

Abstract
Background
Sarcoplasmic reticulum calcium ATPase 2a (SERCA2a) gene therapy improves mechanical function in heart failure, and is under evaluation in a clinical trial. A critical question is whether SERCA2a gene therapy predisposes to increased sarcoplasmic reticulum calcium (SR Ca2+) leak, cellular triggered activity and ventricular arrhythmias in the failing heart.
Methods and Results
We studied the influence of SERCA2a gene therapy upon ventricular arrhythmogenesis in a rat chronic heart failure model. ECG telemetry studies revealed a significant antiarrhythmic effect of SERCA2a gene therapy with reduction of both spontaneous and catecholamine-induced arrhythmias in vivo. SERCA2a gene therapy also reduced susceptibility to reentry arrhythmias in ex vivo programmed electrical stimulation studies. Subcellular Ca2+ homeostasis and spontaneous SR Ca2+ leak characteristics were measured in failing cardiomyocytes transfected in vivo with a novel AAV9.SERCA2a vector. SR Ca2+ leak was reduced following SERCA2a gene therapy, with reversal of the greater spark mass observed in the failing myocytes, despite normalisation of SR Ca2+ load. SERCA2a reduced ryanodine receptor phosphorylation, thereby resetting SR Ca2+ leak threshold, leading to reduced triggered activity in vitro. Both indirect effects of reverse remodelling and direct SERCA2a effects appear to underlie the antiarrhythmic action.
Conclusions
SERCA2a gene therapy stabilizes SR Ca2+ load, reduces ryanodine receptor phosphorylation and decreases SR Ca2+ leak, reduces cellular triggered activity in vitro and spontaneous and catecholamine-induced ventricular arrhythmias in vivo in failing hearts. SERCA2a gene therapy did not therefore predispose to arrhythmias, and may even represent a novel antiarrhythmic strategy in heart failure.
Keywords: Arrhythmia, Gene Therapy, Calcium, Heart Failure, SERCA2a
Introduction
Sarcoplasmic reticulum calcium ATPase 2a (SERCA2a) gene therapy restores mechanical, contractile and energetic function in failing human cardiomyocytes1 and animal models of heart failure (HF).2-4 These beneficial effects form the scientific foundation for a clinical trial of SERCA2a gene therapy for patients with chronic HF,5 a growing clinical epidemic where morbidity and mortality remain high despite current conventional therapy.
One concern raised is whether SERCA2a gene transfer to the failing myocardium may predispose to increased arrhythmic potential, despite the improvements in inotropic and lusitropic function. However, the studies of SERCA2a expression and arrhythmogenesis which raise these concerns have been performed on a normal, rather than a failing, myocardial background.6, 7 The sarcoplasmic reticulum (SR) of the failing ventricular myocardium is characterised by increased spontaneous diastolic calcium (Ca2+) release, frequently measured as Ca2+ sparks in isolated cardiomyocytes, reflecting an increased opening probability of the ryanodine receptors (RyR2).8, 9 As luminal SR Ca2+ levels contribute to RyR2 gating,10, 11 and provide the electrochemical driving force for SR Ca2+ release,12 elevating SR Ca2+ by restoring SERCA2a function could hypothetically increase spontaneous Ca2+ release and trigger arrhythmias. This would be a particular concern given the increased sympathetic drive seen in heart failure patients, as β1adrenoceptor (β1AR) activation increases both SR Ca2+ loading by SERCA2a and may increase the risk of RyR2-mediated diastolic leak.9 In the first clinical SERCA2a gene therapy trials, all patients have mandatory implantable defibrillators or left ventricular assist device support which will offer protection against malignant arrhythmias. However if SERCA2a gene therapy is to be extended to the wider heart failure population, are these precautions against proarrhythmia required?
In this study we sought to address these specific concerns relating to SR Ca2+ leak and arrhythmogenesis in the failing heart following SERCA2a gene transfer. We studied spontaneous and catecholamine-induced arrhythmia generation in vivo in a chronic HF model before and after heterogeneous SERCA2a gene transfer using ECG telemetry and ex vivo with provocation protocols aimed at producing reentry arrhythmias. A significant antiarrhythmic effect of SERCA2a gene transfer was detected and therefore we studied the potential mechanisms for the antiarrhythmic effect. Using a novel AAV9.SERCA2a vector, which yields more homogeneous (∼90%) transfection efficiency four weeks after systemic venous delivery,13 and lacks the confounding effects of green fluorescent protein, we studied changes in cardiomyocyte SR Ca2+ content and spontaneous SR Ca2+ release events in freshly isolated failing cardiomyocytes following in vivo SERCA2a gene transfer.
Methods
Extended methods are included in the online data supplement.
Study Protocols
Two heart failure plus SERCA2a gene therapy cohorts were studied using a previously described rat model of post infarction chronic heart failure (Figure 1).14 Sham ligated (SHAM) or age matched unoperated (AMC) animals served as non-failing controls.
Figure 1.
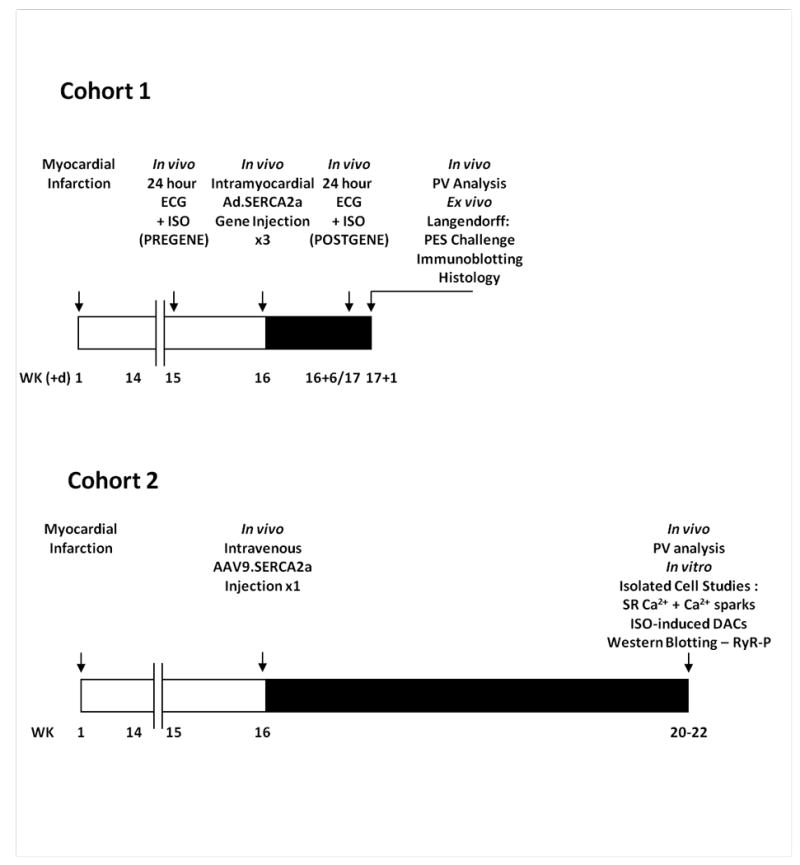
Study protocols. Schematic timelines (weeks (+/- days)) summarising the protocols for the two study cohorts evaluating the influence of SERCA2a gene therapy upon arrhythmia generation in a post infarction chronic heart failure model. Cohort 1 – whole heart in vivo and ex vivo studies with Ad.SERCA2a.GFP; Cohort 2 – in vitro cellular and subcellular studies with AAV9.SERCA2a.
Cohort 1: In vivo and Ex vivo Arrhythmia Protocol
In vivo arrhythmia assessment was performed using implantable ECG telemetry with continuous 24 hour ECG recording for spontaneous ventricular arrhythmias, followed by an in vivo intraperitoneal injection of 0.5mg/kg isoproterenol (ISO) for arrhythmia provocation. Recordings were performed in HF animals at baseline (PREGENE), and six days after receiving three 50μL intramyocardial injections of Ad.SERCA.GFP or Ad.GFP (POSTGENE). SHAM cases received three location-matched Ad.GFP injections. Twenty-four hours later Langendorff perfused hearts were studied for susceptibility to reentry arrhythmias using programmed electrical stimulation. In a subgroup left ventricular function was assessed one week post Ad.SERCA2a injection by pressure-volume (PV) analysis.14
Cohort 2: HF + AAV9.SERCA study for Isolated Cardiomyocyte Studies
Sixteen HF rats received a 300μL tail vein injection of AAV9.SERCA2a (2×1011 drp) at least sixteen weeks post infarction. Four-six weeks later, hearts were harvested for either isolated cardiomyocyte studies (n=12) or western blotting (n=4). A subgroup (n=6) underwent in vivo PV analysis prior to sacrifice. HF rats without gene transfer and AMC served as controls.
