis are of interest and previously undescribed. We found that in vivo, after CLP, resiniferatoxin treatment significantly downregulated TNF gene expression and in vitro, capsazepine downregulates TNF, CCL3, and IL10 in lipopolysaccharide-treated splenocytes. These findings add to the previously reported inhibitory effects of capsazepine and resiniferatoxin on activation of inducible nitric oxide synthase and activation of key mediators of the host's immune response, the transcription factors NF-κB and AP-134–35. Therefore taken together our findings and that of others suggest that TRPV1 has an important role in the host immune response to bacterial infection and lipopolysaccharide-induced toxicity that can impact on the expression of pro-inflammatory and anti-inflammatory cytokines.
In summary, disruption of TRPV1, genetically, in Trpv1-KO mice, or pharmacologically (with intrathecal resiniferatoxin or capsazepine), can impact mortality, decrease bacterial clearance from the blood, and alter cytokine gene expression during sepsis. As therapeutic agents targeting the TRPV1 receptor are being developed to treat a number of pain syndromes and inflammatory condition in animals and humans, these findings are relevant. While one must be circumspect about extrapolating findings in rodent to other species, our results suggest that therapeutic strategies that disrupt or inhibit TRPV1 and TRPV1-expressing neurons warrant careful monitoring.
Acknowledgments
This research was supported by the Intramural Research Program of the National Institutes of Health Clinical Center, National Institutes of Health, Bethesda, Maryland and by The Sheikh Zayed Institute for Pediatric Surgical Innovation, Children's National Medical Center, Washington, District of Columbia.
The authors are grateful for the expert technical support provided by Yuka Uehara, BS (Fellow), Julie Horowitz BS (Fellow), and LaShon Middleton ALATR (Technician) from the Department of Perioperative Medicine, National Institutes of Health Clinical Center, National Institutes of Health, Bethesda, MD, USA.
Footnotes
Parts of this work were presented at the American Society of Anesthesiologists annual meeting on October 21, 2009 in New Orleans, Louisiana.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Okajima K, Harada N. Regulation of inflammatory responses by sensory neurons: Molecular mechanism(s) and possible therapeutic applications. Curr Med Chem. 2006;13:2241–51. doi: 10.2174/092986706777935131. [DOI] [PubMed] [Google Scholar]
- 2.Alawi K, Keeble J. The paradoxical role of the transient receptor potential vanilloid 1 receptor in inflammation. Pharmacol Ther. 2010;125:181–95. doi: 10.1016/j.pharmthera.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 3.Richardson JD, Vasko MR. Cellular mechanisms of neurogenic inflammation. J Pharmacol Exp Ther. 2002;302:839–45. doi: 10.1124/jpet.102.032797. [DOI] [PubMed] [Google Scholar]
- 4.Karai L, Brown DC, Mannes AJ, Connelly ST, Brown J, Gandal M, Wellisch OM, Neubert JK, Olah Z, Iadarola MJ. Deletion of vanilloid receptor 1-expressing primary afferent neurons for pain control. J Clin Invest. 2004;113:1344–52. doi: 10.1172/JCI20449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tender GC, Walbridge S, Olah Z, Karai L, Iadarola M, Oldfield EH, Lonser RR. Selective ablation of nociceptive neurons for elimination of hyperalgesia and neurogenic inflammation. J Neurosurg. 2005;102:522–5. doi: 10.3171/jns.2005.102.3.0522. [DOI] [PubMed] [Google Scholar]
- 6.Davis JB, Gray J, Gunthorpe MJ, Hatcher JP, Davey PT, Overend P, Harries MH, Latcham J, Clapham C, Atkinson K, Hughes SA, Rance K, Grau E, Harper AJ, Pugh PL, Rogers DC, Bingham S, Randall A, Sheardown SA. Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature. 2000;405:183–7. doi: 10.1038/35012076. [DOI] [PubMed] [Google Scholar]
