

Abstract
Health-related stigma is increasingly becoming a major public health issue that is receiving more attention. Young adults with sickle cell disease (SCD) are at risk for health-related stigmatization due to the many challenges of the disease. SCD includes the lifelong challenges of managing the chronic illness while accessing and navigating the health care system. The burdens of the disease can affect all aspects of the lives of individuals with SCD to include physiological, psychological, and social well-being. Although others may be involved in the process of stigmatization, the purpose of this paper was to support the need to develop patient-oriented interventions to prevent and treat health-related stigma in young adults with SCD, as these individuals may face health-related stigma throughout their lives, but especially immediately after transitioning from pediatric to adult care. Additionally, the Revised Theory of Self-Care Management for Sickle Cell Disease is offered as a framework from which theory-based interventions can be derived.
Keywords: sickle cell anemia, stigma
INTRODUCTION
Stigmatization is the process of identifying an attribute of a person or group and associating the attribute with a stereotype that negatively labels or brands another in a way that is perceived as disgraceful by society.1-3 More specifically, health-related stigma refers to a form of devaluation, judgment, or social disqualification of individuals based on a health-related condition.4 Health-related stigma is increasingly becoming a major public health issue that is receiving more attention as stigmatization adds to the burden of individuals and families affected by sickle cell disease (SCD). Despite this, there is minimal information in the literature about SCD and health-related stigma. SCD refers to a family of inherited autosomal recessive genetic disorders that affects about 1 in 365 African Americans, with approximately 89 079 having the disease in the United States.5,6 Individuals who have sickle cell disease produce abnormal hemoglobin S molecules. Many of the clinical manifestations of SCD are due to deoxygentation, vaso-occlusion, and tissue necrosis. Upon deoxygenation or extremes in temperature, the hemoglobin S molecules cause the red blood cells to assume a sickled shape and adhere to the vascular walls as a result of polymerization. Consequently, the sickled cells can lead to occlusion within the capillaries and small vessels, causing pain, tissue necrosis, and eventually anemia and ischemic organ conditions.7-10 The clinical manifestations of SCD often lead to unpredictable episodes of pain and feelings of inadequacy regarding their care,11 which may be due to health-related stigmatization.
Much of the literature on health-related stigma originated from infectious diseases such as human immunodeficiency virus (HIV)/AIDS, tuberculosis, and leprosy as well as from mental health conditions such as schizophrenia.12,13 HIV/AIDS-related stigma has had a significant impact on the health and well-being of individuals around the world for 2 decades.14 According to Sartorius,15 “stigma evokes negative attitudes and feelings and usually results in discrimination of the person or institution in various walks of life.” This level of stigma can lead to unjust disadvantages for the stigmatized, including direct discrimination on the job, in schools, and within families, and may impact the receipt of timely and quality health care. Health-related stigma can have a deleterious affect across the lifespan on individuals being stigmatized.
Individuals with chronic diseases such as chronic obstructive pulmonary disease and chronic pain also face health-related stigmatization. They may feel exiled from the healthier world and feel disgraced due to health encounters and lack of support by providers, friends, family, the community, and the workplace.16,17 The purpose of this paper was to support the need to develop patient-oriented interventions to prevent and treat health-related stigma in young adults with SCD, as these individuals may face health-related stigma throughout their lives as they live with a chronic illness, but especially as they transition from pediatric to adult care. Additionally, a theory is offered from which interventions can be derived.
HEALTH-RELATED STIGMA DURING CHILDHOOD IN INDIVIDUALS WITH SICKLE CELL DISEASE
Many children and adolescents with SCD are challenged by a myriad of complex psychosocial issues, which may be triggered or exacerbated by stigmatization. The magnitude of childhood stigma and the potential for associated psychosocial adjustment issues in children and adolescents with SCD can be explained within the context of psychological, social, and cultural implications.