Isolated Cardiomyocyte Aftercontraction Provocation Studies
The incidence of Ca2+-mediated delayed aftercontractions (DACs) was recorded in freshly isolated cardiomyocytes at baseline and after perfusion with 1 nmol/L ISO and 100 nmol/L ISO, with drug washout between doses.
Calcium Sparks, SR Calcium Load and Leak
Spark studies were performed as previously described by our group,14 with spark analysis based on the standard algorithm developed by Cheng et al.15 Spark mass was calculated using the following equation: spark amplitude × 1.206 × full width at half maximal amplitude (FWHM)3 as previously reported.16 Spark-mediated SR leak was the product of spark mass and frequency from individual myocytes. Total SR Ca2+leak was measured as the tetracaine-dependent SR Ca2+ leak using the protocol described by Shannon et al,17 with caffeine-sensitive SR Ca2+ load measured in series allowing SR leak/load calculation for individual myocytes.
Ryanodine Receptor and Phospholamban Phosphorylation
Phosphorylation of RyR2 and phospholamban (PLB) at both protein kinase A (PKA) (RyR2809, PLB-Ser16) and calmodulin-dependent protein kinase II (CaMKII) (RyR2815, PLB-Thr17) target sites were measured by Western blotting.
Statistical Analysis
Results were compared using 1 way ANOVA with Tukey's posttest comparison (quantitative arrhythmia and HF variables), Kruskal-Wallis test with Dunn's posttest comparison (Ca2+ spark parameters), paired Student's t-test (in vivo PREGENE and POSTGENE arrhythmias) and Fishers exact test (arrhythmia incidence). A p value <0.05 determined statistical significance.
Results
SERCA2a Protein Levels after SERCA2a Gene Therapy
Focal GFP fluorescence in injected regions was demonstrated in all hearts receiving Ad.SERCA2a.GFP or Ad.GFP gene transfer in Cohort 1 seven days after intramyocardial gene injection (Figure 2A). Ad.SERCA2a gene transfer significantly increased SERCA2a protein levels in the injected and remote uninjected regions compared with both fluorescent and non-fluorescent regions from HF+GFP hearts (p=0.01) (Figures 2B and S1). Intravenous AAV9.SERCA2a gene transfer also successfully increased myocardial SERCA2a protein levels in failing rats in cohort 2 at 4-6 weeks post injection (Figure 2C).
Figure 2.
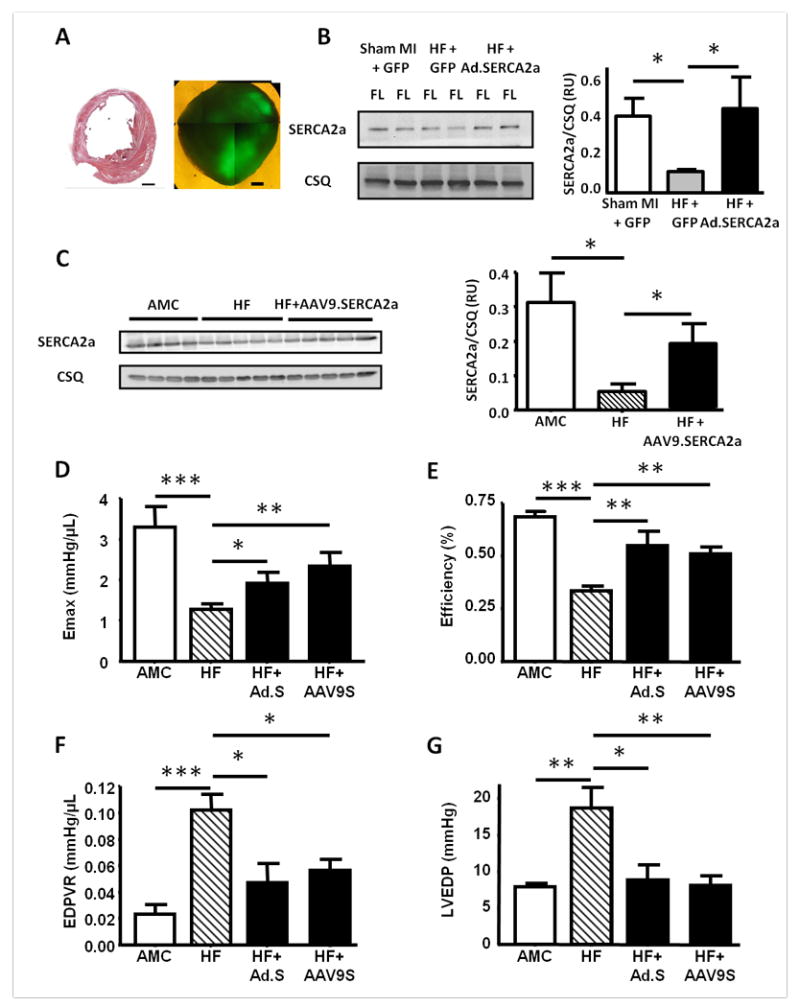
Restoration of SERCA2a protein levels after either Ad.SERCA2a or AAV9.SERCA2a gene transfer, with associated improvements in left ventricular function assessed by pressure-volume (PV) analysis in vivo. A. Post infarction heart failure model and SERCA2a gene transfer: (a) Midventricular 10μm section from a chronically infarcted rat heart after staining with haematoxylin and eosin. Scale bar 2mm. (b) A 2mm thick transverse apical section under fluorescence (488nm) microscopy demonstrating GFP expression in two Ad.SERCA2a.GFP injected regions. Scale bar 1mm. B and C. Western blots of SERCA2a and calsequestrin (CSQ) protein in the fluorescent left ventricular myocardium from Ad.SERCA2a gene injected hearts (B), or myocardium from AAV9.SERCA2a injected HF rats (C). D-G. PV measurement of left ventricular Emax (D), energetic efficiency (E), end diastolic PV relationship (EDPVR) (F) and end diastolic pressure (LVEDP) (G). AMC n=6, HF n=8, HF+Ad.SERCA2a (HF+Ad.S) n=5, HF+AAV9SERCA2a (HF+AAV9.S) n=6. *p<0.05, **p<0.01 and ***p<0.001 vs HF.
In Vivo Left Ventricular Function (cohorts 1 and 2)
Parameters of systolic and diastolic function, and energetic efficiency, as measured by PV analysis, were significantly improved in HF rats after treatment with either Ad.SERCA2a.GFP or AAV9.SERCA2a gene transfer compared with untreated HF controls, confirming previous observations that SERCA2a gene transfer acts as a positive inotrope and lusitrope in the failing heart (Figures 2D-G).2-4
Arrhythmia Studies: Cohort 1
In Vivo Arrhythmias
Spontaneous Ventricular Arrhythmias
This HF model is characterised by a high frequency of spontaneous ventricular arrhythmias (mean 4269 ventricular arrhythmias per heart per day (n=16)), with the majority being isolated ventricular ectopic (VE) beats (mean 4025 VEs per heart per day (94% of total ventricular arrhythmias)) (Figure 3A-B). Heterogeneous Ad.SERCA2a injection significantly reduced both the total number of spontaneous ventricular arrhythmias (Figure 3A) and the number of complex ventricular arrhythmias (Figure 3B), compared to HF+GFP and the HF (PREGENE) phenotype. There was no difference in ventricular arrhythmia incidence in PREGENE HF cohorts between rats receiving Ad.SERCA2a and those receiving control Ad.GFP injections. Daily arrhythmia analysis after gene injection in a subgroup of cases showed a reduction in the arrhythmia burden with the predicted onset of SERCA2a transgene expression, and no evidence for an early proarrhythmic effect upon spontaneous ventricular arrhythmogenesis (Figure S2). Spontaneous malignant ventricular arrhythmias (sustained VT (>30s) and VF (reversible or fatal)) were rare, recorded in 2/16 HF rats (PREGENE), 2/8 HF+GFP rats, 1/8 HF+SERCA2a rats and absent in SHAM MI cases. Analogous to in vitro studies of APD with SERCA2a overexpression,18 heterogeneous SERCA2a transduction significantly shortened the abnormally prolonged QT90 (ms) in this HF model in vivo: SHAM MI + GFP 60±2, HF+GFP 89±2, HF+SERCA 77±2 (p=0.005 vs HF+GFP).
