

Abstract
The normal liver is characterized by immunologic tolerance. Primary mediators of hepatic immune tolerance are liver sinusoidal endothelial cells (LSECs). LSECs block adaptive immunogenic responses to Ag and induce the generation of T regulatory cells. Hepatic fibrosis is characterized by both intense intrahepatic inflammation and altered hepatic immunity. We postulated that, in liver fibrosis, a reversal of LSEC function from tolerogenic to proinflammatory and immunogenic may contribute to both the heightened inflammatory milieu and altered intrahepatic immunity. We found that, after fibrotic liver injury from hepatotoxins, LSECs become highly proinflammatory and secrete an array of cytokines and chemokines. In addition, LSECs gain enhanced capacity to capture Ag and induce T cell proliferation. Similarly, unlike LSECs in normal livers, in fibrosis, LSECs do not veto dendritic cell priming of T cells. Furthermore, whereas in normal livers, LSECs are active in the generation of T regulatory cells, in hepatic fibrosis LSECs induce an immunogenic T cell phenotype capable of enhancing endogenous CTLs and generating potent de novo CTL responses. Moreover, depletion of LSECs from fibrotic liver cultures mitigates the proinflammatory milieu characteristic of hepatic fibrosis. Our findings offer a critical understanding of the role of LSECs in modulating intrahepatic immunity and inflammation in fibro-inflammatory liver disease.
Liver sinusoidal endothelial cells (LSECs) are an integral part of the hepatic reticuloendothelial system (1). LSECs are morphologically identified by their fenestrations, which are transcytoplasmic canals arranged in sieve plates (2). In addition, LSECs express the CD45−CD146+ surface phenotype that makes their ex vivo purification from bulk hepatic nonparenchymal cells (NPCs) feasible (3, 4). Primary roles of LSECs include scavenger function and blood clearance. LSECs are equipped with specialized receptors, including those for endocytosis of acetylated low density lipoprotein and mannosylated proteins, and are uniquely equipped to eliminate viruses, colloids, and macromolecular waste from the circulation (1).
In addition to their role in clearance of toxins, a unique immunological role has been attributed to LSECs. They express modest levels of MHC class I and class II and costimulatory molecules (CD40, CD80, and CD86), and can induce Ag-restricted T cell proliferation (5). However, rather than mediate immunogenic T cell responses to Ag, LSECs induce hepatic immunologic tolerance (6). In fact, the phenomenon of oral tolerance to Ag introduced via the oral route or by portal venous injection has been attributed in part to LSEC-mediated tolerogenicity (7). In multiple experimental models, including presentation of oral Ag, cross-presentation of soluble protein, and uptake of apoptotic tumor cells, T cells that have been engaged by LSECs deviate either toward a CD4+ T regulatory cell (Treg) phenotype or toward CD8+ T cell tolerance, respectively (6–8). In addition, beyond actively inducing T cell anergy via Ag presentation, LSECs have been shown to veto T cell activation by other professional APCs (9). The cellular and molecular mechanisms of LSEC-mediated T cell anergy are not entirely understood. However, Diehl et al. (10) recently reported that B7-H1 signaling on LSECs is required for induction of CD8+ T cell tolerance via PD-1 ligation.
Whereas the hallmark of LSEC immune function in the unperturbed liver is the induction of tolerance, LSECs function has not been studied in disease states. Hepatic fibrosis is characterized by intense intrahepatic inflammation following liver insult, ultimately resulting in activation of hepatic stellate cells (HSCs) that produce extracellular matrix and fibrillar collagen (11). We have previously shown that the immunogenicity of liver APCs is a primary determinant of the intrahepatic inflammatory milieu in fibrotic liver disease (12). We reported that liver dendritic cells (DCs) transform from mediators of hepatic tolerance in the normal liver to purveyors of potent immunogenicity in hepatic fibrosis. The newly immunogenic DCs govern the hepatic inflammatory environment in fibrosis by inducing proinflammatory innate and adaptive immune responses in the liver (12). However, the changes in LSEC immune function in hepatic fibrosis, and their contributions toward the hepatic inflammatory milieu, have not been previously studied. In the current study, we report that, in fibrosis, LSECs become highly proinflammatory and are a critical component of intrahepatic inflammation. Moreover, in the fibrotic liver, LSECs transform from mediators of tolerance into potent stimulators of adaptive immunity. Our findings have important relevance to the understanding of intrahepatic immunity and inflammation in hepatic fibrosis.
Materials and Methods
Animals and procedures
Male C57BL/6, OT-I (B6.Cg-RAG2tm1Fwa-TgN), OT-II (B6.Cg-RAG2tm1Alt-TgN), and RAG−/− mice were purchased from Taconic Farms (Germantown, NY). To induce hepatic fibrosis, mice were treated with thrice-weekly injections of thioacetamide (250 mg/kg; Sigma-Aldrich, St. Louis, MO) and leptin (1.5 mg/kg; Biomyx, San Diego, CA) for 6 wk, as described (12, 13). Alternatively, in selected experiments, mice received biweekly injections of CCl4 (0.5 ml/kg; Sigma-Aldrich) for 12 wk, as described (12). Animal procedures were approved by the New York University Institutional Animal Care and Use Committee.