Pinckney and Stuart18 highlighted the significance of understanding the psychosocial implications of living with SCD and adaptation threats in children and adolescents:
Some of those psychosocial factors are interpersonal skills (eg, self-esteem, assertive communication difficulties), stress-producing (eg, decreased coping strategies, decreased knowledge about SCD), and family factors (eg, cohesion, organization and control, family support, parent-child relationship problems).18
The onset of the stigmatization may begin with the mother of the affected child. In a study of Canadian mothers of children with SCD, mothers reported that SCD stigma was exacerbated for racial and ethnic minority groups.19 These mothers reported daily coping challenges such as fear of their children dying; separation anxiety; and feelings of helplessness, loneliness, and isolation. Trzepacz, Vannatta, Gerhardt, Ramey, and Noll found that relative to the primary caregivers of their peers, primary caregivers of children with SCD perceived their children as having more total problems, particularly emotional problems, and less total competence.20 These daily challenges and perceptions of mothers of children with SCD may lead to heightened levels of overprotective behaviors. During life review interviews with middle-aged and older adults with SCD, participants identified 2 primary factors that may negatively influence self-care behaviors and overall health outcomes: the experience of painful crises and parental behaviors, specifically behaviors characterized as overprotective.21
Sociodemographic variables such as race, gender, age, socioeconomic status, and education also affect psychosocial adjustment in children and adolescents with SCD.22,23 As children grow and progress through the stages of human development, complications related to SCD can become even more challenging. Children and adolescents are often forced to deal with issues related to school absenteeism, increased use of the health care system, decreased school and social activities, as well as poor adaptation.24-28 Children with SCD can also be affected by health-related stigma that originates from teachers as well as peers.29 Children and adolescents may experience low self-esteem, embarrassment, and other complications as a result of stigma. In a study conducted by Patel and Pathan, due to the stigma of SCD, children with sickle cell trait as well as children with sickle cell anemia were concerned about their illness and the perception of looking different compared to their peers.30 They also expressed concern that the illness was burdensome to their families and siblings. Adolescents may be challenged by issues related to body image such as delayed growth and late sexual maturation as well as bone age retardation, small body mass, delayed menarche, hypogonadism, and delayed secondary sex characteristics among adolescents with sickle cell disease.31-33 Mainstream society may impose negative stereotypes on children and adolescents from racial and ethnic minority groups as a result of these factors, which may result in stigmatization.1,34 The increased use of the health care system only negatively impacts the sickle cell disease–stigmatization cycle that continues for the young adult with SCD.
HEALTH-RELATED STIGMATIZATION FOR YOUNG ADULTS WITH SCD
Problems that begin in childhood are continued into adulthood, creating opportunities for young adults with SCD to be stigmatized. In 1973, life expectancy was 14 years.35 Because persons with SCD were not expected to live into adulthood, many may have been overprotected during childhood. Notably, older adults with SCD have frequently indicated that they viewed their parents’ overprotective behavior as being demonstrations of caring.21 In addition, these chronically ill children suffered the effects of social deprivation as a result of prolonged hospitalizations. Adolescents with SCD acknowledge the importance of transition but demonstrate poor preparation for transition to adult-oriented care.36,37 They often leave pediatric care without adequate transfer preparation, and their readiness to transfer is not a major consideration in the decision to transfer them.38 Without adequate preparation to seek health care as an adult patient, young adults may become stigmatized, especially when seeking care for acute pain. The extent of treatment of a painful SCD crisis depends on the health care provider, who assesses the SCD patient’s presentation and ultimately decides whether the individual’s report of pain is credible and deserving of treatment.21,39-41 In a cohort study of 940 subjects, it was found that most children with SCD now survive the childhood years, but young adults who transition to adult medical care are at high risk for early death, especially shortly after transition.42 As children age out of pediatric services, they are more susceptible to changes such as loss of an established primary medical home, access to ambulatory care, and health insurance.43 The lack of adequate transition can lead to recurrent hospitalizations, poor trust, and worse outcomes for this vulnerable population.44 Thus, the transition to adult care is critical for young adults to appropriately access the health care system. Even today, persons with SCD may not develop the skills to fully participate in taking care of their own health needs, and some are not prepared to assume the responsibilities of adult health care seekers.
People afflicted with SCD often experience a life punctuated by unpredictable painful crises and feel inadequate to influence the quality of their care.45 For these reasons, the majority (80%) of adults with SCD avoid the health care system whenever possible and manage their pain at home.46 However, for individuals with SCD who do seek acute care, the majority of visits are related to painful crises.47,48
The credibility or trustworthiness of young African American adults with SCD is often questioned by health care providers, who label patients as malingerers or manipulators, or even drug seekers.40,41,45,49,50 Indeed when individuals with SCD request specific pain medications, clinicians often interpret this as drug-seeking behavior,51 though no scientific evidence suggests that people with SCD will become drug dependent if their pain is treated with narcotics.52
Age plays a role in utilization of health care services and coping with SCD. Of those individuals with SCD seeking acute care and rehospitalizations, those aged 18 to 30 years are the most frequent.6 Older individuals tend to use outpatient services and prayer, while younger individuals are more likely to use the emergency department and cope by ignoring pain and using heat and cold or massage.53, 54
Currently, adults with SCD who present in emergency departments with complaints of an acute pain episode may wait an average of 90 minutes for the first analgesic to be given.55 The delay may be due in part to the fact that the pain of SCD is poorly understood and it is difficult to objectively assess a pain crisis. Additional barriers to adequate pain management include the fact that most individuals with SCD in the United States are African American, and many are of lower socioeconomic status.56 In a study to determine whether differential opioid prescription by race in emergency departments has diminished since 2000, it was found that white patients were significantly more likely to receive opioid prescription than black patients.57 Thus, when young adults with SCD seek treatment for acute pain in an emergency department, there is great potential for racial stereotyping, mistrust, and problematic physician-patient communication.58 These factors may result in a negative pain management experience.
