

Abstract
Barrett’s esophagus is a condition in which the normal stratified squamous epithelium of the distal esophagus is replaced by intestinal metaplasia. For more than three decades the prevailing clinical paradigm has been that Barrett’s esophagus is a complication of symptomatic reflux disease that predisposes to esophageal adenocarcinoma, yet no clinical strategy for cancer prevention or early detection based on this paradigm has been proven to reduce esophageal adenocarcinoma mortality in a randomized clinical trial in part because only about 5-10% of individuals with Barrett’s esophagus develop esophageal adenocarcinoma. Recent research indicates that Barrett’s metaplasia is an adaptation for mucosal defense in response to chronic reflux in most individuals. The risk of progressing to esophageal adenocarcinoma is determined by development of genomic instability and dynamic clonal evolution in the distal esophagus modulated by host and environmental risk and protective factors, including inherited genotype. The challenge in Barrett’s esophagus lies in integrating knowledge about genomic instability and clonal evolution into clinical management to increase the lifespans and quality of life of individuals with this condition.
Background
Prevailing paradigm and clinical management
In individuals with Barrett’s esophagus, the distal portion of the normal esophageal stratified squamous epithelium is replaced by specialized intestinal metaplasia(1). The paradigm that Barrett’s esophagus arises as a complication of chronic symptomatic gastroesophageal reflux disease and predisposes to esophageal adenocarcinoma has dominated clinical thought for more than three decades(2, 3). Practice guidelines have endorsed endoscopic screening of individuals with symptomatic reflux to detect Barrett’s(4), endoscopic biopsy surveillance of Barrett’s for early detection of esophageal adenocarcinoma(1, 4), and intervention typically reserved for individuals with high-grade dysplasia or cancer(1, 4). Yet, the rate of progression from Barrett’s esophagus to esophageal adenocarcinoma is low, and as a consequence 90-95% of individuals diagnosed with Barrett’s esophagus follow a benign course, living out their lives without developing or dying of esophageal adenocarcinoma(5-9). Current strategies to decrease the cancer risk in Barrett’s esophagus did not anticipate these low rates of progression to, and death from, esophageal adenocarcinoma. These strategies have been further compromised by use of morphological assessment of dysplasia for risk assessment in Barrett’s esophagus. Although dysplasia is frequently used as a surrogate endpoint in Barrett’s esophagus research, neither high-grade dysplasia nor any other grade of dysplasia fulfill criteria for valid surrogates since dysplasia does not accurately represent the true endpoint, esophageal adenocarcinoma, because dysplasia classification is subjective, not reproducible, and because it does not provide robust discrimination between individuals who will regress or remain stable for prolonged periods and those who will develop life-threatening disease(10-12).
Although some data support endoscopic biopsy surveillance for early detection(1, 12, 13), aspirin and other nonsteroidal anti-inflammatory drugs (NSAIDs) for chemoprevention(14, 15), and medical surgical interventions including ablation for cancer control in individuals with Barrett’s esophagus(16-19), there are as yet no randomized control trials that have convincingly demonstrated reduction in the incidence and mortality of esophageal adenocarcinoma(1, 12). In summary, no management strategy for early detection or prevention based on the prevailing clinical paradigm has yet been proven to reduce mortality of esophageal adenocarcinoma or all cause mortality.
On the Horizon
Multilevel evolution in Barrett’s esophagus
In this manuscript, we will explore an overarching evolutionary theory of the development of Barrett’s esophagus and its progression to esophageal adenocarcinoma that incorporates inherited changes in the constitutive genome and clonal somatic genomic instability in Barrett’s epithelium that predispose to esophageal adenocarcinoma. In other words, there has been natural selection at the level of individuals in our ancestors, and there is ongoing selection at the level of cells in the Barrett’s epithelium. As part of the overarching evolutionary theory, we will examine a relatively new theory that specialized intestinal metaplasia is a successful adaptation to gastroesophageal reflux that remains stable for the lifetimes of most individuals. This theory suggests that the propensity to develop specialized metaplasia in response to gastroesophageal reflux is the product of natural selection at the level of individuals and natural selection at the level of cells in a reflux environment.
Defining the constitutive genome from which somatic clones evolve
At the level of the individual, there is substantial evidence for an inherited component to Barrett’s esophagus and esophageal adenocarcinoma based on case reports, twin studies, familial clusters and clinical series(20-22). For example, families have been described that show strong predispositions to esophageal adenocarcinoma, Barrett’s esophagus and reflux disease(21, 23). Twin studies of reflux disease suggest heritability of 30%-40%, and twins have been reported to develop Barrett’s esophagus suggesting a role for genetic susceptibility in these conditions(24-26). Larger studies support a role for genetic susceptibility for Barrett’s esophagus and esophageal adenocarcinoma(27). In one study, 7.3% of individuals presenting with Barrett’s esophagus or esophageal adenocarcinoma were reported to have a familial component(22). In clinical practice, a family history is now recommended for physicians seeing individuals with Barrett’s esophagus and esophageal adenocarcinoma(28). Advances in whole genome sequencing of family members(29) will greatly accelerate discovery of inherited genetic alterations that predispose to Barrett’s esophagus, esophageal adenocarcinoma or both. Once identified, these individuals could be enrolled in trials to prevent esophageal adenocarcinoma or detect it when early and curable.
In 1976, Nowell described another level of evolution when he hypothesized that “Acquired genetic lability permits stepwise selection of variant sublines and underlies tumor progression”(30). At this level, clonal evolutionary theory focuses on the genetics of the evolving clonal populations in the distal esophagus, their interactions with each other and their relationship to the native esophageal squamous epithelium. There is substantial evidence that evolution of esophageal adenocarcinoma is associated with potentially modifiable host and environmental risk (e.g., obesity and cigarette smoke) and protective (e.g., aspirin and other NSAIDs) factors in the population(12).
