

Abstract
Many endocrine disrupting chemicals (EDCs) adversely impact estrogen signaling by interacting with two estrogen receptors (ERs): ERα and ERβ. Though the receptors have similar ligand binding and DNA binding domains, ERα and ERβ have some unique properties in terms of ligand selectivity and target gene regulation. EDCs that target ER signaling can modify genomic and non-genomic ER activity through direct interactions with ERs, indirectly through transcription factors like the aryl hydrocarbon receptor (AhR), or through modulation of metabolic enzymes that are critical for normal estrogen synthesis and metabolism. Many EDCs act through multiple mechanisms as exemplified by chemicals that bind both AhR and ER, such as 3-methylcholanthrene. Other EDCs that target ER signaling include phytoestrogens, bisphenolics, and organochlorine pesticides and many alter normal ER signaling through multiple mechanisms. EDCs can also display tissue-selective ER agonist and antagonist activities similar to selective estrogen receptor modulators (SERMs) designed for pharmaceutical use. Thus, biological effects of EDCs need to be carefully interpreted because EDCs can act through complex tissue-selective modulation of ERs and other signaling pathways in vivo. Current requirements by the U.S. Environmental Protection Agency require some in vitro and cell-based assays to identify EDCs that target ER signaling through direct and metabolic mechanisms. Additional assays may be useful screens for identifying EDCs that act through alternative mechanisms prior to further in vivo study.
Keywords: Endocrine disrupting chemicals, estrogen receptor alpha, estrogen receptor beta, aryl hydrocarbon receptor, estrogen synthesis, high-throughput screening
1. Introduction
Endocrine disrupting chemicals (EDCs1) are compounds in the environment or diet that interfere with normal hormone biosynthesis, signaling, or metabolism (1). Many EDCs display estrogenic activity and interfere with normal estrogen signaling which is mediated by two estrogen receptors (ERs): ERα and ERβ. ERα and ERβ have both unique and overlapping physiological roles in mediating estrogen signaling which are dependent on the cellular context, the availability of cofactors, and the ligand. EDCs that target ER signaling can modify genomic and non-genomic ER activity through direct interactions with ERs, indirectly through transcription factors like the aryl hydrocarbon receptor (AhR), or through modulation of metabolic enzymes that are critical for normal estrogen synthesis and metabolism. Though the receptors bind endogenous estrogen, 17β-estradiol (E2), with similar affinities, many exogenous ligands have been shown to display selectivity for ERα or ERβ (2). ERs have relatively large ligand binding pockets and are relatively promiscuous nuclear receptors in terms of binding exogenous chemicals. Despite broad specificity for ligands, both ER subtypes have ligand binding pockets that determine common structural features of estrogenic ligands. Though EDCs function through multiple mechanisms, many display impacts on ER signaling by directly binding with the ER ligand binding pocket. Such direct acting EDCs include pharmaceutical chemicals, bisphenolics, phytoestrogens, and organochlorine pesticides. Often, EDCs act through multiple mechanisms, including indirect action through activation of other transcription factors, most notably the aryl hydrocarbon receptor, or through modification of estrogen metabolism.
In vitro and cell-based assays for detecting estrogenic EDCs are valuable because they are targeted tests that can often be performed in a high-throughput manner with lower costs than in vivo animal studies. Current guidelines for detecting direct acting estrogenic EDCs in vitro are aimed at identifying chemicals that bind ER and initiate transcription of ER targets. Several additional high-throughput strategies are available to identify EDCs that target ERs but most are limited to the identification of direct acting EDCs. Transcriptional reporter assays, bioluminescence or fluorescence resonance energy transfer (BRET or FRET), and fluorescence polarization assays can be used to successfully identify chemicals that directly interact with ERs. It is much more difficult to identify EDCs that interfere with normal ER signaling indirectly, but in vitro and cell-based assays aimed at identifying chemicals that interfere with estrogen metabolism are currently available. As our understanding of ER signaling expands, the development of targeted in vitro assays will aid in the identification of EDCs prior to further in vivo study.
2. Estrogen receptor signaling
Two estrogen receptors, ERα and ERβ, regulate gene expression in response to estrogen exposure. ER signaling may occur in a ligand dependent or ligand independent manner. Estrogen target genes are regulated through genomic pathways, either by direct ER-DNA interactions or through a tethering mechanism in which ERs tether to DNA through other transcription factors, and non-genomic pathways in which estrogen exposure leads to rapid activation of kinase signaling cascades. The multiple signaling pathways mediated by the two ER subtypes complicate the effects of EDCs on estrogen signaling and each subtype may mediate unique responses to ligands. Importantly, the tissue specific effects of ER signaling are complex and EDCs can display selective modulation of ER signaling.
2.1 ERα and ERβ subtypes
For decades, ERα was thought to be the only ER regulating estrogen signaling and the vast majority of studies regarding the effects of EDCs have been conducted with a focus on ERα. In 1996, a second ER, ERβ, was cloned from rat prostate and ovary thereby shifting our understanding of ER signaling (3). ERα and ERβ are encoded by distinct genes on separate chromosomes (4) and display unique and overlapping physiological roles that are highly dependent on the tissue and cell type. ERs mediate estrogen signaling in reproductive tissues and non-reproductive tissues including the brain, lungs, colon, prostate, and cardiovascular system. Both receptors are expressed in the brain, lung, uterus, breast, heart, and intestine. ERα shows unique expression in hepatocytes and the hippocampus while ERβ is the prominent subtype expressed in the prostate, vagina, and cerebellum (5). Based on observations of ERα and ERβ knockout mice, ERα is the dominant receptor regulating normal mammary development while ERβ is less critical for normal mammary and reproductive development (6, 7). ERβ is thought to oppose the proliferative action of ERα in mammary cells (8–11), but the physiological role of ERβ in many tissues is still under debate.