Stigmatization or stigma-related behaviors and depression have been linked in the literature. A study of 232 adults with SCD59 found that respondents reported higher levels of depressive symptoms (32%) than the overall US population (9.5%). Depression in combination with episodes of poorly managed pain may lead to feelings of inadequacy and even suicidal ideations in adults with SCD.60 Anie and Green found that adults with SCD commonly reported low self-esteem and feelings of hopelessness as a result of frequent pain, hospitalizations, and subsequent loss of employment.61 An adult with SCD said, “It’s devastating for a person to be in pain but not to be believed. It makes you feel less than human. The trust is broken when the person you come to for help reacts negatively.”62 Negative pain management experiences may result in psychological disturbances, such as depression and anxiety, are often associated with a diminished ability to cope with pain and further perpetuate the cycle of pain intensity with significant impact on health outcomes.63
STIGMA REDUCTION INTERVENTIONS
Strategies have been developed to reduce stigma and inequities related to health at multiple levels. Interventions have been developed for conditions such as HIV/AIDS, leprosy, tuberculosis, epilepsy, and mental illness.64,65 Goals in implementing stigma reduction strategies are to prevent delays in health services, to decrease risk of transmission, and to raise awareness and sensitivity about stigma-related issues for many people, as well as the affected individuals. Heijnders and Van Der Meij conducted a literature review that identified several levels for implementing stigma reduction strategies and interventions and found that strategies aimed at individual and community levels proved to be the most effective.64 Individual or intrapersonal strategies include treatment, counseling, cognitive-behavioral therapy, empowerment, group counseling and self-help, advocacy, or support groups. Community-level strategies include education, contact, advocacy, and protests. Currently, there are no published SCD-specific theory-based interventions aimed at preventing or treating health-related stigma.
THEORY-BASED STIGMA INTERVENTION FOR SICKLE CELL DISEASE
In individuals with SCD, a severe SCD crisis that results in the need for hospitalization usually evolves through 4 distinct physiologic phases: (1) prodromal; (2) the initial evolving, infarctive phase; (3) the established phase, and (4) the resolving, healing, recovery, postcrisis phase.66,67 During the first, the prodromal phase (pre crisis), patients develop symptoms of numbness, aches, and paresthesia in the sites subsequently affected by pain. Other reported signs and symptoms of the prodromal phase include fatigue, jaundice of sclera, nausea and vomiting, change of appetite, and stiffness in joints.68 This phase can last up to 4 days, and individuals describe the pain as a low-intensity ache. The ability of an adult with SCD to recognize and respond to the cues of onset of an SCD crisis as well as the transition through differing phases of the SCD crisis may make a significant difference in patient presentation and treatment—that is, if adults with SCD present for treatment during the 2-to 4-day period of low pain intensity, they may be able to avoid the need for aggressive pain management and inpatient hospitalization and have their prodromal phase pain adequately addressed.
Increased cue recognition and seeking care early during the prodromal, or precrisis, phase would be more effective for adults with SCD in preventing the evolution of an acute pain crisis or the necessity of an inpatient admission via the emergency department. Unfortunately, there have been few studies about the sickle cell crisis prodrome.69,71 Murray and May reported that 58% of 102 patients experienced a prodromal phase of an impending painful crisis up to 24 hours before developing features typical of their usual crisis.70 Akinola and colleagues69 reported that 12 of 14 patient premonitions were followed by a typical painful crisis that required either home treatment (n = 4) or hospitalization (n = 8). It has been established that therapeutic interventions are more likely to be effective during the earlier phases of a crisis.71 Ballas describes the SCD prodrome as “an intriguing entity that needs further study.”67 Moreover, it may provide an avenue for the initiation of proactive or anticipatory analgesia72,73 in order to abort an evolving crisis and to prevent the evolution of painful crises.
Young adults with SCD who are admitted through emergency departments must be intensively treated to be stabilized prior to admission.74 Emergency department visits could be avoided if patients presented earlier to a primary care setting that provided both medical and psychological support to interdict pain crises and prevent severe crises.75,76 The earlier therapy is introduced, the better the clinical outcome for adults with SCD.