Cellular isolation and culture
Liver NPCs were isolated, as we have previously described (14). Briefly, the portal vein of mice was infused with 3 ml 1% collagenase IV (Sigma-Aldrich), followed by hepatectomy and mechanical digestion. Low speed (30 × g) centrifugation of liver suspensions was performed to exclude hepatocytes, followed by high speed (400 × g) centrifugation to isolate the NPCs. NPCs were further enriched over an Optiprep (Sigma-Aldrich) gradient. Leukocytes, including liver NK cells, T cells, NKT cells, B cells, macrophages, Kupffer cells, and neutrophils, were excluded using anti-CD45 immunomagnetic beads (Miltenyi Biotec, Bergisch-Gladbach, Germany) and passage through depletion columns. LSECs were then purified using anti-CD146 immunomagnetic beads and passage through positive selection columns or by FACS sorting CD45−CD146+ cells. This technique resulted in a >95% pure LSEC population (Supplemental Fig. 1). Other liver NPC subsets were similarly isolated either by FACS sorting or using immunomagnetic beads and passage through positive selection columns. HSCs were isolated by density centrifugation over a three-layer Optiprep gradient, as we have described (12). Splenocytes were harvested by mechanical disruption of spleens. Splenic T cells were purified using anti-CD90, anti-CD8, or anti-CD4 immunomagnetic beads and passage through positive selection columns.
Flow cytometry and cytokine analysis
Flow cytometry was performed using the FACSCalibur (BD Biosciences, Franklin Lakes, NJ) after incubating 5 × 105 cells/tube with 1 μg anti-FcγRIII/II Ab (2.4G2, Fc block; mAb Core, Sloan-Kettering Institute, New York, NY) and then labeling with 1 μg fluorescently conjugated Ab to CD4 (RM4-5), CD8α (53-6.7), CD11b (M1/70), CD11c (N418), CD25 (PC61.5), CD31 (390), CD40 (HM40-3), CD44 (IM7), CD45 (Ly5.2), CD62L (MEL-14), CD86 (GL1), CD105 (MJ7/18), CD146 (ME-9F1), CD178 (MFL3), MHC class II (I-Ab), and Foxp3 (FJK-16s) (all from eBiosciences, San Diego, CA, except CD146 [Miltenyi Biotec]). For cytokine analysis, cell suspensions were cultured in complete RPMI (RPMI 1640 with 10% heat-inactivated FBS, 2 mM l-glutamine, and 0.05 mM 2-ME) at a concentration of 1 × 106 cells/ml for 24 h before supernatant harvest and analysis using either a cytometric bead array (BD Biosciences) or the Milliplex immunoassay (Millipore, Billerica, MA). Intracellular cytokine staining was performed using an intracellular cytokine detection kit, according to the manufacturer’s protocol (BD Biosciences). TGF-β was assayed by ELISA (Bender MedSystems, Vienna, Austria).
In vitro T cell assays
For CD4+ T cell proliferation assays, LSECs were pulsed with OVA323–339 peptide (Abcam, Cambridge, MA; LSEC.OVA323) for 90 min before washing and plating in various concentration with OT-II T cells for 72 h in 96-well plates. For the last 24 h, 1 μCi [3H]thymidine was added to each well and radioactivity uptake was measured using a MicroBeta counter (PerkinElmer, Waltham, MA). To assess LSEC induction of Tregs, LSECs were cultured in equal numbers with syngeneic spleen CD4+ T cells before assay of T cell coexpression of CD25 and Foxp3 at 96 h. To examine LSEC-mediated activation of CD8+ T cells, LSECs were loaded with OVA257–264 (Abcam; LSEC.OVA257) before coculture with OT-I T cells and analysis of T cell surface phenotype at 24 h.
In vivo T cell assays
To test LSEC induction of T cell proliferation in vivo, C57BL/6 mice were administered 2 × 106 OT-I T cells i.v., followed by footpad immunization with LSEC.OVA257 (2 × 105) at 24 h. T cell proliferation in the draining ipsilateral popliteal lymph node was measured at 96 h by counting the number of CD8+OVAtetramer+ T cells by flow cytometry. To determine whether LSECs can enhance endogenous CTL responses, Rag−/− mice were reconstituted with 1 × 107 CD8+ OT-I T cells. On day 10, reconstituted mice were immunized with 1 × 106 LSEC.OVA257. On day 17, splenocytes were harvested from recipient mice, restimulated in vitro with OVA257-264, and tested for Th1 and Th2 cytokine production. To determine the ability of LSECs to produce de novo CTL, C57BL/6 mice were immunized i.p. twice at weekly intervals with 1 × 106 LSEC.OVA257 or mock immunized. One week after the second immunization, mice were challenged i.v. with a 1:1 ratio of CFSEhigh-labeled splenocytes (Invitrogen, Carlsbad, CA) loaded with OVA257–264 and CFSElow-labeled splenocytes that were mock loaded. CTL-specific lysis in the spleen or liver was determined at 4 h, according to the following formula:100 – [ratio (CFSEhigh cells/CFSElow cells)OVA immunized × 100/ratio(CFSEhigh cells/CFSElow cells)mock immunized].
