

Key Points
The early virological factors in HIV-1 infection, including transmission and the nature of the founder virus, can affect the time course of viraemia through the early peak to set point.
The identification of patients within the first few weeks of HIV-1 infection has provided early evidence of immune system damage, including massive apoptosis of CD4+ T cells, which is associated with the presence of apoptotic microparticles and TRAIL (tumour necrosis factor-related apoptosis-inducing ligand) in the blood, and damage to germinal centres in mucosal lymphoid tissues.
The first innate immune responses include the appearance of acute-phase proteins, early cytokine storm and activation of natural killer (NK) cells. An innate immune response to HIV-1 can be damaging, however, as it can draw susceptible T cells to the infection foci.
The first T cell response controls the founder virus by killing infected T cells. However, the T cell response also selects mutational changes in the founder virus, allowing immune evasion.
The first B cell response consists of early immune complexes, followed by non-neutralizing antibodies against the founder virus and then the slow development of broadly acting neutralizing antibodies. Development of vaccines that rapidly induce broadly acting neutralizing antibodies might be beneficial in preventing HIV infection.
Understanding the early events and immune responses is crucial to devising vaccine strategies that can improve the weak protection offered by current HIV vaccines that are being trialled, such as the RV144 (Thai) efficacy trial.
Unprecedented insight into the early stages of HIV-1 infection has provided important clues for vaccine design. Here, the authors discuss how early virological and immunological events, including transmission by a single founder virus and marked CD4+T cell loss, might influence the course of disease.
Abstract
The early immune response to HIV-1 infection is likely to be an important factor in determining the clinical course of disease. Recent data indicate that the HIV-1 quasispecies that arise following a mucosal infection are usually derived from a single transmitted virus. Moreover, the finding that the first effective immune responses drive the selection of virus escape mutations provides insight into the earliest immune responses against the transmitted virus and their contributions to the control of acute viraemia. Strong innate and adaptive immune responses occur subsequently but they are too late to eliminate the infection. In this Review, we discuss recent studies on the kinetics and quality of early immune responses to HIV-1 and their implications for developing a successful preventive HIV-1 vaccine.
Main
Recent advances that enable the identification of patients within the first few weeks of HIV-1 infection1,2 have provided researchers access to samples from acutely infected patients earlier and in higher numbers than previously available. This has advanced our understanding of the nature of the transmitted virus and the first immune responses in the period before establishment of stable viraemia (the viral set point), which occurs 3–6 months after infection. The first weeks following HIV-1 transmission are extremely dynamic: they are associated with rapid damage to generative immune cell microenvironments, caused by direct viral cytopathicity and bystander effects, and with immune responses that partially control the virus.
In this Review, we focus our discussion on the early host or viral factors that are crucial for determining the outcome of HIV-1 infection. These include the nature of the transmitted virus, or founder virus, suppression of the initial infection by genetically influenced immune responses, and the rate of virus mutation and viral fitness of selected mutants. In addition, we review what is known about the nature of innate and adaptive immune responses during this early phase of infection, drawn from studies of humans and macaques infected with HIV-1 and simian immunodeficiency virus (SIV), respectively. Finally, we discuss how our knowledge of the events of early HIV-1 infection can improve the design of a preventive vaccine (Box 1).
The biology of early HIV-1 infection
Transmission. Most HIV-1 infections occur by sexual exposure through the genital tract or rectal mucosa. Although it is not possible to study the very first events following HIV-1 transmission in humans in vivo, we have gained some understanding from studies in which mucosal tissue explants were infected in vitro3,4,5. Further understanding of the first stages of infection in vivo has been obtained from studies in which macaques were inoculated intrarectally or intravaginally with SIV6,7. It is still uncertain whether HIV-1 is transmitted as a free or a cell-bound virus, but SIV can be transmitted in either form8. In addition, the mechanism by which HIV-1 crosses the genital mucosal epithelium is unclear. Diffusion of HIV-1 across the vaginal mucosa is slowed by cervicovaginal mucus9. It is possible that virus that reaches the mucosal epithelium crosses this barrier by transcytosis or by making direct contact with dendrites of intraepithelial dendritic cells (DCs). Preliminary unpublished findings suggest that virions may also move through intercellular spaces in the epithelium to make initial cell contact with underlying mucosal Langerhans cells and CD4+ T cells (T. Hope and S. McCoombe, personal communication). Given that multiple sexual exposures are usually needed for infection to occur, crossing of the epithelial cell barrier by the virus is probably a rare event, although it is more common if the genital mucosa is damaged by physical trauma or co-existing genital infections10,11.
Eclipse phase. Following transmission of the virus, there is a period of ∼10 days, known as the eclipse phase, before viral RNA becomes detectable in the plasma (Fig. 1). Single-genome amplification and sequencing of the first detectable virus has shown that ∼80% of mucosally transmitted HIV-1 clade B and C infections are initiated by a single virus12,13,14. Infectious molecular clones derived from these primary founder viruses could infect CD4+ T cells with greater efficiency than they could infect monocytes and macrophages14, which differs from the virus quasispecies that arise later in the infection and can infect lymphoid and myeloid cell types with equal efficiency. Studies in rhesus macaques inoculated intrarectally with a complex SIV quasispecies also showed that productive infection arises from a single infecting virus15, which supports the use of SIV infection of rhesus macaques as a model for HIV-1 transmission and vaccine studies. In other studies7,16 in which macaques were infected experimentally, the first cells to be infected in the vaginal mucosa were found in foci of resident memory T cells that expressed the virus receptors CD4 and CC-chemokine receptor 5 (CCR5), which is consistent with the cell tropism of cloned HIV-1 founder virus14.
Figure 1. Definition of acute HIV-1 infection.
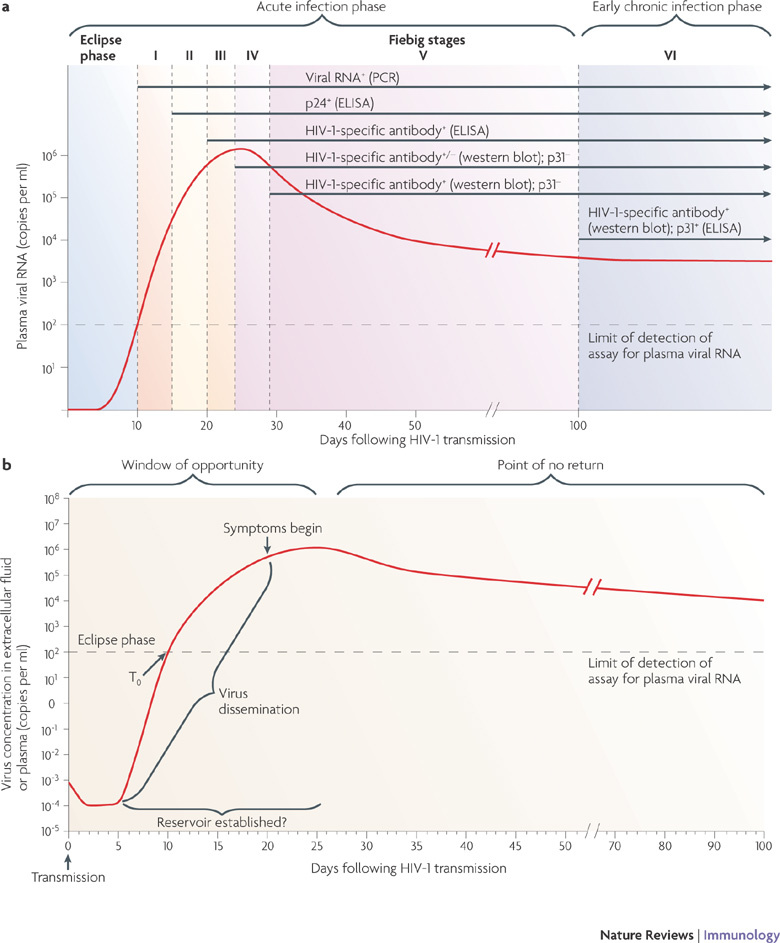
a | Recent analysis of samples from individuals early after infection with HIV-1 has revealed that the first weeks following infection can be divided into clinical stages that are defined by a stepwise gain in positivity for the detection of HIV-1 antigens and HIV-1-specific antibodies in diagnostic assays (in brackets)82. The time between infection and the first detection of viral RNA in the plasma is referred to as the eclipse phase. Plasma virus levels then increase exponentially, peaking at 21–28 days after infection, and this is followed by a slower decrease in plasma viral RNA levels. Patients can be categorized into Fiebig stages I–VI, which are based on a sequential gain in positive HIV-1 clinical diagnostic assays (viral RNA measured by PCR, p24 and p31 viral antigens measured by enzyme-linked immunosorbent assay (ELISA), HIV-1-specific antibody detected by ELISA and HIV-1-specific antibodies detected by western blot). Patients progress from acute infection through to the early chronic stage of infection at the end of Fiebig stage V, approximately 100 days following infection, as the plasma viral load begins to plateau. b | Fundamental events in acute HIV-1 infection. Following HIV-1 infection, the virus first replicates locally in the mucosa and is then transported to draining lymph nodes, where further amplification occurs. This initial phase of infection, until systemic viral dissemination begins, constitutes the eclipse phase. The time when virus is first detected in the blood is referred to as T0; after this there is an exponential increase in plasma viraemia to a peak 21–28 days after infection. By this time, significant depletion of mucosal CD4+ T cells has already occurred. Around the time of peak viraemia, patients may become symptomatic and reservoirs of latent virus are established in cells that have a slower rate of decay than CD4+ T cells. The 'window of opportunity' between transmission and peak viraemia, prior to massive CD4+ T cell destruction and the establishment of viral reservoirs, is the narrow but crucial period in which an HIV-1 vaccine must control viral replication, prevent extensive CD4+ T cell depletion and curb generalized immune activation. Part a is modified, with permission, from Ref. 12 © (2008) National Academy of Sciences, USA. Part b is modified from Ref. 160.
Homogeneity of the founder virus indicates that the established infection probably arises from a single focus of infected mucosal CD4+ T cells. Virus replication at this focus might in fact be supported by early innate immune responses that lead to the recruitment of additional susceptible T cells to the site16. The failure of most infected foci to become established may be explained by the high error rate in reverse transcription that occurs during HIV-1 replication and the effects of the host antiviral apolipoprotein B mRNA-editing enzyme, catalytic polypeptide-like (APOBEC) cytidine deaminases APOBEC3G and APOBEC3F, which cause many viruses produced in infected CD4+ T cells to be defective17.
Peak viraemia. At the end of the eclipse phase, virus and/or virus-infected cells reach the draining lymph node, where they meet activated CD4+CCR5+ T cells, which are targets for further infection. This process is augmented by DCs that bind and internalize virus through DC-specific ICAM3-grabbing non-integrin (DC-SIGN; also known as CD209) and carry the virus to activated T cells18. B cells may also be involved in the early spread of infection by binding the virus through the complement receptor CD21 (also known as CR2)19. The virus then replicates rapidly and spreads throughout the body to other lymphoid tissues, particularly gut-associated lymphoid tissue (GALT), where activated CD4+CCR5+ memory T cells are present in high numbers20,21,22. Approximately 20% of CD4+ T cells in the GALT are infected in both humans with acute HIV-1 infection and SIV-infected macaques. Up to 60% of uninfected CD4+ T cells at this site become activated and die by apoptosis, resulting in the release of apoptotic microparticles that can suppress immune function23. Therefore, ∼80% of CD4+ T cells in the GALT can be depleted in the first 3 weeks of HIV-1 infection20,24,25. While HIV-1 is replicating in the GALT and other lymphoid tissues, the plasma viraemia increases exponentially to reach a peak, usually more than a million RNA copies per ml of blood, at 14–21 days after SIV infection in macaques and at 21–28 days after HIV-1 infection in humans (Fig. 1). CD4+ T cell numbers are low at the time of peak viraemia but later return to near normal levels in the blood but not in the GALT20,24,25. Although B cells are not depleted during early HIV-1 infection, B cell responses are impaired owing to the destruction of other cell types that are important for the development of germinal centres. Up to 50% of germinal centres in the gut are lost within the first 80 days of infection26.