To increase the potential for appropriate and timely treatment, adults with SCD must be able to communicate substantive information to the health care provider in order for the provider to arrive at the conclusion that this is a well-informed, credible individual presenting in the prodromal phase of a sickle cell crisis. The adult with SCD needs to know what information the health care provider needs and how to communicate that information. Improved positive self-presentation is important for African Americans to get the best medical care.77 The more information the adult with SCD provides, the greater the likelihood that the provider can devise an individualized treatment plan in collaboration with the patient while avoiding stereotypes of adults with SCD and their consumption of opioid analgesics.78 Positive self-presentation can be improved through communication skills and, therefore, adults with SCD may be more likely to receive individualized, proactive pain strategies to improve the quality of their pain management experience.77
The Revised Theory of Self-Care Management for Sickle Cell Disease, developed by the first author (Figure 1), focuses on vulnerability factors and self-care management resources that influence health outcomes and can be used to design a theory-based intervention for young adults with SCD. In the model, vulnerability factors (lack of sickle cell crisis cue recognition/response, number of complication, number of sickle cell crises per year, and overprotection) negatively impact health outcomes (pain management experience, depressive symptoms, self-esteem, and perceived health-related stigma). However, self-care management resources (self-efficacy, coping behaviors, social support, self-care ability, self-care actions, and assertive communications skills) can positively mediate the relationship between vulnerability factors and health outcomes. A subset of the theory can be used to design interventions to reduce health-related stigma, a health outcome, by focusing on specific antecedents—lack of cue recognition/response and ineffective communication skills—which contribute to satisfaction with the pain management experience and health-related stigmatization. An intervention that focuses on cue recognition/response and communication skills is hypothesized to improve these outcomes by reducing the potential for health-related stigmatization and improving the pain management experience. In vulnerability management, the factor of focus in the intervention is lack of sickle cell crisis cue recognition/response, the self-care management resource is assertive communication skills, and the health outcomes are the pain management experience and perceived health-related stigma.
Figure 1.
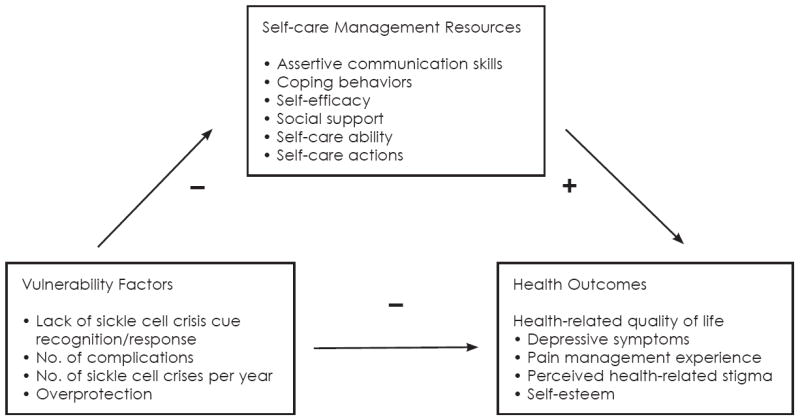
The Theory of Self-care Management for Sickle Cell Disease (Revised)
SUMMARY
Health-related stigma is a challenge for young adults with SCD. The stigmatization may begin in childhood, but it is often most evident as young adults transition from pediatric care to adult care. Moreover, the stigmatization may hinder care seeking for the acute pain exacerbations of pain—the hallmark of the disease. Health-related stigma can have detrimental effects on the lives of individuals and families living with SCD. It has been supported that improving provider communication improves the health care encounter—in particular, trust—in individuals with SCD.79 However, there have been no published studies examining the impact of improving the communication skills of the individual with SCD. One way of improving communication is presenting for care before pain impacts communication. It is important to design interventions that improve communication skills and, thus, care seeking in individuals with SCD as a means to eliminate or reduce health-related stigma.
Theory-based interventions to impact health-related stigma should be designed considering the context from which the stigma emerged in order to assists individuals to overcome the challenges of health-related stigma. The Revised Theory of Self-Care Management for Sickle Cell Disease offers a framework to develop interventions to prevent and reduce health-related stigma. Theory-based interventions will empower young adults with SCD with the skills to advocate for themselves and communicate their needs in a timely and effective manner.
Acknowledgments
Funding/Support: This research was supported by a grant from the National Institute of Nursing Research (1K23NR011061).