The structural features of ERα and ERβ have been reviewed extensively (12, 13) so the discussion of ERα and ERβ is limited to key differences within the activation function (AF) regions and ligand binding and DNA binding domains of the receptors. Two activation functions, AF-1 and AF-2, interact with cofactors to down- or up-regulate transcription of target genes. The N terminal AF-1 domain can regulate transcription independent of ligand while AF-2, found in the ligand binding domain (LBD), regulates transcription in a ligand dependent manner. ERα and ERβ have strikingly different AF-1 domains; they share only 17% similarity and the AF-1 domain of ERα enhances estrogen-induced expression of reporter genes to a greater extent than that of ERβ (14, 15). The AF-2 domain consists of four alpha helices (H3, H4, H5, and H12) that form a hydrophobic groove to which cofactors with an LXXLL motif, known as a nuclear receptor (NR) box, can bind. The LBDs of ERα and ERβ share 59% similarity but the receptors only differ in two amino acids in the ligand binding pocket: Leu 384 of ERα corresponds to Met 336 of ERβ and Met421 of ERα corresponds to Ile373 of ERβ. Such slight differences within the ligand binding pockets contribute subtype selectivity to some chemicals though the receptors bind E2 with similar affinities (2). The DNA binding domains (DBDs) of ERα and ERβ share 97% similarity and the receptors bind DNA at estrogen response elements (EREs), 13 base pair inverted repeats (GGTCAnnnTGACC) to regulate target gene expression. Many ER target genes do not contain perfect EREs in their promoters and chromatin immunoprecipitation coupled with gene expression microarrays has been used to map ERα and ERβ genomic binding sites. In MCF7 breast cancer cells, 71% of ERα binding sites contained full EREs with at most 2 base pair deviations from the consensus ERE sequence; 25% of binding sites contained half ERE sequences (16). The majority of binding sites occured in distal regions or intragenic regions and only 5% of binding sites fall within the proximal promoter regions of target genes (16, 17). Genomic mapping of ERβ binding sites in MCF7 cells in which ERβ was inducibly overexpressed revealed that ERβ binding regions mapped more closely to transcription start sites compared to those of ERα (18). Of all ERβ binding sites, only 5% contained an isolated ERE or ERE-like site and approximately 60% of binding sites contained ERE-like sequences and activator protein 1 (AP-1) binding sequences. In a comparison of the ERα and ERβ binding sites observed in MCF7 cells, over 75% of ERβ binding sites were also identified as ERα binding sites (19). Since the DNA binding domains of ERα and ERβ display high homology and there is significant overlap in their genomic binding sites, it is not surprising that these receptors can regulate common targets; however, unique gene regulation has been observed in U2OS osteosarcoma cells and Hs578T breast cancer cells engineered to express ERα or ERβ (20, 21). It should be noted that ERα and ERβ form both homodimers and heterodimers in response to ligand binding and, like ER homodimers, heterodimers are capable of binding EREs (22, 23). The biological role of ER heterodimers is still unclear and thus the impacts of EDCs on heterodimer activity are largely unknown.
2.2 ER signaling pathways
Estrogen signaling occurs through multiple pathways in which ERs regulate transcription of target genes directly or indirectly. Ultimately, the impacts of EDCs on ER signaling are best understood in a ligand dependent context. Inappropriate ER signaling can lead to increased risk of hormone-dependent cancer, impaired fertility, abnormal fetal growth and development, and altered metabolism in white adipose tissue (1). The effects of EDCs are not limited to the ligand dependent activity of ERs but it is the best studied aspect of ER-targeted endocrine disruption. The mechanisms of ligand independent ER signaling are complex and involve activation of multiple signaling pathways (13). The discussion of ER signaling mechanisms herein is limited to the ligand dependent activation pathways.
ERs signal through both genomic (nuclear) and non-genomic (extra-nuclear) pathways in response to ligand (Scheme 1). The mechanisms of ER signaling have been discussed extensively in the literature (12, 13) and genomic and non-genomic ER signaling mechanisms are presented here in brief. In the genomic pathway, ERs mediate target gene regulation through binding directly to EREs or tethering to EREs through transcription factors like specificity protein 1 (Sp1) or AP-1 at their cognate response elements (24, 25). In classical genomic signaling, ligands bind the receptor in the LBD which induces conformational changes allowing dimerization and DNA binding. Crystal structures of the LBD of ERα and ERβ bound to agonists, compounds that lead to transcriptional activation, or antagonists, compounds that inhibit transcriptional activation, reveal similar structural features of ERα and ERβ that contribute to cofactor recruitment and transcriptional output (26–28). When bound to agonists like the endogenous estrogen E2, conformational changes in the receptors reveal a cofactor binding site. Ligands may induce conformations that preferentially recruit specific cofactors thereby inducing differential responses. The best described nuclear receptor cofactors are the p160 family of coactivators, namely SRC-1, SRC-2, and SRC-3 but the cofactor complexes that mediate the ultimate outcome of ER signaling are complicated; more that 300 cofactors have been described in the literature (29).
Scheme 1.

Ligand dependent ER signaling pathways. Genomic signaling occurs when ligands enter the cell and bind ER to induce dimerization. ER dimers bind DNA directly at ERE sequences or indirectly by tethering to DNA through other transcriptions factors like Sp1 or AP-1. Non-genomic signaling occurs when ligands bind membrane bound receptors, which leads to activation of kinase signaling cascades.
The non-genomic pathway involves activation of other signal transduction pathways that leads to rapid responses, within minutes, to estrogen exposure. The mechanism of non-genomic ER signaling is not clear and is thought to be mediated by membrane-associated full length ERs or a receptor distinct from ERα and ERβ (30). A G-protein coupled receptor known as GPR30 mediates rapid estrogen signaling independent of ERs (31) which can lead to activation of the MAPK or PI3 kinase signaling cascades (32, 33), fluctuations in intracellular calcium (34), or stimulation of cAMP production (35). Known EDCs like bisphenol A (BPA) and diethylstilbestrol (DES) also induce rapid estrogen signaling (36), indicating that EDCs can exert effects on non-genomic ER signaling.
As mentioned previously, ER signaling can occur non-classically through AP-1 and Sp1 transcription factors in response to estrogenic ligands including known EDCs. The mechanisms of ER/AP-1 and ER/Sp1 mediated transactivation of target genes are complex and depend on ERα or ERβ subtype, cell context, and the type of ligand (37). Transient transfections with a reporter construct containing a GC-rich Sp1 binding site in the promoter demonstrated that ERα can mediate estrogen responsive reporter expression in breast and prostate cancer cells but not in HeLa cervical cancer cells (25). Though ICI 182,780 and tamoxifen are considered anti-estrogenic ligands, treatments with either compound led to transcriptional activation of the Sp1 reporter similar to E2, demonstrating the importance of promoter context in the expression of target genes in response to ligand binding. Transcription of the Sp1 reporter construct in response to E2 treatment did not occur in cells transfected with ERβ indicating ER subtypes display unique actions through classical Sp1 binding sites. EDCs can also induce ERα/Sp1 transactivation. In MCF7 cells transfected with wild-type ERα and a Sp1 reporter construct, known EDCs including octylphenol, nonylphenol, bisphenol A, and other xenoestrogens activated expression of the Sp1 reporter similar to E2 (38).