Genomic instability, clonal evolution, and clonal evolutionary parameters in Barrett’s esophagus
The genomes of esophageal adenocarcinomas contain complex alterations that disrupt regulatory pathways and are associated with profound changes in the transcriptome and proteome(12, 31-34). Several lines of evidence indicate that these abnormalities arise by a process similar to that postulated by Nowell, including genomic instability, generation of genetic variants, natural selection and dynamic evolution of clones(12). Most esophageal adenocarcinomas, on the order of 90-95%, arise in association with chromosome instability that leads to gains, losses or loss of heterozygosity (LOH) of large regions of chromosomes(31, 35, 36). Studies suggest that evaluation of the observable patterns generated by clonal evolution – chromosomal alterations and instability, temporal order of mutation events, clonal genetic diversity and clonal expansions – may facilitate risk assessment in Barrett’s esophagus using novel approaches that can be translated to the clinic and potentially to other cancers.
Spatial data from Barrett’s esophagus and esophageal adenocarcinoma from the same patient at the same time have been used to develop models of clonal evolution, leading to the hypothesis that 9p LOH (and CDKN2A mutation and methylation) are early events, occurring before 17p LOH (and TP53 mutations), which predispose to development of DNA content abnormalities (tetraploidy, aneuploidy) during evolution to esophageal adenocarcinoma(37).
9p and 17p LOH, CDKN2A mutations and methylation and TP53 mutations have been found to be highly associated with clonal expansions, suggesting that these alterations provide a selective advantage to a Barrett’s clone(38). However, 9q LOH and 17q LOH as well as microsatellite shifts behave as neutral genetic abnormalities. Although neutral chromosome changes are occasionally detected in clonal expansions, these typically represent hitchhikers (“passengers”) on known selected genomic abnormalities (“drivers”)(38). Increasing sizes of clones with 17p (TP53) LOH, tetraploidy and aneuploidy are associated with increasing risk of progression to esophageal adenocarcinoma, but sizes of clones with CDKN2A abnormalities are not after controlling for 17p LOH(39). This result supports the hypothesis that expansion of a genetically unstable clone increases risk of progression to esophageal adenocarcinoma. One prediction of this hypothesis is that the genetically unstable clone produces viable variants during an expansion that increase the probability of evolving to cancer. This hypothesis was evaluated in another study which reported that clonal diversity, as assessed by number of clones, Shannon Index, and mean pairwise divergence, were all associated with an increased rate of progression to esophageal adenocarcinoma even when 17p LOH and DNA content abnormalities were controlled for in the model(40). A follow-up study evaluated a wide range of diversity metrics from the ecology literature as well as defining clones based on different sets of loci and lesions(41). It found that every diversity measure and every method for defining a clone produced biomarkers that were highly statistically significant (p < 0.001) predictors of progression to esophageal adenocarcinoma in this cohort. This suggests that diversity measures will be robust biomarkers of progression in Barrett’s esophagus. Clonal genetic diversity has also been reported at the crypt level in Barrett’s esophagus(42), and these preexisting genetic variants could be a source of development of resistance to interventions to prevent cancer. In summary, several elements of Nowell’s theory, including manifestations of genomic (chromosomal) instability, expansion of genetically unstable clones, and generation of viable clonal variants, contribute to clonal evolution from Barrett’s esophagus to esophageal adenocarcinoma.
Specialized intestinal metaplasia and mucosal defense
As important as the genomic results are in identifying individuals at increased risk for neoplastic clonal evolution to esophageal adenocarcinoma, they provide little insight into the 90-95% of individuals who will not progress to esophageal adenocarcinoma during their lifetimes. Barrett’s esophagus arises in an environment of chronic injury associated with acid and bile reflux into the esophagus(12). A novel theory has been recently proposed that specialized intestinal metaplasia of the distal esophagus is a successful adaptation for mucosal defense that persists for the lifetime of most individuals(43) (Figure 1). One combined expression and proteomic study concluded that “…Barrett’s metaplasia may be regarded as a specific microevolution allowing for accumulation of mucosal morphological and physiological changes that better protect against reflux injury(44).” These results suggest that specialized intestinal metaplasia provides a barrier against reflux injury that confers a selective advantage over the native esophageal epithelium, leading to expansion of intestinal metaplasia in the distal esophagus.
Figure 1. Barrett’s specialized intestinal metaplasia and mucosal defense.