References
- 1.Sankar P, Cho MK, Wolpe PR, Schairer C. What is in a cause? Exploring the relationship between genetic cause and felt stigma. Genet Med. 2006;8(1):33–42. doi: 10.1097/01.gim.0000195894.67756.8b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Link BG, Phelan JC. Conceptualizing stigma. Annu Rev Sociol. 2001;27:363–385. [Google Scholar]
- 3.Link BG, Phelan JC. Stigma and its public health implications. Lancet. 2006;367(9509):528–529. doi: 10.1016/S0140-6736(06)68184-1. [DOI] [PubMed] [Google Scholar]
- 4.Weiss MG, Ramakrishna J, Somma D. Health-related stigma: Rethinking concepts and interventions. Psychol Health Med. 2006;11(3):277–287. doi: 10.1080/13548500600595053. [DOI] [PubMed] [Google Scholar]
- 5.Hassell KL. Population estimates of sickle cell disease in the U.S. Am J Prev Med. 2010;38(4):S512–521. doi: 10.1016/j.amepre.2009.12.022. [DOI] [PubMed] [Google Scholar]
- 6.Brousseau DC, Owens PL, Mosso AL, Panepinto JA, Steiner CA. Acute care utilization and rehospitalizations for sickle cell disease. JAMA. 2010;303(13):1288–1294. doi: 10.1001/jama.2010.378. [DOI] [PubMed] [Google Scholar]
- 7.Edwards CL, Scales MT, Loughlin C, Bennett GG, Harris-Peterson S, Castro LM, et al. A brief review of the pathophysiology, associated pain, and psychosocial issues in sickle cell disease. Int J Behav Med. 2005;12(3):171–179. doi: 10.1207/s15327558ijbm1203_6. [DOI] [PubMed] [Google Scholar]
- 8.Newcombe P. Pathophysiology of sickle cell disease crisis. Emerg Nurse. 2002;9(9):19–22. doi: 10.7748/en2002.02.9.9.19.c1404. [DOI] [PubMed] [Google Scholar]
- 9.Redding-Lallinger R, Knoll C. Sickle cell disease—pathophysiology and treatment. Curr Probl Pediatr Adolesc Health Care. 2006;36(10):346–376. doi: 10.1016/j.cppeds.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 10.Schnog JB, Duits AJ, Muskiet FA, ten Cate H, Rojer RA, Brandjes DP. Sickle cell disease; a general overview. Neth J Med. 2004;62(10):364–374. [PubMed] [Google Scholar]
- 11.Strickland OL, Jackson G, Gilead M, McGuire DB, Quarles S. Use of focus groups for pain and quality of life assessment in adults with sickle cell disease. J Natl Black Nurses Assoc. 2001;12(2):36–43. [PubMed] [Google Scholar]
- 12.Switaj P, Wciorka J, Smolarska-Switaj J, Grygiel P. Extent and predictors of stigma experienced by patients with schizophrenia. Eur Psychiatry. 2009;24(8):513–520. doi: 10.1016/j.eurpsy.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 13.van Zelst C. Stigmatization as an environmental risk in schizophrenia: A user perspective. Schizophr Bull. 2009;35(2):293–296. doi: 10.1093/schbul/sbn184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parker R, Aggleton P. HIV and AIDS-related stigma and discrimination: A conceptual framework and implications for action. Soc Sci Med. 2003;57(1):13–24. doi: 10.1016/s0277-9536(02)00304-0. [DOI] [PubMed] [Google Scholar]
- 15.Sartorius N. Lessons from a 10-year global programme against stigma and discrimination because of an illness. Psychol Health Med. 2006;11(3):383–388. doi: 10.1080/13548500600595418. [DOI] [PubMed] [Google Scholar]
- 16.Slade SC, Molloy E, Keating JL. Stigma experienced by people with nonspecific chronic low back pain: A qualitative study. Pain Med. 2009;10(1):143–154. doi: 10.1111/j.1526-4637.2008.00540.x. [DOI] [PubMed] [Google Scholar]
- 17.Halding AG, Heggdal K, Wahl A. Experiences of self-blame and stigmatisation for self-infliction among individuals living with COPD. Scand J Caring Sci. 2010 May 31; doi: 10.1111/j.1471-6712.2010.00796.x. [DOI] [PubMed] [Google Scholar]
- 18.Pinckney RB, Stuart GW. Adjustment difficulties of adolescents with sickle cell disease. J Child Adolesc Psychiatr Nurs. 2004;17(1):5–12. doi: 10.1111/j.1744-6171.2004.00005.x. [DOI] [PubMed] [Google Scholar]
- 19.Burnes DPR, Antle BJ, Williams CC, Cook L. Mothers raising children with sickle cell disease at the intersection of race, gender, and illness stigma. Health Soc Work. 2008;33(3):211–220. doi: 10.1093/hsw/33.3.211. [DOI] [PubMed] [Google Scholar]
- 20.Trzepacz AM, Vannatta K, Gerhardt CA, Ramey C, Noll RB. Emotional, social, and behavioral functioning of children with sickle cell disease and comparison peers. J Pediatr Hematol Oncol. 2004;26(10):642–648. doi: 10.1097/01.mph.0000139456.12036.8d. [DOI] [PubMed] [Google Scholar]