Figure 3.
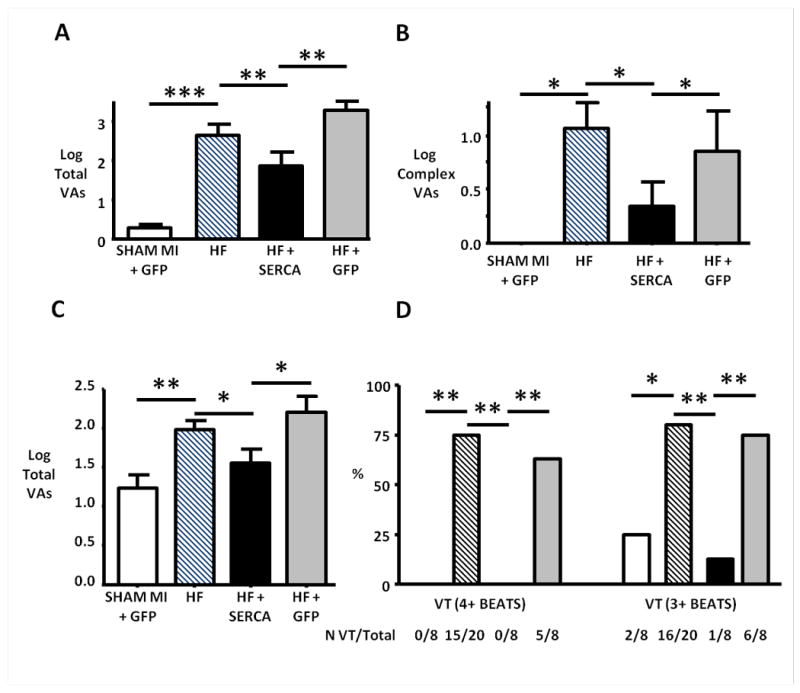
Reduction of spontaneous and catecholamine-provoked ventricular arrhythmias in failing hearts in vivo after SERCA2a gene transfer. A and B. Spontaneous ventricular arrhythmias during a 24 hour study period. HF rats showed significantly increased spontaneous ventricular arrhythmic events compared with non-failing controls (SHAM MI + GFP). SERCA2a gene transfer significantly reduced the total number of spontaneous ventricular arrhythmias (predominantly ventricular ectopy) (A) and complex ventricular arrhythmias (couplets, triplets and ventricular tachycardia) (B) compared to both untreated HF and HF+GFP cases. C and D. Isoproterenol-induced ventricular arrhythmias during the sixty minute recording period post ISO injection. SERCA2a gene transfer reduced the total number of ISO-induced ventricular arrhythmias (C), and the proportion of cases (%) with ISO-induced ventricular tachycardia (VT) defined as either 4+ or 3+ consecutive ventricular cycles (D).
*p<0.05, **p<0.01 and ***p<0.001.
Isoproterenol-Induced Ventricular Arrhythmias
The in vivo ISO injection protocol (0.5mg/kg), which produced matched heart rate responses in the three study groups (Figure S3), induced a high incidence of malignant arrhythmias in the HF model. Fatal ventricular fibrillation was triggered in 4/20 HF cases, with reversible VF observed in a further two HF (PREGENE) cases. ISO-induced VT was present in 11/16 surviving cases. Heterogeneous SERCA2a transfection significantly reduced the total number of ISO-induced ventricular arrhythmias during the sixty minute period post ISO injection (Figure 3C). Interestingly SERCA2a transfection completely abolished ISO-induced VT and VF in the failing hearts, whereas ISO-induced VT was present in 15/20 (75%) of HF (PREGENE) and 5/8 (63%) of HF+GFP animals, but absent in SHAM MI + GFP cases (Figure 3D).
Ex Vivo PES-Induced Ventricular Arrhythmias
SERCA2a transfection of the failing heart had an antiarrhythmic effect, significantly reducing both the incidence of PES-induced VT (37±16% vs 86±14% p=0.04), and shortening the S2 interval (ms) at which VT was triggered (62±3 vs 73±3 p=0.04) compared to HF+GFP cases.
Arrhythmia Studies: Cohort 2
Isolated Cardiomyocyte Studies
Cardiomyocytes isolated from infarcted hearts displayed a typical failing phenotype, with slower Ca2+ transient decay times (Figure 4A), reduced SR Ca2+ content (Figure 4B) and increased spark frequency (Figure 5). As noted above AAV9.SERCA2a gene transfer increased myocardial SERCA2a protein levels in failing rats (Figure 2C), leading to restoration of SR Ca2+ content and transient decay times toward normal levels (Figure 4A-B).
Figure 4.
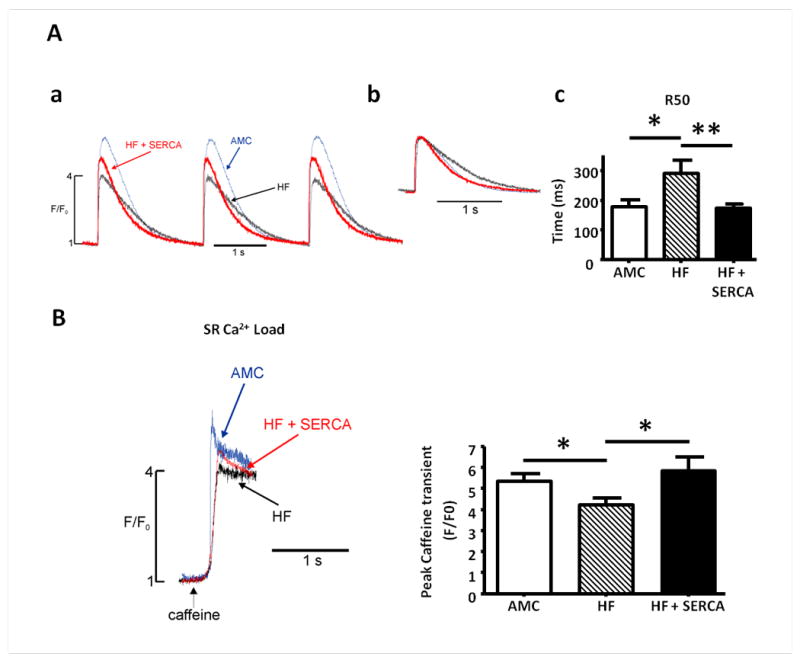
Improvement in cytoplasmic Ca2+ transients, myocyte relaxation kinetics, SR Ca2+ content and basal cytoplasmic Ca2+ levels in failing cardiomyocytes following in vivo AAV9.SERCA2a gene transfer. A. Cytoplasmic Ca2+ transients (AMC (blue), HF (black), HF+SERCA2a (red)) measured from baseline corrected change in emitted fluorescence in fluo4-loaded myocytes stimulated at 0.5Hz (a), with overlain amplitude corrected traces demonstrating difference in Ca2+ transient decay kinetics (b), and quantitative comparison of time (ms) to 50% reduction from peak amplitude (R50) in myocytes paced at 0.5Hz (c). B. SR Ca2+ content in failing myocytes following SERCA2a gene therapy. Representative examples of traces of emitted fluorescence following application of caffeine to determine SR Ca2+ content, and quantitative measurement of SR Ca2+. Number of myocytes (n) studied: AMC n=9, HF n=12, HF+SERCA n=15. *p<0.05 *** p<0.001.
Figure 5.