Establishing viral set point. At the point of peak viraemia the immune response has not affected the amino acid sequence of the virus, despite the extensive activation of innate immune cells (see below). Thereafter, the viral load decreases over 12–20 weeks to reach a more stable level, known as the viral set point27,28,29 (Fig. 1). Virus diversification occurs during this decrease in viral load, and multiple escape mutants are selected under the pressure of adaptive immune responses that are first detectable just before peak viraemia30,31,32,33. In the absence of antiretroviral drug therapy (ART), the set point is maintained by a balance between virus turnover and the immune responses.
The death rate of infected cells has been calculated from decay curves of viraemia after ART initiation34. For most infected memory T cells, the half-life is less than a day34. However, other cell populations have slower rates of decay35, and cell populations other than CD4+ T cells maintain latent pools of HIV-1 (Ref. 36). Cells are probably latently infected within days of HIV-1 transmission and are unlikely to be removed by natural or vaccine-stimulated anti-HIV-1 immune responses, given that they cannot be eliminated by ART37.
Immune activation. Activation of innate cells and B and T cells is a striking feature of acute HIV-1 infection of humans and SIV infection of rhesus macaques, and it persists to a varying degree into chronic infection. The dysregulation of immune cells is not limited to cells that are infected by, or are specific for, HIV-1 (Ref. 38). Chronic immune activation is not observed in naturally SIV-infected sooty mangabeys, in which the infections rarely progress to AIDS. This is despite high levels of virus replication and acute CD4+ T cell depletion39, suggesting a role for immune activation in AIDS development. Indeed, there is a positive correlation between markers of CD8+ T cell activation and HIV disease progression40,41,42.
Immune activation is associated with early and extensive apoptosis of B and T cells, leading to the release of apoptotic microparticles into the blood (Fig. 2), and increased expression of tumour necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL; also known as TNFSF10) and FAS ligand (also known as CD95L), which kill bystander cells and are immunosuppressive23. The causes of HIV-associated immune activation established in early HIV-1 infection are not clearly defined42,43. Multiple related events (reviewed in Ref. 44) probably contribute to such activation, including direct viral infection of immune cells, pro-inflammatory cytokine production by innate cells (which drives both direct and bystander activation of other immune cells), translocation of microbial products into the blood through damaged intestinal epithelium21,22, loss of virally infected regulatory T (TReg) cells and chronic mycobacterial and viral co-infections.
Figure 2. Early events in acute HIV-1 infection.
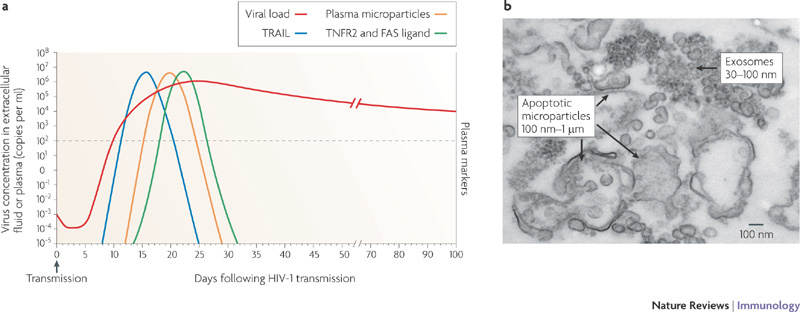
a | The timing of the appearance of soluble proteins and apoptotic microparticles in the blood during acute HIV-1 infection. An increase in the level of soluble tumour necrosis factor-related apoptosis-inducing ligand (TRAIL; also known as TNFSF10) in the plasma is the first evidence of infection-induced apoptosis and/or immune activation and it occurs before the peak in viraemia. This increase in TRAIL also coincides with the appearance of high levels of interferon-α (IFNα; see Fig. 3). Soon thereafter increases in the number of apoptotic microparticles are observed, followed by increased levels of soluble TNF receptor 2 (TNFR2; also known as TNFRSF1B) and soluble FAS ligand. A portion of the microparticles express CC-chemokine receptor 5, suggesting that they originate from cellular targets of HIV-1 infection. The appearance of these soluble components in the plasma early during HIV-1 infection probably represents a pathological rather than a protective cascade of events that is a cause of, or is associated with, virus-induced CD4+ T cell death. b | Electron micrograph of apoptotic microparticles and exosomes in the plasma from a patient with acute HIV-1 infection. There are ∼700-fold more microparticles than virions present in acute HIV-1 infection. Part a is based on data from Ref. 23. Part b is reproduced, with permission, from Ref. 23 © (2008) American Society for Microbiology.
Genetic control of HIV-1 set point
In contrast to other pathogens that have infected and selected humans for millennia, HIV-1 is a new pathogen to humans45,46,47. Therefore, the influence of the host's genetics on the immune response to HIV-1 infection may be more evident. The most dramatic finding in this regard is that homozygosity for a 32 base pair deletion in CCR5, which abrogates its expression, protects almost completely from HIV-1 infection48. Furthermore, the HLA alleles HLA-B*5701, HLA-B*5703, HLA-B*5801, HLA-B27 and HLA-B51 are all associated with good control of the virus and a slower progression to AIDS49, partly because the epitopes recognized by the T cells in these individuals are focused on conserved regions of the viral Gag protein (see below). A genome-wide association study50 found a strong protective influence for a single nucleotide polymorphism (SNP) located 35 kilobases upstream of the HLA-C locus and confirmed the association of HLA-B57 with a low viral set point. This HLA-C-linked SNP may be associated with low-level expression of HLA-C50, which might in turn affect T cell or natural killer (NK) cell function during HIV-1 infection. By contrast, some subtypes of HLA-B35 are associated with rapid disease progression, especially if homozygous51, although the mechanism is not understood.
It has been shown that the expression of the killer immunoglobulin-like receptors KIR3DS1 and KIR3DL1 — which deliver activating and inhibitory signals to NK cells, respectively — delays progression to AIDS in individuals with HLA class I allotypes containing the 80Ile variant of the Bw4 motif52, which are thought to be ligands for these receptors53,54. Expansion of NK cells that express KIR3DS1 and/or KIR3DL1 during acute HIV-1 infection has been observed but only if the HLA-B Bw4 80Ile motif is present55, which is supported by in vitro data demonstrating that NK cells expressing KIR3DS1 control HIV-1 replication efficiently in HLA-B Bw4 80Ile-expressing target cells56. It is possible that KIR3DS1 mediates specific recognition of HIV-infected cells by NK cells, although the exact nature of the ligand is elusive. These observations probably reflect an influence of interactions between KIR3DS1 and/or KIR3DL1 and HLA-B Bw4 80Ile on the development and/or functions of NK cells, and possibly CD8+ T cells, which may help to control viral set point.
Early innate immune responses to HIV-1
Acute-phase proteins and cytokines. Insight into the earliest systemic immune responses to HIV-1 infection has been gained by studying plasma donors who acquired HIV-1 infection. Frequent samples were taken before infection, through peak viraemia and seroconversion57,58. Samples from different donors were aligned relative to the time that viral RNA was first detectable (100 copies per ml) (T0). The first detectable innate immune response, occurring sometimes just before T0, was an increase in the levels of some acute-phase proteins, such as serum amyloid A (H. Kramer and B. Kessler, personal communication). A further wave of acute-phase protein production coincided with a cytokine response (described below) and a rapid increase in plasma viraemia. The production of acute-phase proteins can be triggered by pro-inflammatory cytokines (such as interleukin-1 (IL-1)) and also by extrinsic factors such as lipopolysaccharide (LPS). LPS is detectable in the plasma during chronic infection with HIV-1 or SIV and may be derived from commensal bacteria that translocate from the gut lumen following depletion of HIV-1-infected intestinal CCR5+ T helper 17 cells21,22,59. Immunostaining of GALT biopsies collected from acutely infected patients showed higher levels of pro-inflammatory cytokines than healthy tissues60.
As viraemia increases, so do the levels of cytokines and chemokines in the plasma (Fig. 3). Levels of IL-15, type I interferons (IFNs) and CXC-chemokine ligand 10 (CXCL10) increase rapidly but transiently. IL-18, TNF, IFNγ and IL-22 also increase rapidly but are sustained at high levels, whereas the increase in IL-10 is slightly delayed58 (Fig. 3). Some of these cytokines have antiviral activity; for example, type I IFNs inhibit HIV replication in severe combined immunodeficient mice reconstituted with human lymphocytes61. Also, type I IFNs, IL-15 and IL-18 enhance innate and adaptive immune responses. However, the intense cytokine response during acute HIV infection may also promote viral replication and mediate immunopathology (discussed below).
Figure 3. The cytokine storm in acute HIV-1 infection.
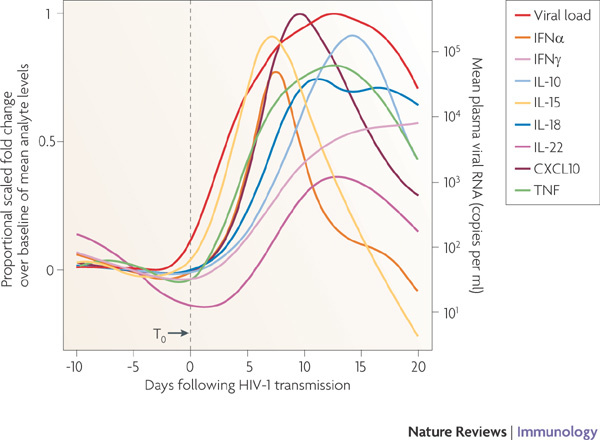
The relative kinetics of elevation of acute-phase proteins, cytokines and chemokines in the plasma during acute HIV-1 infection. There are two initial waves of cytokines: interleukin-15 (IL-15) and interferon-α (IFNα), followed by tumour necrosis factor (TNF), IL-18 and IL-10. CXCL10, CXC-chemokine ligand 10. Figure is reproduced, with permission, from Ref. 58 © (2009) American Society for Microbiology.
The cellular sources of the acute-phase cytokines and chemokines during early HIV-1 infection have not been definitively identified, but probably include infected CD4+CCR5+ T cells, activated DCs16, monocytes, macrophages21, NK cells, NKT cells and, subsequently, HIV-specific T cells. The cytokine storm observed during early HIV-1 infection is much greater than that observed in acute hepatitis B and hepatitis C virus infections58, indicating that a systemic cytokine response of this magnitude is not a pre-requisite for viral clearance. The intense cytokine response in acute HIV infection may instead fuel viral replication and mediate immunopathology (discussed below). High-level systemic cytokine responses during acute infections with avian influenza virus and severe acute respiratory syndrome-associated coronavirus are likewise associated with immunopathological consequences62,63.
DCs. DCs are markedly reduced in number during acute HIV-1 infection64 (N. Bhardwaj and P.B., unpublished observations). This rapid decline in circulating DCs, particularly plasmacytoid DCs (pDCs), may be due to activation-induced cell death or to the migration of activated DCs into lymph nodes, where an increase in DC numbers is observed65,66. In vitro, pDCs become activated by the binding of viral envelope proteins to CD4 expressed by the pDCs followed by virion endocytosis and by the triggering of Toll-like receptor 7 by viral RNA67. However, HIV-exposed conventional DCs do not become fully activated and show defective IL-12 production68, which is consistent with the low levels of IL-12 observed during acute HIV infection58. In addition, HIV-exposed pDCs produce IFNα, which enhances adaptive immune responses. However, HIV-exposed pDCs also produce indoleamine 2,3-dioxygenase (IDO), which induces the differentiation of CD4+ T cells into TReg cells that might suppress HIV-specific immune responses65,69,70. Conventional DCs can prime virus-specific CD4+ and CD8+ T cell responses following in vitro exposure to HIV71.
NK and NKT cells. As with most viral infections, NK cells and NKT cells become activated during acute HIV infection56,72,73. Prior to the peak in viraemia, blood NK cells proliferate and show enhanced activity when tested ex vivo56. The NK cell population expressing KIR3DS1 and/or KIR3DL1 expands during acute infection in individuals that also express HLA-B Bw4 80Ile55. NK and NKT cells can control HIV replication through cytolysis of virally infected cells and the production of antiviral cytokines and chemokines. In addition, they can interact with DCs and thereby influence T cell responses. HIV-1 has evolved a strategy to reduce the expression of ligands for NK cell receptors by infected cells74. This finding, and the clear role of KIR3D molecules in determining the viral set point52,54, supports the involvement of NK cells in the control of HIV-1. However, the timing of NK cell antiviral effects remains uncertain. NK cells do not contribute to the selection of virus escape mutants before peak viraemia, although it is possible, but not proved, that they account for some of the unexplained mutations that appear together with those that are selected by early T cell responses as viraemia decreases to reach the set point75. Alternatively, the antiviral effects of NK (and/or NKT) cells might have a greater influence at later time points.