- 21.Jenerette CM, Lauderdale G. Successful aging with sickle cell disease: Using qualitative methods to inform theory. J Theor Construct Test. 2008;12(1):16. [PMC free article] [PubMed] [Google Scholar]
- 22.Edwards CL, Scales MT, Loughlin C, Bennett GG, Harris-Peterson S, DeCastro LM, et al. A brief review of the pathophysiology, associated pain, and psychosocial issues in sickle cell disease. Int J Behav Med. 2005;12(3):171–179. doi: 10.1207/s15327558ijbm1203_6. [DOI] [PubMed] [Google Scholar]
- 23.Kell RS, Kliewer W, Erickson MT, Ohene-Frempong K. Psychological adjustment of adolescents with sickle cell disease: Relations with demographic, medical, and family competence variables. J Pediatr Psychol. 1998;23(5):301–312. doi: 10.1093/jpepsy/23.5.301. [DOI] [PubMed] [Google Scholar]
- 24.Broome ME, Maikler V, Kelber S, Bailey P, Lea G. An intervention to increase coping and reduce health care utilization for school-age children and adolescents with sickle cell disease. J Natl Black Nurses Assoc. 2001;12(2):6–14. [PubMed] [Google Scholar]
- 25.Maikler VE, Broome ME, Bailey P, Lea G. Childrens’ and adolescents’ use of diaries for sickle cell pain. J Soc Pediatr Nurs. 2001;6(4):161–169. doi: 10.1111/j.1744-6155.2001.tb00240.x. [DOI] [PubMed] [Google Scholar]
- 26.Burlew K, Telfair J, Colangelo L, Wright EC. Factors that influence adolescent adaptation to sickle cell disease. J Pediatr Psychol. 2000;25(5):287–299. doi: 10.1093/jpepsy/25.5.287. [DOI] [PubMed] [Google Scholar]
- 27.Day S, Chismark E. The cognitive and academic impact of sickle cell disease. J Sch Nurs. 2006;22(6):330–335. doi: 10.1177/10598405060220060401. [DOI] [PubMed] [Google Scholar]
- 28.Gil KM, Carson JW, Sedway JA, Porter LS, Schaeffer JJ, Orringer E. Follow-up of coping skills training in adults with sickle cell disease: Analysis of daily pain and coping practice diaries. Health Psychol. 2000;19(1):85–90. doi: 10.1037//0278-6133.19.1.85. [DOI] [PubMed] [Google Scholar]
- 29.Hurtig AL, White LS. Children and adolescents: The unexpected terrain of emotional development. In: Hurly L, Viera T, editors. Sickle cell disease: Psychological and psychosocial issues. University of Illinois Press; Urbana: 1986. pp. 24–40. [Google Scholar]
- 30.Patel AB, Pathan HG. Quality of life in children with sickle cell hemoglobinopathy. Indian J Pediatr. 2005;72(7):567–571. doi: 10.1007/BF02724180. [DOI] [PubMed] [Google Scholar]
- 31.Graham C, Maude GH, Serjeant GR. Delayed menarche in homozygous sickle cell disease. West Indian Med J. 1986;35(1):18–22. [PubMed] [Google Scholar]
- 32.Wethers DL. Problems and complications in the adolescent with sickle cell disease. Am J Pediatr Hematol Oncol. 1982;4(1):47–53. [PubMed] [Google Scholar]
- 33.Singhal A, Thomas P, Cook R, Wierenga K, Serjeant G. Delayed adolescent growth in homozygous sickle cell disease. Arch Dis Child. 1994;71(5):404–408. doi: 10.1136/adc.71.5.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burnes DP, Antle BJ, Williams CC, Cook L. Mothers raising children with sickle cell disease at the intersection of race, gender, and illness stigma. Health Soc Work. 2008;33(3):211–220. doi: 10.1093/hsw/33.3.211. [DOI] [PubMed] [Google Scholar]
- 35.Diggs LM. Anatomic lesions in sickle cell disease. In: Abramson H, Bertles JF, Wethers DL, editors. Sickle cell disease: Diagnosis, management, education, and research. St Louis: C.V Mosby; 1973. pp. 189–229. [Google Scholar]
- 36.WisonSchaeffer JJ, Gil KM, Burchinal M, Kramer KD, Nash KB, Orringer E, et al. Depression, disease severity, and sickle cell disease. J Behav Med. 1999;22(2):115–126. doi: 10.1023/a:1018755831101. [DOI] [PubMed] [Google Scholar]
- 37.McPherson M, Thaniel L, Minniti CP. Transition of patients with sickle cell disease from pediatric to adult care: Assessing patient readiness. Pediatr Blood Cancer. 2009;52(7):838–841. doi: 10.1002/pbc.21974. [DOI] [PubMed] [Google Scholar]
- 38.Wojciechowski EA, Hurtig A, Dorn L. A natural history study of adolescents and young adults with sickle cell disease as they transfer to adult care: A need for case management services. J Pediatr Nurs. 2002;17(1):18. doi: 10.1053/jpdn.2002.30930. [DOI] [PubMed] [Google Scholar]
- 39.Jacob E. Pain management in sickle cell disease. Pain Manag Nurs. 2001;2(4):121–131. doi: 10.1053/jpmn.2001.26297. [DOI] [PubMed] [Google Scholar]