Ag capture assays
For in vitro Ag uptake assays, DCs or LSECs (5 × 105) were incubated with FITC-dextran, FITC-albumin, or FITC-mannose albumin (1 mg/ml; all Sigma-Aldrich) at 37°C for various time intervals. Ag uptake was determined by flow cytometry. Assays were performed in triplicate.
Histology and immunohistochemistry
For histological analysis, livers were fixed with 10% buffered formalin, dehydrated in ethanol, embedded with paraffin, and stained with H&E or picric acid-Sirius red. For immunohistochemical studies, paraffin-embedded liver sections were stained with anti-CD45 (BD Biosciences) or anti–α-smooth muscle actin (SMA; Sigma-Aldrich). Images were captured using an Axiovert 40 microscope (Zeiss, Thornwood, NY) and a Jenoptik digital camera (Jena, Germany).
Statistics
Data are presented as mean ± SEM. Statistical significance (p < 0.05) was determined by the Student t test and the log rank test.
Results
Model of fibrosis
Livers of mice treated with hepatotoxin demonstrated an irregular surface contour on morphologic examination (Fig. 1A). Histologic analysis revealed that in treated animals the organized hepatic architecture was replaced by regenerative nodules bounded by fibrous septa (Fig. 1B). On picric acid-Sirius red staining, collagen deposition was seen surrounding and bridging the central portal triads. α-SMA staining confirmed HSC activation in vivo in livers of treated mice. Fibrosis was accompanied by a modest, but distinct CD45+ leukocytic infiltrate (Fig. 1B).
FIGURE 1.

Model of liver fibrosis. A, The liver surface contour of thioacetamide/leptin-treated mice at 8 wk is markedly irregular and exhibits diffuse nodularity. B, H&E examination (original magnification×10) of the livers of treated mice reveals a distorted hepatic architectural pattern. Regenerative nodules are seen (asterisk) bounded by fibrous septa (arrows). Picric acid-Sirius red (original magnification×10) staining highlights the collagen network both surrounding and bridging the portal triads. α-SMA staining (original magnification×10) is prominent in the livers of treated mice, suggesting HSC activation. CD45 (original magnification×20) immunohistochemistry confirms the presence of a lymphocytic infiltrate in the livers of treated animals.
LSEC surface phenotype in fibrosis
Because liver fibrosis was accompanied by intrahepatic leukocytosis, the fraction of CD45−CD146+ LSECs among all NPCs was lower in the fibrotic liver compared with the normal liver (Fig. 2A). However, the actual number of LSECs was similar in both normal and fibrotic mice. Overall, ~2–4 × 106 LSECs were isolated per mouse. We compared the surface phenotype of normal LSECs (N-LSECs) and fibrotic LSECs (F-LSECs) by flow cytometry. Surface phenotype was similar for both groups (Fig. 2B). In particular, CD31 and CD105 were expressed highly on LSEC populations regardless of the presence of fibrosis. Conversely, there was low expression of MHC class II, CD40, CD86, and CD11b.
FIGURE 2.

LSEC surface phenotype in fibrosis. A, Live NPCs from normal and fibrotic livers were analyzed for expression of both CD45 and CD146. Dead cells were excluded using 7-aminoactinomycin D (data not shown). B, CD45−CD146+ LSECs were gated and further analyzed for cell surface marker expression. Isotype staining, shown in shaded histograms for N-LSEC, was similar for both groups. Mean fluorescence index is indicated for each marker below their respective histograms. Data are representative of experiments repeated more than three times using three to four mice per experiment. FSC, forward light scatter; SSC, side light scatter.
F-LSECs are capable of enhanced Ag capture in vitro
A primary function of LSECs is Ag capture, both for blood clearance and for the initiation of immune responses (2). We tested the capacity of LSECs from normal or fibrotic livers to capture Ag by both generalized macropinocytosis and via specialized mannose receptors. F-LSECs had greater capacity to capture dextran (Fig. 3A), albumin (Fig. 3B), and mannose-albumin (Fig. 3C) compared with N-LSECs. In contrast to LSECs, DCs from fibrotic livers were deficient at capturing albumin (Fig. 3D) and mannosylated albumin (Fig. 3E), consistent with the mature phenotype that DCs acquire in liver fibrosis (12).
FIGURE 3.

Ag capture by LSECs is altered in hepatic fibrosis. LSECs (5 × 105) were tested for their ability to capture dextran (A), albumin (B), and mannose-albumin (C) in vitro at various time points of incubation. Similarly, DCs (5 × 105) from normal and fibrotic livers were tested for their ability to capture albumin (D) and mannose-albumin (E). Experiments were repeated more than three times and performed in triplicate. *p < 0.05.