Some EDCs could potentially display pathway selective actions. For example, WAY-169916 is a non-steroidal selective inhibitor of NFκB activity that works in an ER-dependent manner (39). ERα and ERβ activation can antagonize the functional activity of NFκB in response to estrogen treatment in a wide variety of cell lines (reviewed in (40)). WAY-169916 is unique in that the compound inhibits NFκB action only in the presence of ERα or ERβ and is devoid of classical estrogenic activity as it does not induce expression of ERE reporter constructs in the presence of ERα or ERβ. The pathway selective inhibitory effects of WAY-169916 are maintained in vivo as measured by a reduction in inflammatory gene expression in the absence of classical estrogenic activity including uterotrophic side-effects (39). The pathway selective action of WAY-169916 demonstrates the potential for EDCs to elicit very specific effects mediated by ERs in the absence of classical ER signaling.
2.3 Selective estrogen receptor modulators (SERMs) and EDCs
For decades, the pharmaceutical industry has worked to develop SERMs that act as agonists and antagonists in a tissue selective manner. In the context of EDCs, SERMs provide a useful framework for understanding specific signaling elicited by ER ligands. The SERM tamoxifen has been used as an effective therapy for treating ERα positive breast cancers for the past 30 years and has contributed to a decline in breast cancer mortality rates (41). Tamoxifen antagonizes ERα in mammary cells, thereby inhibiting ERα-mediated breast cancer progression and is an ERα agonist in the endometrium which contributes to increased risk for endometrial cancers after extended exposure (41). The tissue specific effects of tamoxifen are due in part to coactivator availability and cell context. In Ishikawa endometrial cells, tamoxifen induces cell cycle progression and the proliferative effect of tamoxifen is lost when SRC-1 expression is knocked down (42). SRC-1 expression is higher in Ishikawa cells compared to MCF7 breast cancer cells, a trend observed across a number of endometrial and breast cancer cell lines, suggesting SRC-1 may mediate some of the tissue specific effects of tamoxifen.
In vitro assays for investigating SERMs have been extensively applied to EDCs and the results suggest that many EDCs are SERMs. For example, experiments with ER-negative MDA-MB-231 breast cancer cells transfected with wild-type or a truncated ERα variant that cannot recruit cofactors through AF-2 demonstrate the SERM-like properties of some EDCs (43). In cells transfected with wild-type ERα, treatment with E2, tamoxifen, and xenoestrogens including BPA, HPTE, and nonylphenol induce expression of an ERE-luciferase reporter demonstrating the estrogenic activity of all the ligands. In cells transfected with truncated ERα, E2 and the alkylphenols could not activate expression of the reporter while tamoxifen, BPA, and HPTE were able to activate ER-mediated luciferase expression indicating BPA and HPTE have SERM-like properties similar to tamoxifen. Pharmaceutical SERMs demonstrate estrogenic ligands can modulate ER signaling in a tissue-selective manner and EDCs can have similar selective properties.
3. Direct action of EDCs on ER signaling
Many EDCs directly impact ER signaling by competing with endogenous estrogen for ER binding sites. EDCs that show estrogenic activity through ER binding may have structural features shared with E2 that are determined by the ligand binding pockets of ERα and ERβ. Though ERα and ERβ have very similar ligand binding pockets, subtle differences in the amino acids that line the pocket contribute to ligand selectivity between the subtypes (44). EDCs that display direct impacts on ER signaling through interaction with ER LBDs include pharmaceutical chemicals, industrial bisphenolics, organochlorine pesticides, and phytoestrogens.
3.1 Structural features of estrogenic chemicals
Crystal structures of the LBDs of ERα and ERβ reveal features of the ligand binding pockets that are critical for understanding which compounds may display estrogenic activity through direct interaction with ERα or ERβ. ERs are fairly promiscuous nuclear receptors in terms of ligand selectivity; the ligand binding pockets of ERα and ERβ are significantly larger, 450 and 390 Å3 respectively, than E2, just 245 Å3, allowing a diverse set of small molecules access to the LBD (26, 28). Both receptors display similarly high affinities for E2 due to hydrophobic interactions and a network of hydrogen bonds between the hydroxyl groups on E2, a water molecule, and amino acids that line the ligand binding pocket. Glu353 and Arg394 of ERα and Glu305 and Arg346 of ERβ share hydrogen bonds with a water molecule and the hydroxyl in the A ring of E2. On the other side of the E2 molecule, the hydroxyl of the D ring shares a hydrogen bond with His524 of ERα, corresponding to His475 in ERβ (Figure 1A). Ligands that bind ERs directly in the LBD share structural similarities with E2 in that they typically have hydroxyls that undergo hydrogen bonding with the Glu, Arg, and His residues in the ligand binding pocket. Though ERα and ERβ have similar affinities for E2, there are many ligands that display selectivity for ERα or ERβ, including known EDCs. Differences in two amino acids within the ligand binding pocket are the major determinants of the subtype selectivity of some ligands. In helix 5, Leu384 of ERα corresponds to Met336 of ERβ and in loop 6–7 Met421 of ERα corresponds to Ile373 of ERβ (Figure 1B). In light of the potential anti-proliferative role of ERβ, significant efforts have focused on developing ERβ selective ligands. Based on structural similarities of known ERβ selective compounds, structural determinants of ERβ selectivity and binding affinity can be useful for predicting chemicals that will directly bind ERs and potentially display subtype selectivity (45). Binding affinities for the receptors are often expressed relative to E2 and the relative binding affinities (RBAs) of many estrogenic ligands have been characterized (2, 44).
Figure 1.