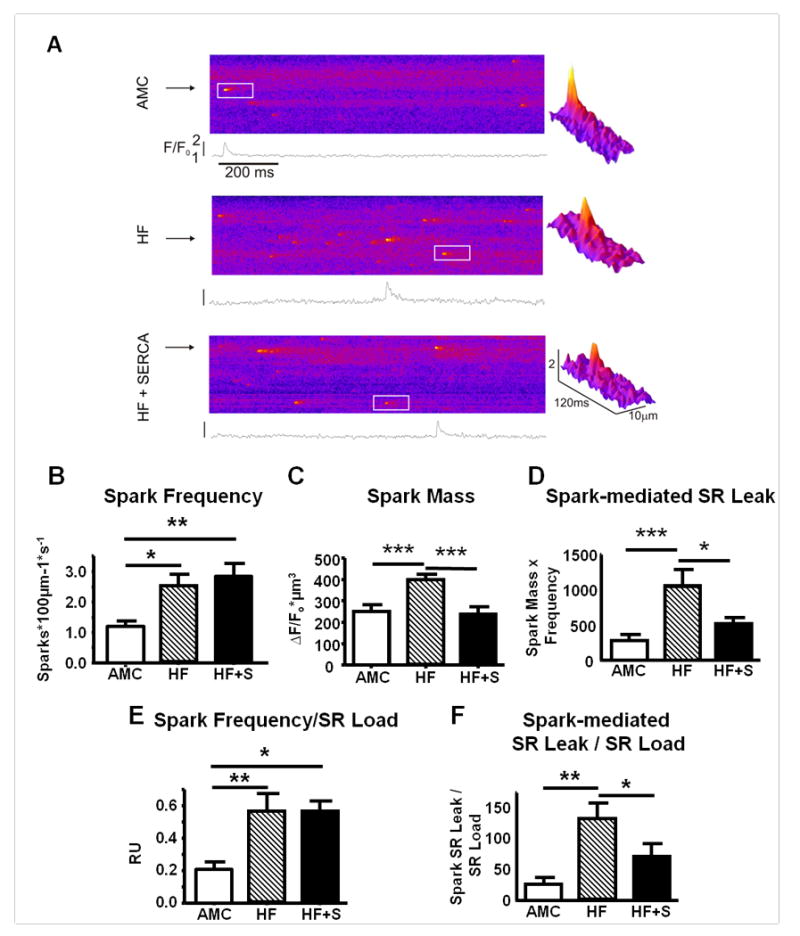
Spontaneous SR Ca2+ release events (sparks) in failing myocytes after SERCA2a gene transfer. A. Representative confocal line scan images demonstrating spontaneous Ca2+ sparks from AMC (upper), HF (middle), and HF+SERCA2a gene transfer (lower) cardiomyocytes. The white inset region is expanded to generate a three dimensional spark representation (shown right), demonstrating the differences in spark morphology after SERCA2a gene transfer. B. Mean spark frequency (number of sparks100μm-1s-1). C. Mean spark mass. D. Spark-mediated SR Ca2+ leak (spark mass × frequency). E-F. Spark frequency (E) and spark-mediated SR Ca2+ leak (F) corrected for matched SR Ca2+ load (mean per heart). Number of sparks/cells/hearts studied: AMC=247/19/7, HF=853/30/11, HF+SERCA=445/10/5.
*p<0.05, **p<0.01, ***p<0.001.
Ca2+ Sparks
Ca2+ spark frequency, and the ratio of Ca2+ sparks:SR load, was higher in the failing myocytes but did not increase further following SERCA2a gene therapy (Figure 5B and E), suggesting that the increased SR Ca2+ content after SERCA2a treatment did not promote additional increases in RyR2 opening frequency in failing cardiomyocytes with a baseline ‘leaky’ SR. Ca2+ spark mass, a parameter reflecting the amount of calcium released with each spark, was significantly increased in failing myocytes, and reduced to normal levels in failing myocytes after SERCA2a gene transfer (Figure 5C). The total spark-mediated SR Ca2+ leak, the product of spark mass and frequency, was also reduced by SERCA2a gene transfer (Figure 5D). Interestingly, after SERCA2a gene therapy the degree of spark-mediated SR Ca2+ leak per unit SR Ca2+ load was significantly reduced (Figure 5F).
Total SR Ca2+ Leak
It has been recently reported that SR Ca2+ leak may also result from spark independent mechanisms.19 To account for this, ‘total’ SR Ca2+ leak was measured using tetracaine perfusion to inhibit RyR2 opening in cardiomyocytes isolated from a separate cohort of study hearts (Figure 6). Tetracaine-dependent SR Ca2+ leak was increased in cardiomyocytes from the HF model, and was lowered to normal levels in myocytes from HF+SERCA2a hearts (Figure 6Bi). The SR Ca2+ content in this group of failing cardiomyocytes was reduced, and elevated back towards normal levels after SERCA2a gene transfer (Figure 6Bii (see reproducible change matched in figure 4B)). Similar to the spark-mediated leak, total SR Ca2+ leak, when corrected for matched SR Ca2+ load, showed that rescue by SERCA2a gene therapy restored a normal SR Ca2+ load/leak relationship (Figure 6Biii). These results confirmed that SR Ca2+ leak threshold is lowered in heart failure (consistent with previously reported studies) and this threshold is raised back towards normal levels after rescue by SERCA2a gene therapy.
Figure 6.
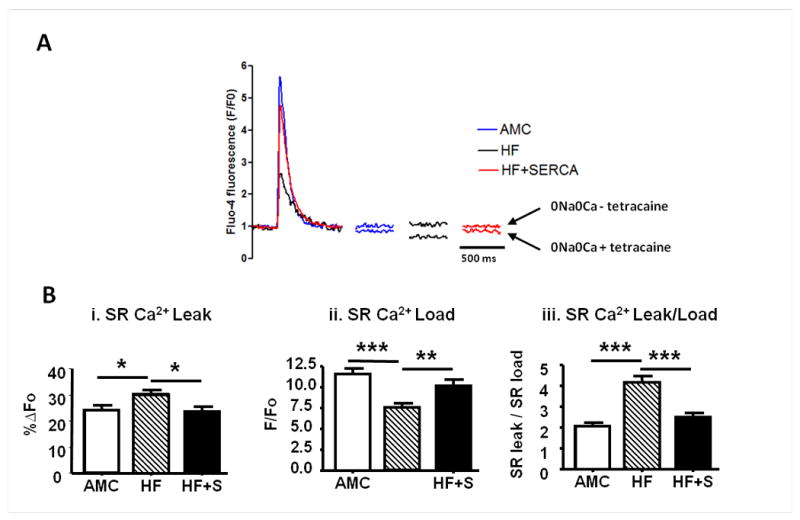
SERCA2a gene transfer reduces SR Ca2+ leak and normalises the SR Load-Leak relationship in failing cardiomyocytes. A-B. Measurement of tetracaine-dependent SR Ca2+ leak (using method from Shannon et al17). A. Representative examples of Ca2+ transients with paired basal resting levels pre (above) and post (below) 1mM tetracaine (both recorded in 0Na+ 0 Ca2+). B(i). SR Ca2+ leak. B(ii). SR Ca2+ load. B(iii). SR Ca2+ leak per unit load (AMC n=16, HF n=24, HF+S n=13). *p<0.05, ** p<0.01, ***p<0.001.
RyR2 Phosphorylation
Potential RyR2-dependent mechanisms for resetting of the SR Ca2+ leak threshold were explored by measurement of RyR2 phosphorylation status. Phosphorylation at serine 2815, the CaMKII site, after correction for total RyR2 levels, was increased in the HF model, and reduced to normal levels after SERCA2a gene therapy (Figure 7). Phosphorylation of PLB at the CaMKII site Thr17 was also increased in failing myocytes. SERCA2a demonstrated a trend to reduced phosphorylation at PLB-Thr17, particularly in the PLB monomer fraction (p=0.06 vs HF). Both RyR and PLB phosphorylation by PKA were increased in the HF model, but unchanged following SERCA2a gene therapy (Figure 7).
Figure 7.
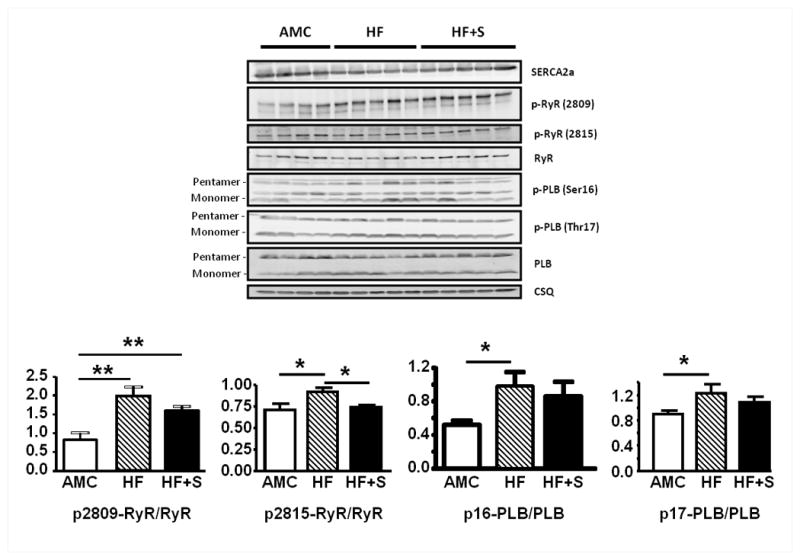
Protein immunoblotting for SERCA2a, RyR, PLB and CSQ from cohort 2, with levels of PKA- and CaMKII-mediated phosphorylation of RyR and PLB, and quantitative comparison after correction for total RyR and PLB respectively (AMC n=4 HF n=5 HF+S n=5). *p<0.05, ** p<0.01.