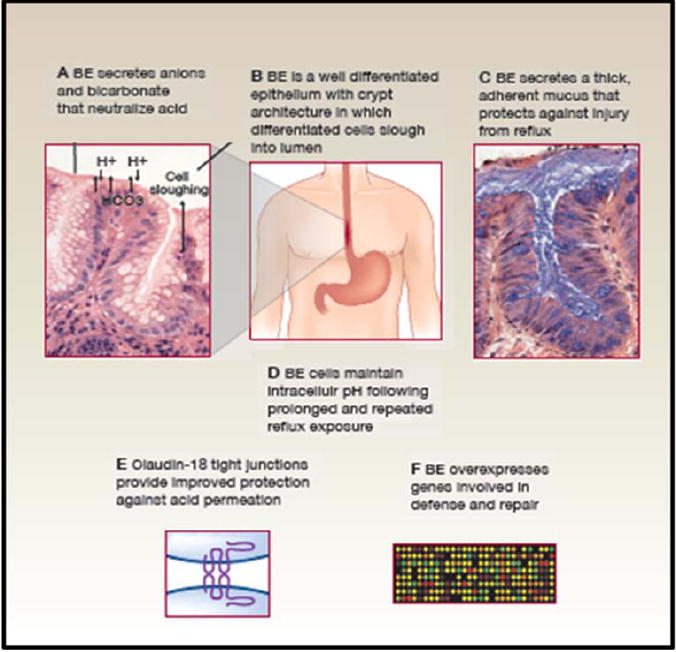
Barrett’s metaplasia arises in an environment of chronic reflux in which the distal esophagus is exposed to high levels of local and systemic damage from acid, bile, and tobacco products as well as the inflammatory responses to the injury(12, 55-60). All are mutagenic. Barrett’s metaplasia has a number of defenses against this mutagenic environment that are not found in esophageal squamous epithelium(12, 43). A. Barrett’s metaplasia secretes anions, including bicarbonate, that participate in buffering acid reflux (61). B. Barrett’s metaplasia is a well differentiated epithelium with crypt architecture in which putative stem cells residing at the base give rise to proliferating transient amplifying cells and differentiated cells that are sloughed into the lumen. This architecture has been proposed to be tumor suppressive because mutations in transient amplifying or differentiated non-stem cells would be shed from the body before they could accumulate the serial mutations that lead to cancer(62). C Barrett’s metaplasia secretes a thick adherent mucus not present in squamous esophageal epithelium for defense against acid and bile reflux(45, 46, 63)(64). D. Barrett’s esophageal cells maintain physiological intracellular pH following prolonged and repeated reflux exposure(65). E. The tight junctions of Barrett’s metaplasia overexpress claudin 18 and several other claudins, including 1, 4, 12 and 23, that provide protection against acid permeation(66). F. A combined expression and proteomics study of Barrett’s metaplasia reported overexpression of genes involved in mucosal defense and repair(44).
Chromosomal instability is associated with disruption of mucosal defense
Two manuscripts published in 1989 evaluated mucous secretion in Barrett’s esophagus by transmission electron microscopy. The first reported that specialized intestinal metaplasia typically has active intracellular mechanisms for synthesis and transport of mucous, including glycogen aggregates, rough endoplasmic reticulum, Golgi apparatus and mucous secretory granules(45). Barrett’s esophagus thus has a spectrum of morphologic features thought to participate in mucosal defense that overlaps with normal gastric and small intestinal mucosa(45). The second manuscript reported that biopsies of individuals with Barrett’s esophagus who had flow cytometric DNA content abnormalities (tetraploidy or aneuploidy), including some who progressed to esophageal adenocarcinoma, had reduced mucous content as well striking ultrastructural abnormalities that included distended rough endoplasmic reticulum, increased cytoplasmic glycogen aggregates, small or dysmorphic Golgi apparatus, atypical mucous granules and a “simplified cytoplasm” with fewer organelles and mucous granules(46). Currently available data are not sufficient to determine whether genomic instability develops as a consequence of loss of the barrier function with genotoxic injury to stem cells at the base of the crypts, whether chromosome instability leads to disruption of the barrier function of Barrett’s metaplasia, whether they are independent of each other or whether both interact in a vicious cycle that leads to selfish cell proliferation and evolution to esophageal adenocarcinoma.
If the goals are to reduce the mortality of esophageal adenocarcinoma and improve the quality of life of individuals with Barrett’s esophagus who will not progress to cancer, then more accurate risk assessment than currently exists will be required to distinguish between benign early adaptations that will not shorten the lifespan of an individual and clonal evolution of life threatening early disease. A phased approach has recently been proposed using a series of risk models, involving four strong population attributable risk factors, obesity, tobacco, reflux and diet low in fruits and vegetables for the general population; history, physical exam and blood tests for primary care; and blood and tissue based biomarkers for secondary care(12). Although reflux disease and Barrett’s esophagus may inform several of the risk models, this approach differs from the previous paradigm in that it does not depend on a symptomatic reflux → Barrett’s esophagus → esophageal adenocarcinoma paradigm that currently guides clinical management. Here, we focus on the transition from Barrett’s esophagus to esophageal adenocarcinoma as a process of dynamic clonal evolution modulated by both inherited genotype and environmental risk and protective factors.
Converting measures of chromosome instability, chromosomal mutation rate and clonal diversity to risk assessment tools
A ten year prospective cohort study reported that a panel of 9p LOH, 17p LOH and DNA content abnormalities (tetraploidy and/or aneuploidy) was a strong predictor of progression from Barrett’s esophagus to esophageal adenocarcinoma (relative risk = 38.7; 95% CI = 10.8-138.5; p<0.001)(47). Individuals with all three manifestations of chromosome instability at baseline had a 79.1% five-year cumulative incidence of cancer compared to a zero percent cumulative incidence of cancer to nearly eight years in individuals who had none of the markers.
Rapidly advancing technologies create opportunities for evaluating somatic genomic evolution on a single platform(31), making it more suitable for the clinical laboratory. Single nucleotide polymorphism (SNP) arrays provide flexible platforms for a variety of approaches to investigate cancer, including GWAS, customized platforms to assess specific regions of the genome and a uniform platform for genome-wide assessment of somatic LOH, copy change and aneuploidy(31, 35, 36).
Indentifying evolutionary mechanisms by which candidate chemoprevention agents act and anticipating evolution of resistance for clinical trials
Importantly, current use of aspirin and other NSAIDs in the ten year prospective cohort study above was associated with marked risk reduction in patients with two or more chromosome instability biomarkers at baseline. NSAID non-users had a 79% 10-year cumulative incidence of esophageal adenocarcinoma compared to 30% for current NSAID users (p<0.001)(47). In a separate study of the same cohort, current use of aspirin and other NSAIDs was associated with decreased progression to DNA content abnormalities (tetraploidy, aneuploidy) as well as esophageal adenocarcinoma(48). These results support development of clinical trials using aspirin or other NSAIDs. Further studies in observational cohorts could inform the trials by elucidating the evolutionary mechanisms of the NSAID protective association and identifying genomic abnormalities associated with NSAID resistance and evolution to cancer for early stopping criteria in trials.