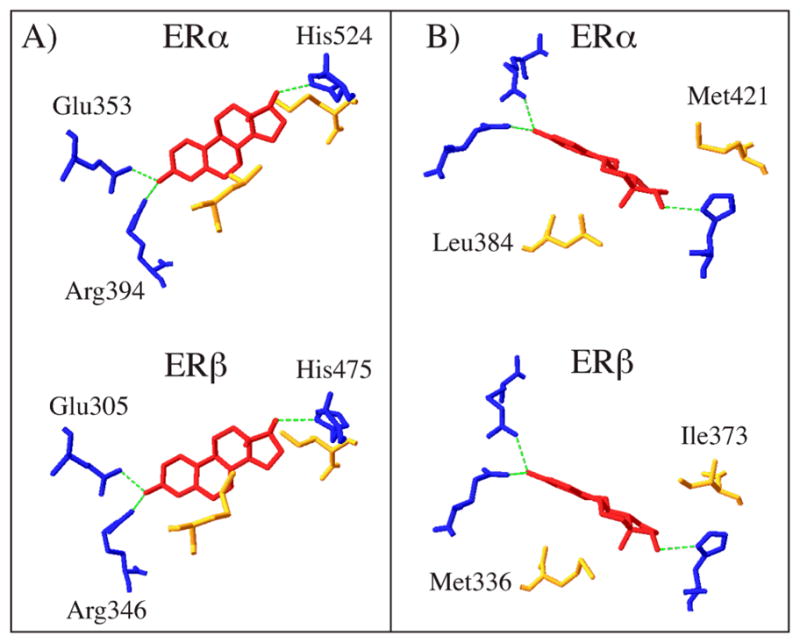
Crystal structures of ERα and ERβ LBD bound to E2 (shown in red) modified from PDB 1ERE and PDB 2J7X. A) Comparison of ERα and ERβ hydrogen bond network with E2 (H bonds shown in green); B) ERα and ERβ differ in only two amino acids in the ligand binding pocket (shown in yellow).
3.2 Known estrogenic EDCs
EDCs can impact estrogen signaling through interaction with the LBDs of ERα and ERβ, thereby activating the receptors inappropriately. Examples of such direct acting EDCs include pharmaceuticals, phytoestrogens, pesticides, and industrial chemicals. Here, we highlight known EDCs that impact ER signaling in part through interaction with the ligand binding pockets (Figure 2). It must be noted that EDCs often act through multiple mechanisms and can display tissue specific effects. Phytoestrogens can bind ERs with relatively low affinities, but display ERβ selectivity suggesting EDCs may have impacts on ER subtype specific signaling pathways. Other estrogenic EDCs, such as the pharmaceutical DES, can interact with ligand binding pockets of ERs with very high affinity and impact both genomic and non-genomic ER signaling. The complex responses to EDCs like DES highlight the complexity of predicting the effects of EDCs on the multiple signaling pathways mediated by ERs. This review will provide examples of known EDCs and additional recent reviews have more comprehensive information regarding EDCs (1, 46–48).
Figure 2.

Chemical structures of estrogenic ligands and EDCs that target ER signaling.
3.2.1 High affinity pharmaceuticals: DES
One of the first examples of endocrine disruption occurred when women were exposed to DES (Figure 2, 2) as a therapy for preventing miscarriages during pregnancy; prenatal exposure to DES was later linked to vaginal cancer in daughters of mothers taking DES (49) and structural, functional, and cellular abnormalities in the reproductive system of males exposed to DES in utero (50). DES has been a model for EDC action and has been extensively studied because of its significant adverse impacts on humans in utero (51). DES appears to act through both genomic and non-genomic ER signaling to induce adverse ER signaling. It is structurally quite similar to E2 and crystal structures of ER LBD bound to DES show the hydroxyl groups of DES are similarly positioned as those of E2. DES displays even higher affinity for ERs due to additional hydrophobic interactions that further stabilize the ligand (2, 27). Because it has high affinity for the receptors, it is a potent transcriptional activator through genomic signaling. High incidence of uterine tumors occurs in mice after neonatal treatment with DES, and similar effects are observed for other estrogenic ligands including E2 and genistein (51). More recent evidence suggests DES can impact non-genomic estrogen signaling as well (36, 52). DES treatment of MCF7 breast cancer cells leads to rapid activation of PI3 kinase signaling and phosphorylation of AKT. Further, activation of the signaling cascade leads to phosphorylation of EZH2, a histone methyl transferase, and modification of the chromatin structure which may contribute to the epigenetic effects of DES (52).
3.2.2 Organochlorine pesticides: Methoxychlor and DDT
Methoxychlor and 1,1,1-trichloro-2,2-bis(p-chlorophenyl)ethane (DDT) are organochlorine pesticides that can exhibit estrogenic activity through interaction with ERα and ERβ LBDs (44). DDT and methoxychlor adversely affect the female reproductive tract by stimulating uterine proliferation and impairing normal follicle development (reviewed in (53)). Though DDT was banned in the 1970’s, it remains an EDC of interest due to its persistence in the environment and its accumulation in adipose tissue. DDT and the dechlorination metabolite dichlorodiphenyldichloroethylene (DDE) have been detected in the adipose tissue of humans throughout the world (54). DDT occurs as a mixture of three isomers: p,p′-DDT, o,p′-DDT, and o,o′-DDT. The estrogenicity of DDT arises primarily from o,p′-DDT (Figure 2, 3) which can bind ERα and ERβ with RBAs of 0.01 and 0.02, respectively; p,p′-DDT (Figure 2, 4) has RBAs <0.01 for both receptors (44).
Methoxychlor (Figure 2, 5) was developed as an alternative to DDT but its use was also banned by the United States Environmental Protection Agency in 2003 due to its endocrine disrupting properties (55). Methoxychlor induces a variety of adverse physiological effects (reviewed in (56)) but most notably it stimulates uterotrophic activity and impairs overall fertility in rat models. Though methoxychlor has relatively low binding affinities for ERα and ERβ (RBAs of <0.01 for ERα and ERβ (44)), the major metabolite of methoxychlor, 2,2-bis(p-hydroxyphenyl)-1,1,1-trichloroethane (HPTE) (Figure 2, 6), exhibits unique estrogenic activity which likely mediates the endocrine disrupting properties of methoxychlor. HPTE can act as an agonist for ERα and an antagonist for ERβ (57). In HepG2 liver carcinoma cells transfected with ERα or ERβ and a luciferase reporter linked to an estrogen responsive complement 3 promoter, HPTE induced maximal reporter expression relative to E2 only in cells expressing ERα. In cells transfected with ERβ, maximal HPTE-induced luciferase expression only reached 13% that of the maximal E2 response indicating its agonistic properties are ERα selective. HPTE selectively antagonizes E2-mediated ERβ activation. HPTE co-treatment antagonized E2 induction of luciferase in HepG2 cells transfected with ERβ but had no effect on E2 induced luciferase expression in cells transfected with ERα (57). ERα selective agonistic properties were also observed for similar chemicals with bishydroxyphenyl core structures indicating many structurally related compounds may impact ER signaling primarily through ERα (58). HPTE may also mediate effects through non-classical ER signaling mechanisms similar to E2. HPTE and E2 treatment led to similar gene expression profiles in uteri from mice expressing an ERα mutant deficient in DNA binding which limits ERα mediated gene regulation to the pathway in which ERα tethers to DNA through transcription factors like AP-1 and Sp1 (59). Additionally, ERα knockout mice did not respond to HPTE treatment indicating the effects of HPTE on gene regulation in the mouse uterus are dependent on ERα.