Implications for vaccine design. Can the protective potential of innate immune responses be harnessed by vaccination? Because NK cells share some characteristics with memory cells after their initial activation76,77, it may be possible to prime their antiviral activity through vaccination. However, the activation of innate immunity should be attempted with caution, as innate immune responses can also be harmful. For example, induction of mucosal inflammatory responses by some microbicides has led to increased acquisition of HIV-1 infection (reviewed in Ref. 78). Furthermore, as discussed earlier, activated DCs can transmit virus to CD4+ T cells and, during the eclipse phase of infection, chemokines produced by pDCs can recruit susceptible CD4+ T cells to the foci of infection16,79. Immune activation induced by innate immune cells and the resulting production of pro-inflammatory cytokines and chemokines can promote HIV-1 replication. Type I IFNs and TNF also have pro-apoptotic effects and can thereby contribute to a loss of activated DCs and the bystander destruction of CD4+ T cells and B cells. The opposing effects of innate immune activation were highlighted in a study in which IL-15 was administered to treat acute SIV infection in rhesus macaques: NK cell and SIV-specific CD8+ T cell numbers were increased, resulting in fewer SIV-infected cells in lymph nodes, but the activation and proliferation of CD4+ T cells was enhanced and a higher viral load was established80. Therefore, vaccine-induced activation of innate immune responses will have to be thoroughly tested in the macaque SIV model and used with caution in humans.
Early T cell responses in HIV-1 infection
CD8+ T cell responses. A few studies have measured HIV-1-specific CD8+ T cell responses during early HIV-1 infection, before the first antibodies are detectable30,31,33,81 (Fiebig stages I or II82). Similar to SIV infection in macaques, the first T cell responses to HIV-1 infection arise as viraemia approaches its peak, and the T cell response peaks 1–2 weeks later, as viraemia declines. The homogeneity of the founder virus at the time of the peak of viraemia12,13,14 indicates that there is no immune-driven selection of escape mutants as viraemia increases.
Following the peak in the CD8+ T cell response, the virus sequence starts to change dramatically. Rapid selection of mutations occurs at discrete sites in the virus genome as viraemia declines to the viral set point14,83 (Fig. 4). Detailed analysis of four patients during the very early stages of infection75 indicated that most of the amino acid changes in the virus were selected by CD8+ T cells that recognize epitopes expressed by the founder virus but not by the escape mutant virus. Mutations in the viral envelope protein that were selected by neutralizing antibodies appeared later, at ∼12 weeks. A minority of virus escape mutants were not associated with demonstrable T cell responses: a few mutations were probably reversions from the sequence of the transmitted virus that was selected by T cells in the patient's sexual partner; others may have been selected by antibody-dependent cell-mediated virus inhibition or by NK cells. Notably, T cell- and antibody-mediated selection of viral escape mutants rarely involved a single amino acid change in the epitope; most mutants involved multiple changes such that various mutants were 'tested' until the fittest were selected75. The first T cell-selected mutations could replace the original sequence of the founder virus within 10 days, and were then followed by sequential selection of escape mutations at different epitopes. This pattern continues throughout the course of HIV infection75. Changes in sequence could involve amino acids that are upstream of the T cell epitope and are probably important for antigen processing84,85,86.
Figure 4. Early T cell selection of virus escape mutations in acute HIV-1 infection.
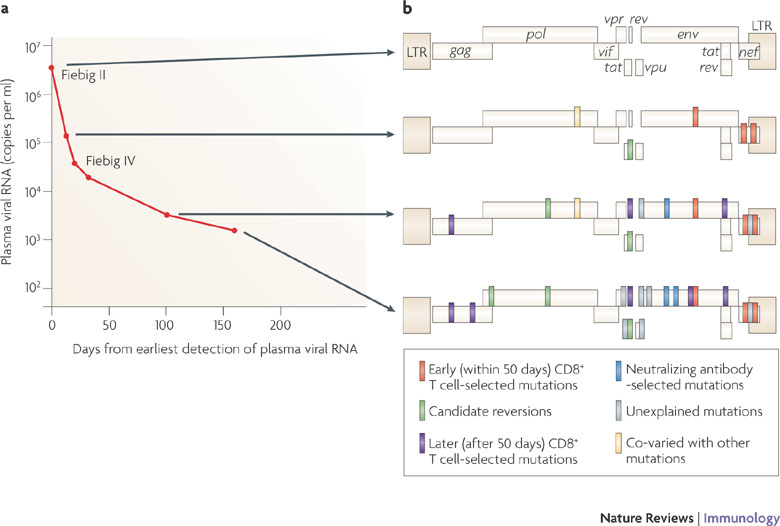
The virus escape mutations occurring in a single representative patient during acute HIV-1 infection. The changes in plasma virus load (a) and the emergence of amino acid changes (b) are shown. At the first time point, when the patient was virus positive but seronegative (Fiebig stage II), the founder virus (which was clade B) showed no evidence of immune selection. Thereafter there is an increasing number of selected sites at which the founder virus sequence is completely altered by, usually a cluster of, amino acid changes. Those marked in red were selected early (within 50 days from peak viraemia) by demonstrable CD8+ T cell responses. Those in purple were selected later by CD8+ T cells. Those in light green are single amino acid reversions to the clade B virus consensus sequence. Those in blue were mutations in V1 and V3 of the env gene selected by neutralizing antibodies. Those in grey were selected through undefined means, possibly by T cells, natural killer cells or antibodies. Yellow represents changes that co-varied with another mutation. LTR, long terminal repeat. Figure is based on data from Ref. 75.
The earliest T cell responses are often specific for Env and Nef75,87. Responses to other viral proteins, including the conserved Gag p24 and Pol proteins, tended to arise during later waves of T cell responses and may be more important for maintaining the viral load at the set point than for controlling early viraemia75,87,88. Often, the first T cell responses decline rapidly when the escape mutations are selected, or they may decline through exhaustion75,87. The loss of T cells after virus mutation implies complete loss of the epitope and no tendency for the virus to revert to the original sequence because of loss of fitness.
The finding that escape mutants appeared so rapidly raises questions regarding the effectiveness of the early T cell response. A mathematical model has provided some answers75. The rapid loss of the founder virus sequence and its replacement by escape mutant viruses implies complete CD8+ T cell-mediated inhibition of virus production by infected cells. From the rate of loss of founder virus sequence, the fraction of cells killed per day was calculated to be 0.15–0.35 for the earliest T cell responses75. As a virus-infected cell has a lifespan of 1 day in vivo, this means that 15–35% of infected cells must be killed prematurely by a single T cell response, which must reduce virus production. Therefore, CD8+ T cells curb viraemia in acute HIV-1 infection. However, selection of escape mutants would minimize this beneficial effect if the mutants were as fit as the founder virus and if the earliest responses were not immediately succeeded by new T cell responses to new (mutated) epitopes, which in turn may select further escape mutants75 (Fig. 4). Ultimately, responding T cells target epitopes that are more highly conserved and in which escape occurs at a cost to the fitness of the virus. Such immunodominant responses to more highly conserved epitopes are more likely to result in a lower level of viraemia at the set point89. When a virus that has undergone such escape mutations is transmitted, its set point is also lower in the new host90. The level of set point viraemia is therefore influenced by the nature of the transmitted virus and the specificity of early CD8+ T cell responses. Immunodominant T cell responses to the more conserved immunodominant virus epitopes are likely to result in a lower viral set point89.
CD8+ T cells are also important for the maintenance of viral set point. There have been many reports of virus escape mutations from around the time the set point is reached30,32,84,85,91,92,93,94,95,96,97,98,99. Using the same mathematical models as described earlier, CD8+ T cells are thought to make only a small contribution (killing 4–6% of virus-infected cells per day) to infected-cell death during chronic infection100, the rest being due to virus cytopathicity or infected-cell activation. However, this may be an underestimate of the T cell contribution because of the fitness costs of the escape mutations on the virus, such that mutant viruses grow more slowly than the founder virus. Some of the epitopes that are recognized by the T cells during later stages of infection are so highly conserved that the virus must undergo compensating mutations at other sites for escape to occur75,94,97,98,101, which slows the outgrowth of the mutant viruses. The calculation is further confounded by the difficulty of simultaneous virus escape from more than one T cell response13,85,91. In contrast to the earliest stages of HIV-1 infection when the range of epitopes recognized by the T cell response is narrow, the later response is broad, often directed against more than 10 epitopes102. Responses to conserved epitopes are probably important in the long-term control of viral load, because patients with HLA-B27, HLA-B*5701, HLA-B*5703 or HLA-B*5801 that do well clinically have CD8+ T cells that recognize less-variable regions of the virus, particularly in Gag. The HIV-1 quasispecies in these patients do escape slowly during long-term infection, but each escape mutant incurs a proved fitness cost to the virus101. The time it takes for the first T cell responses to become targeted to conserved epitopes might be important in determining long-term control of viral infection75,88,99. It is not clear what features determine which CD8+ T cell epitopes will become immunodominant; it is clear that HLA type is important, but the precursor frequency of naive T cells that are specific for HIV proteins is also likely to be a factor that is probably influenced both by genetics and a history of previous (cross-reactive) antigen exposure. Vaccines could influence this.
The CD4+ T cell response. HIV-1 infects and significantly depletes memory CD4+ T cells25,79, and HIV-1-specific CD4+ T cells are particularly susceptible to HIV-1 infection103. CD4+ T cell responses to HIV proteins have always been difficult to show, and there is a disparity between the measurements of CD4+ T cell responses to antigen when observing cytokine production versus proliferation104. Nevertheless, several epitopes for CD4+ T cells have been identified, particularly in Gag105. Expansion of HIV-specific CD4+ T cell responses occurs in acute HIV-1 infection, but such responses decline rapidly106,107; although, very early administration of ART, to control viraemia and prevent the killing of CD4+ T cells, can rescue strong HIV-1 CD4+ T cell responses108,109. However, even with the probably suboptimal help from the weakened CD4+ T cell repertoire, the first CD8+ T cell responses are strong, although their progression into long-term memory cells could be impaired. The rapid decline of CD8+ T cell responses observed after the founder epitope is eliminated from the virus in the plasma, owing to escape mutations75, is consistent with the impaired long-term CD8+ T cell memory that has been observed in a model in which mice were depleted of CD4+ T cells110.
Implications for vaccine development. The findings described above suggest a role for CD8+ T cells in the earliest immune control of acute HIV-1 infection. CD8+ T cells develop abnormally111 and become dysfunctional as HIV-1 infection progresses (reviewed in Ref. 112), but the early HIV-1-specific CD8+ T cell response seems to be functionally normal109 (G. Ferrari, personal communication). Although not all the factors that contribute to a low virus set point and good long-term prognosis (without ART) are known, it is clear that CD8+ T cells are important components. If a vaccine cannot completely prevent infection, there should be a benefit from stimulating appropriate CD8+ T cell responses, as shown recently in the macaque SIV model113. An effective vaccine would need to stimulate CD8+ T cell responses to multiple epitopes, especially to those that are highly conserved. It would also be favourable to stimulate a broad T cell response that recognizes common variants of the founder virus epitope sequence, which would limit escape options114,115.
Antibody responses during acute HIV-1 infection
Early neutralizing and non-neutralizing antibody responses. Antibodies that neutralize autologous virus develop slowly, arising ∼12 weeks or longer after HIV-1 transmission116,117,118. Antibodies that show some degree of neutralization of heterologous virus eventually arise in ∼20% of patients years after infection117,119,120. To determine the specificity and kinetics of antibody production after HIV-1 transmission and to understand why broadly reactive neutralizing antibodies are not made during acute HIV infection, it is important to study the earliest B cell responses to the transmitted virus117,118.