Triggered Activity in Isolated Cardiomyocytes
Consistent with the observation of reduced spontaneous SR Ca2+ leak, and despite both increased SR content and unaltered spark frequency, cardiomyocytes from HF+SERCA hearts were more stable in vitro, with significantly fewer basal and ISO-induced Ca2+-mediated DACs compared to HF controls (Figure 8).
Figure 8.
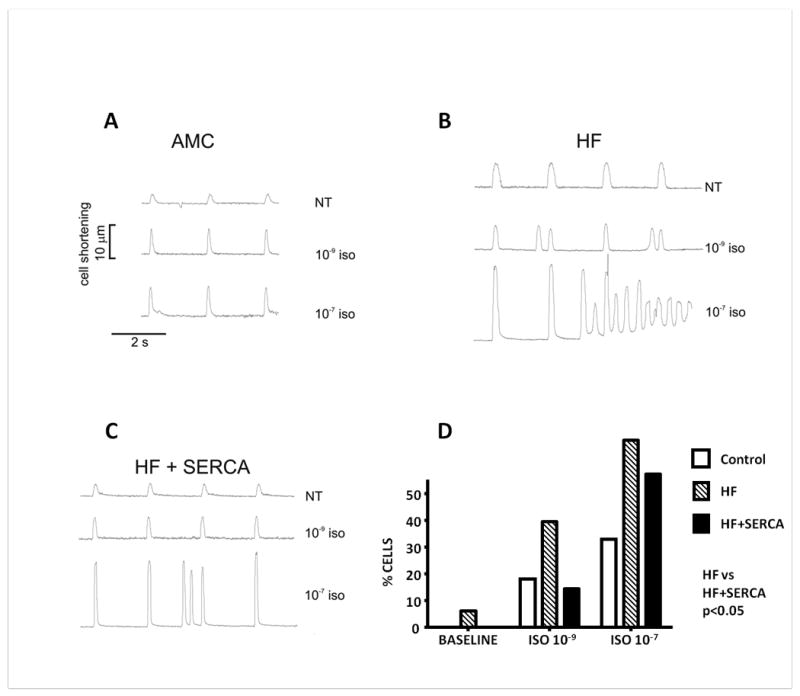
Reduction of catecholamine-induced triggered activity in failing cardiomyocytes after SERCA2a gene transfer. A-C. Representative tracings demonstrating contraction profiles of freshly isolated cells field stimulated at 0.5Hz at baseline perfusion with normal Tyrode's solution (NT), and after perfusion with 1 nmol/L and 100 nmol/L Isoproterenol (ISO). A. Age matched controls. B. Failing cardiomyocytes. C. Failing cardiomyocytes four-six weeks post AAV9.SERCA2a gene transfer. D. Proportion of cardiomyocytes from AMC, HF and HF + AAV9.SERCA2a rat hearts demonstrating Ca2+-mediated delayed aftercontractions at baseline, and after treatment with ISO. Number of myocytes studied: AMC=33, HF=33, HF+SERCA=14. Two way ANOVA p<0.05.
Discussion
A number of pre-clinical studies have shown that restoration of SERCA2a protein levels and activity by gene therapy improves cardiac function in failing ventricular myocardium.1-4 SERCA2a gene therapy represents the first therapeutic strategy specifically targeting abnormal Ca2+ cycling in heart failure to be tested in a clinical trial.5 However concerns regarding the potential for SERCA2a gene transfer to increase ventricular arrhythmia susceptibility via disruption of the SR load-leak equilibrium in favor of increased leak have been raised.6 Failing ventricular cardiomyocytes are characterised by increased spontaneous SR Ca2+ release events (sparks) despite reduced SR Ca2+ stores and, intuitively, reloading a leaky SR by restoring SERCA2a activity could be proarrhythmic, given the mechanistic links between SR Ca2+ leak, triggered activity and ventricular arrhythmogenesis.20 Furthermore, heterogeneous restoration of SERCA2a levels using the current vectors and delivery techniques may increase the dispersion of repolarisation, a biophysical property which supports continuous wavefront propagation through the failing ventricular myocardium.21
This study was designed to investigate the influence of SERCA2a gene transfer upon arrhythmia generation and specifically the potential to influence Ca2+-induced arrhythmias in the failing heart. The main findings of this study were: 1) SERCA2a gene transfer significantly reduced spontaneous and ISO-induced ventricular arrhythmic events in vivo, completely abolishing ISO-induced VT and VF (Figure 3). 2) SERCA2a gene transfer significantly reduced the degree of QT prolongation, the incidence of PES-induced VT and shortened the S2 cycle length at which VT was initiated compared to failing hearts. 3) SERCA2a gene transfer normalised Ca2+ transient decay kinetics and reduced the incidence of ISO-induced Ca2+-mediated delayed aftercontractions in failing cardiomyocytes (Figures 4 and 8). 4) SERCA2a gene therapy did not lower (or raise) spontaneous Ca2+ spark frequency, but significantly reduced spark mass which had been raised in the HF model. Both spark-mediated and total SR Ca2+ leak were reduced after SERCA2a gene therapy, despite the normalised SR Ca2+ load. (Figures 5-6). 5) SERCA2a reduced RyR2 phosphorylation at CaMKII sites to normal levels, suggesting that molecular remodelling underlies resetting of the SR Ca2+ leak threshold (Figure 7).
What underpins this significant antiarrhythmic effect of SERCA2a gene transfer in the failing heart? Several potential direct and indirect mechanisms may contribute, and it is important to note that these are not mutually exclusive.
Reduced Arrhythmogenic SR Ca2+ Leak
1. Increased Ca2+ Buffering by SERCA2a
Results from the isolated cell studies support a direct antiarrhythmic effect upon intracellular Ca2+ homeostasis. SERCA2a gene therapy reduced SR Ca2+ leak compared to failing cardiomyocytes: since the leak is recognised to be proarrhythmic,8, 20 a reduction will be beneficial. Restored SERCA2a levels may directly influence spark morphology and arrhythmic potential by limiting the spatial spread of Ca2+ and preventing local Ca2+ elevation to the threshold levels that initiate and propagate internal Ca2+-induced Ca2+-release, spontaneous Ca2+ waves and delayed afterdepolarisations (DADs). Inhibiting SERCA2a activity pharmacologically has previously been reported to prolong Ca2+ spark decay and increase the space constant of spread for the released Ca2+ ions.22 Reduced DAD frequency in failing cells after SERCA2a gene transfer was evident both spontaneously and particularly in the presence of βAR activation. Conceivably this may reflect the increased Ca2+ sink resulting directly from increased SERCA2a activity, and indirectly via the restored capacity of other cytoplasmic Ca2+ buffers which may be saturated in the failing heart. Modelling of intracellular Ca2+ release and Ca2+ waves in ventricular cardiomyocytes demonstrated the importance of resting cytoplasmic Ca2+ levels and buffering capacity to influence the probability of wave propagation.23 In summary, the buffering effect of restored SERCA2a levels and activity in the failing cardiomyocyte following gene transfer acts to stabilise the cell, reducing the magnitude of the SR Ca2+ leak, and preventing the conversion of the residual Ca2+ leak into arrhythmogenic DADs.