Clonal evolution is a dynamic, stochastic process
Because somatic evolution is a stochastic process that is dominated by outliers with greatest fitness in the cell population, biomarkers for risk stratification and prediction, based on the presence or absence of a phenotypic or molecular lesion in a population of cells, are inherently unstable. The clone with the marker may not have yet evolved, it may be a small minority that is difficult to detect, or it may go extinct in the future (Figure 2). An alternative is to measure the rate of somatic evolution, and to develop interventions that slow the rate of progression(49). Using high density SNP arrays we have found evidence for large clonal expansions, clonal extinction, and long periods of relative stasis lasting over 12 years in which the clonal composition of the Barrett’s segment was relatively stable with the exception of the accumulation of small copy number and LOH lesions (Figure 2). This can be the prototype for large, well designed studies using a uniform platform to determine the extent to which chromosome instability, clonal expansions, and clonal diversity predict progression to esophageal adenocarcinoma as well as identifying benign subsets of individuals who require no clinical intervention because of their low risk. While genomic biomarkers characterize somatic alterations at snapshots of time during patient evaluation, clonal evolutionary biomarkers have the potential to summarize changes of genomic state temporally that may be widely applicable to a broad range of neoplasms because genomic instability, selection of variants, and clonal expansions are believed to occur during progression in most, if not all, cancers.
Figure 2. Benign clonal evolution in one patient with Barrett’s esophagus studied longitudinally over 16 years.
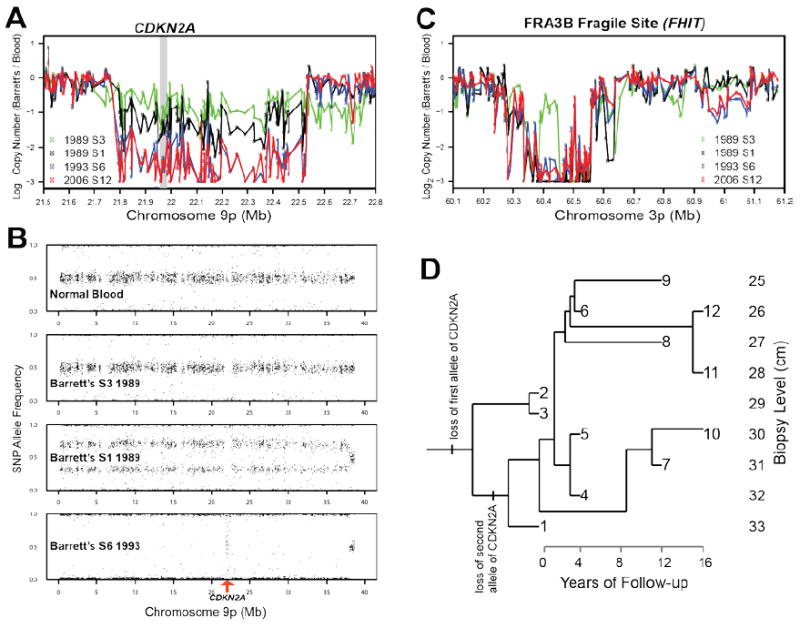
Purified Barrett’s epithelium from endoscopic biopsies was assayed with Illumina 317K SNP arrays and compared to a blood sample control. (A) Copy number analysis, normalized by SNP intensities from blood, reveal a single copy loss at CDKN2A in samples 2 (data not shown) and 3 in 1989, but homozygous deletion in CDKN2A in sample 1 and all samples from following years. At first endoscopy, in 1989, two clones were detected, one with a small deletion of one allele at the CDKN2A locus, and the other with copy neutral LOH of the entire 9p arm with the CDKN2A deleted allele, generating biallelic deletion at CDKN2A. (B) The SNP allele frequencies reveal a focal deletion in the CDKN2A locus in samples 2 and 3 in 1989, but sample 1 included a mixture of the clone from samples 2 and 3 with a new clone with copy neutral LOH of 9p and biallelic deletion of CDKN2A. All samples from 1993 and later show that the clone with biallelic deletion of CDKN2A went to fixation, leading to random noise in the allele frequencies for the SNPs in that region, seen in the vertical (“waterfall”) band in the bottom panel of B. The fact that the rest of the 9p arm remains diploid can be seen in the copy number data (A). The clone with deletion of the single allele of CDKN2A, which extends past 22.5Mb on chromosome 9p, also had a single deletion in fragile site FRA3B at 60.42Mb that distinguished it from the other clones (C). This and other lesions of the clone in samples 2 and 3 were never observed again after 1989, suggesting that that clone was driven extinct by the clone from sample 1, with biallelic deletion of CDKN2A. A Camin-Sokal maximum parsimony reconstruction of the genealogy of clones (D), based on polymorphic copy number lesions in 283 loci across the entire genome in the Barrett’s biopsies, shows that there was only one large clonal expansion, between 1989 and 1993. After 1993, the Barrett’s segment remained stable, with the accumulation of small interstitial lesions but no clonal expansions, no aneuploidy and no progression to cancer.