Env-specific antibody responses to autologous, consensus Env epitopes were determined in the same plasma donor cohort as described earlier for innate immunity57. The first detectable B cell response was found to occur 8 days after T0 in the form of immune complexes, whereas the first free antibody in the plasma was specific for Env glycoprotein (gp)41 and appeared 13 days after T0. By contrast, the appearance of Env gp120-specific antibodies was delayed an additional 14 days, as was the production of other non-neutralizing Env-specific antibodies57,118,121,122,123 (Fig. 5; Table 1). The first HIV-1-specific IgA responses in mucosal secretions, which were detected within the first 3 weeks after T0, also recognized gp41 during acute HIV infection (N. L. Yates and G.D.T., unpublished observations). A study that applied mathematical modelling to early viral dynamics indicated that the initial gp41-specific IgG and IgM responses did not significantly affect the early dynamics of plasma viral load57. These acute gp41- and gp120-specific antibodies did not select escape mutations, indicating that these early arising antibodies are ineffective against HIV-1. Similar analyses of the effect of the initial immune complexes and gp41-specific IgA responses on viral dynamics are needed to understand the interplay between the initial host antibody responses and virus replication. It is not known why the initial antibody response to Env is non-neutralizing; it may relate to the immunodominance of denatured or non-functional Env forms124,125. The first antibodies to induce escape mutants are autologous-virus-neutralizing antibodies that develop ∼12 or more weeks after transmission (Table 1). Fc receptor (FcR)-mediated and complement-associated anti-HIV effector functions have also been reported during primary infection126,127,128; however, further studies are required to define their role and capacity to select escape mutations.
Figure 5. Composite alignment of the earliest innate and adaptive immune responses detected after HIV-1 transmission.
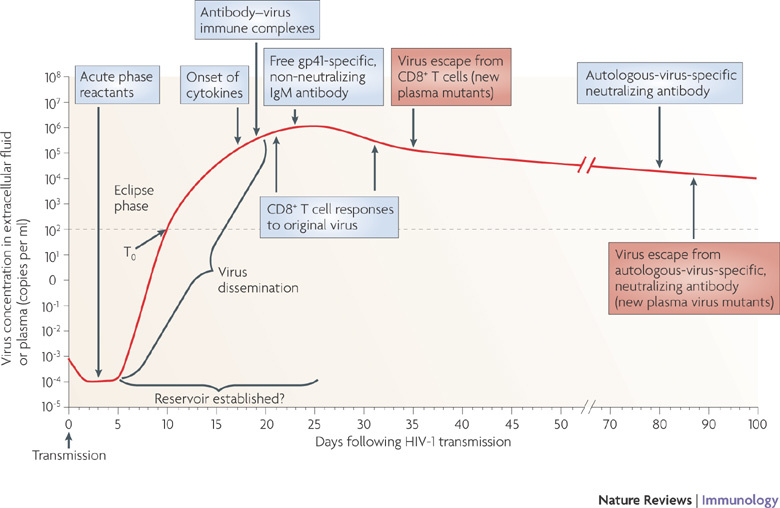
The first systemically detectable immune responses to HIV-1 infection are the increases in levels of acute-phase proteins in the plasma, which are observed when virus replication is still largely restricted to the mucosal tissues and draining lymph nodes (eclipse phase). When virus is first detected in the plasma (T0), broad and dynamic increases in plasma cytokine levels are also observed. Within days, as plasma viraemia is still increasing exponentially, the first antibody–virus immune complexes are detected. Expansion of the earliest HIV-1-specific CD8+ T cell responses also commences prior to peak viraemia, followed by detection of the first free glycoprotein (gp)41-specific but non-neutralizing IgM antibodies. Complete virus escape from the first CD8+ T cell responses can occur rapidly, within 10 days of T cell expansion. By this time, viral reservoirs exist, possibly becoming established within days of infection. The earliest autologous-virus-neutralizing antibodies are detected around day 80 following infection, as viral loads are still declining prior to the onset of the viral set point. Antibody escape virus mutants emerge in the plasma within the following week.
Table 1.
Env-specific antibody responses in acute HIV-1 infection
| Antibody specificity | Time of onset after transmission (days) |
|---|---|
| gp41 | 23 |
| gp120 | 38 |
| Non-neutralizing to CD4-binding site, MPER and CD4-inducible epitopes | 40–70 |
| Autologous-virus neutralizing antibodies | Earliest ∼84 |
| Broad-specificity and neutralizing to CD4-binding site, carbohydrate and MPER | Not usually made, but when they are they arise ∼30 months after transmission in chronic infection |
| gp, glycoprotein; MPER, membrane-proximal external region. | |
The range of epitopes bound by the first (specific to autologous virus) neutralizing antibodies in HIV-1 clade C infection is narrow and epitopes are often restricted to certain virus isolates129. Similar to infections with clade B HIV-1, the initial autologous-virus-neutralizing antibodies induced in clade C infections are induced with similar kinetics and are usually specific only for the initially transmitted Env variant116,117. Although the autologous-virus-neutralizing antibody response can control the virus quasispecies present when these antibodies appear in infections with HIV-1 clade B or C viruses, the narrowness of the response allows rapid viral escape116,117 (K. Bar and G. Shaw, personal communication).
Broad-specificity, neutralizing antibodies to conserved Env regions are rare. Interestingly, antibodies specific for the conserved regions of HIV-1 Env — such as the carbohydrate epitope recognized by the unique broad-specificity neutralizing monoclonal antibody 2G12 (Ref. 130), the CD4-binding site recognized by the monoclonal antibody 1b12 (Ref. 131) and the membrane-proximal region recognized by the monoclonal antibodies 2F5, Z13 and 4E10 (Refs 132, 133, 134 — are rarely generated during HIV-1 infection; when they do occur, they develop only after ∼20–30 months of infection57,119,120,135. These observations indicate that both genetic factors and maturation of the antibody response to HIV-1 are necessary for the generation of this rare, late, broad-specificity, neutralizing antibody response. Affinity maturation through somatic hypermutation may be crucial for the generation of these neutralizing antibodies and may be delayed because of impaired CD4+ T cell help. Because the 2F5, 4E10 and 1b12 monoclonal antibodies have long hydrophobic complementarity-determining region 3 (CDR3) sequences and show polyreactivity for autologous molecules, it has been suggested that B cell regulatory mechanisms such as self tolerance may control their production136.
Early damage to mucosal B cell generative microenvironments. Acute HIV-1 infection profoundly affects blood and tissue B cells123. HIV-1 induces early class switching in polyclonal B cells and is associated with marked increases in the number of blood and tissue memory B cells and plasma cells, as well as a decrease in the number of naive B cells123. In the mucosal B cell inductive microenvironments, such as Peyer's patches, where HIV and SIV replicate at high levels during acute infection137,138,139, both HIV-1 (Refs 123, 139) and SIV140 can induce the lysis of follicular B cells, massive B cell apoptosis and loss of ∼50% of germinal centres within the first 80 days of infection. Early loss of germinal centres may result in defects in the ability to rapidly generate high-affinity HIV-1 antibodies and lead to a delay in the induction of autologous-virus-neutralizing antibodies.
Implications for vaccine design. The finding that the generation of potentially protective antibodies is delayed until after initial control of viraemia ∼12 weeks after transmission and then focused on only a few epitopes implies that it will be important to develop a vaccine that primes a very early and broad antibody response that targets multiple neutralizing epitopes for effective control of early viral expansion; the natural process is too little, too late. The early perturbations to B cells by the virus similarly indicate the need for a vaccine that either has high levels of durable protective antibody responses or primes in order to induce a rapid secondary response. The rarity of broad-specificity, neutralizing antibody responses to conserved epitopes in Env emphasizes the need to search for and find those small B cell subsets that can make broad-specificity, neutralizing antibodies: immunogens and adjuvants are needed that target those specific B cells.
Outlook
A clear picture of the earliest immune responses to HIV-1 (Fig. 5) has major implications for HIV-1 prevention in general and for vaccine design. After transmission, there is probably only a 5–10 day window during the eclipse phase in which the virus-infected cells could be eradicated, before the virus spreads widely and integrates to generate long-lasting and non-eradicable reservoirs of latent virus. True sterilizing immunity can be attained only if the virus is prevented from infecting any host cells. This could be achieved only through broad-specificity neutralizing antibodies that are already present in the plasma and at mucosal sites before virus transmission. In support of this hypothesis, it has been shown that local application, or intravenous injection, of neutralizing monoclonal antibodies against SIV in macaques is protective against subsequent challenge with the virus141,142,143,144.
If neutralizing antibodies cannot be generated in sufficient quantity, affinity and breadth, other immune mechanisms could abort the infection by attacking the founder virus and/or the first infected cells. CD8+ T cell-mediated killing, antibody-mediated mechanisms dependent on FcRs (including antibody-dependent cell-mediated cytotoxicity (ADCC)), NK cell-mediated lysis and β-chemokine release all have the potential to prevent early infection. However, to prevent infection these effector mechanisms would have to be ready primed, as there is not time to activate and expand central memory CD8+ T cells145, for example, before chronic infection is established.
Harnessing NK cells and NKT cells might be an effective strategy to control the increase of virally infected cells during the eclipse phase or during the increase in viraemia of early HIV-1 infection. Although it may be hazardous to induce chronic hyperactivation of these cells as a means to inhibit virus infection, it may be possible to immunize subjects with HIV-1 antigens such as peptides that specifically expand potentially protective NK and NKT cell subpopulations, thereby altering the cell repertoire to contain a higher proportion of protective NK cells76,77.
The modest protection offered by the vaccines used in the recent RV144 clinical trial carried out in Thailand with volunteers at low risk of HIV-1 infection146 (Box 1) may be an example of weak immune responses combining to raise the threshold for infection — a rare event.
Once infection starts to spread, enhancing the natural containment processes might be the only immunological option to benefit infected individuals. CD8+ T cell responses, which are clearly effective in reducing the peak viraemia during acute infection, could be enhanced through vaccination by increasing their breadth of epitope recognition so that, rather than mediating sequential responses to single epitopes, there would be a simultaneous multi-epitope-specific CD8+ T cell response to the virus113,147,148. Focusing this response on conserved epitopes, for which escape incurs a fitness cost to the virus, would be desirable. Strategies for enhancing or preserving CD4+ T cell help would also be of benefit for supporting the CD8+ T cells. However, it is important to recognize that CD8+ T cells are highly sensitive to single amino acid variation in epitope peptides75,149, so even minor mismatches between vaccine-encoded epitopes and viral epitopes could be a serious problem and could diminish the effectiveness of any vaccine-stimulated T cell response.
If a vaccine can induce greater breadth in early T and B cell responses to HIV-1 than occurs naturally during acute infection, then the use of a combination of protective epitopes in a preventative vaccine may control the early dissemination of HIV-1, resulting in a lower viral set point and better long-term immune control. Preliminary unpublished results with experimental vaccines that include multiple common variants of HIV-1 proteins such as Gag (mosaic vaccines) have been shown to enhance the breadth and magnitude of T cell responses in animal studies150,151,152. This approach and other novel strategies for expanding the breadth of induced Env-specific B cell responses are also central to improving the prospects of vaccine success.
It can be thought to be good news that most HIV-1 transmissions that result in productive infection are mediated by only one virion, indicating a vulnerability of the virus to immune attack during the eclipse phase. This suggests that a well-designed vaccine strategy might have a chance of achieving good (if not perfect) control around the time of acute peak viraemia, preventing the onset of damaging chronic immune activation and damage to generative immune cell environments. However, vaccine strategies must be developed that potentiate what is clearly a qualitatively and quantitatively insufficient immune response in the first few weeks of HIV-1 infection. It is hoped that both the innate and adaptive arms of the immune system can be harnessed to develop an HIV-1 vaccine that ensures that adequate immune protection is in place before transmission, enabling earlier, broader and more effective secondary responses for preventing or controlling acute HIV-1 infection.
Box 1 | Problems facing the development of an HIV vaccine.