2. Stabilisation of RyR2 Gating
Stabilisation of RyR2 gating also represents an antiarrhythmic mechanism. The SR leak phenotype in this HF model is reminiscent of that observed under other circumstances where RyR2 opening probability is increased, such as after application of FK506 to normal myocytes,24 or in mice overexpressing CaMKII.25 RyR2 mutations leading to increased Ca2+ sensitivity and opening probability also increase spark frequency despite reduced SR Ca2+ load.26 Recently it has been reported that aldosterone, which is increased in the serum and myocardium in chronic heart failure patients, stimulates increased spark frequency, reflecting increased RyR2 opening probability and SR Ca2+ leak.27
Reduced Ca2+ leak following SERCA2a gene transfer to failing cardiomyocytes may reflect stabilised RyR2 gating. Phosphorylation of RyR2 at both PKA and CaMKII sites, both elevated in this HF model, is known to increase RyR2 opening probability.8, 9 Phosphorylation at serine 2815, the putative CaMKII site, was reduced to normal levels in the SERCA2a treated failing hearts (Figure 7). A recent report highlights the importance of the S2815 CaMKII RyR2 phosphorylation site in the generation of arrhythmias in heart failure.28 Transgenic mice in which S2814 (the murine residue analogous to S2815 in the rat) is constitutively activated via mutagenesis (S2814D) developed heart failure, increased SR Ca2+ leak and catecholamine-induced ventricular arrhythmias. In contrast, genetic ablation of this RyR2 phosphorylation site (S2814A) protected against catecholamine-induced arrhythmias in an aortic constriction model.28
Analogous phospholamban phosphorylation is also consistent with reduced functional CaMKII activity in the failing hearts after SERCA2a gene therapy. CaMKII activation in hypertrophy and heart failure may result from either higher basal Ca2+ levels, with increased formation of calmodulin-Ca2+ complexes, or increased levels of oxidative stress via Ca2+-independent activation.29, 30 SERCA2a gene transfer to failing cardiomyocytes lowers basal Ca2+ levels,1 and improves myocardial energetics, a possible surrogate for lower levels of oxidative stress (figure 2E),2 both changes lowering CaMKII activation. RyR2 stabilisation may be further enhanced by lower basal cytoplasmic Ca2+ secondary to enhanced SERCA2a function, as well as better local mitochondrial function resulting in improved local redox status with reduced RyR2 oxidation by reactive oxygen species.31
One proposed mechanism for increased SR Ca2+ leak in failing cells is a lowering of the luminal Ca2+ threshold to trigger spontaneous opening, a process termed store overload-induced Ca2+ release (SOICR).11, 32 When evaluated in terms of spark frequency, no change in spark frequency:SR load ratio after SERCA2a gene transfer was observed (Figure 5E), suggesting the restored SR Ca2+ levels via this intervention do not alter RyR2 channel opening probability. However placing both the reduced spark-mediated and total SR leak in the context of the normalised SR Ca2+ load, it appears likely that RyR2 gating by luminal Ca2+ may also have been altered to a more stable configuration, increasing the threshold for SOICR towards normal levels (Figure 5F and 6Biii).32 This is supported by previous studies demonstrating that as SR leak increased the SR Ca2+ load was reduced.17 Our results suggest that in the chronically failing heart there is a lower SOICR threshold with increased RyR2 phosphorylation and opening, greater leak, and hence lower SR Ca2+ load. These changes are reversed after rescue by SERCA2a gene therapy, with lower levels of RyR2-2815 phosphorylation and reduced SR Ca2+ leak, and simultaneously the capacity to achieve the increased SR Ca2+ load observed in this and previous studies after SERCA2a gene transfer.1
3. Indirect Antiarrhythmic effects from Reverse Remodelling of the Failing Ventricle
Recovery of mechanical left ventricular function is reported in various animal models following SERCA2a gene therapy including this study.2-4 By acting as a ‘molecular left ventricular assist device’ the improved function of SERCA2a-treated hearts may translate to a reduction in arrhythmia generation via a number of potential indirect mechanisms. These include reduced wall tension and the detrimental effects of mechanical-electrical feedback,33 reduction of adverse gene expression profiles,34 improved mitochondrial function and cellular energetics.35, 36 The significant improvement of diastolic function, as well as some measures of systolic function, would argue for a degree of functional recovery. The increased SERCA2a levels in the remote myocardium would be consistent with the hypothesis that reduction of arrhythmias is part of a global improvement, albeit produced by the focal delivery (cohort 1).
Several of lines of evidence exist from this study to suggest it is not purely the indirect benefits of reverse remodelling. Firstly in the model used (chronic post infarction heart failure) the persistent presence of a large infarct (39-44% of the left ventricle (see table S1)) limits potential for full recovery of systolic function in either treated cohort. Secondly we found no evidence of an acute proarrhythmic effect early in the SERCA2a treated hearts when assessing spontaneous arrhythmias during the first six days after Ad.SERCA2a injection (Figure S2). Indeed the in vivo antiarrhythmic effect appeared during the first 48 hours after transgene delivery in the Ad.SERCA2a treated cohort, at a time when significant structural reverse remodelling is unlikely to have occurred.
SERCA2a gene transfer initiates reverse remodelling in an energetically favourable manner (Figure 2E and ref.2), and this may further accelerate the progression from an electrically unstable substrate to one with higher thresholds for arrhythmia inducibility and propagation. Even if an acute proarrhythmic effect were present (and we have shown evidence against this), then conceivably it could become balanced by the benefits of reverse remodelling of the failing myocardium.
It is also plausible that the slower kinetics for restoration of SERCA2a activity using the viral gene therapy approach may allow molecular reverse remodelling of SR-cytoplasmic Ca2+ cycling and SR leak to be generated in a safer manner, compared with an acute activation of SERCA2a activity in the failing heart to immediate high levels using a pharmacological approach. However again we have no evidence to support this concern, and no purely selective SERCA2a activators are currently available to test this hypothesis.
Reduced Susceptibility to Reentry Arrhythmias
Several lines of evidence suggest that repolarisation was normalised and vulnerability to reentry arrhythmias were reduced in the failing rat ventricle after SERCA2a gene transfer, which may contribute to the antiarrhythmic effect. We have previously shown that SERCA2a expression shortens action potential duration in isolated cardiomyocytes.18 In these studies SERCA2a gene transfer reduced the QT interval, which was prolonged in this HF model. Restoring SERCA2a protein levels and activity in the failing heart resulted in reduced susceptibility to PES-induced reentry arrhythmias, and a narrowing of the time window of vulnerability to premature electrical extrastimuli, demonstrated by both shortening of the S2 interval for VT inducibility and reduction of QT prolongation. Improving cardiomyocyte Ca2+ cycling by SERCA2a gene therapy may therefore stabilise electrical heterogeneities in the failing myocardium, an influence also observed after overexpression in the normal heart.37
Failing myocardial substrate is critical for beneficial effects of SERCA2a gene therapy
It is interesting to contrast the findings of this study with those from SERCA2a overexpression studies in normal myocytes, which in some settings have demonstrated the potential for adverse effects with respect to excessive buffering 38 or proarrhythmia.6 The context is critical. In these studies supraphysiological SERCA2a levels were introduced on a background of normal myocyte physiology, and excessive SR-cytoplasmic Ca2+ cycling in normal myocytes has detrimental effects, as observed in other models.39, 40 In contrast, this paper highlights the beneficial effects of rescuing the failing myocyte, where basal SERCA2a levels are low and contribute to both the mechanical dysfunction and arrhythmic substrate. The results of this study highlight that in this context (heart failure), restoring SERCA2a to normal levels via gene transfer stabilizes the SR Ca2+ physiology and has a potent antiarrhythmic effect.
A limitation of this study is that rodent myocardial Ca2+ physiology differs from human, and is more SERCA2a dependent. However the majority of the findings concerning SR Ca2+ load-leak, which gave rise to the potential concerns regarding SERCA2a gene therapy in the ‘leaky’ failing heart addressed in this paper, are derived from studies in rodent hearts or isolated rat cardiomyocytes. The lack of increased leak or proarrhythmic effect in this sensitive model which shows high susceptibility to Ca2+-dependent arrhythmias is encouraging and reassuring, whilst accepting that only results from randomised controlled, double blinded trials will answer the question regarding arrhythmogenesis in the human heart. To date the twelve month follow up of 39 patients with chronic heart failure receiving AAV1.SERCA2a gene therapy has been presented, and no proarrhythmic concerns have been reported (AHA Scientific Sessions 2010).41 This would support our findings that SERCA2a gene therapy can impart the benefits of a positive inotrope and induce reverse remodelling while avoiding a proarrhythmic effect.