LSECs produce elevated levels of inflammatory mediators in fibrosis
Because liver NPCs produce intense intrahepatic inflammation in liver fibrosis (11) and LSECs constitute a high fraction of NPCs, we postulated that F-LSECs were proinflammatory. To test this, freshly isolated N-LSECs and F-LSECs were cultured for 24 h, and supernatants were assayed for chemokines and cytokines with established import in fibrogenesis. F-LSECs produced markedly elevated levels of TNF-α as well as a number of chemokines, including MIP-1α, MIP-1β, MIP-2, MCP-1, RANTES, and KC compared with N-LSECs (Fig. 4A). LSECs also accounted for a high fraction of inflammatory mediators produced in liver fibrosis when compared with the contribution of other hepatic NPCs (Supplemental Fig. 2). However, GM-CSF, G-CSF, IFN-γ–inducible protein-10, IL-1α, IL-1β, IL-9, IL-10, IL-17, and TGF-β were produced at similar levels in both F-LSECs and N-LSECs (data not shown). To investigate whether F-LSECs can also secondarily account for elevations in cytokine and chemokine levels not directly produced by F-LSECs, via interaction with neighboring cells, we cultured freshly isolated NPCs from normal or fibrotic livers for 24 h before supernatant harvest (Fig. 4B). In parallel, we cultured equal numbers of NPCs from fibrotic livers that had been depleted of F-LSECs by FACS. Remarkably, F-LSEC depletion lowered the elevated levels of a number of inflammatory mediators in fibrosis NPC cultures toward normal, suggesting that LSECs may secondarily account for elevations in these cytokines and chemokines by interaction with neighboring cell types (Fig. 4B). Similar findings were obtained in experiments using the CCL4 model of hepatic fibrosis (Supplemental Fig. 3).
FIGURE 4.

LESC are proinflammatory in hepatic fibrosis. A, Freshly isolated LSECs were cultured for 24 h, and inflammatory mediators were measured in cell culture supernatant. p < 0.05 for each comparison. B, NPCs were harvested from the livers of normal and fibrotic mice and cultured for 24 h. Depletion of LSECs from fibrotic NPC concentrates by FACS abrogated their elevated cytokine and chemokine production. Experiments were repeated more than three times and performed in triplicate. p < 0.05 for each inflammatory mediator.
F-LSECs produce enhanced T cell activation
Because LSECsd become proinflammatory in fibrosis, we postulated that they would have greater immunogenic potential. To test this, LSEC.OVA257 from normal or fibrotic mice were cocultured with equal numbers of CD8+ OT-I T cells for 24 h before analysis of T cell activation by flow cytometry. F-LSECs induced greater downregulation of CD62L and upregulation of CD25 and CD178 expression on OT-I T cells and modest upregulation of CD44 compared with the effects of N-LSECs (Fig. 5A). Similar enhancement of LSEC ability to activate CD8+ T cells was seen using the CCL4 model of liver fibrosis (Supplemental Fig. 4).
FIGURE 5.

LSECs from fibrotic livers are immunogenic. Freshly isolated LSECs (2 × 105) from normal or fibrotic livers were either A, loaded with OVA257–264 and cocultured with CD8+ OT-I T cells (2 × 105) before measurement of T cell activation markers or B, coincubated with CD4+ T cells (2 × 105) from C57BL/6 mice before determination of CD25 and Foxp3 coexpression. Data are representative of experiments repeated three times.
In normal livers, LSECs are associated with immunologic tolerance via the generation of Tregs (5). To test these effects in fibrosis, LSECs were coincubated with spleen CD4+ T cells before assessment of T cell coexpression of CD25 and Foxp3. Notably, after coculture with N-LSECs, 15% of T cells were CD25+Foxp3+, indicative of Tregs. Conversely, only 1% of CD4+ T cells incubated with F-LSECs exhibited the Treg phenotype (Fig. 5B).
In fibrosis, LSECs induce higher T cell proliferation
To test the ability of LSECs from fibrotic livers to induce T cell proliferation in vitro, F-LSECs or N-LSECs were loaded with OVA323–339 and plated in various concentrations with OT-II T cells. F-LSECs induced higher T cell proliferation at the two highest concentrations tested (Supplemental Fig. 5). To test LSEC induction of T cell proliferation in vivo, LSECs from normal or fibrotic livers were loaded with OVA257–264 and administered into the footpad of mice that had been given an i.v. injection of OT-I T cells 24 h earlier. Treatment with F-LSEC.OVA257–264 induced more robust Ag-restricted T cell proliferation than N-LSECs (Fig. 6A, 6B). In addition, CD44 was more highly expressed on CD8+OVA tetramer+ T cells after transfer of F-LSEC.OVA257 compared with N-LSEC.OVA257 or mock adoptive transfer (Fig. 6C). Immunization using LSECs that had not been Ag loaded did not affect either tetramer cell number or activation status (data not shown).
FIGURE 6.

F-LSECs produce enhanced T cell proliferation in vivo. Mice were adoptively transferred with OT-I T cells (2 × 106) before receiving footpad immunization with LSEC. OVA257 (2 × 105) or mock immunized. At 96 h, ipsilateral popliteal lymph nodes were harvested and costained for CD8 and OVA tetramer. The fraction of OVA tetramer+ cells among all CD8+ cells (A), the total number of Ova tetramer+ cells (B), and their expression of CD44 (C) were determined by flow cytometry. Experiments were repeated three times using two to four mice per experiment. *p < 0.05.