Characterizing phenotypes of benign and dangerous clonal populations
Although expression and proteomic studies are largely in the discovery phase in Barrett’s esophagus and esophageal adenocarcinoma, their potential to be translated to the clinic to identify a population of individuals who will follow a benign course should not be underestimated, especially in view of the results of the combined expression and proteomic study described above(44). As another example, a recent study of glioblastoma and glioma from The Cancer Genome Atlas Research Network reported that DNA methylation analysis using Illumina Golden Gate and Infinium arrays combined with expression analysis identified a subset of patients with significantly improved prognosis(50). DNA methylation, expression and protein profiling may be an approach to distinguishing specialized intestinal metaplasia that has adapted to reflux and will remain stable for prolonged periods from a genetically unstable clone with greater risk of esophageal adenocarcinoma.
Ongoing randomized trials
A large chemoprevention trial is being conducted in the UK in individuals with Barrett’s esophagus without high grade dysplasia, using two doses of aspirin (0 vs. 300 mg) with an all cause mortality endpoint(51). The same trial also includes randomization to low- and high-dose proton pump inhibitor therapy. This trial may provide useful information about the role of aspirin and proton pump therapy for chemoprevention in individuals without high grade dysplasia in Barrett’s esophagus. A randomized trial of endoscopic surveillance with biopsies every two years for about 10 years versus endoscopy for symptoms is in the recruitment stage in the UK(52).
Potential game changer: Identification of infectious agents that modulate clonal evolution in the distal esophagus
Helicobacter pylori has been reported to be associated with an increased risk of gastric adenocarcinoma and decreased risk of esophageal adenocarcinoma(53). Recent research suggests that the microbiome of the distal esophagus is different in health and disease(54). If further research discovers an infectious agent for esophageal adenocarcinoma, then the approach to prevention could change dramatically with the potential use of vaccines or antibiotics.
Summary
New research suggests that specialized intestinal metaplasia contributes to mucosal defense in most individuals, and the vast majority of these individuals will live out their lives unaffected by esophageal adenocarcinoma. However, a small number of individuals in the population develop profound changes in their genomes that lead to esophageal adenocarcinoma(12, 31, 35-37). Advances in esophageal genomics, and detection of individuals with inherited mutations placing them at risk for esophageal adenocarcinoma will inform increasingly sophisticated risk models to improve identification of patients at risk for esophageal adenocarcinoma, provide opportunity for advances in prevention and guide selection of interventions appropriate to risk.
Acknowledgments
Supported by NIH P01 CA91955, RC1 CA146973, R01 CA140657, R03 CA137811 and Research Scholar Grant #117209-RSG-09-163-01-CNE from the American Cancer Society
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
References
- 1.Wang KK, Sampliner RE. Updated guidelines 2008 for the diagnosis, surveillance and therapy of Barrett’s esophagus. Am J Gastroenterol. 2008;103:788–97. doi: 10.1111/j.1572-0241.2008.01835.x. [DOI] [PubMed] [Google Scholar]
- 2.Barrett N. Chronic peptic ulcer of the oesophagus and ‘oesophagitis’. Br J Surg. 1950;38:175–82. doi: 10.1002/bjs.18003815005. [DOI] [PubMed] [Google Scholar]
- 3.Naef AP, Savary M, Ozzello L. Columnar-lined lower esophagus: an acquired lesion with malignant predisposition. Report on 140 cases of Barrett’s esophagus with 12 adenocarcinomas. Journal of Thoracic and Cardiovascular Surgery. 1975;70:826–35. [PubMed] [Google Scholar]
- 4.Sampliner RE. Practice guidelines on the diagnosis, surveillance, and therapy of Barrett’s esophagus. Am J Gastroenterol. 1998;93:1028–32. doi: 10.1111/j.1572-0241.1998.00362.x. [DOI] [PubMed] [Google Scholar]
- 5.Thomas T, Abrams KR, De Caestecker JS, Robinson RJ. Meta analysis: Cancer risk in Barrett’s oesophagus. Aliment Pharmacol Ther. 2007;26:1465–77. doi: 10.1111/j.1365-2036.2007.03528.x. [DOI] [PubMed] [Google Scholar]
- 6.Yousef F, Cardwell C, Cantwell MM, Galway K, Johnston BT, Murray L. The incidence of esophageal cancer and high-grade dysplasia in Barrett’s esophagus: a systematic review and meta-analysis. Am J Epidemiol. 2008;168:237–49. doi: 10.1093/aje/kwn121. [DOI] [PubMed] [Google Scholar]
- 7.Anderson LA, Murray LJ, Murphy SJ, Fitzpatrick DA, Johnston BT, Watson RG, et al. Mortality in Barrett’s oesophagus: results from a population based study. Gut. 2003;52:1081–4. doi: 10.1136/gut.52.8.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Solaymani-Dodaran M, Logan RF, West J, Card T. Mortality associated with Barrett’s esophagus and gastroesophageal reflux disease diagnoses-a population-based cohort study. Am J Gastroenterol. 2005;100:2616–21. doi: 10.1111/j.1572-0241.2005.00340.x. [DOI] [PubMed] [Google Scholar]