Conclusions
In summary, our study demonstrates significant antiarrhythmic effects of SERCA2a gene transfer in the rat post myocardial infarction model of heart failure. Isolated ventricular myocytes from the untreated failing hearts demonstrated increased spontaneous SR Ca2+ leak and ISO-induced triggered activity in vitro, and animals with heart failure had increased spontaneous and catecholamine-induced arrhythmias in vivo. Following SERCA2a gene transfer SR Ca2+ levels were restored, RyR phosphorylation at serine 2815 was normalised, spontaneous Ca2+ sparks were smaller and decayed faster, and total SR leak was reduced, allowing the beneficial positive inotropic and lusitropic effects of SERCA2a gene therapy to occur without an associated proarrhythmic tendency. This suggests that SERCA2a gene therapy represents a novel therapeutic strategy with positive inotropic, lusitropic and antiarrhythmic benefits, as opposed to the proarrhythmic effects observed with other positive inotropes (e.g. βAR agonists and phosphodiesterase inhibitors). These antiarrhythmic effects of SERCA2a gene therapy appear to be the combination of both the indirect benefits of electrical reverse remodelling in the recovering ventricle and direct effect of restored SERCA2a protein levels upon intracellular calcium homeostasis, allowing the positive inotropic benefits to be obtained without a proarrhythmic cost. In contrast to the previous a priori concern, SERCA2a gene transfer does not predispose to arrhythmias and may even represent a novel antiarrhythmic strategy in heart failure.
Supplementary Material
Acknowledgments
Funding Sources: ARL was supported by a UK Medical Research Council Clinical Research Training Fellowship. This work was also in part funded by the Fondation Leducq. The telemetry system was funded by the British Heart Foundation (BHF RG/05/009).
Footnotes
Conflict of Interest Disclosures:RJH is the scientific founder of Celladon Inc which is developing AAV1.SERCA gene therapy for therapeutic purposes.
References
- 1.del Monte F, Harding SE, Schmidt U, Matsui T, Kang ZB, Dec GW, Gwathmey JK, Rosenzweig A, Hajjar RJ. Restoration of contractile function in isolated cardiomyocytes from failing human hearts by gene transfer of SERCA2a. Circulation. 1999;100:2308–11. doi: 10.1161/01.cir.100.23.2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.del Monte F, Williams E, Lebeche D, Schmidt U, Rosenzweig A, Gwathmey JK, Lewandowski ED, Hajjar RJ. Improvement in survival and cardiac metabolism after gene transfer of sarcoplasmic reticulum Ca(2+)-ATPase in a rat model of heart failure. Circulation. 2001;104:1424–9. doi: 10.1161/hc3601.095574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kawase Y, Ly HQ, Prunier F, Lebeche D, Shi Y, Jin H, Hadri L, Yoneyama R, Hoshino K, Takewa Y, Sakata S, Peluso R, Zsebo K, Gwathmey JK, Tardif JC, Tanguay JF, Hajjar RJ. Reversal of Cardiac Dysfunction After Long-Term Expression of SERCA2a by Gene Transfer in a Pre-Clinical Model of Heart Failure. J Am Coll Cardiol. 2008;51:1112–9. doi: 10.1016/j.jacc.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 4.Byrne MJ, Power JM, Preovolos A, Mariani JA, Hajjar RJ, Kaye DM. Recirculating cardiac delivery of AAV2/1SERCA2a improves myocardial function in an experimental model of heart failure in large animals. Gene Ther. 2008;15:1550–7. doi: 10.1038/gt.2008.120. [DOI] [PubMed] [Google Scholar]
- 5.Hajjar RJ, Zsebo K, Deckelbaum L, Thompson C, Rudy J, Yaroshinsky A, Ly H, Kawase Y, Wagner K, Borow K, Jaski B, London B, Greenberg B, Pauly DF, Patten R, Starling R, Mancini D, Jessup M. Design of a Phase 1/2 Trial of Intracoronary Administration of AAV1/SERCA2a in Patients With Heart Failure. J Cardiac Failure. 2008;14:355–67. doi: 10.1016/j.cardfail.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 6.Chen Y, Escoubet B, Prunier F, Amour J, Simonides WS, Vivien B, Lenoir C, Heimburger M, Choqueux C, Gellen B, Riou B, Michel JB, Franz WM, Mercadier JJ. Constitutive cardiac overexpression of sarcoplasmic/endoplasmic reticulum Ca2+-ATPase delays myocardial failure after myocardial infarction in rats at a cost of increased acute arrhythmias. Circulation. 2004;109:1898–903. doi: 10.1161/01.CIR.0000124230.60028.42. [DOI] [PubMed] [Google Scholar]
- 7.Prunier F, Kawase Y, Gianni D, Scapin C, Danik SB, Ellinor PT, Hajjar RJ, del MF. Prevention of ventricular arrhythmias with sarcoplasmic reticulum Ca2+ ATPase pump overexpression in a porcine model of ischemia reperfusion. Circulation. 2008;118:614–24. doi: 10.1161/CIRCULATIONAHA.108.770883. [DOI] [PubMed] [Google Scholar]
- 8.Shannon TR, Pogwizd SM, Bers DM. Elevated sarcoplasmic reticulum Ca2+ leak in intact ventricular myocytes from rabbits in heart failure. Circulation Research. 2003;93:592–4. doi: 10.1161/01.RES.0000093399.11734.B3. [DOI] [PubMed] [Google Scholar]
- 9.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates fkbp12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–76. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 10.Satoh H, Blatter LA, Bers DM. Effects of [Ca2+]i, SR Ca2+ load, and rest on Ca2+ spark frequency in ventricular myocytes. Am J Physiol. 1997;272:H657–H668. doi: 10.1152/ajpheart.1997.272.2.H657. [DOI] [PubMed] [Google Scholar]
- 11.Gyorke I, Gyorke S. Regulation of the cardiac ryanodine receptor channel by luminal Ca2+ involves luminal Ca2+ sensing sites. Biophys J. 1998;75:2801–10. doi: 10.1016/S0006-3495(98)77723-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trafford AW, Diaz ME, Sibbring GC, Eisner DA. Modulation of CICR has no maintained effect on systolic Ca2+: simultaneous measurements of sarcoplasmic reticulum and sarcolemmal Ca2+ fluxes in rat ventricular myocytes. J Physiology. 2000;522Pt 2:259–70. doi: 10.1111/j.1469-7793.2000.t01-2-00259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suckau L, Fechner H, Chemaly E, Krohn S, Hadri L, Kockskamper J, Westermann D, Bisping E, Ly H, Wang X, Kawase Y, Chen J, Liang L, Sipo I, Vetter R, Weger S, Kurreck J, Erdmann V, Tschope C, Pieske B, Lebeche D, Schultheiss HP, Hajjar RJ, Poller WC. Long-Term Cardiac-Targeted RNA Interference for the Treatment of Heart Failure Restores Cardiac Function and Reduces Pathological Hypertrophy. Circulation. 2009;119:1241–52. doi: 10.1161/CIRCULATIONAHA.108.783852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lyon AR, MacLeod KT, Zhang Y, Garcia E, Kanda GK, Lab MJ, Korchev YE, Harding SE, Gorelik J. Loss of T-tubules and other changes to surface topography in ventricular myocytes from failing human and rat heart. Proc Natl Acad Sci U S A. 2009;106:6854–9. doi: 10.1073/pnas.0809777106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng H, Song LS, Shirokova N, Gonzalez A, Lakatta EG, Rios E, Stern MD. Amplitude Distribution of Calcium Sparks in Confocal Images: Theory and Studies with an Automatic Detection Method. Biophy J. 1999;76:606–17. doi: 10.1016/S0006-3495(99)77229-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hilliard FA, Steele DS, Laver D, Yang Z, Le Marchand SJ, Chopra N, Piston DW, Huke S, Knollmann BC. Flecainide inhibits arrhythmogenic Ca2+ waves by open state block of ryanodine receptor Ca2+ release channels and reduction of Ca2+ spark mass. J Mol Cell Cardiol. 2010;48:293–301. doi: 10.1016/j.yjmcc.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shannon TR, Ginsburg KS, Bers DM. Quantitative assessment of the SR Ca2+ leak-load relationship. Circ Res. 2002;91:594–600. doi: 10.1161/01.res.0000036914.12686.28. [DOI] [PubMed] [Google Scholar]