In liver fibrosis, LSECs enhance Ag-specific CTL
In the normal liver, LSECs inhibit Ag-restricted CTL (7). To test whether LSECs from fibrotic livers either prevent or augment CTL, we reconstituted Rag mice with OT-I T cells and subsequently used either N-LSEC.OVA257 or F-LSEC.OVA257 for immunization. One week after i.p. immunization, splenocytes were harvested from recipient mice, restimulated in vitro with OVA257–264 for 4 d, before assay of CTL supernatant for immunogenic cytokines. CTL supernatant derived from reconstituted Rag mice that had been immunized with F-LSEC.OVA257 produced significantly elevated levels of IFN-γ, TNF-α, MCP-1, and IL-6 (Fig. 7A–D). Conversely, N-LSEC.OVA257–264 immunization failed to augment production of immunogenic cytokines in CTL cultures. Moreover, immunization with peptide-pulsed F-LSECs, but not N-LSECs, resulted in decreased levels of IL-10, an inhibitory cytokine, in CTL cultures (Fig. 7E).
FIGURE 7.

F-LSECs enhance endogenous CTL. Rag mice were reconstituted with OT-I T cells (1 × 107) before either mock immunization or immunization using LSEC.OVA257 (1 × 106). One week postimmunization, splenocytes from treated animals were restimulated in vitro with OVA257–264 before measurement of IFN-γ (A), TNF-α (B), IL-6 (C), MCP-1 (D), and IL-10 (E) in day 5 CTL cultures. Experiments were repeated twice using two to four mice per experiment. *p < 0.05.
F-LSECs induce de novo CTL responses
To determine the relative capacity of F-LSECs to generate de novo CTL responses in vivo, naive C57BL/6 mice were immunized twice with N-LSEC.OVA257 or F-LSEC.OVA257. One week after the second immunization, mice were challenged with CFSE-labeled splenocytes either loaded with OVA257–264 (CFSEhigh) or mock loaded (CFSElow). Ag-restricted CTL lysis was measured in both the liver and spleen. Immunization with N-LSEC.OVA257–264 did not induce CTL lysis of OVA-loaded splenocytes consistent with their tolerogenic immunophenotype. Conversely, immunization with F-LSECs induced high CTL lysis of OVA257–264-presenting targets in both the spleen (Fig. 8A) and liver (Fig. 8B). Consistent with the cytotoxicity data, day 5 spleen CTL cultures from mice immunized with F-LSEC.OVA257–264 produced the elevated IFN-γ (Fig. 8C) and IL-6 (Fig. 8D). Immunization with nonpeptide-pulsed F-LSECs was nonactivating (data not shown), confirming the specificity of the assay.
FIGURE 8.

F-LSECs induce potent de novo CTL responses. Naive C57BL/6 mice were either mock immunized or twice immunized using LSEC.OVA257 (1 × 106). One week later, in vivo CTL lysis of OVA257-presenting cellular targets was measured in the spleen (A) and the liver (B), and splenocyte CTL cultures were assayed for production of IFN-γ (C) and IL-6 (D). Experiments were repeated three times and performed in triplicate using two to four mice per group. *p < 0.05.
F-LSECs do not inhibit DC immunogenicity
The tolerogenic effects of LSECs have recently been extended to active disruption of T cell stimulation by professional APCs. In particular, LSECs from normal livers have been shown to prevent Ag-restricted DC stimulation of T cells in vitro (9). To determine whether F-LSECs have similar inhibitory effects on DCs, freshly isolated spleen DCs were loaded with OVA257–264 and used to stimulate OT-I T cells. In selected wells, nonpeptide-pulsed LSECs were added in equal numbers to DCs. Notably, whereas N-LSECs inhibited T cell proliferation, as previously reported (9), F-LSECs were noninhibitory of DC induction of T cell proliferation (Fig. 9).
FIGURE 9.
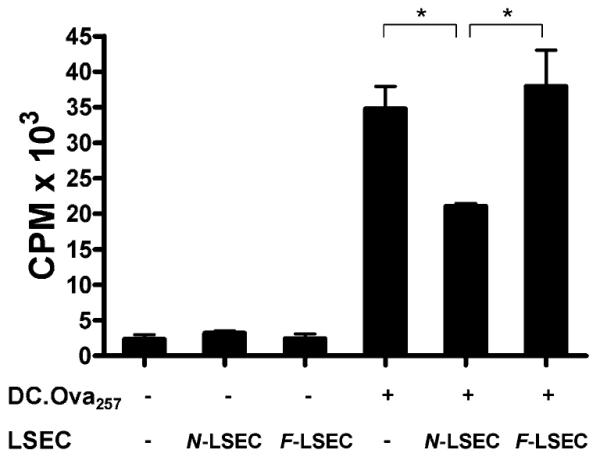
F-LSECs do not mitigate Ag presentation by DC. OT-I T cells (1 × 105) were cultured alone or coincubated with DC.OVA257–264 (1 × 104) and LSECs (1 × 104), separately or in combination, harvested from normal or fibrotic livers. DC.OVA257 induced potent Ag-specific T cell proliferation that was mitigated by coculture with N-LSECs. Conversely, coculture with F-LSECs did not lower DC-mediated T cell proliferation. Experiments were repeated three times and performed in triplicate. *p < 0.05.