- 9.Moayyedi P, Burch N, Akhtar-Danesh N, Enaganti SK, Harrison R, Talley NJ, et al. Mortality rates in patients with Barrett’s oesophagus. Aliment Pharmacol Ther. 2008;27:316–20. doi: 10.1111/j.1365-2036.2007.03582.x. [DOI] [PubMed] [Google Scholar]
- 10.Prentice RL. Surrogate endpoints in clinical trials: definition and operational criteria. Stat Med. 1989;8:431–40. doi: 10.1002/sim.4780080407. [DOI] [PubMed] [Google Scholar]
- 11.Fleming TR, Prentice RL, Pepe MS, Glidden D. Surrogate and auxiliary endpoints in clinical trials, with potential applications in cancer and AIDS research. Statistics in Medicine. 1994;13:955–68. doi: 10.1002/sim.4780130906. [DOI] [PubMed] [Google Scholar]
- 12.Reid BJ, Li X, Galipeau PC, Vaughan TL. Barrett’s oesophagus and oesophageal adenocarcinoma: time for a new synthesis. Nat Rev Cancer. 2010;10:87–101. doi: 10.1038/nrc2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corley DA, Levin TR, Habel LA, Weiss NS, Buffler PA. Surveillance and survival in Barrett’s adenocarcinomas: a population-based study. Gastroenterology. 2002;122:633–40. doi: 10.1053/gast.2002.31879. [DOI] [PubMed] [Google Scholar]
- 14.Corley DA, Kerlikowske K, Verma R, Buffler P. Protective association of aspirin/NSAIDs and esophageal cancer: a systematic review and meta-analysis. Gastroenterology. 2003;124:47–56. doi: 10.1053/gast.2003.50008. [DOI] [PubMed] [Google Scholar]
- 15.Abnet CC, Freedman ND, Kamangar F, Leitzmann MF, Hollenbeck AR, Schatzkin A. Non-steroidal anti-inflammatory drugs and risk of gastric and oesophageal adenocarcinomas: results from a cohort study and a meta-analysis. Br J Cancer. 2009;100:551–7. doi: 10.1038/sj.bjc.6604880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Overholt BF, Lightdale CJ, Wang KK, Canto MI, Burdick S, Haggitt RC, et al. Photodynamic therapy with porfimer sodium for ablation of high-grade dysplasia in Barrett’s esophagus: international, partially blinded, randomized phase III trial. Gastrointest Endosc. 2005;62:488–98. doi: 10.1016/j.gie.2005.06.047. [DOI] [PubMed] [Google Scholar]
- 17.Overholt BF, Wang KK, Burdick JS, Lightdale CJ, Kimmey M, Nava HR, et al. Five-year efficacy and safety of photodynamic therapy with Photofrin in Barrett’s high-grade dysplasia. Gastrointest Endosc. 2007;66:460–8. doi: 10.1016/j.gie.2006.12.037. [DOI] [PubMed] [Google Scholar]
- 18.Shaheen NJ, Sharma P, Overholt BF, Wolfsen HC, Sampliner RE, Wang KK, et al. Radiofrequency ablation in Barrett’s esophagus with dysplasia. N Engl J Med. 2009;360:2277–88. doi: 10.1056/NEJMoa0808145. [DOI] [PubMed] [Google Scholar]
- 19.Spechler SJ, Fitzgerald RC, Prasad GA, Wang KK. History, molecular mechanisms, and endoscopic treatment of Barrett’s esophagus. Gastroenterology. 2010;138:854–69. doi: 10.1053/j.gastro.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Romero Y, Cameron AJ, Locke GR, 3rd, Schaid DJ, Slezak JM, Branch CD, et al. Familial aggregation of gastroesophageal reflux in patients with Barrett’s esophagus and esophageal adenocarcinoma. Gastroenterology. 1997;113:1449–56. doi: 10.1053/gast.1997.v113.pm9352846. [DOI] [PubMed] [Google Scholar]
- 21.Groves C, Jankowski J, Barker F, Holdstock G. A family history of Barrett’s oesophagus: another risk factor? Scand J Gastroenterol. 2005;40:1127–8. doi: 10.1080/00365520510023189. [DOI] [PubMed] [Google Scholar]
- 22.Chak A, Ochs-Balcom H, Falk G, Grady WM, Kinnard M, Willis JE, et al. Familiality in Barrett’s esophagus, adenocarcinoma of the esophagus, and adenocarcinoma of the gastroesophageal junction. Cancer Epidemiol Biomarkers Prev. 2006;15:1668–73. doi: 10.1158/1055-9965.EPI-06-0293. [DOI] [PubMed] [Google Scholar]
- 23.Munitiz V, Parrilla P, Ortiz A, Martinez-de-Haro LF, Yelamos J, Molina J. High risk of malignancy in familial Barrett’s esophagus: presentation of one family. J Clin Gastroenterol. 2008;42:806–9. doi: 10.1097/MCG.0b013e3180329015. [DOI] [PubMed] [Google Scholar]
- 24.Cameron AJ, Lagergren J, Henriksson C, Nyren O, Locke GR, 3rd, Pedersen NL. Gastroesophageal reflux disease in monozygotic and dizygotic twins. Gastroenterology. 2002;122:55–9. doi: 10.1053/gast.2002.30301. [DOI] [PubMed] [Google Scholar]
- 25.Mohammed I, Cherkas LF, Riley SA, Spector TD, Trudgill NJ. Genetic influences in gastro-oesophageal reflux disease: a twin study. Gut. 2003;52:1085–9. doi: 10.1136/gut.52.8.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gelfand MD. Barrett esophagus in sexagenarian identical twins. J Clin Gastroenterol. 1983;5:251–3. doi: 10.1097/00004836-198306000-00011. [DOI] [PubMed] [Google Scholar]