- 18.Terracciano CM, Hajjar RJ, Harding SE. Overexpression of SERCA2a accelerates repolarisation in rabbit ventricular myocytes. Cell Calcium. 2002;31:299–305. doi: 10.1016/s0143-4160(02)00058-1. [DOI] [PubMed] [Google Scholar]
- 19.Zima AV, Bovo E, Bers DM, Blatter LA. Ca(2)+ spark-dependent and -independent sarcoplasmic reticulum Ca(2)+ leak in normal and failing rabbit ventricular myocytes. J Physiol. 2010;588:4743–57. doi: 10.1113/jphysiol.2010.197913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Venetucci LA, Trafford AW, O'Neill SC, Eisner DA. The sarcoplasmic reticulum and arrhythmogenic calcium release. Cardiovascular Research. 2008;77:285–92. doi: 10.1093/cvr/cvm009. [DOI] [PubMed] [Google Scholar]
- 21.Akar FG, Rosenbaum DS. Transmural electrophysiological heterogeneities underlying arrhythmogenesis in heart failure. Circ Res. 2003;93:638–45. doi: 10.1161/01.RES.0000092248.59479.AE. [DOI] [PubMed] [Google Scholar]
- 22.Gomez AM, Cheng H, Lederer WJ, Bers DM. Ca2+ diffusion and sarcoplasmic reticulum transport both contribute to [Ca2+]i decline during Ca2+ sparks in rat ventricular myocytes. J Physiology. 1996;496:575–81. doi: 10.1113/jphysiol.1996.sp021708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Izu LT, Means SA, Shadid JN, Chen-Izu Y, Balke CW. Interplay of Ryanodine Receptor Distribution and Calcium Dynamics. Biophy J. 2006;91:95–112. doi: 10.1529/biophysj.105.077214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gomez AM, Schuster I, Fauconnier J, Prestle J, Hasenfuss G, Richard S. FKBP12.6 overexpression decreases Ca2+ spark amplitude but enhances [Ca2+]i transient in rat cardiac myocytes. Am J Physiol Heart Circ Physiol. 2004;287:H1987–H1993. doi: 10.1152/ajpheart.00409.2004. [DOI] [PubMed] [Google Scholar]
- 25.Maier LS, Zhang T, Chen L, Desantiago J, Brown JH, Bers DM. Transgenic CaMKIIdeltaC overexpression uniquely alters cardiac myocyte Ca2+ handling: reduced SR Ca2+ load and activated SR Ca2+ release. Circ Res. 2003;92:904–11. doi: 10.1161/01.RES.0000069685.20258.F1. [DOI] [PubMed] [Google Scholar]
- 26.Fernandez-Velasco M, Rueda A, Rizzi N, Benitah JP, Colombi B, Napolitano C, Priori SG, Richard S, Gomez AM. Increased Ca2+ sensitivity of the ryanodine receptor mutant RyR2R4496C underlies catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2009;104:201–9. doi: 10.1161/CIRCRESAHA.108.177493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gomez AM, Rueda A, Sainte-Marie Y, Pereira L, Zissimopoulos S, Zhu X, Schaub R, Perrier E, Perrier R, Latouche C, Richard S, Picot MC, Jaisser F, Lai FA, Valdivia HH, Benitah JP. Mineralocorticoid Modulation of Cardiac Ryanodine Receptor Activity Is Associated With Downregulation of FK506-Binding Proteins. Circulation. 2009;119:2179–87. doi: 10.1161/CIRCULATIONAHA.108.805804. [DOI] [PubMed] [Google Scholar]
- 28.van Oort RJ, McCauley MD, Dixit SS, Pereira L, Yang Y, Respress JL, Wang Q, De Almeida AC, Skapura DG, Anderson ME, Bers DM, Wehrens XH. Ryanodine Receptor Phosphorylation by Calcium/Calmodulin-Dependent Protein Kinase II Promotes Life-Threatening Ventricular Arrhythmias in Mice With Heart Failure. Circulation. 2010;122:2669–79. doi: 10.1161/CIRCULATIONAHA.110.982298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang R, Khoo MS, Wu Y, Yang Y, Grueter CE, Ni G, Price EE, Jr, Thiel W, Guatimosim S, Song LS, Madu EC, Shah AN, Vishnivetskaya TA, Atkinson JB, Gurevich VV, Salama G, Lederer WJ, Colbran RJ, Anderson ME. Calmodulin kinase II inhibition protects against structural heart disease. Nat Med. 2005;11:409–17. doi: 10.1038/nm1215. [DOI] [PubMed] [Google Scholar]
- 30.Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, Bartlett RK, Lowe JS, O'Donnell SE, Aykin-Burns N, Zimmerman MC, Zimmerman K, Ham AJ, Weiss RM, Spitz DR, Shea MA, Colbran RJ, Mohler PJ, Anderson ME. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. 2008;133:462–74. doi: 10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Terentyev D, Gyorke I, Belevych AE, Terentyeva R, Sridhar A, Nishijima Y, Carcache de Blanco E, Khanna S, Sen CK, Cardounel AJ, Carnes CA, Gyorke S. Redox Modification of Ryanodine Receptors Contributes to Sarcoplasmic Reticulum Ca2+ Leak in Chronic Heart Failure. Circ Res. 2008;103:1466–72. doi: 10.1161/CIRCRESAHA.108.184457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang D, Wang R, Xiao B, Kong H, Hunt DJ, Choi P, Zhang L, Chen SRW. Enhanced Store Overload-Induced Ca2+ Release and Channel Sensitivity to Luminal Ca2+ Activation Are Common Defects of RyR2 Mutations Linked to Ventricular Tachycardia and Sudden Death. Circ Res. 2005;97:1173–81. doi: 10.1161/01.RES.0000192146.85173.4b. [DOI] [PubMed] [Google Scholar]
- 33.Lab MJ. Mechanosensitive-mediated interaction, integration, and cardiac control. Ann N Y Acad Sci. 2006;1080:282–300. doi: 10.1196/annals.1380.022. [DOI] [PubMed] [Google Scholar]
- 34.Wu X, Zhang T, Bossuyt J, Li X, McKinsey TA, Dedman JR, Olson EN, Chen J, Brown JH, Bers DM. Local InsP3-dependent perinuclear Ca2+ signaling in cardiac myocyte excitation-transcription coupling. Journal of Clinical Investigation. 2006;116:675–82. doi: 10.1172/JCI27374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Szabadkai G, Duchen MR. Mitochondria: the hub of cellular Ca2+ signaling. Physiology. 2008;23:84–94. doi: 10.1152/physiol.00046.2007. [DOI] [PubMed] [Google Scholar]
- 36.Liu T, O'Rourke B. Enhancing Mitochondrial Ca2+ Uptake in Myocytes From Failing Hearts Restores Energy Supply and Demand Matching. Circ Res. 2008;103:279–88. doi: 10.1161/CIRCRESAHA.108.175919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cutler MJ, Wan X, Laurita KR, Hajjar RJ, Rosenbaum DS. Targeted SERCA2a gene expression identifies molecular mechanism and therapeutic target for arrhythmogenic cardiac alternans. Circ Arrhythm Electrophysiol. 2009;2:686–94. doi: 10.1161/CIRCEP.109.863118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Teucher N, Prestle J, Seidler T, Currie S, Elliott EB, Reynolds DF, Schott P, Wagner S, Kogler H, Inesi G, Bers DM, Hasenfuss G, Smith GL. Excessive sarcoplasmic/endoplasmic reticulum Ca2+-ATPase expression causes increased sarcoplasmic reticulum Ca2+ uptake but decreases myocyte shortening. Circulation. 2004;110:3553–9. doi: 10.1161/01.CIR.0000145161.48545.B3. [DOI] [PubMed] [Google Scholar]
- 39.Yuan Q, Fan GC, Dong M, Altschafl B, Diwan A, Ren X, Hahn HH, Zhao W, Waggoner JR, Jones LR, Jones WK, Bers DM, Dorn GW, Wang HS, Valdivia HH, Chu G, Kranias EG. Sarcoplasmic reticulum calcium overloading in junctin deficiency enhances cardiac contractility but increases ventricular automaticity. Circulation. 2007;115:300–9. doi: 10.1161/CIRCULATIONAHA.106.654699. [DOI] [PubMed] [Google Scholar]
- 40.Chopra N, Kannankeril PJ, Yang T, Hlaing T, Holinstat I, Ettensohn K, Pfeifer K, Akin B, Jones LR, Franzini-Armstrong C, Knollmann BC. Modest Reductions of Cardiac Calsequestrin Increase Sarcoplasmic Reticulum Ca2+ Leak Independent of Luminal Ca2+ and Trigger Ventricular Arrhythmias in Mice. Circ Res. 2007;101:617–26. doi: 10.1161/CIRCRESAHA.107.157552. [DOI] [PubMed] [Google Scholar]
- 41.Mancini D, CUPID T, I. Abstract 19470: Sustained Improvement in Clinical Outcomes with MYDICAR(R) Compared to Optimal Therapy 1-year after Initial Gene Therapy in Patients with Advanced Heart Failure-The CUPID Trial. Circulation. 2010;122:A19470. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.