- 40.Maxwell K, Streetly A, Bevan D. Experiences of hospital care and treatment seeking for pain from sickle cell disease: Qualitative study. BMJ. 1999;318(7198):1585–1590. doi: 10.1136/bmj.318.7198.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maxwell KS, Streetly A. Living with sickle cell pain. Nurs Stand. 1998;13(9):33. [PubMed] [Google Scholar]
- 42.Quinn CT, Rogers ZR, McCavit TL, Buchanan GR. Improved survival of children and adolescents with sickle cell disease. Blood. 2010;115(17):3447–3452. doi: 10.1182/blood-2009-07-233700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lotstein DS, Inkelas M, Hays RD, Halfon N, Brook R. Access to care for youth with special health care needs in the transition to adulthood. J Adolesc Health. 2008;43(1):23–29. doi: 10.1016/j.jadohealth.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 44.Hunt SE, Sharma N. Transition from pediatric to adult care for patients with sickle cell disease. JAMA. 2010;304(4):408–409. doi: 10.1001/jama.2010.1026. author reply 409. [DOI] [PubMed] [Google Scholar]
- 45.Strickland O, Jackson G, Gilead M, McGuire DB, Quarles S. Use of focus groups for pain and quality of life assessment in adults with sickle cell disease. Journal of the National Black Nurses Association: JNBMA. 2001;12(2):36–43. [PubMed] [Google Scholar]
- 46.Ely B, Dampier C, Gilday M, O’Neal P, Brodecki D. Caregiver report of pain in infants and toddlers with sickle cell disease: Reliability and validity of a daily diary. J Pain. 2002;3(1):50–57. doi: 10.1054/jpai.2002.xb30064. [DOI] [PubMed] [Google Scholar]
- 47.Waters J, Thomas V. Pain from sickle cell crisis. Nurs Times. 1995;91(16):29–31. [PubMed] [Google Scholar]
- 48.Yale S, Nagib N, Guthrie T. Approach to the vaso-occlusive crisis in adults with sickle cell disease. Am Fam Physician. 2000;61(5):1349, 1356, 1363–1364. [PubMed] [Google Scholar]
- 49.Armstrong F, Pegelow CH, Gonzalez JC, Martinez A. Impact of children’s sickle cell history on nurse and physician ratings of pain and medication decisions. J Pediatr Psychol. 1992;17(5):651–664. doi: 10.1093/jpepsy/17.5.651. [DOI] [PubMed] [Google Scholar]
- 50.Gorman K. Sickle cell disease: Do you doubt your patient’s pain? Am J Nurs. 1999;99(3):38–44. [PubMed] [Google Scholar]
- 51.Jacob E. Pain management in sickle cell disease. Pain Manage Nurs. 2001;2(4):121–131. doi: 10.1053/jpmn.2001.26297. [DOI] [PubMed] [Google Scholar]
- 52.Alao AO, Westmoreland N, Jindal S. Drug addiction in sickle cell disease: Case report. Int J Psychiatry Med. 2003;33(1):97–101. doi: 10.2190/7XMD-L45D-47DH-7MEC. [DOI] [PubMed] [Google Scholar]
- 53.Sanders KA, Labott SM, Molokie R, Shelby SR, Desimone J. Pain, coping and health care utilization in younger and older adults with sickle cell disease. J Health Psychol. 2010;15(1):131–137. doi: 10.1177/1359105309345554. [DOI] [PubMed] [Google Scholar]
- 54.Jenerette CM, Phillips RCS. An examination of differences in intra-personal resources, self-care management, and health outcomes in older and younger adults with sickle cell disease. Southern Online Journal of Nursing Research. 2006;7(3):1–24. [Google Scholar]
- 55.Tanabe P, Myers R, Zosel A, Brice J, Ansari AH, Evans J, et al. Emergency department management of acute pain episodes in sickle cell disease. Acad Emerg Med. 2007;14(5):419–425. doi: 10.1197/j.aem.2006.11.033. [DOI] [PubMed] [Google Scholar]
- 56.Ely B, Dampier C, Gilday M, O’Neal P, Brodecki D. Caregiver report of pain in infants and toddlers with sickle cell disease: Reliability and validity of a daily diary. J Pain. 2002;3(1):50–57. doi: 10.1054/jpai.2002.xb30064. [DOI] [PubMed] [Google Scholar]
- 57.Pletcher MJ, Kertesz SG, Kohn MA, Gonzales R. Trends in opioid prescribing by race/ethnicity for patients seeking care in US emergency departments. JAMA. 2008;299(1):70–78. doi: 10.1001/jama.2007.64. [DOI] [PubMed] [Google Scholar]
- 58.Todd KH, Green C, Bonham VL, Jr, Haywood C, Jr, Ivy E. Sickle cell disease related pain: Crisis and conflict. J Pain. 2006;7(7):453–458. doi: 10.1016/j.jpain.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 59.Jenerette C, Funk M, Murdaugh C. Sickle cell disease: A stigmatizing condition that may lead to depression. Issues Ment Health Nurs. 2005;26(10):1081–1101. doi: 10.1080/01612840500280745. [DOI] [PubMed] [Google Scholar]