3.2.3 Industrial phenolics: BPA and alkylphenols
Chemicals used for industrial purposes are often used in large quantities and exposure risks are high due to the large volume and application of such chemicals. BPA (Figure 2, 7) is a monomer of polycarbonate plastics that is one of the highest volume chemicals used in industry. Exposure to BPA is thought to occur primarily through ingestion since polycarbonate plastics are used in food and water containers, though it is also present in medical tubing and epoxy resin. A recent study by the Center for Disease Control estimates over 90% of the United States population have significant levels of BPA in the urine (60). The estrogenic effects of BPA have been demonstrated with in vitro and in vivo experiments but there is significant controversy surrounding the potential for BPA to significantly impact normal endocrine function (reviewed in (61)). BPA has two phenolic rings, similar to DES, but displays much lower binding affinities and transcriptional potencies for ERα and ERβ compared to DES or E2 (RBA of 0.01 for ERα and ERβ) (44, 62). BPA can induce a uterotrophic response in immature CD-1 mice after 3 days of exposure but only at high doses (100 mg/kg) (63), though human daily intake is estimated to be around 1 μg/kg (61). BPA can also elicit signaling through non-classic ER signaling pathways at high concentrations. In mice expressing a mutant form of ERα that is deficient in DNA binding which limits ER mediated responses to pathways in which ERα is tethered to DNA through other transcription factors, relatively high doses of BPA elicit similar uterine gene expression profiles as E2 (59). Despite the evidence that BPA elicits estrogenic responses in many experimental systems, there is uncertainty as to whether such evidence can be extrapolated to human exposure levels considering the relatively high doses of BPA required to elicit such responses.
Given the low affinity of BPA for ERs, it is likely that the estrogenic effects of BPA are due to non-genomic ER signaling. BPA can bind membrane-associated ERα or GPR30 and initiate rapid signaling (64, 65). BPA has higher affinity for GPR30 compared to ERα and ERβ (RBA for GPR30 of 2.83) (65). Nanomolar doses of BPA stimulated a calcium flux in GH3/B6 pituitary cells in which ERα is membrane-associated indicating rapid signaling can be triggered at environmentally relevant concentrations of BPA (64). At higher concentrations of BPA, in the micromolar range, non-genomic signaling is activated in MCF7 cells as indicated by detection of phosphorylated AKT and phosphorylated ERK proteins and expression of transiently transfected reporter constructs specifically responsive to MAPK and PI3K activation (66). The ability of BPA to induce genomic ER mediated transcription is dependent on the availability of cofactors. In HeLa cells transfected with ERα or ERβ and the coactivators TIF2 or SRC-1, BPA had greater effects on gene expression in cells expressing ERβ when TIF-2 was the available cofactor but similar effects in cells expressing ERα or ERβ when SRC-1 was present (67). The complex mechanisms through which BPA can initiate signaling contribute to the controversy surrounding BPA and its role as an EDC.
Other industrial chemicals that can directly interact with ER LBDs and elicit estrogenic responses include alkyl phenols such as 4-t-octylphenol (Figure 2, 8) and nonylphenol (Figure 2, 9) (44, 68). Nonylphenol has a relatively weak affinity for ERs (RBAs for ERα and ERβ of 0.05 and 0.09, respectively) as does 4-tert-octylphenol (RBAs for ERα and ERβ of 0.02 and 0.07, respectively) (44), but alkylphenols still induce estrogenic responses. In a comparison of gene expression profiles in MCF7 breast cancer cells after treatment with E2 or a variety of alkylphenols, a high correlation was observed in profiles from cells treated with E2 and nonylphenol or 4-tert-octylphenol (69). Like BPA, nonylphenol also shows a higher binding affinity for GPR30 compared to ERα and ERβ RBA of 2.15) (65) and can invoke non-genomic responses such as ERK phosphorylation and rapid calcium flux through membrane associated ERα (70). Comparisons of the effects elicited by alkylphenols and the hydrophobicity of the chemicals revealed that more hydrophobic alkylphenols like nonylphenol and 4-tert-octylphenol were more potent activators of calcium flux and less potent inducers of ERK phosphorylation in GH3/B6/F10 pituitary tumor cells (70).
3.2.4 Phytoestrogens: Liquiritigenin and genistein
Plants produce a wide variety of secondary metabolites some of which are phytoestrogens, chemicals that have similar structures as E2 and display estrogenic activity through ER signaling pathways. Phytoestrogens have poly-phenolic structures and can be classified as flavonoids, coumestans, and lignans (44). Exposure occurs primarily through dietary intake of beverages and foods containing fruits, herbs, and vegetables, most notably soy, which have high levels of phytoestrogens (71). Flavonoids are one of the most prevalent classes of phytoestrogens found in dietary sources and are further classified as chalcones, flavones, flavonols, flavanones, flavanols, anthocyanins, and isoflavones. Most studies have focused on resveratrol, quercetin, daidzein, and genistein as they are some of the most commonly ingested phytoestrogens (72).
Though phytoestrogens are often discussed in the context of cancer prevention and do not typically elicit physiological abnormalities like DES or other environmental EDCs, phytoestrogens cannot be neglected as EDCs due to widespread exposure to plant material in the diet and the profound physiological effects mediated by ERs (72). Daily phytoestrogen intake ranges from 0.15–3 mg per day in the United States (72–74). Phytoestrogens have relatively weak affinities for ERα and ERβ but serum levels can reach near micromolar concentrations after a soy-rich meal (75), well above the concentration of endogenously circulating estrogens (20–200 pg/mL). Phytoestrogens can act as endocrine disruptors by affecting estrogen biosynthesis and menstrual cycle (72). Menstrual cycles were longer in women consuming 40 mg of isoflavones per day for 3 months indicating phytoestrogen can perturb normal estrous cycle (76). Phytoestrogen exposure in infants can also be a concern given that 25% of infant formulas are soy-based and concentrations of the phytoestrogens genistein and daidzein were nearly 500 times higher in urine of infants given soy-based formula compared to infants given formula derived from cow milk (77). Many animal studies also indicate that phytoestrogens can compete for ER binding and modulate normal ER action in target tissues. In transgenic estrogen reporter mice, genistein treatment inhibited the estrogenic response elicited by E2 in the liver indicating phytoestrogen exposure can modify the effects of endogenous estrogens (78).