- 7.Arnalich F, Hernanz A, Jimenez M, Lopez J, Tato E, Vazquez JJ, Montiel C. Relationship between circulating levels of calcitonin gene-related peptide, nitric oxide metabolites and hemodynamic changes in human septic shock. Regul Pept. 1996;65:115–21. doi: 10.1016/0167-0115(96)00080-8. [DOI] [PubMed] [Google Scholar]
- 8.Berg RM, Strauss GI, Tofteng F, Qvist T, Edvinsson L, Fahrenkrug J, Qvist J, Fonsmark L, Skinhoj P, Moller K. Circulating levels of vasoactive peptides in patients with acute bacterial meningitis. Intensive Care Med. 2009;35:1604–8. doi: 10.1007/s00134-009-1515-3. [DOI] [PubMed] [Google Scholar]
- 9.Beer S, Weighardt H, Emmanuilidis K, Harzenetter MD, Matevossian E, Heidecke CD, Bartels H, Siewert JR, Holzmann B. Systemic neuropeptide levels as predictive indicators for lethal outcome in patients with postoperative sepsis. Crit Care Med. 2002;30:1794–8. doi: 10.1097/00003246-200208000-00020. [DOI] [PubMed] [Google Scholar]
- 10.Orliac ML, Peroni RN, Abramoff T, Neuman I, Podesta EJ, Adler-Graschinsky E. Increases in vanilloid TRPV1 receptor protein and CGRP content during endotoxemia in rats. Eur J Pharmacol. 2007;566:145–52. doi: 10.1016/j.ejphar.2007.03.032. [DOI] [PubMed] [Google Scholar]
- 11.Clark N, Keeble J, Fernandes ES, Starr A, Liang L, Sugden D, de Winter P, Brain SD. The transient receptor potential vanilloid 1 (TRPV1) receptor protects against the onset of sepsis after endotoxin. FASEB J. 2007;21:3747–55. doi: 10.1096/fj.06-7460com. [DOI] [PubMed] [Google Scholar]
- 12.Wang Y, Novotny M, Quaiserova-Mocko V, Swain GM, Wang DH. TRPV1-mediated protection against endotoxin-induced hypotension and mortality in rats. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1517–23. doi: 10.1152/ajpregu.00005.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ang SF, Moochhala SM, Bhatia M. Hydrogen sulfide promotes transient receptor potential vanilloid 1-mediated neurogenic inflammation in polymicrobial sepsis. Crit Care Med. 2010;38:619–28. doi: 10.1097/CCM.0b013e3181c0df00. [DOI] [PubMed] [Google Scholar]
- 14.Liu L, Simon SA. Capsazepine, a vanilloid receptor antagonist, inhibits nicotinic acetylcholine receptors in rat trigeminal ganglia. Neurosci Lett. 1997;228:29–32. doi: 10.1016/s0304-3940(97)00358-3. [DOI] [PubMed] [Google Scholar]
- 15.Docherty RJ, Yeats JC, Piper AS. Capsazepine block of voltage-activated calcium channels in adult rat dorsal root ganglion neurones in culture. Br J Pharmacol. 1997;121:1461–7. doi: 10.1038/sj.bjp.0701272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Szolcsanyi J, Szallasi A, Szallasi Z, Joo F, Blumberg PM. Resiniferatoxin. An ultrapotent neurotoxin of capsaicin-sensitive primary afferent neurons. Ann N Y Acad Sci. 1991;632:473–5. doi: 10.1111/j.1749-6632.1991.tb33161.x. [DOI] [PubMed] [Google Scholar]
- 17.Olah Z, Szabo T, Karai L, Hough C, Fields RD, Caudle RM, Blumberg PM, Iadarola MJ. Ligand-induced dynamic membrane changes and cell deletion conferred by vanilloid receptor 1. J Biol Chem. 2001;276:11021–30. doi: 10.1074/jbc.M008392200. [DOI] [PubMed] [Google Scholar]
- 18.Brown DC, Iadarola MJ, Perkowski SZ, Erin H, Shofer F, Laszlo KJ, Olah Z, Mannes AJ. Physiologic and antinociceptive effects of intrathecal resiniferatoxin in a canine bone cancer model. Anesthesiology. 2005;103:1052–9. doi: 10.1097/00000542-200511000-00020. [DOI] [PubMed] [Google Scholar]
- 19.Gunthorpe MJ, Szallasi A. Peripheral TRPV1 receptors as targets for drug development: new molecules and mechanisms. Curr Pharm Des. 2008;14:32–41. doi: 10.2174/138161208783330754. [DOI] [PubMed] [Google Scholar]