- 60.Ohaeri JU, Shokunbi WA, Akinlade KS, Dare LO. The psychosocial problems of sickle cell disease sufferers and their methods of coping. Soc Sci Med. 1995;40(7):955–960. doi: 10.1016/0277-9536(94)00154-l. [DOI] [PubMed] [Google Scholar]
- 61.Anie KA, Green J. Psychological therapies for sickle cell disease and pain. Cochrane Database Syst Rev. 2002;2(2):CD001916. doi: 10.1002/14651858.CD001916. [DOI] [PubMed] [Google Scholar]
- 62.Schreiber C. Redefining pain: New guidelines challenge misconception about sickle cell disease. Nurseweek (CALIF) 2000;13(2):25. [Google Scholar]
- 63.Edwards CL, Scales MT, Loughlin C, Bennett GG, Harris-Peterson S, Castro LM, et al. A brief review of the pathophysiology, associated pain, and psychosocial issues in sickle cell disease. Int J Behav Med. 2005;12(3):171–179. doi: 10.1207/s15327558ijbm1203_6. [DOI] [PubMed] [Google Scholar]
- 64.Heijnders M, Van Der Meij S. The fight against stigma: An overview of stigma-reduction strategies and interventions. Psychol Health Med. 2006;11(3):353–363. doi: 10.1080/13548500600595327. [DOI] [PubMed] [Google Scholar]
- 65.Scambler G, Heijnders M, van Brakel WH ICRAAS. Understanding and tackling health-related stigma. Psychol Health Med. 2006;11(3):269–270. doi: 10.1080/13548500600594908. [DOI] [PubMed] [Google Scholar]
- 66.Ballas SK, Smith ED. Red blood cell changes during the evolution of the sickle cell painful crisis. Blood. 1992;79(8):2154–2163. [PubMed] [Google Scholar]
- 67.Ballas SK. The sickle cell painful crisis in adults: Phases and objective signs. Hemoglobin. 1995;19(6):323–333. doi: 10.3109/03630269509005824. [DOI] [PubMed] [Google Scholar]
- 68.Jacob E, Beyer JE, Miaskowski C, Savedra M, Treadwell M, Styles L. Are there phases to the vaso-occlusive painful episode in sickle cell disease? J Pain Symptom Manage. 2005;29(4):392–400. doi: 10.1016/j.jpainsymman.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 69.Akinola NO, Stevens SM, Franklin IM, Nash GB, Stuart J. Rheological changes in the prodromal and established phases of sickle cell vasoocclusive crisis. Br J Haematol. 1992;81(4):598–602. doi: 10.1111/j.1365-2141.1992.tb02998.x. [DOI] [PubMed] [Google Scholar]
- 70.Murray N, May A. Painful crises in sickle cell disease—patients’ perspectives. BMJ. 1988;297(6646):452–454. doi: 10.1136/bmj.297.6646.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stuart J, Stone PC, Akinola NO, Gallimore JR, Pepys MB. Monitoring the acute phase response to vaso-occlusive crisis in sickle cell disease. J Clin Pathol. 1994;47(2):166–169. doi: 10.1136/jcp.47.2.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Katz J, Kavanagh BP, Sandler AN, Nierenberg H, Boylan JF, Friedlander M, et al. Preemptive analgesia. Clinical evidence of neuroplasticity contributing to postoperative pain. Anesthesiology. 1992;77(3):439–446. doi: 10.1097/00000542-199209000-00006. [DOI] [PubMed] [Google Scholar]
- 73.McQuay HJ. Pre-emptive analgesia. Br J Anaesth. 1992;69(1):1–3. doi: 10.1093/bja/69.1.1. [DOI] [PubMed] [Google Scholar]
- 74.Mayer ML, Konrad TR, Dvorak CC. Hospital resource utilization among patients with sickle cell disease. J Health Care Poor Underserved. 2003;14(1):122–135. [PubMed] [Google Scholar]
- 75.Epstein K, Yuen E, Riggio JM, Ballas SK, Moleski SM. Utilization of the office, hospital and emergency department for adult sickle cell patients: A five-year study. J Natl Med Assoc. 2006;98(7):1109–1113. [PMC free article] [PubMed] [Google Scholar]
- 76.Givens M, Rutherford C, Joshi G, Delaney K. Impact of an emergency department pain management protocol on the pattern of visits by patients with sickle cell disease. J Emerg Med. 2007;32(3):239–243. doi: 10.1016/j.jemermed.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 77.Malat JR, van Ryn M, Purcell D. Race, socioeconomic status, and the perceived importance of positive self-presentation in health care. Soc Sci & Med. 2006;62(10):2479–2488. doi: 10.1016/j.socscimed.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 78.Ballas SK. Pain management of sickle cell disease. Hematol Oncol Clin North Am. 2005;19(5):785–802. doi: 10.1016/j.hoc.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 79.Haywood C, Jr, Lanzkron S, Ratanawongsa N, Bediako SM, Lattimer L, Powe NR, et al. The association of provider communication with trust among adults with sickle cell disease. J Gen Intern Med. 2010;25(6):543–548. doi: 10.1007/s11606-009-1247-7. [DOI] [PMC free article] [PubMed] [Google Scholar]