All attempts to make a vaccine against HIV-1 have failed. Three vaccine approaches have been tested in clinical trials for efficacy. The AIDSVAX glycoprotein (gp)120 vaccine stimulated the production of non-neutralizing antibody to the virus envelope proteins and failed to protect vaccinated individuals from infection153. The STEP vaccine, comprised of three recombinant attenuated adenovirus serotype 5 viruses expressing HIV-1 Gag, Pol and Nef, stimulated CD8+ T cell responses to the viral proteins but again showed no protective effect154,155. Similar virus vector-based vaccines have been shown to stimulate simian immunodeficiency virus (SIV)-specific CD8+ T cell responses in rhesus macaques, and an adenovirus serotype 5 vector expressing Gag protected against challenge with a chimeric SIV–HIV (SHIV89.6p) virus but was not protective against challenge with the more natural SIVmac239 (Ref. 156). More recent data show that recombinant vaccines that stimulate much broader and stronger CD8+ T cell responses can partially protect against SIVmac239 and SIVmac251 virus challenge, resulting in more attenuated infection with low virus load and prolonged survival of rhesus macaques113,157.
A third efficacy trial, in Thailand using a canary pox viral vector expressing gp120, Gag and Pol to prime immune responses followed by the AIDSVAX gp120 vaccine to boost the immune response, has been reported recently146. This showed for the first time a small protective effect, with 30% fewer vaccine recipients becoming infected with HIV-1 than controls; the result was statistically significant in one of the three analyses made. The volunteer cohort was low risk (annual incidence of infection ∼0.3%) and this may be relevant as it may be easier to protect such people than those at high risk. It is not clear whether protection was mediated by antibody, T cells, innate cells or some combination of the three, but those who did become infected did not have reduced virus levels, which is usually seen for protection mediated by T cells in SIV models113,157.
There is a general consensus in the field that future vaccine approaches should be less empirical and that a deeper understanding of the earliest immune responses to HIV-1 and SIV infection is needed. It will also be important to understand why broad-specificity, neutralizing antibodies are not routinely induced and to determine ways to safely induce them, and to identify what immune responses lead to a better outcome — as in the rare individuals, known as 'elite controllers', who successfully control HIV-1 infection for decades without needing antiretroviral drug therapy158,159.
Acknowledgements
This work was supported by the Center for HIV/AIDS Vaccine Immunology (CHAVI) grant A1067854-03. Additional support came from the Medical Research Human Immunology Unit, the National Institute for Health Research, Oxford Biomedical Research Centre and grants 38643 and 37874 from the Bill and Melinda Gates Foundation. A.J.M. and P.B. are Jenner Investigators and P.B. is supported by a Jenner Fellowship. G.D.T is supported by the US National Institutes of Health, grants RO1AI052779, U19AI067854, AI068618 and AI64518 and the Bill and Melinda Gates Foundation (grant 38619). We are grateful to members of the CHAVI Scientific Leadership Group, N. Letvin, M. Cohen, J. Sodroski, D. Goldstein and G. Shaw for scientific input and for reviewing this manuscript, and to the CHAVI Clinical Core Investigators and their patients for participation in the CHAVI studies cited in this review.
Glossary
- Viral set point
The time at which plasma viraemia settles to a stable level (within approximately 3–6 months from the onset of HIV infection). Viral set point is partially predictive of both how quickly HIV infection will progress and the risk of HIV transmission.
- Founder virus
A transmitted virus or a virus that gives rise to all virus quasispecies in an infected individual.
- Viral fitness
The ability of a virus to replicate in a given environment. By definition in in vitro studies, a drug-resistant virus has greater ability to replicate than wild-type virus when measured in the presence of a drug, similarly a T cell escape mutant will replicate better than wild-type virus when co-cultured with specific T cells. The T cell-resistant or drug-resistant virus may replicate less well than the wild type when the selective force is withdrawn.
- Langerhans cell
A type of dendritic cell that is resident in the epithelial layer of the skin and mucosa.
- Single-genome amplification
A method of DNA sequencing that uses high-fidelity polymerase and minimizes PCR amplification, thereby excluding sequence errors and recombination events that may be introduced during amplification.
- Clade
HIV is subdivided, based on degree of sequence divergence, into three major groups, M, N and O; group M is subdivided into 10 subtypes or clades, of which clade C is the predominant subtype worldwide (prevalent in subSaharan Africa and India) and clade B is the most studied subtype (prevalent in North America and Eastern Europe).
- Quasispecies
A distribution of non-identical but closely related viral genomes. The entire distribution forms an organized cooperative structure, which acts like (quasi) a single unit (species).
- APOBEC cytidine deaminases
A family of host antiviral proteins that introduce multiple mutations, including stop codons, in retroviruses by deaminating cytosine residues in nascent retroviral cDNA.
- Germinal centre
A highly specialized and dynamic microenvironment located in peripheral lymphoid tissues (for example, the spleen or lymph nodes). It is the main site of B cell maturation, leading to the generation of memory B cells and plasma cells that produce high-affinity antibody.
- Regulatory T (TReg) cell
A type of CD4+ T cell that is characterized by its expression of forkhead box P3 (FOXP3) and high levels of CD25. TReg cells can suppress many types of immune responses.
- Genome-wide association study
An approach that involves rapidly scanning single nucleotide polymorphism markers across the complete genomes of many individuals to find genetic variations associated with a particular disease.
- Single nucleotide polymorphisms
(SNPs). Single nucleotide variations in genomic DNA sequences in which one of the four nucleotides is substituted for another. SNPs are the most frequent type of polymorphism in the genome and can be used to map genes connected by linkage disequilibrium.
- Bw4 motif
The amino acid sequences at positions 77–83 in the α1 domain of HLA class I heavy chains. There are four Bw4 motif sequences, which differ only at positions 77 (Asn, Asp or Ser) and 80 (Ile or Thr). The Bw4 motif is involved in the recognition of certain HLA alleles by killer immunoglobulin-like receptors.
- Seroconversion
Development of a detectable concentration of pathogen-specific antibodies in the serum as a result of infection or immunization. Seroconversion normally occurs in patients with HIV-1 infection 3–4 weeks following infection, around Fiebig stage III, typically when plasma virus loads are reaching their peak.
- Acute-phase proteins
A group of proteins, including C-reactive protein, serum amyloid A, complement components and fibrinogen, that are secreted into the blood in increased or decreased quantities by hepatocytes in response to trauma, inflammation or disease. These proteins can be inhibitors or mediators of inflammatory processes.
- T helper 17 cells
A subset of CD4+ T helper cells that produce interleukin-17 (IL-17) and that are thought to be important in antibacterial and antifungal immunity and may also have a role in autoimmune diseases. Their generation involves IL-23 and IL-21, as well as the transcription factors RORγt (retinoic acid-related orphan receptor-γt) and STAT3 (signal transducer and activator of transcription 3).
- Envelope proteins
Envelope proteins of HIV are initially produced as a precursor glycoprotein of 160 kDa (gp160) that is cleaved to generate gp120 and gp41. Three gp120 molecules and three gp41 molecules are thought to combine in a trimer to form the envelope spike.
- Fiebig stages
Stages of HIV-1 infection. They are defined by a series of laboratory tests that measure the emergence of HIV-1-specific antibodies, viral RNA and viral antigens.
- Class switching
The somatic-recombination process by which the class of immunoglobulin is switched from IgM to IgG, IgA or IgE.
- Peyer's patches
Specialized lymphoid follicles localized in the submucosa of the small intestine and appendix.
- Antibody-dependent cell-mediated cytotoxicity
(ADCC). A cytotoxic mechanism by which an antibody-coated target cell is directly killed by a leukocyte that expresses FcRs, such as an NK cell, macrophage or neutrophil.
Biographies
Andrew J. McMichael completed his M.B. at the University of Cambridge, UK, and his Ph.D. at the Medical Research Council (MRC) National Institute of Medical Research, London, UK. He has worked on HIV since 1987, focusing on T cell immunity and vaccine development. He has directed the MRC Human Immunology Unit, Oxford, UK, since 1998 and the Weatherall Institute of Molecular Medicine, Oxford, UK, since 2000. He is a Scientific Leadership Group member of the Center for HIV/AIDS Vaccine Immunology (CHAVI). He is a member of the European Molecular Biology Organization and a Fellow of the UK Academy of Medical Science, London, UK, and of The Royal Society, London, UK.
Persephone Borrow received her Ph.D. from the University of Cambridge, UK, in 1989. She subsequently carried out postdoctoral research at The Scripps Research Institute, La Jolla, USA, becoming an assistant member there in 1995. In 1997 she took up the post of Viral Immunology Group Leader at the newly established Edward Jenner Institute for Vaccine Research, UK, and was promoted to Senior Group Leader in 2003. She joined the University of Oxford, UK, in 2005, where she is currently a Reader in the Nuffield Department of Clinical Medicine. Her research programme focuses on innate and T cell responses to viruses that cause persistent infections, aiming to understand the roles of different types of response in protection and pathogenesis.
Georgia D. Tomaras received her Ph.D. from Syracuse Upstate Medical University, New York, USA, in 1998. She completed her postdoctoral fellowship at Duke University, Durham, USA, in 2001 and is Assistant Professor of Surgery, Immunology, Molecular Genetics and Microbiology and Associate Research Director, Duke Human Vaccine Institute, at Duke University School of Medicine. Her research programme focuses on understanding the cellular and humoral immune responses to HIV-1 infection and experimental HIV-1 vaccination.
Nilu Goonetilleke received her Ph.D. from the Open University, UK. She is a senior immunologist responsible for coordinating all CHAVI studies within the Weatherall Institute of Molecular Medicine. Her research focus is on the immunology of infectious diseases and vaccine design, with a particular focus on T cell immunology.
Barton F. Haynes received his M.D. from Baylor College of Medicine, Houston, USA, in 1973. After completing infectious diseases and allergy and immunology training at the US National Institutes of Health (NIH), he went to Duke University in 1980. Here, he is Director of the Human Vaccine Institute, where teams of investigators are working on vaccines for emerging infections, including HIV-1, tuberculosis and pandemic influenza virus. He is currently funded by the US NIH as the leader of CHAVI, and the Bill and Melinda Gates Foundation funds his leadership of a Collaboration for AIDS Vaccine Discovery Center. He is a member of the Institute of Medicine of the National Academy of Sciences, USA, and a Fellow of the American Academy of Arts and Sciences, Cambridge, USA.
Related links
FURTHER INFORMATION
Competing interests
The authors declare no competing financial interests.