- 27.Chak A, Faulx A, Kinnard M, Brock W, Willis J, Wiesner GL, et al. Identification of Barrett’s esophagus in relatives by endoscopic screening. Am J Gastroenterol. 2004;99:2107–14. doi: 10.1111/j.1572-0241.2004.40464.x. [DOI] [PubMed] [Google Scholar]
- 28.Ochs-Balcom HM, Falk G, Grady WM, Kinnard M, Willis J, Elston R, et al. Consortium approach to identifying genes for Barrett’s esophagus and esophageal adenocarcinoma. Transl Res. 2007;150:3–17. doi: 10.1016/j.trsl.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 29.Roach J, Glusman G, Smit AFA, Huff CD, Hubley R, Shannon PT, et al. Analysis of Genetic Inheritance in a Family Quartet by Whole-Genome Sequencing. Science Express. 2010:636–9. doi: 10.1126/science.1186802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–8. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 31.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Baal JW, Milana F, Rygiel AM, Sondermeijer CM, Spek CA, Bergman JJ, et al. A comparative analysis by SAGE of gene expression profiles of esophageal adenocarcinoma and esophageal squamous cell carcinoma. Cell Oncol. 2008;30:63–75. doi: 10.1155/2008/328529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peng D, Sheta EA, Powell SM, Moskaluk CA, Washington K, Goldknopf IL, et al. Alterations in Barrett’s-related adenocarcinomas: a proteomic approach. Int J Cancer. 2008;122:1303–10. doi: 10.1002/ijc.23258. [DOI] [PubMed] [Google Scholar]
- 34.Zhao J, Chang AC, Li C, Shedden KA, Thomas DG, Misek DE, et al. Comparative proteomics analysis of Barrett metaplasia and esophageal adenocarcinoma using two-dimensional liquid mass mapping. Mol Cell Proteomics. 2007;6:987–99. doi: 10.1074/mcp.M600175-MCP200. [DOI] [PubMed] [Google Scholar]
- 35.Li X, Galipeau PC, Sanchez CA, Blount PL, Maley CC, Arnaudo J, et al. Single nucleotide polymorphism-based genome-wide chromosome copy change, loss of heterozygosity, and aneuploidy in Barrett’s esophagus neoplastic progression. Cancer Prev Res (Phila Pa) 2008;1:413–23. doi: 10.1158/1940-6207.CAPR-08-0121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paulson TG, Maley CC, Li X, Li H, Sanchez CA, Chao DL, et al. Chromosomal instability and copy number alterations in Barrett’s esophagus and esophageal adenocarcinoma. Clin Cancer Res. 2009;15:3305–14. doi: 10.1158/1078-0432.CCR-08-2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barrett MT, Sanchez CA, Prevo LJ, Wong DJ, Galipeau PC, Paulson TG, et al. Evolution of neoplastic cell lineages in Barrett oesophagus. Nat Gen. 1999;22:106–9. doi: 10.1038/8816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maley CC, Galipeau PC, Li X, Sanchez CA, Paulson TG, Reid BJ. Selectively advantageous mutations and hitchhikers in neoplasms: p16 lesions are selected in Barrett’s esophagus. Cancer Res. 2004;64:3414–27. doi: 10.1158/0008-5472.CAN-03-3249. [DOI] [PubMed] [Google Scholar]
- 39.Maley CC, Galipeau PC, Li X, Sanchez CA, Paulson TG, Blount PL, et al. The combination of genetic instability and clonal expansion predicts progression to esophageal adenocarcinoma. Cancer Res. 2004;64:7629–33. doi: 10.1158/0008-5472.CAN-04-1738. [DOI] [PubMed] [Google Scholar]
- 40.Maley CC, Galipeau PC, Finley JC, Wongsurawat VJ, Li X, Sanchez CA, et al. Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat Genet. 2006;38:468–73. doi: 10.1038/ng1768. [DOI] [PubMed] [Google Scholar]
- 41.Merlo LM, Shah NA, Li X, Blount PL, Vaughan TL, Reid BJ, et al. A Comprehensive Survey of Clonal Diversity Measures in Barrett’s Esophagus as Biomarkers of Progression to Esophageal Adenocarcinoma. Cancer Prev Res (Phila) 2010 doi: 10.1158/1940-6207.CAPR-10-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leedham SJ, Preston SL, McDonald SA, Elia G, Bhandari P, Poller D, et al. Individual crypt genetic heterogeneity and the origin of metaplastic glandular epithelium in human Barrett’s oesophagus. Gut. 2008;57:1041–8. doi: 10.1136/gut.2007.143339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Orlando RC. Mucosal Defense in Barrett’s Esophagus. In: Sharma P, S R, editors. Barrett’s Esophagus and Esophageal Adenocarcinoma. 2. Oxford, UK: Blackwell Publishing, Ltd; 2006. pp. 60–72. [Google Scholar]
- 44.Ostrowski J, Mikula M, Karczmarski J, Rubel T, Wyrwicz LS, Bragoszewski P, et al. Molecular defense mechanisms of Barrett’s metaplasia estimated by an integrative genomics. J Mol Med. 2007;85:733–43. doi: 10.1007/s00109-007-0176-3. [DOI] [PubMed] [Google Scholar]
- 45.Levine DS, Rubin CE, Reid BJ, Haggitt RC. Specialized metaplastic columnar epithelium in Barrett’s esophagus. A comparative transmission electron microscopic study. Laboratory Investigation. 1989;60:418–32. [PubMed] [Google Scholar]
- 46.Levine DS, Reid BJ, Haggitt RC, Rubin CE, Rabinovitch PS. Correlation of ultrastructural aberrations with dysplasia and flow cytometric abnormalities in Barrett’s epithelium. Gastroenterology. 1989;96:355–67. doi: 10.1016/s0016-5085(89)91559-x. [DOI] [PubMed] [Google Scholar]