Many phytoestrogens, including flavonoids, display selectivity for ERβ based on competitive binding assays and transcriptional assays (44), although the magnitude of selectivity in binding assays does not always correspond with selectivity in transcriptional assays. For example, liquiritigenin (Figure 2, 10) is a flavone found in Glycyrrhizae uralensis that shows a 20-fold selectivity for ERβ in competitive binding assays (RBAs not determined) (79). Transcriptional assays in 3 different cell types reveal that although liquiritigenin binds ERα in binding assays, it induces minimal transcriptional activation of reporter genes through ERα at concentrations as high as 2.5 μM (79). Though the crystal structure of the ERβ LBD bound to liquiritigenin is not yet resolved, it is possible that the ligand induces conformational changes that selectively recruit coactivators to ERβ. Indeed, SRC-2 recruitment to ER target genes selectively occurred in cells expressing ERβ after liquiritigenin treatment (79), suggesting ligand binding and transcriptional potency are not always correlated.
Genistein (Figure 2, 11), an isoflavone found in soy that shows high binding affinity and selectivity for ERβ (44), has been shown to have more potent transcriptional activity in cells transfected with ERβ (80). Crystal structures of genistein indicate that coactivator binding can stabilize the ligand in the ligand binding pocket. Genistein is an agonist in transcriptional assays, but initial crystal structure analysis of genistein and the ERβ LBD showed a shift in H12 towards the antagonist conformation in which the coactivator binding site is partially blocked (28). Crystal structures and computational modeling of the ERβ LBD with genistein in the presence of a coactivator LXXLL peptide fragment show H12 in the agonist orientation suggesting the coactivator stabilizes the complex in an active conformation (81). Genistein also selectively recruits SRC-2 to ERβ which may contribute to the transcriptional selectivity for ERβ (82). Given the importance of coactivators in stimulating transcription and the potential role of coactivators in stabilizing ligand-bound ER in an active conformation, the estrogenic effects of potential EDCs will be highly dependent on the availability of coactivators which can vary among cell types thereby contributing to tissue specific effects.
4. Indirect actions of EDCs on ER signaling
4.1 Aryl hydrocarbon receptor ligands and functional cross-talk with ERs
EDCs can impact ER signaling indirectly through interactions with the aryl hydrocarbon receptor (AhR) which is activated by a wide variety of hydrophobic ligands, most notably polycyclic aromatic hydrocarbons (PAHs) or halogenated aromatic hydrocarbons (HAHs) (83, 84). AhR regulates expression of metabolic enzymes in response to ligand binding and the mechanism of AhR signaling shares many similarities with that of ER signaling (85). The AhR contains a basic-helix-loop-helix DNA binding domain and a PER-ARNT-SIM (PAS) homology domain to which ligands bind (86). AhR is maintained in an inactive cytosolic complex that includes Hsp90 and p23 in the absence of ligand (87–89). Similarly, ERs are maintained in a complex that includes Hsp90 and p23 in the absence of E2 (90). Upon ligand binding, AhR undergoes a conformational change that reveals a nuclear localization sequence, dissociates from the inactive cytosolic complex, and migrates to the nucleus where it heterodimerizes with aryl hydrocarbon receptor nuclear translocator (ARNT), another bHLH-PAS protein (91). The heterodimer binds DNA sequences known as dioxin response elements (DREs) and recruits coactivators, including SRC-1, SRC-2, and SRC-3 (92), to stimulate transcription of target genes. One of the most studied AhR target genes is cytochrome P450-dependent monooxygenase (CYP) 1A1 (93). The high affinity AhR ligand 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) is the most potent inducer of Cyp1A1 expression and is a known EDC (93).
Dioxins and other AhR ligands are primarily anti-estrogenic (94, 95) but can also exhibit weak estrogenic activity (94, 96–98). AhR and ERs are promiscuous receptors and bind a diverse array of chemicals, most of which are hydrophobic ligands. The most notable AhR ligands are environmental chemicals and an endogenous ligand has yet to be identified (83). There is some overlap in the chemicals that can bind AhR and ER. It has recently been shown that pharmaceutical anti-estrogens such as tamoxifen and many other estrogenic ligands activate AhR and induce CYP1A1 expression in breast cancer cells that lack ERα or ERβ expression (99). Dietary flavonoids that are known estrogenic ligands can also activate AhR such as daidzein and genistein (44, 100, 101), demonstrating ER ligands can act through multiple mechanisms. To illustrate the complexity of promiscuous ligands, we return to the discussion of WAY-169916, the non-steroidal ER-dependent selective inhibitor of NFκB activity discussed in section 2.2. WAY-169916 is considered a SERM that shows pathway selectivity in terms of ER-mediated signaling (39) but it has also recently been shown that WAY-169916 can act as a selective AhR modulator (SAhRM) (102). WAY-169916 can bind AhR with approximately three orders of magnitude lower affinity relative to ER and weakly activates expression of CYP1A1 in an ER-independent manner. Despite its relatively low activity in terms of classical DRE driven gene expression, WAY-169916 treatment has anti-inflammatory effects in a variety of mouse models of inflammatory diseases and represses expression of inflammatory acute phase response genes in an AhR/ARNT dependent manner (102). WAY-169916 demonstrates that AhR ligands may selectively modify AhR signaling pathways, thereby acting as a SAhRM, and ER signaling pathways, thereby acting as a SERM. Experiments conducted with 3-methylcholanthrene (3-MC) demonstrate the role of cellular context in determining which pathways are likely to be affected by promiscuous ligands. 3-MC can interact with AhR and ER to stimulate transcription of DRE or ERE driven reporter constructs. In HepG2 liver carcinoma cells, 3-MC acts as an ER agonist and induces expression of ERE-luciferase in a dose dependent manner when cells are transfected with ERα. In contrast, 3-MC cannot stimulate expression of ERE-luciferase in HC11 mouse mammary epithelial cells that express endogenous ERα but rather inhibits E2-stimulated expression of the ERE reporter (103). Further analysis of the phenomenon revealed that HepG2 cells treated with 3-MC produced estrogenic metabolites while HC11 cells did not. Cell context specific effects have also been observed for TCDD in MCF7 and HepG2 cells (104).