References
- 1.Pilcher CD, et al. Detection of acute infections during HIV testing in North Carolina. N. Engl. J. Med. 2005;352:1873–1883. doi: 10.1056/NEJMoa042291. [DOI] [PubMed] [Google Scholar]
- 2.Powers KA, et al. Improved detection of acute HIV-1 infection in sub-Saharan Africa: development of a risk score algorithm. AIDS. 2007;21:2237–2242. doi: 10.1097/QAD.0b013e3282f08b4d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hu Q, et al. Blockade of attachment and fusion receptors inhibits HIV-1 infection of human cervical tissue. J. Exp. Med. 2004;199:1065–1075. doi: 10.1084/jem.20022212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Veazey RS, et al. Protection of macaques from vaginal SHIV challenge by vaginally delivered inhibitors of virus-cell fusion. Nature. 2005;438:99–102. doi: 10.1038/nature04055. [DOI] [PubMed] [Google Scholar]
- 5.Margolis L, Shattock R. Selective transmission of CCR5-utilizing HIV-1: the 'gatekeeper' problem resolved? Nature Rev. Microbiol. 2006;4:312–317. doi: 10.1038/nrmicro1387. [DOI] [PubMed] [Google Scholar]
- 6.Li Q, et al. Visualizing antigen-specific and infected cells in situ predicts outcomes in early viral infection. Science. 2009;323:1726–1729. doi: 10.1126/science.1168676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller CJ, et al. Propagation and dissemination of infection after vaginal transmission of simian immunodeficiency virus. J. Virol. 2005;79:9217–9227. doi: 10.1128/JVI.79.14.9217-9227.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sodora DL, Gettie A, Miller CJ, Marx PA. Vaginal transmission of SIV: assessing infectivity and hormonal influences in macaques inoculated with cell-free and cell-associated viral stocks. AIDS Res. Hum. Retroviruses. 1998;14:S119–S123. doi: 10.1089/aid.1998.14.171. [DOI] [PubMed] [Google Scholar]
- 9.Lai SK, et al. Human immunodeficiency virus type 1 is trapped by acidic but not by neutralized human cervicovaginal mucus. J. Virol. 2009;83:11196–11200. doi: 10.1128/JVI.01899-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Galvin SR, Cohen MS. The role of sexually transmitted diseases in HIV transmission. Nature Rev. Microbiol. 2004;2:33–42. doi: 10.1038/nrmicro794. [DOI] [PubMed] [Google Scholar]
- 11.Haaland RE, et al. Inflammatory genital infections mitigate a severe genetic bottleneck in heterosexual transmission of subtype A and C HIV-1. PLoS Pathog. 2009;5:e1000274. doi: 10.1371/journal.ppat.1000274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keele BF, et al. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc. Natl Acad. Sci. USA. 2008;105:7552–7557. doi: 10.1073/pnas.0802203105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abrahams MR, et al. Quantitating the multiplicity of infection with human immunodeficiency virus type 1 subtype C reveals a non-poisson distribution of transmitted variants. J. Virol. 2009;83:3556–3567. doi: 10.1128/JVI.02132-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Salazar-Gonzalez JF, et al. Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J. Exp. Med. 2009;206:1273–1289. doi: 10.1084/jem.20090378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keele BF, et al. Low-dose rectal inoculation of rhesus macaques by SIVsmE660 or SIVmac251 recapitulates human mucosal infection by HIV-1. J. Exp. Med. 2009;206:1117–1134. doi: 10.1084/jem.20082831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Q, et al. Glycerol monolaurate prevents mucosal SIV transmission. Nature. 2009;458:1034–1038. doi: 10.1038/nature07831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bishop KN, Holmes RK, Sheehy AM, Malim MH. APOBEC-mediated editing of viral RNA. Science. 2004;305:645. doi: 10.1126/science.1100658. [DOI] [PubMed] [Google Scholar]
- 18.Geijtenbeek TB, et al. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell. 2000;100:587–597. doi: 10.1016/S0092-8674(00)80694-7. [DOI] [PubMed] [Google Scholar]
- 19.Moir S, et al. B cells of HIV-1-infected patients bind virions through CD21–complement interactions and transmit infectious virus to activated T cells. J. Exp. Med. 2000;192:637–646. doi: 10.1084/jem.192.5.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brenchley JM, et al. CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J. Exp. Med. 2004;200:749–759. doi: 10.1084/jem.20040874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brenchley JM, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nature Med. 2006;12:1365–1371. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- 22.Brenchley JM, et al. Differential Th17 CD4 T-cell depletion in pathogenic and nonpathogenic lentiviral infections. Blood. 2008;112:2826–2835. doi: 10.1182/blood-2008-05-159301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gasper-Smith N, et al. Induction of plasma (TRAIL), TNFR-2, Fas ligand, and plasma microparticles after human immunodeficiency virus type 1 (HIV-1) transmission: implications for HIV-1 vaccine design. J. Virol. 2008;82:7700–7710. doi: 10.1128/JVI.00605-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Veazey RS, et al. Gastrointestinal tract as a major site of CD4+ T cell depletion and viral replication in SIV infection. Science. 1998;280:427–431. doi: 10.1126/science.280.5362.427. [DOI] [PubMed] [Google Scholar]
- 25.Mattapallil JJ, et al. Massive infection and loss of memory CD4+ T cells in multiple tissues during acute SIV infection. Nature. 2005;434:1093–1097. doi: 10.1038/nature03501. [DOI] [PubMed] [Google Scholar]
- 26.Levesque MC, et al. Polyclonal B cell differentiation and loss of gastrointestinal tract germinal centers in the earliest stages of HIV-1 infection. PLoS Med. 2009;6:e1000107. doi: 10.1371/journal.pmed.1000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ho DD, et al. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature. 1995;373:123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- 28.Schacker TW, Hughes JP, Shea T, Coombs RW, Corey L. Biological and virologic characteristics of primary HIV infection. Ann. Intern. Med. 1998;128:613–620. doi: 10.7326/0003-4819-128-8-199804150-00001. [DOI] [PubMed] [Google Scholar]
- 29.Rodriguez B, et al. Predictive value of plasma HIV RNA level on rate of CD4 T-cell decline in untreated HIV infection. JAMA. 2006;296:1498–1506. doi: 10.1001/jama.296.12.1498. [DOI] [PubMed] [Google Scholar]
- 30.Borrow P, Lewicki H, Hahn BH, Shaw GM, Oldstone MB. Virus-specific CD8+ cytotoxic T-lymphocyte activity associated with control of viremia in primary human immunodeficiency virus type 1 infection. J. Virol. 1994;68:6103–6110. doi: 10.1128/jvi.68.9.6103-6110.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koup RA, et al. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J. Virol. 1994;68:4650–4655. doi: 10.1128/jvi.68.7.4650-4655.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Price DA, et al. Positive selection of HIV-1 cytotoxic T lymphocyte escape variants during primary infection. Proc. Natl Acad. Sci. USA. 1997;94:1890–1895. doi: 10.1073/pnas.94.5.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilson JD, et al. Direct visualization of HIV-1-specific cytotoxic T lymphocytes during primary infection. AIDS. 2000;14:225–233. doi: 10.1097/00002030-200002180-00003. [DOI] [PubMed] [Google Scholar]
- 34.Markowitz M, et al. A novel antiviral intervention results in more accurate assessment of human immunodeficiency virus type 1 replication dynamics and T-cell decay in vivo. J. Virol. 2003;77:5037–5038. doi: 10.1128/JVI.77.8.5037-5038.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sedaghat AR, Siliciano JD, Brennan TP, Wilke CO, Siliciano RF. Limits on replenishment of the resting CD4+ T cell reservoir for HIV in patients on HAART. PLoS Pathog. 2007;3:e122. doi: 10.1371/journal.ppat.0030122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brennan TP, et al. Analysis of human immunodeficiency virus type 1 viremia and provirus in resting CD4+ T cells reveals a novel source of residual viremia in patients on antiretroviral therapy. J. Virol. 2009;83:8470–8481. doi: 10.1128/JVI.02568-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dinoso JB, et al. Treatment intensification does not reduce residual HIV-1 viremia in patients on highly active antiretroviral therapy. Proc. Natl Acad. Sci. USA. 2009;106:9403–9408. doi: 10.1073/pnas.0903107106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Finkel TH, et al. Apoptosis occurs predominantly in bystander cells and not in productively infected cells of HIV- and SIV-infected lymph nodes. Nature Med. 1995;1:129–134. doi: 10.1038/nm0295-129. [DOI] [PubMed] [Google Scholar]
- 39.Silvestri G, et al. Nonpathogenic SIV infection of sooty mangabeys is characterized by limited bystander immunopathology despite chronic high-level viremia. Immunity. 2003;18:441–452. doi: 10.1016/S1074-7613(03)00060-8. [DOI] [PubMed] [Google Scholar]
- 40.Liu Z, et al. Elevated CD38 antigen expression on CD8+ T cells is a stronger marker for the risk of chronic HIV disease progression to AIDS and death in the Multicenter AIDS Cohort Study than CD4+ cell count, soluble immune activation markers, or combinations of HLA-DR and CD38 expression. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1997;16:83–92. doi: 10.1097/00042560-199710010-00003. [DOI] [PubMed] [Google Scholar]
- 41.Giorgi JV, et al. Predictive value of immunologic and virologic markers after long or short duration of HIV-1 infection. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 2002;29:346–355. doi: 10.1097/00126334-200204010-00004. [DOI] [PubMed] [Google Scholar]
- 42.Deeks SG, et al. Immune activation set point during early HIV infection predicts subsequent CD4+ T-cell changes independent of viral load. Blood. 2004;104:942–947. doi: 10.1182/blood-2003-09-3333. [DOI] [PubMed] [Google Scholar]
- 43.Li Q, et al. Microarray analysis of lymphatic tissue reveals stage-specific, gene expression signatures in HIV-1 infection. J. Immunol. 2009;183:1975–1982. doi: 10.4049/jimmunol.0803222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sodora DL, Silvestri G. Immune activation and AIDS pathogenesis. AIDS. 2008;22:439–446. doi: 10.1097/QAD.0b013e3282f2dbe7. [DOI] [PubMed] [Google Scholar]
- 45.Gao F, et al. Origin of HIV-1 in the chimpanzee Pan troglodytes troglodytes. Nature. 1999;397:436–441. doi: 10.1038/17130. [DOI] [PubMed] [Google Scholar]
- 46.Korber B, et al. Timing the ancestor of the HIV-1 pandemic strains. Science. 2000;288:1789–1796. doi: 10.1126/science.288.5472.1789. [DOI] [PubMed] [Google Scholar]
- 47.Keele BF, et al. Chimpanzee reservoirs of pandemic and nonpandemic HIV-1. Science. 2006;313:523–526. doi: 10.1126/science.1126531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu R, et al. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell. 1996;86:367–377. doi: 10.1016/S0092-8674(00)80110-5. [DOI] [PubMed] [Google Scholar]
- 49.Carrington M, O'Brien SJ. The influence of HLA genotype on AIDS. Annu. Rev. Med. 2003;54:535–551. doi: 10.1146/annurev.med.54.101601.152346. [DOI] [PubMed] [Google Scholar]
- 50.Fellay J, et al. A whole-genome association study of major determinants for host control of HIV-1. Science. 2007;317:944–947. doi: 10.1126/science.1143767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gao X, et al. Effect of a single amino acid change in MHC class I molecules on the rate of progression to AIDS. N. Engl. J. Med. 2001;344:1668–1675. doi: 10.1056/NEJM200105313442203. [DOI] [PubMed] [Google Scholar]
- 52.Qi Y, et al. KIR/HLA pleiotropism: protection against both HIV and opportunistic infections. PLoS Pathog. 2006;2:e79. doi: 10.1371/journal.ppat.0020079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Martin MP, et al. Epistatic interaction between KIR3DS1 and HLA-B delays the progression to AIDS. Nature Genet. 2002;31:429–434. doi: 10.1038/ng934. [DOI] [PubMed] [Google Scholar]
- 54.Martin MP, et al. Innate partnership of HLA-B and KIR3DL1 subtypes against HIV-1. Nature Genet. 2007;39:733–740. doi: 10.1038/ng2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alter G, et al. HLA class I subtype-dependent expansion of KIR3DS1+ and KIR3DL1+ NK cells during acute human immunodeficiency virus type 1 infection. J. Virol. 2009;83:6798–6805. doi: 10.1128/JVI.00256-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alter G, et al. Evolution of innate and adaptive effector cell functions during acute HIV-1 infection. J. Infect. Dis. 2007;195:1452–1460. doi: 10.1086/513878. [DOI] [PubMed] [Google Scholar]