Discussion
Intrahepatic immunity has been studied extensively in the normal liver. The liver is the paradigm for immunologic tolerance and, in that capacity, is the center for important physiologic processes, such as oral tolerance to ingested Ag (15). Similarly, the acceptance of hepatic allografts with minimal immune suppression, hepatic autoimmune disease, and the disproportional occurrence of metastatic disease to the liver have all been linked, in part, to tolerogenic immune processes within the liver (16). Both professional APCs, such as plasmacytoid DCs, and nonprofessional APCs, such as LSECs, are primary immunologic mediators underlying hepatic tolerance (7, 17). However, whereas intrahepatic immunity within the normal liver is the subject of intense study, there is little information regarding adaptive immunity in chronic fibroinflammatory liver disease. In particular, the immune-stimulatory capabilities of both professional and nonprofessional hepatic APCs in liver fibrosis are poorly understood. This is a very relevant area of study as our group and others have recently shown that primary events in hepatic fibrosis, such as HSC activation, and even the degree of fibrosis, may be contingent on intrahepatic immunity (11, 12).
In the current study, we found that the immunogenic function of LSECs is reversed in fibrosis compared with the properties of N-LSECs. F-LSECs produce an array of proinflammatory mediators (Fig. 4), capture Ag at a higher rate (Fig. 3A–C), and induce higher Ag-specific T cell proliferation (Fig. 6). F-LSECs also induce an activated T cell surface phenotype (Fig. 5A) while preventing the generation of Tregs (Fig. 5B). Moreover, in contrast to their counterparts in the normal liver, LSECs from the fibrotic liver do not veto DC-mediated T cell activation (Fig. 9) and are capable of enhancing CTL responses (Fig. 7) and generating de novo CTL in vivo (Fig. 8). We have previously reported that, in hepatotoxin-induced models of liver fibrosis, DCs also acquire enhanced immunogenic capacity and produce potent T cell responses to Ag (12). The mechanism for enhanced immunogenicity of APCs in fibrosis is not fully understood. We have found that higher DC production of TNF-α in the fibrotic liver is, in part, responsible for their enhanced immunogenic capabilities (12). As such, TNF-α blockade reverts DC function to a tolerogenic phenotype. For example, blockade of TNF-α prevented fibrotic liver DCs from inducing higher allogeneic or Ag-restricted T cell proliferation or inducing NK cell production of IFN-γ and cytotoxicity (12). However, determining the mechanism of enhanced LSEC function in fibrosis has been elusive. Blockade of LSEC production of TNF-α or RANTES, for example, did not mitigate the higher immunogenicity of LSECs (data not shown). However, given the wide array of mediators produced at elevated levels by LSECs in fibrosis (Fig. 4A), it is likely that undetermined factors produced by F-LSECs are responsible for their altered functional profile.
Liver fibrosis is a complexwound healing-type response to hepatic injury. After hepatic insult, an array of inflammatory mediators and reactive oxygen species is produced in the liver by both hepatic leukocytes, including T cells, as well as hepatocytes and biliary epithelial cells (11). The end result of the intense intrahepatic inflammation is activation of HSCs, which further produce high levels of cytokines (IL-1α, TNF-α, IL-6) and chemokines (RANTES, MCP-1, MIP-1α) and lay down extracellular matrix proteins, including fibrillar collagen. The physiologic consequences of higher APC-related immunogenicity in fibrosis relate to the elevated intrahepatic inflammatory milieu in fibrosis. For example, we found that depletion of proinflammatory F-LESCs significantly lowers the levels of multiple cytokines (IL-1α, IL-1β, IL-9, IL-10, IL-17, IFN-γ–inducible protein-10, G-CSF, GM-CSF) and chemokines (RANTES, MCP-1, MIP-1α, MIP-1β) in fibrotic liver suspensions (Fig. 4B). Because LSECs alone do not produce many of the aforementioned inflammatory mediators, this effect most likely results from LSEC interactions with other cell types. Consistent with this notion, we have shown that F-LSECs can induce T cells to produce an array of proinflammatory mediators, including IL-6, IFN-γ, TNF-α, and MCP-1 (Figs. 7A–D,8C, 8D). Similarly, we have previously shown that in hepatic fibrosis, DCs control the inflammatory milieu within the fibrotic liver by activation of immune effector cells, including NK and T cells (12). Therefore, heightened APC immunogenicity in both LSECs and DCs appears to profoundly affect the very inflammatory environment in the liver, which is characteristic of hepatic fibrosis. Our data do not prove, but raise the specter that, given their prominent role in intrahepatic inflammation, F-LSECs may affect the actual fibrotic process within the liver. Consistent with this notion, Deleve et al. (18) reported that whereas in the unperturbed liver LSECs actively promote HSC quiescence, in fibrosis, capillarized LSECs allow for HSC activation. However, definitive determination of the importance of LSECs in fibrogenesis is hampered by the inability to deplete LSECs in vivo in experimental models of liver fibrosis.