Though ligand promiscuity strongly influences cross-talk between AhR and ER, additional mechanisms may also mediate such cross-talk (85). First, induction of CYPs may enhance E2 metabolism, thereby reducing estrogenic response. Hydroxylation of E2 increases more than 10 fold in MCF7 breast cancer cells treated with TCDD, possibly due to induction of CYP1A1 and CYP1B1 (105, 106). Increased metabolic turnover of estrogen may not be the primary mechanism of AhR mediated inhibition of ER signaling given the observation that TCDD inhibits E2-stimulated cathepsin D expression within 60 minutes, preceding the induction of CYP1A1 protein (107). As an alternative mechanism of AhR-ER cross-talk, AhR may impair ER mediated transcription through direct binding to ER target gene promoters (108). DRE sequences that are required for the inhibitory effects of dioxin on estrogen stimulation of gene expression have been identified in the promoter regions of c-fos (109), pS2 (110), and heat shock protein 27 (111). A third mechanism of AhR-ER crosstalk may be that activated AhR competes for available cofactors thereby inhibiting the transcriptional potential of ER. Activated AhR can recruit coregulators shared by activated ER, including RIP140 (112), SRC-1, and SRC-2 (113). It has recently been shown that ARNT can also act a coactivator of ERα and ERβ (114) and is a more potent coactivator for ERβ mediated transcription (115) suggesting AhR activation may impair ER mediated transcriptional activation by sequestering ARNT and this effect may be more profound for ERβ signaling. The competition for cofactors will depend on the amounts and types of cofactors expressed in the cell which will be highly dependent on cell context and tissue type.
4.2 Metabolic modulators of estrogen metabolism
EDCs can impact ER signaling indirectly through modification of E2 metabolism thereby reducing or enhancing ER signaling in target tissues (116). Estrogen synthesis is catalyzed by a set of CYPs and hydroxysteroid dehydrogenases. Cholesterol is the universal precursor of de novo synthesis of steroidal hormones and undergoes a side chain cleavage by CYP11A to form pregnenolone which is further modified to form androgens (117, 118). Androgens are the direct precursors of estrogens and the conversion of androgens to estrogens is catalyzed by aromatase, also known as CYP19A1 (119). Aromatase catalyzes the formation of two estrogens: estrone and E2. Conversion of estrone to the more potent E2 is catalyzed by 17β-hydroxysteroid dehydrogenase (118). In pre-menopausal women, estrogen synthesis occurs primarily in the ovaries where it is tightly regulated by the expression of aromatase which is up regulated by follicle stimulating hormone (FSH) and leutinizing hormone (LH) during follicle maturation in the preovulatory stage. Estrogens stimulate expression of LH receptors which promotes a surge of LH and FSH thereby triggering ovulation. During this critical window of estrogen synthesis, impairment of aromatase expression or activity could prevent ovulation (116). Estrogen metabolism occurs primarily in the liver through oxidation by CYP enzymes, glucuronidation by UDP-glucuronosyl transferases, sulfation by sulfotransferases, or O-methylation by catechol O-methyltransferase, all of which generate inactive metabolites that are excreted from the body (120).
Estrogen synthesis and metabolism may be targets for some EDCs that inhibit normal estrogen signaling. Atrazine, a triazine herbicide that is widely present in the environment, is a known EDC that was shown to induce mammary tumors in F344 female rats (121) and increase plasma levels of estradiol in female Sprague-Dawley rats(122). Atrazine and other triazine herbicides were later shown to induce aromatase activity and gene expression (123). Pesticides including chlordane or methoxychlor also induce aromatase in vitro (124). Enhancing the activity or expression of aromatase may increase rates of estrogen synthesis, thereby mediating estrogenic effects. Conversely, dietary soy supplementation rich in flavonoids decreased estrogen synthesis in premenopausal women, which may explain the cancer preventive effects of phytoestrogens (125). Data from in vitro experiments suggest that phytoestrogens competitively bind aromatase preventing normal estrogen synthesis from androgen substrates. Flavones can bind aromatase with low micromolar Ki values (126) and, in general, flavones are more potent inhibitors compared to isoflavones (127). The flavonoids apigenin, chrysin, and naringenin inhibit aromatase in human placental microsomes and human breast fibroblasts (128).
Estrogen metabolism and excretion may also be impacted by EDCs through modified expression of enzymes required for estrogen clearance in the liver. E2 is initially metabolized by CYPs including CYP1B1 and CYP1A1 which form 4-hydroxyestradiol and 2-hydroxyestradiol, respectively (120). In the breast and uterus, CYP1B1 is highly expressed and the prominent metabolite is 4-hydroxyestradiol (129) while 2-hydroxyestradiol is the most prominent metabolite in the liver, most likely due to oxidation by a member of the CYP3A family (130). CYP1B1 is induced by activation of AhR and AhR agonists which can increase E2 hydroxylation in MCF-7 breast cancer cells (129). Further metabolism of the hydroxylated estradiol may produce reactive metabolites that can induce DNA damage (131) which is thought to be the primary mechanism of ER-independent estrogen-mediated carcinogenesis (132).
5. In vitro and cell-based approaches to identify EDCs that impact ER signaling
Because of concerns associated with EDCs, national and international programs have developed guidelines for screening chemicals with endocrine disrupting properties (133, 134). The guidelines focus on a multi-tiered approach consisting of: Tier 1) In vitro and in vivo assays for identifying and classifying chemicals based on their potential interaction with the endocrine system; Tier 2) Developing dose-response data in animal models (134). In 2009, the U.S. Environmental Protection Agency announced assays for use in Tier 1 screening for EDCs (135). In vitro and cell-based screens designed to identify estrogenic compounds include: 1) ER binding assays using rat uterine cytosol preparations, 2) ER transcriptional assays using hERα-HeLa-9903 cells which are human cervical tumor cells with stable expression of ERα and stable integration of a 5xERE luciferase reporter, 3) steroidogenesis assays with the adrenocarcinoma cell line H295R, and 4) aromatase inhibition assays using recombinant aromatase (135). Additional in vitro and cell-based assays are available to identify potential EDCs including alternative approaches to identify compounds that bind the receptors and initiate ER mediated transcription as well as proliferation assays that can sensitively detect estrogenic chemicals (Table 1). In vivo assays using rodent models are required to effectively determine if chemicals will act as EDCs and elucidate tissue-specific effects of EDCs. In vitro assays cannot replace in vivo assays due to limitations in considering effects of metabolism, clearance, and tissue specific ER agonist and antagonist activities, but in vitro and cell-based assays have the advantage of typically being high-throughput, requiring less time and costs. Additionally, in vitro assays allow much more stringent control which allows determination of effects on specific mechanistic processes of ER signaling. In vivo identification of EDCs that target ER signaling is complicated by rat or mouse strain and dietary exposure to estrogenic compounds which can have profound effects on experiments measuring subtle changes in endocrine function (136). Here, we limit the discussion to the available in vitro assays useful for identifying EDCs that target ER signaling in a relatively high-throughput manner. In vivo experiments for identifying EDCs are required for a complete understanding of the endocrine disrupting properties of chemicals but in vitro pre-screening allows initial identification of chemicals that target ER signaling and can correlate with in vivo data for a large range of chemicals (137). All the assays discussed herein are summarized in Table 1.