- 57.Tomaras GD, et al. Initial B-cell responses to transmitted human immunodeficiency virus type 1: virion-binding immunoglobulin M (IgM) and IgG antibodies followed by plasma anti-gp41 antibodies with ineffective control of initial viremia. J. Virol. 2008;82:12449–12463. doi: 10.1128/JVI.01708-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stacey AR, et al. Induction of a striking systemic cytokine cascade prior to peak viremia in acute human immunodeficiency virus type 1 infection, in contrast to more modest and delayed responses in acute hepatitis B and C virus infections. J. Virol. 2009;83:3719–3733. doi: 10.1128/JVI.01844-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Raffatellu M, et al. Simian immunodeficiency virus-induced mucosal interleukin-17 deficiency promotes Salmonella dissemination from the gut. Nature Med. 2008;14:421–428. doi: 10.1038/nm1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nilsson J, et al. Early immune activation in gut-associated and peripheral lymphoid tissue during acute HIV infection. AIDS. 2007;21:565–574. doi: 10.1097/QAD.0b013e3280117204. [DOI] [PubMed] [Google Scholar]
- 61.Lapenta C, et al. Type I interferon is a powerful inhibitor of in vivo HIV-1 infection and preserves human CD4+ T cells from virus-induced depletion in SCID mice transplanted with human cells. Virology. 1999;263:78–88. doi: 10.1006/viro.1999.9869. [DOI] [PubMed] [Google Scholar]
- 62.de Jong MD, et al. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nature Med. 2006;12:1203–1207. doi: 10.1038/nm1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cameron MJ, et al. Interferon-mediated immunopathological events are associated with atypical innate and adaptive immune responses in patients with severe acute respiratory syndrome. J. Virol. 2007;81:8692–8706. doi: 10.1128/JVI.00527-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Killian MS, Fujimura SH, Hecht FM, Levy JA. Similar changes in plasmacytoid dendritic cell and CD4 T-cell counts during primary HIV-1 infection and treatment. AIDS. 2006;20:1247–1252. doi: 10.1097/01.aids.0000232231.34253.bd. [DOI] [PubMed] [Google Scholar]
- 65.Malleret B, et al. Primary infection with simian immunodeficiency virus: plasmacytoid dendritic cell homing to lymph nodes, type I interferon, and immune suppression. Blood. 2008;112:4598–4608. doi: 10.1182/blood-2008-06-162651. [DOI] [PubMed] [Google Scholar]
- 66.Lore K, et al. Accumulation of DC-SIGN+CD40+ dendritic cells with reduced CD80 and CD86 expression in lymphoid tissue during acute HIV-1 infection. AIDS. 2002;16:683–692. doi: 10.1097/00002030-200203290-00003. [DOI] [PubMed] [Google Scholar]
- 67.Beignon AS, et al. Endocytosis of HIV-1 activates plasmacytoid dendritic cells via Toll-like receptor-viral RNA interactions. J. Clin. Invest. 2005;115:3265–3275. doi: 10.1172/JCI26032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Granelli-Piperno A, Golebiowska A, Trumpfheller C, Siegal FP, Steinman RM. HIV-1-infected monocyte-derived dendritic cells do not undergo maturation but can elicit IL-10 production and T cell regulation. Proc. Natl Acad. Sci. USA. 2004;101:7669–7674. doi: 10.1073/pnas.0402431101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Boasso A, et al. HIV inhibits CD4+ T-cell proliferation by inducing indoleamine 2, 3-dioxygenase in plasmacytoid dendritic cells. Blood. 2007;109:3351–3359. doi: 10.1182/blood-2006-07-034785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Manches O, et al. HIV-activated human plasmacytoid DCs induce Tregs through an indoleamine 2, 3-dioxygenase-dependent mechanism. J. Clin. Invest. 2008;118:3431–3439. doi: 10.1172/JCI34823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sabado RL, et al. In vitro priming recapitulates in vivo HIV-1 specific T cell responses, revealing rapid loss of virus reactive CD4+ T cells in acute HIV-1 infection. PLoS ONE. 2009;4:e4256. doi: 10.1371/journal.pone.0004256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Borrow P, Bhardwaj N. Innate immune responses in primary HIV-1 infection. Curr. Opin. HIV AIDS. 2008;3:36–44. doi: 10.1097/COH.0b013e3282f2bce7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Clay CC, et al. Neuroinvasion of fluorescein-positive monocytes in acute simian immunodeficiency virus infection. J. Virol. 2007;81:12040–12048. doi: 10.1128/JVI.00133-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ward J, et al. HIV modulates the expression of ligands important in triggering natural killer cell cytotoxic responses on infected primary T-cell blasts. Blood. 2007;110:1207–1214. doi: 10.1182/blood-2006-06-028175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Goonetilleke N, et al. The first T cell response to transmitted/founder virus contributes to the control of acute viremia in HIV-1 infection. J. Exp. Med. 2009;206:1253–1272. doi: 10.1084/jem.20090365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cooper MA, et al. Cytokine-induced memory-like natural killer cells. Proc. Natl Acad. Sci. USA. 2009;106:1915–1919. doi: 10.1073/pnas.0813192106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sun JC, Beilke JN, Lanier LL. Adaptive immune features of natural killer cells. Nature. 2009;457:557–561. doi: 10.1038/nature07665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cummins JE, Jr, Doncel GF. Biomarkers of cervicovaginal inflammation for the assessment of microbicide safety. Sex. Transm. Dis. 2009;36:S84–S91. doi: 10.1097/OLQ.0b013e3181994191. [DOI] [PubMed] [Google Scholar]
- 79.Li Q, et al. Peak SIV replication in resting memory CD4+ T cells depletes gut lamina propria CD4+ T cells. Nature. 2005;434:1148–1152. doi: 10.1038/nature03513. [DOI] [PubMed] [Google Scholar]
- 80.Mueller YM, et al. IL-15 treatment during acute simian immunodeficiency virus (SIV) infection increases viral set point and accelerates disease progression despite the induction of stronger SIV-specific CD8+ T cell responses. J. Immunol. 2008;180:350–360. doi: 10.4049/jimmunol.180.1.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pantaleo G, et al. Major expansion of CD8+ T cells with a predominant Vβ usage during the primary immune response to HIV. Nature. 1994;370:463–467. doi: 10.1038/370463a0. [DOI] [PubMed] [Google Scholar]
- 82.Fiebig EW, et al. Dynamics of HIV viremia and antibody seroconversion in plasma donors: implications for diagnosis and staging of primary HIV infection. AIDS. 2003;17:1871–1879. doi: 10.1097/00002030-200309050-00005. [DOI] [PubMed] [Google Scholar]
- 83.Bernardin F, Kong D, Peddada L, Baxter-Lowe LA, Delwart E. Human immunodeficiency virus mutations during the first month of infection are preferentially found in known cytotoxic T-lymphocyte epitopes. J. Virol. 2005;79:11523–11528. doi: 10.1128/JVI.79.17.11523-11528.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Draenert R, et al. Immune selection for altered antigen processing leads to cytotoxic T lymphocyte escape in chronic HIV-1 infection. J. Exp. Med. 2004;199:905–915. doi: 10.1084/jem.20031982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jones NA, et al. Determinants of human immunodeficiency virus type 1 escape from the primary CD8+ cytotoxic T lymphocyte response. J. Exp. Med. 2004;200:1243–1256. doi: 10.1084/jem.20040511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tenzer S, et al. Antigen processing influences HIV-specific cytotoxic T lymphocyte immunodominance. Nature Immunol. 2009;10:636–646. doi: 10.1038/ni.1728. [DOI] [PubMed] [Google Scholar]
- 87.Turnbull EL, et al. Kinetics of expansion of epitope-specific T cell responses during primary HIV-1 infection. J. Immunol. 2009;182:7131–7145. doi: 10.4049/jimmunol.0803658. [DOI] [PubMed] [Google Scholar]
- 88.Wang YE, et al. Protective HLA class I alleles that restrict acute-phase CD8+ T-cell responses are associated with viral escape mutations located in highly conserved regions of human immunodeficiency virus type 1. J. Virol. 2009;83:1845–1855. doi: 10.1128/JVI.01061-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Streeck H, et al. Human immunodeficiency virus type 1-specific CD8+ T-cell responses during primary infection are major determinants of the viral set point and loss of CD4+ T cells. J. Virol. 2009;83:7641–7648. doi: 10.1128/JVI.00182-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Goepfert PA, et al. Transmission of HIV-1 Gag immune escape mutations is associated with reduced viral load in linked recipients. J. Exp. Med. 2008;205:1009–1017. doi: 10.1084/jem.20072457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Koup RA. Virus escape from CTL recognition. J. Exp. Med. 1994;180:779–782. doi: 10.1084/jem.180.3.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Borrow P, et al. Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nature Med. 1997;3:205–211. doi: 10.1038/nm0297-205. [DOI] [PubMed] [Google Scholar]
- 93.O'Connor DH, et al. Acute phase cytotoxic T lymphocyte escape is a hallmark of simian immunodeficiency virus infection. Nature Med. 2002;8:493–499. doi: 10.1038/nm0502-493. [DOI] [PubMed] [Google Scholar]
- 94.Leslie AJ, et al. HIV evolution: CTL escape mutation and reversion after transmission. Nature Med. 2004;10:282–289. doi: 10.1038/nm992. [DOI] [PubMed] [Google Scholar]
- 95.Allen TM, et al. Selective escape from CD8+ T-cell responses represents a major driving force of human immunodeficiency virus type 1 (HIV-1) sequence diversity and reveals constraints on HIV-1 evolution. J. Virol. 2005;79:13239–13249. doi: 10.1128/JVI.79.21.13239-13249.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fernandez CS, et al. Rapid viral escape at an immunodominant simian-human immunodeficiency virus cytotoxic T-lymphocyte epitope exacts a dramatic fitness cost. J. Virol. 2005;79:5721–5731. doi: 10.1128/JVI.79.9.5721-5731.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Martinez-Picado J, et al. Fitness cost of escape mutations in p24 Gag in association with control of human immunodeficiency virus type 1. J. Virol. 2006;80:3617–3623. doi: 10.1128/JVI.80.7.3617-3623.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schneidewind A, et al. Escape from the dominant HLA-B27-restricted cytotoxic T-lymphocyte response in Gag is associated with a dramatic reduction in human immunodeficiency virus type 1 replication. J. Virol. 2007;81:12382–12393. doi: 10.1128/JVI.01543-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Brumme ZL, et al. Marked epitope- and allele-specific differences in rates of mutation in human immunodeficiency type 1 (HIV-1) Gag, Pol, and Nef cytotoxic T-lymphocyte epitopes in acute/early HIV-1 infection. J. Virol. 2008;82:9216–9227. doi: 10.1128/JVI.01041-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Asquith B, Edwards CT, Lipsitch M, McLean AR. Inefficient cytotoxic T lymphocyte-mediated killing of HIV-1-infected cells in vivo. PLoS Biol. 2006;4:e90. doi: 10.1371/journal.pbio.0040090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Crawford H, et al. Compensatory mutation partially restores fitness and delays reversion of escape mutation within the immunodominant HLA-B*5703-restricted Gag epitope in chronic human immunodeficiency virus type 1 infection. J. Virol. 2007;81:8346–8351. doi: 10.1128/JVI.00465-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Addo MM, et al. Comprehensive epitope analysis of human immunodeficiency virus type 1 (HIV-1)-specific T-cell responses directed against the entire expressed HIV-1 genome demonstrate broadly directed responses, but no correlation to viral load. J. Virol. 2003;77:2081–2092. doi: 10.1128/JVI.77.3.2081-2092.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Douek DC, et al. HIV preferentially infects HIV-specific CD4+ T cells. Nature. 2002;417:95–98. doi: 10.1038/417095a. [DOI] [PubMed] [Google Scholar]
- 104.Pitcher CJ, et al. HIV-1-specific CD4+ T cells are detectable in most individuals with active HIV-1 infection, but decline with prolonged viral suppression. Nature Med. 1999;5:518–525. doi: 10.1038/8400. [DOI] [PubMed] [Google Scholar]
- 105.Kaufmann DE, et al. Comprehensive analysis of human immunodeficiency virus type 1-specific CD4 responses reveals marked immunodominance of gag and nef and the presence of broadly recognized peptides. J. Virol. 2004;78:4463–4477. doi: 10.1128/JVI.78.9.4463-4477.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Oxenius A, et al. Variable fate of virus-specific CD4+ T cells during primary HIV-1 infection. Eur. J. Immunol. 2001;31:3782–3788. doi: 10.1002/1521-4141(200112)31:12<3782::AID-IMMU3782>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 107.Gloster SE, et al. Association of strong virus-specific CD4 T cell responses with efficient natural control of primary HIV-1 infection. AIDS. 2004;18:749–755. doi: 10.1097/00002030-200403260-00005. [DOI] [PubMed] [Google Scholar]
- 108.Rosenberg ES, et al. Vigorous HIV-1-specific CD4+ T cell responses associated with control of viremia. Science. 1997;278:1447–1450. doi: 10.1126/science.278.5342.1447. [DOI] [PubMed] [Google Scholar]