These findings offer a critical understanding of the role of LSECs in intrahepatic immunity and inflammation in hepatic fibrosis and may also have implications to liver allograft tolerance (19, 20). However, a limitation to this study is that modeling LSEC function in vitro may not reflect their function in the hepatic milieu. LSEC immune phenotype may also be unavoidably modified upon isolation. The study of LSECs is thus hampered by the inability to deplete LSECs in vivo and thereby distinguish their contributions to hepatic function from other NPCs in vivo. Furthermore, the physiologic correlate to LSEC-mediated tolerance in the human liver and its reversal in human liver fibrosis are uncertain. Karrar et al. (21) reported that sinusoidal endothelial cells from the normal human liver induce apoptosis in activated T cells after direct cellular contact, consistent with their role in tolerance induction in rodents. Neutralizing Abs to LSECs have also been surmised to facilitate acute liver allograft rejection by downregulating the immune regulatory cytokine TGF-β and thus upregulating alloreactive T cell proliferation (19). However, very little is known about the immunogenicity of human LSECs in disease states, such as fibrosis. Yeligar et al. (22) recently reported that chronic alcohol consumption increases RANTES production in human endothelial cells via activation of NF-κB, hypoxia-inducible factor-1α, and AP-1. An important area for investigation would be to isolate LSECs from the fibrotic human liver and determine their role in hepatic immunity and inflammation. However, any potential investigation into LSEC immune function in cirrhosis is confounded by the fact that the majority of cases of hepatic cirrhosis in humans are related to hepatitis C virus infection, which alone modulates intrahepatic immunity, even in the absence of parenchymal fibrosis (23, 24).
Supplementary Material
Acknowledgments
This work was supported in part by a Liver Scholar Award from the American Liver Foundation (to G.M.), a Whitehead Fellowship Award for Junior Faculty in Biomedical and Biological Sciences (to G.M.), and National Institutes of Health Awards DK085278 (to G.M.) and CA108573 (to A.B.F.).
Abbreviations used in this paper
- DC
dendritic cell
- F-LSEC
fibrotic liver sinusoidal endothelial cell
- FSC
forward light scatter
- HSC
hepatic stellate cell
- LSEC
liver sinusoidal endothelial cell
- N-LSEC
normal LSEC
- NPC
nonparenchymal cell
- SMA
smooth muscle actin
- SSC
side light scatter
- Treg
T regulatory cell
Footnotes
Disclosures The authors have no financial conflicts of interest.
The online version of this article contains supplemental material.
References
- 1.Elvevold K, Smedsrød B, Martinez I. The liver sinusoidal endothelial cell: a cell type of controversial and confusing identity. Am. J. Physiol. Gastrointest. Liver Physiol. 2008;294:G391–G400. doi: 10.1152/ajpgi.00167.2007. [DOI] [PubMed] [Google Scholar]
- 2.Braet F, Wisse E. Structural and functional aspects of liver sinusoidal endothelial cell fenestrae: a review. Comp. Hepatol. 2002;1:1. doi: 10.1186/1476-5926-1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Katz SC, Pillarisetty VG, Bleier JI, Shah AB, DeMatteo RP. Liver sinusoidal endothelial cells are insufficient to activate T cells. J. Immunol. 2004;173:230–235. doi: 10.4049/jimmunol.173.1.230. [DOI] [PubMed] [Google Scholar]
- 4.Schurich A, Böttcher JP, Burgdorf S, Penzler P, Hegenbarth S, Kern M, Dolf A, Endl E, Schultze J, Wiertz E, et al. Distinct kinetics and dynamics of cross-presentation in liver sinusoidal endothelial cells compared to dendritic cells. Hepatology. 2009;50:909–919. doi: 10.1002/hep.23075. [DOI] [PubMed] [Google Scholar]
- 5.Knolle PA, Limmer A. Neighborhood politics: the immunoregulatory function of organ-resident liver endothelial cells. Trends Immunol. 2001;22:432–437. doi: 10.1016/s1471-4906(01)01957-3. [DOI] [PubMed] [Google Scholar]
- 6.Limmer A, Ohl J, Kurts C, Ljunggren HG, Reiss Y, Groettrup M, Momburg F, Arnold B, Knolle PA. Efficient presentation of exogenous antigen by liver endothelial cells to CD8+ T cells results in antigen-specific T-cell tolerance. Nat. Med. 2000;6:1348–1354. doi: 10.1038/82161. [DOI] [PubMed] [Google Scholar]
- 7.Limmer A, Ohl J, Wingender G, Berg M, Jüngerkes F, Schumak B, Djandji D, Scholz K, Klevenz A, Hegenbarth S, et al. Cross-presentation of oral antigens by liver sinusoidal endothelial cells leads to CD8 T cell tolerance. Eur. J. Immunol. 2005;35:2970–2981. doi: 10.1002/eji.200526034. [DOI] [PubMed] [Google Scholar]