- 47.Galipeau PC, Li X, Blount PL, Maley CC, Sanchez CA, Odze RD, et al. NSAIDs modulate CDKN2A, TP53, and DNA content risk for future esophageal adenocarcinoma. PLoS Med. 2007;4:e67. doi: 10.1371/journal.pmed.0040067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vaughan TL, Dong LM, Blount PL, Ayub K, Odze RD, Sanchez CA, et al. Non-steroidal anti-inflammatory drugs and risk of neoplastic progression in Barrett’s oesophagus: a prospective study. Lancet Oncol. 2005;6:945–52. doi: 10.1016/S1470-2045(05)70431-9. [DOI] [PubMed] [Google Scholar]
- 49.Pepper JW, Findlay CS, Kassen R, Spencer SL, Maley CC. Cancer research meets evolutionary biology. Evolutionary Applications. 2009;2:62–70. doi: 10.1111/j.1752-4571.2008.00063.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510–22. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jankowski J, Sharma P. Review article: approaches to Barrett’s oesophagus treatment-the role of proton pump inhibitors and other interventions. Aliment Pharmacol Ther. 2004;19(Suppl 1):54–9. doi: 10.1111/j.0953-0673.2004.01839.x. [DOI] [PubMed] [Google Scholar]
- 52.Jankowski J, Barr H. Improving surveillance for Barrett’s oesophagus: AspECT and BOSS trials provide an evidence base. BMJ. 2006;332:1512. doi: 10.1136/bmj.332.7556.1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Blaser MJ. Disappearing microbiota: Helicobacter pylori protection against esophageal adenocarcinoma. Cancer Prev Res (Phila Pa) 2008;1:308–11. doi: 10.1158/1940-6207.CAPR-08-0170. [DOI] [PubMed] [Google Scholar]
- 54.Yang L, Lu X, Nossa CW, Francois F, Peek RM, Pei Z. Inflammation and intestinal metaplasia of the distal esophagus are associated with alterations in the microbiome. Gastroenterology. 2009;137:588–97. doi: 10.1053/j.gastro.2009.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.von Zglinicki T. Oxidative stress shortens telomeres. Trends Biochem Sci. 2002;27:339–44. doi: 10.1016/s0968-0004(02)02110-2. [DOI] [PubMed] [Google Scholar]
- 56.Jenkins GJ, Cronin J, Alhamdani A, Rawat N, D’Souza F, Thomas T, et al. The bile acid deoxycholic acid has a non-linear dose response for DNA damage and possibly NF-kappaB activation in oesophageal cells, with a mechanism of action involving ROS. Mutagenesis. 2008;23:399–405. doi: 10.1093/mutage/gen029. [DOI] [PubMed] [Google Scholar]
- 57.Grisham MB, Jourd’heuil D, Wink DA. Review article: chronic inflammation and reactive oxygen and nitrogen metabolism--implications in DNA damage and mutagenesis. Aliment Pharmacol Ther. 2000;14(Suppl 1):3–9. doi: 10.1046/j.1365-2036.2000.014s1003.x. [DOI] [PubMed] [Google Scholar]
- 58.Sihvo EI, Salminen JT, Rantanen TK, Ramo OJ, Ahotupa M, Farkkila M, et al. Oxidative stress has a role in malignant transformation in Barrett’s oesophagus. Int J Cancer. 2002;102:551–5. doi: 10.1002/ijc.10755. [DOI] [PubMed] [Google Scholar]
- 59.Trayhurn P, Bing C, Wood IS. Adipose tissue and adipokines--energy regulation from the human perspective. J Nutr. 2006;136:1935S–9S. doi: 10.1093/jn/136.7.1935S. [DOI] [PubMed] [Google Scholar]
- 60.Turker MS, Gage BM, Rose JA, Elroy D, Ponomareva ON, Stambrook PJ, et al. A novel signature mutation for oxidative damage resembles a mutational pattern found commonly in human cancers. Cancer Res. 1999;59:1837–9. [PubMed] [Google Scholar]
- 61.Tobey NA, Argote CM, Vanegas XC, Barlow W, Orlando RC. Electrical parameters and ion species for active transport in human esophageal stratified squamous epithelium and Barrett’s specialized columnar epithelium. Am J Physiol Gastrointest Liver Physiol. 2007;293:G264–70. doi: 10.1152/ajpgi.00047.2007. [DOI] [PubMed] [Google Scholar]
- 62.Cairns J. Mutation Selection and the Natural History of Cancer. Nature. 1975;255:197–200. doi: 10.1038/255197a0. [DOI] [PubMed] [Google Scholar]
- 63.Dixon J, Strugala V, Griffin SM, Welfare MR, Dettmar PW, Allen A, et al. Esophageal mucin: an adherent mucus gel barrier is absent in the normal esophagus but present in columnar-lined Barrett’s esophagus. Am J Gastroenterol. 2001;96:2575–83. doi: 10.1111/j.1572-0241.2001.04159.x. [DOI] [PubMed] [Google Scholar]
- 64.Glickman JN, Blount PL, Sanchez CA, Cowan DS, Wongsurawat VJ, Reid BJ, et al. Mucin core polypeptide expression in the progression of neoplasia in Barrett’s esophagus. Hum Pathol. 2006;37:1304–15. doi: 10.1016/j.humpath.2006.03.023. [DOI] [PubMed] [Google Scholar]
- 65.Lao-Sirieix P, Corovic A, Jankowski J, Lowe A, Triadafilopoulos G, Fitzgerald RC. Physiological and molecular analysis of acid loading mechanisms in squamous and columnar-lined esophagus. Dis Esophagus. 2008;21:529–38. doi: 10.1111/j.1442-2050.2007.00807.x. [DOI] [PubMed] [Google Scholar]
- 66.Jovov B, Van Itallie CM, Shaheen NJ, Carson JL, Gambling TM, Anderson JM, et al. Claudin-18: a dominant tight junction protein in Barrett’s esophagus and likely contributor to its acid resistance. Am J Physiol Gastrointest Liver Physiol. 2007;293:G1106–13. doi: 10.1152/ajpgi.00158.2007. [DOI] [PubMed] [Google Scholar]