- 109.Oxenius A, et al. Early highly active antiretroviral therapy for acute HIV-1 infection preserves immune function of CD8+ and CD4+ T lymphocytes. Proc. Natl Acad. Sci. USA. 2000;97:3382–3387. doi: 10.1073/pnas.97.7.3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sun JC, Williams MA, Bevan MJ. CD4+ T cells are required for the maintenance, not programming, of memory CD8+ T cells after acute infection. Nature Immunol. 2004;5:927–933. doi: 10.1038/ni1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Champagne P, et al. Skewed maturation of memory HIV-specific CD8 T lymphocytes. Nature. 2001;410:106–111. doi: 10.1038/35065118. [DOI] [PubMed] [Google Scholar]
- 112.Streeck H, van Bockel D, Kelleher A. T-cell responses in primary HIV-1 infection. Curr. Opin. HIV AIDS. 2008;3:52–59. doi: 10.1097/COH.0b013e3282f269d6. [DOI] [PubMed] [Google Scholar]
- 113.Liu J, et al. Immune control of an SIV challenge by a T-cell-based vaccine in rhesus monkeys. Nature. 2009;457:87–91. doi: 10.1038/nature07469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Dong T, et al. HIV-specific cytotoxic T cells from long-term survivors select a unique T cell receptor. J. Exp. Med. 2004;200:1547–1557. doi: 10.1084/jem.20032044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Turnbull EL, et al. HIV-1 epitope-specific CD8+ T cell responses strongly associated with delayed disease progression cross-recognize epitope variants efficiently. J. Immunol. 2006;176:6130–6146. doi: 10.4049/jimmunol.176.10.6130. [DOI] [PubMed] [Google Scholar]
- 116.Wei X, et al. Antibody neutralization and escape by HIV-1. Nature. 2003;422:307–312. doi: 10.1038/nature01470. [DOI] [PubMed] [Google Scholar]
- 117.Richman DD, Wrin T, Little SJ, Petropoulos CJ. Rapid evolution of the neutralizing antibody response to HIV type 1 infection. Proc. Natl Acad. Sci. USA. 2003;100:4144–4149. doi: 10.1073/pnas.0630530100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gray ES, et al. Neutralizing antibody responses in acute human immunodeficiency virus type 1 subtype C infection. J. Virol. 2007;81:6187–6196. doi: 10.1128/JVI.00239-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Stamatatos L, Morris L, Burton DR, Mascola JR. Neutralizing antibodies generated during natural HIV-1 infection: good news for an HIV-1 vaccine? Nature Med. 2009;15:866–870. doi: 10.1038/nm.1949. [DOI] [PubMed] [Google Scholar]
- 120.Gray ES, et al. Broad neutralization of human immunodeficiency virus type 1 mediated by plasma antibodies against the gp41 membrane proximal external region. J. Virol. 2009;83:11265–11274. doi: 10.1128/JVI.01359-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Derdeyn CA, et al. Envelope-constrained neutralization-sensitive HIV-1 after heterosexual transmission. Science. 2004;303:2019–2022. doi: 10.1126/science.1093137. [DOI] [PubMed] [Google Scholar]
- 122.Alam SM, et al. The role of antibody polyspecificity and lipid reactivity in binding of broadly neutralizing anti-HIV-1 envelope human monoclonal antibodies 2F5 and 4E10 to glycoprotein 41 membrane proximal envelope epitopes. J. Immunol. 2007;178:4424–4435. doi: 10.4049/jimmunol.178.7.4424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Alam SM, et al. Human immunodeficiency virus type 1 gp41 antibodies that mask membrane proximal region epitopes: antibody binding kinetics, induction, and potential for regulation in acute infection. J. Virol. 2008;82:115–125. doi: 10.1128/JVI.00927-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Crooks ET, et al. A comparative immunogenicity study of HIV-1 virus-like particles bearing various forms of envelope proteins, particles bearing no envelope and soluble monomeric gp120. Virology. 2007;366:245–262. doi: 10.1016/j.virol.2007.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Tobin GJ, et al. Deceptive imprinting and immune refocusing in vaccine design. Vaccine. 2008;26:6189–6199. doi: 10.1016/j.vaccine.2008.09.080. [DOI] [PubMed] [Google Scholar]
- 126.Forthal DN, Landucci G, Daar ES. Antibody from patients with acute human immunodeficiency virus (HIV) infection inhibits primary strains of HIV type 1 in the presence of natural-killer effector cells. J. Virol. 2001;75:6953–6961. doi: 10.1128/JVI.75.15.6953-6961.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Aasa-Chapman MM, et al. Detection of antibody-dependent complement-mediated inactivation of both autologous and heterologous virus in primary human immunodeficiency virus type 1 infection. J. Virol. 2005;79:2823–2830. doi: 10.1128/JVI.79.5.2823-2830.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Burrer R, Haessig-Einius S, Aubertin AM, Moog C. Neutralizing as well as non-neutralizing polyclonal immunoglobulin (Ig)G from infected patients capture HIV-1 via antibodies directed against the principal immunodominant domain of gp41. Virology. 2005;333:102–113. doi: 10.1016/j.virol.2004.12.034. [DOI] [PubMed] [Google Scholar]
- 129.Moore PL, et al. The c3-v4 region is a major target of autologous neutralizing antibodies in human immunodeficiency virus type 1 subtype C infection. J. Virol. 2008;82:1860–1869. doi: 10.1128/JVI.02187-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Calarese DA, et al. Antibody domain exchange is an immunological solution to carbohydrate cluster recognition. Science. 2003;300:2065–2071. doi: 10.1126/science.1083182. [DOI] [PubMed] [Google Scholar]
- 131.Kwong PD, et al. HIV-1 evades antibody-mediated neutralization through conformational masking of receptor-binding sites. Nature. 2002;420:678–682. doi: 10.1038/nature01188. [DOI] [PubMed] [Google Scholar]
- 132.Ofek G, et al. Structure and mechanistic analysis of the anti-human immunodeficiency virus type 1 antibody 2F5 in complex with its gp41 epitope. J. Virol. 2004;78:10724–10737. doi: 10.1128/JVI.78.19.10724-10737.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Cardoso RM, et al. Structural basis of enhanced binding of extended and helically constrained peptide epitopes of the broadly neutralizing HIV-1 antibody 4E10. J. Mol. Biol. 2007;365:1533–1544. doi: 10.1016/j.jmb.2006.10.088. [DOI] [PubMed] [Google Scholar]
- 134.Nelson JD, et al. An affinity-enhanced neutralizing antibody against the membrane-proximal external region of human immunodeficiency virus type 1 gp41 recognizes an epitope between those of 2F5 and 4E10. J. Virol. 2007;81:4033–4043. doi: 10.1128/JVI.02588-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Shen X, et al. In vivo gp41 antibodies targeting the 2F5 monoclonal antibody epitope mediate human immunodeficiency virus type 1 neutralization breadth. J. Virol. 2009;83:3617–3625. doi: 10.1128/JVI.02631-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Haynes BF, Moody MA, Verkoczy L, Kelsoe G, Alam SM. Antibody polyspecificity and neutralization of HIV-1: a hypothesis. Hum. Antibodies. 2005;14:59–67. doi: 10.3233/HAB-2005-143-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Fox CH, et al. Lymphoid germinal centers are reservoirs of human immunodeficiency virus type 1 RNA. J. Infect. Dis. 1991;164:1051–1057. doi: 10.1093/infdis/164.6.1051. [DOI] [PubMed] [Google Scholar]
- 138.Pantaleo G, et al. Lymphoid organs function as major reservoirs for human immunodeficiency virus. Proc. Natl Acad. Sci. USA. 1991;88:9838–9842. doi: 10.1073/pnas.88.21.9838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Muro-Cacho CA, Pantaleo G, Fauci AS. Analysis of apoptosis in lymph nodes of HIV-infected persons. J. Immunol. 1995;154:5555–5566. [PubMed] [Google Scholar]
- 140.Zhang ZQ, et al. Early depletion of proliferating B cells of germinal center in rapidly progressive simian immunodeficiency virus infection. Virology. 2007;361:455–464. doi: 10.1016/j.virol.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 141.Mascola JR, et al. Protection of macaques against vaginal transmission of a pathogenic HIV-1/SIV chimeric virus by passive infusion of neutralizing antibodies. Nature Med. 2000;6:207–210. doi: 10.1038/72318. [DOI] [PubMed] [Google Scholar]
- 142.Hessell AJ, et al. Fc receptor but not complement binding is important in antibody protection against HIV. Nature. 2007;449:101–104. doi: 10.1038/nature06106. [DOI] [PubMed] [Google Scholar]
- 143.Hessell AJ, et al. Effective, low-titer antibody protection against low-dose repeated mucosal SHIV challenge in macaques. Nature Med. 2009;15:951–954. doi: 10.1038/nm.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hessell AJ, et al. Broadly neutralizing human anti-HIV antibody 2G12 is effective in protection against mucosal SHIV challenge even at low serum neutralizing titers. PLoS Pathog. 2009;5:e1000433. doi: 10.1371/journal.ppat.1000433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Engram JC, et al. Vaccine-induced, simian immunodeficiency virus-specific CD8+ T cells reduce virus replication but do not protect from simian immunodeficiency virus disease progression. J. Immunol. 2009;183:706–717. doi: 10.4049/jimmunol.0803746. [DOI] [PubMed] [Google Scholar]
- 146.Rerks-Ngarm Supachai, Pitisuttithum Punnee, Nitayaphan Sorachai, Kaewkungwal Jaranit, Chiu Joseph, Paris Robert, Premsri Nakorn, Namwat Chawetsan, de Souza Mark, Adams Elizabeth, Benenson Michael, Gurunathan Sanjay, Tartaglia Jim, McNeil John G., Francis Donald P., Stablein Donald, Birx Deborah L., Chunsuttiwat Supamit, Khamboonruang Chirasak, Thongcharoen Prasert, Robb Merlin L., Michael Nelson L., Kunasol Prayura, Kim Jerome H. Vaccination with ALVAC and AIDSVAX to Prevent HIV-1 Infection in Thailand. New England Journal of Medicine. 2009;361(23):2209–2220. doi: 10.1056/NEJMoa0908492. [DOI] [PubMed] [Google Scholar]
- 147.Reynolds MR, et al. Macaques vaccinated with live-attenuated SIV control replication of heterologous virus. J. Exp. Med. 2008;205:2537–2550. doi: 10.1084/jem.20081524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Althaus CL, De Boer RJ. Dynamics of immune escape during HIV/SIV infection. PLoS Comput. Biol. 2008;4:e1000103. doi: 10.1371/journal.pcbi.1000103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lee JK, et al. T cell cross-reactivity and conformational changes during TCR engagement. J. Exp. Med. 2004;200:1455–1466. doi: 10.1084/jem.20041251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Santra S, et al. A centralized gene-based HIV-1 vaccine elicits broad cross-clade cellular immune responses in rhesus monkeys. Proc. Natl Acad. Sci. USA. 2008;105:10489–10494. doi: 10.1073/pnas.0803352105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Barouch D, Korber B. Abstr. S06-04 AIDS vaccine conference 2009, Paris. 2009. HIV-1 mosaic antigens expand cellular immune breadth and depth in rhesus monkeys. [Google Scholar]
- 152.Korber B, Gnankaran S, Perkins S. Abstr. S06-03 AIDS vaccine conference 2009, Paris. 2009. New applications for mosaic antigen designs. [Google Scholar]
- 153.Pitisuttithum P, et al. Randomized, double-blind, placebo-controlled efficacy trial of a bivalent recombinant glycoprotein 120 HIV-1 vaccine among injection drug users in Bangkok, Thailand. J. Infect. Dis. 2006;194:1661–1671. doi: 10.1086/508748. [DOI] [PubMed] [Google Scholar]
- 154.McElrath MJ, et al. HIV-1 vaccine-induced immunity in the test-of-concept Step Study: a case-cohort analysis. Lancet. 2008;372:1894–1905. doi: 10.1016/S0140-6736(08)61592-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Buchbinder SP, et al. Efficacy assessment of a cell-mediated immunity HIV-1 vaccine (the Step Study): a double-blind, randomised, placebo-controlled, test-of-concept trial. Lancet. 2008;372:1881–1893. doi: 10.1016/S0140-6736(08)61591-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Casimiro DR, et al. Attenuation of simian immunodeficiency virus SIVmac239 infection by prophylactic immunization with dna and recombinant adenoviral vaccine vectors expressing Gag. J. Virol. 2005;79:15547–15555. doi: 10.1128/JVI.79.24.15547-15555.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Hansen SG, et al. Effector memory T cell responses are associated with protection of rhesus monkeys from mucosal simian immunodeficiency virus challenge. Nature Med. 2009;15:293–299. doi: 10.1038/nm.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Walker BD, Burton DR. Toward an AIDS vaccine. Science. 2008;320:760–764. doi: 10.1126/science.1152622. [DOI] [PubMed] [Google Scholar]
- 159.Baker BM, Block BL, Rothchild AC, Walker BD. Elite control of HIV infection: implications for vaccine design. Expert Opin. Biol. Ther. 2009;9:55–69. doi: 10.1517/14712590802571928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wong SBJ, Siliciano RF. AIDS Vaccine Development: Challenges and Opportunities. 2007. pp. 17–22. [Google Scholar]