- 8.Berg M, Wingender G, Djandji D, Hegenbarth S, Momburg F, Hämmerling G, Limmer A, Knolle P. Cross-presentation of antigens from apoptotic tumor cells by liver sinusoidal endothelial cells leads to tumor-specific CD8+ T cell tolerance. Eur. J. Immunol. 2006;36:2960–2970. doi: 10.1002/eji.200636033. [DOI] [PubMed] [Google Scholar]
- 9.Schildberg FA, Hegenbarth SI, Schumak B, Scholz K, Limmer A, Knolle PA. Liver sinusoidal endothelial cells veto CD8 T cell activation by antigen-presenting dendritic cells. Eur. J. Immunol. 2008;38:957–967. doi: 10.1002/eji.200738060. [DOI] [PubMed] [Google Scholar]
- 10.Diehl L, Schurich A, Grochtmann R, Hegenbarth S, Chen L, Knolle PA. Tolerogenic maturation of liver sinusoidal endothelial cells promotes B7-homolog 1-dependent CD8+ Tcell tolerance. Hepatology. 2008;47:296–305. doi: 10.1002/hep.21965. [DOI] [PubMed] [Google Scholar]
- 11.Bataller R, Brenner DA. Liver fibrosis. J. Clin. Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Connolly MK, Bedrosian AS, Mallen-St Clair J, Mitchell AP, Ibrahim J, Stroud A, Pachter HL, Bar-Sagi D, Frey AB, Miller G. In liver fibrosis, dendritic cells govern hepatic inflammation in mice via TNF-alpha. J. Clin. Invest. 2009;119:3213–3225. doi: 10.1172/JCI37581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dai K, Qi JY, Tian DY. Leptin administration exacerbates thioacetamide-induced liver fibrosis in mice. World J. Gastroenterol. 2005;11:4822–4826. doi: 10.3748/wjg.v11.i31.4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pillarisetty VG, Shah AB, Miller G, Bleier JI, DeMatteo RP. Liver dendritic cells are less immunogenic than spleen dendritic cells because of differences in subtype composition. J. Immunol. 2004;172:1009–1017. doi: 10.4049/jimmunol.172.2.1009. [DOI] [PubMed] [Google Scholar]
- 15.Ilan Y. Oral tolerance: can we make it work? Hum. Immunol. 2009;70:768–776. doi: 10.1016/j.humimm.2009.06.018. [DOI] [PubMed] [Google Scholar]
- 16.Crispe IN, Giannandrea M, Klein I, John B, Sampson B, Wuensch S. Cellular and molecular mechanisms of liver tolerance. Immunol. Rev. 2006;213:101–118. doi: 10.1111/j.1600-065X.2006.00435.x. [DOI] [PubMed] [Google Scholar]
- 17.Goubier A, Dubois B, Gheit H, Joubert G, Villard-Truc F, Asselin-Paturel C, Trinchieri G, Kaiserlian D. Plasmacytoid dendritic cells mediate oral tolerance. Immunity. 2008;29:464–475. doi: 10.1016/j.immuni.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deleve LD, Wang X, Guo Y. Sinusoidal endothelial cells prevent rat stellate cell activation and promote reversion to quiescence. Hepatology. 2008;48:920–930. doi: 10.1002/hep.22351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ge X, Karrar A, Ericzon BG, Broomé U, Sumitran-Holgersson S. Antibodies to liver sinusoidal endothelial cells modulate immune responses in liver transplantation. Transplant. Proc. 2005;37:3335–3337. doi: 10.1016/j.transproceed.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 20.Onoe T, Ohdan H, Tokita D, Shishida M, Tanaka Y, Hara H, Zhou W, Ishiyama K, Mitsuta H, Ide K, Asahara T. Liver sinusoidal endothelial cells tolerize T cells across MHC barriers in mice. J. Immunol. 2005;175:139–146. doi: 10.4049/jimmunol.175.1.139. [DOI] [PubMed] [Google Scholar]
- 21.Karrar A, Broomé U, Uzunel M, Qureshi AR, Sumitran-Holgersson S. Human liver sinusoidal endothelial cells induce apoptosis in activated T cells: a role in tolerance induction. Gut. 2007;56:243–252. doi: 10.1136/gut.2006.093906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yeligar SM, Machida K, Tsukamoto H, Kalra VK. Ethanol augments RANTES/CCL5 expression in rat liver sinusoidal endothelial cells and human endothelial cells via activation of NF-kappa B, HIF-1 alpha, and AP-1. J. Immunol. 2009;183:5964–5976. doi: 10.4049/jimmunol.0901564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kriegs M, Bürckstümmer T, Himmelsbach K, Bruns M, Frelin L, Ahlén G, Sällberg M, Hildt E. The hepatitis C virus non-structural NS5A protein impairs both the innate and adaptive hepatic immune response in vivo. J. Biol. Chem. 2009;284:28343–28351. doi: 10.1074/jbc.M109.038877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tu Z, Pierce RH, Kurtis J, Kuroki Y, Crispe IN, Orloff MS. Hepatitis C virus core protein subverts the antiviral activities of human Kupffer cells. Gastroenterology. 2010;138:305–314. doi: 10.1053/j.gastro.2009.09.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.