- 20.Bates BD, Mitchell K, Keller JM, Chan CC, Swaim WD, Yaskovich R, Mannes AJ, Iadarola MJ. Prolonged analgesic response of cornea to topical resiniferatoxin, a potent TRPV1 agonist. Pain. 2010;149:522–8. doi: 10.1016/j.pain.2010.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cui X, Besch V, Khaibullina A, Hergen A, Quezado M, Eichacker P, Quezado ZM. Neuronal nitric oxide synthase deficiency decreases survival in bacterial peritonitis and sepsis. Intensive Care Med. 2007;33:1993–2003. doi: 10.1007/s00134-007-0814-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hylden JL, Wilcox GL. Intrathecal morphine in mice: A new technique. Eur J Pharmacol. 1980;67:313–6. doi: 10.1016/0014-2999(80)90515-4. [DOI] [PubMed] [Google Scholar]
- 23.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mishra SK, Hoon MA. Ablation of TrpV1 neurons reveals their selective role in thermal pain sensation. Mol Cell Neurosci. 2010;43:157–63. doi: 10.1016/j.mcn.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schumacher MA. Transient Receptor Potential Channels in Pain and Inflammation: Therapeutic Opportunities. Pain Pract. 2010;10:185–200. doi: 10.1111/j.1533-2500.2010.00358.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nassini R, Materazzi S, De Siena G, De Cesaris F, Geppetti P. Transient receptor potential channels as novel drug targets in respiratory diseases. Curr Opin Investig Drugs. 2010;11:535–42. [PubMed] [Google Scholar]
- 27.Wadachi R, Hargreaves KM. Trigeminal nociceptors express TLR-4 and CD14: A mechanism for pain due to infection. J Dent Res. 2006;85:49–53. doi: 10.1177/154405910608500108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barajon I, Serrao G, Arnaboldi F, Opizzi E, Ripamonti G, Balsari A, Rumio C. Toll-like receptors 3, 4, and 7 are expressed in the enteric nervous system and dorsal root ganglia. J Histochem Cytochem. 2009;57:1013–23. doi: 10.1369/jhc.2009.953539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Y, Ji A, Weihe E, Schafer MK. Cell-specific expression and lipopolysaccharide-induced regulation of tumor necrosis factor alpha (TNFalpha) and TNF receptors in rat dorsal root ganglion. J Neurosci. 2004;24:9623–31. doi: 10.1523/JNEUROSCI.2392-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cross A, Asher L, Seguin M, Yuan L, Kelly N, Hammack C, Sadoff J, Gemski P., Jr. The importance of a lipopolysaccharide-initiated, cytokine-mediated host defense mechanism in mice against extraintestinally invasive Escherichia coli. J Clin Invest. 1995;96:676–86. doi: 10.1172/JCI118110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vazquez-Torres A, Vallance BA, Bergman MA, Finlay BB, Cookson BT, Jones-Carson J, Fang FC. Toll-like receptor 4 dependence of innate and adaptive immunity to Salmonella: Importance of the Kupffer cell network. J Immunol. 2004;172:6202–8. doi: 10.4049/jimmunol.172.10.6202. [DOI] [PubMed] [Google Scholar]
- 32.Cross AS, Sadoff JC, Kelly N, Bernton E, Gemski P. Pretreatment with recombinant murine tumor necrosis factor alpha/cachectin and murine interleukin 1 alpha protects mice from lethal bacterial infection. J Exp Med. 1989;169:2021–7. doi: 10.1084/jem.169.6.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Yang H, Ulloa L, Al-Abed Y, Czura CJ, Tracey KJ. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2003;421:384–8. doi: 10.1038/nature01339. [DOI] [PubMed] [Google Scholar]
- 34.Oh GS, Pae HO, Seo WG, Kim NY, Pyun KH, Kim IK, Shin M, Chung HT. Capsazepine, a vanilloid receptor antagonist, inhibits the expression of inducible nitric oxide synthase gene in lipopolysaccharide-stimulated RAW264.7 macrophages through the inactivation of nuclear transcription factor-kappa B. Int Immunopharmacol. 2001;1:777–84. doi: 10.1016/s1567-5769(01)00012-1. [DOI] [PubMed] [Google Scholar]
- 35.Chen CW, Lee ST, Wu WT, Fu WM, Ho FM, Lin WW. Signal transduction for inhibition of inducible nitric oxide synthase and cyclooxygenase-2 induction by capsaicin and related analogs in macrophages. Br J Pharmacol. 2003;140:1077–87. doi: 10.1038/sj.bjp.0705533. [DOI] [PMC free article] [PubMed] [Google Scholar]