Table 1.
In vitro and cell-based assays for identifying estrogenic EDCs.
| ER binding | Reference |
|---|---|
| Rat uterine cytosol* | (134) |
| Fluorescence polarization | (138) |
| ER dimerization | |
| Fluorescence resonance energy transfer | (139, 140)) |
| Bioluminescence resonance energy transfer | (23, 143) |
| ER transcriptional activation | |
| hERα-HeLa-9903 reporter cell line* | (134) |
| T47D-KBLuc reporter cell line | (144) |
| HELN-ERα and HELN-ERβ reporter cell lines | (145, 146) |
| HEK 293 ERα and ERβ ALP reporter cell lines | (147) |
| Recombinant yeast assays | (148) |
| Proliferation assays | |
| E-SCREEN assay | (150, 151) |
| Metabolic assays | |
| H295R steroidogenesis assay* | (134) |
| Aromatase competitive inhibition assay* | (134) |
| H295R steroidogenic enzyme expression assay | (152) |
| JEG-3 steroidogenesis assay | (153) |
Indicates assays required by the U.S. Environmental Protection Agency in Tier 1 screening for potential EDCs (134).
5.1 Assays to measure binding and dimerization
Binding assays are useful for identifying EDCs that directly interact with the LBDs of ERα and ERβ. The U.S. Environmental Protection Agency Tier 1 assays include an ER binding assay utilizing rat uterine cytosol but the assay does not quantitatively measure ER binding affinity or discriminate between binding to ERα or ERβ (135). Several approaches are available for sensitively quantifying receptor binding affinity. Traditional radiolabelled competitive binding assays have been successfully used to characterize the ligand binding affinities for ERα and ERβ of a wide variety of environmental compounds (44). Alternatively, a nonradioactive fluorescence polarization assay has been developed that is more amenable to high-throughput screening, in which changes in polarization of fluorescent E2 are measured with increasing concentrations of competitor compound (138).
After chemicals bind ERs, dimerization must occur in order for ERs to mediate transcriptional activation. Multiple assays to measure dimerization have been developed which are useful for high-throughput screening. Fluorescence resonance energy transfer (FRET) assays can be used to measure protein-protein interactions and may be used to detect ER homodimer and heterodimer formation (139) and ER-coactivator interactions (140) in response to ligand. FRET utilizes proteins fused to fluorophores such as green fluorescent protein and measures the proximity of two proteins; energy transfer from an excited “donor” fluorophore on one protein to the nearby “acceptor” fluorophore of another protein results in a reduction in the emission of the donor which can be measured (141). The drawbacks of FRET assays include autofluorescence, photobleaching, and direct excitation of the acceptor fluorophore. Bioluminescence resonance energy transfer (BRET) is an alternative assay to measure dimerization that overcomes some of the limitations associated with FRET. BRET assays utilize an enzyme-catalyzed bioluminescent donor such as luciferase that emits photons only in the presence of substrate. When the substrate is added, luciferase catalyzes a reaction that emits light which can excite an acceptor, such as yellow fluorescent protein, if it is in close proximity to luciferase. Proteins of interest are fused to luciferase or the acceptor fluorophore, respectively (142). BRET assays have been successfully used to measure ER homodimerization and heterodimerization in live cells and high-throughput screening for chemicals that induce ER dimers is possible using this highly optimized assay (23, 143).
5.2 Transcriptional assays
Transcriptional assays in cell lines using reporter genes such as luciferase or β-galactosidase downstream of an ERE provide a tool to identify compounds that act as ER agonists or antagonists. Transcription of target genes is dependent on a variety of factors, including promoter context, cell context, and the availability of cofactors. Several cell lines have been generated to identify chemicals with estrogenic activity and quantify the relative potency of such compounds. T47D breast cancer cells express both ERα and ERβ and chemicals that activate either receptor can be identified using T47D-KBLuc cells in which a luciferase reporter downstream of three tandem EREs is stably integrated (144). T47D-KBLuc cells were shown to be highly sensitive and stable over many passages making them amenable for use in high-throughput screening (144). Currently, Tier 1 transcriptional assays aimed at detecting chemicals with estrogenic activity are limited to HeLa cells that express ERα alone, neglecting the potential for EDCs to selectively activate ERβ. In order to identify compounds that display selectivity for ERα or ERβ, HeLa cells with stable ERE-luciferase (HELN) were created with stable expression of ERα or ERβ (HELN-ERα and HELN-ERβ, respectively); in this system, genistein was shown to have high selectivity for ERβ (145). HELN-ERα and HELN-ERβ cells have also been used to demonstrate the anti-estrogenic activities of persistent pesticides on ERβ mediated transcription (146). A similar approach has been applied to human embryonic kidney 293 cells by stable expression of ERα or ERβ and stable integration of a human placental alkaline phosphatase reporter downstream of a single ERE (147). Alternative reporter systems include recombinant yeast assays in which a β-galactosidase reporter is expressed after ligand-dependent activation of ER (148) but mammalian systems provide a better model given the role of coactivators and interactions of signaling pathways in mediating the transcriptional activation of ERs.
5.3 Proliferation assays
The E-SCREEN is a is a highly sensitive cell-based assay for detecting estrogenic compounds and has been utilized for detecting estrogenic activity in wastewater and environmental samples (149, 150). MCF7 breast cancer cells are estrogen-dependent and proliferate rapidly after exposure to estrogenic chemicals. The E-SCREEN assay measures proliferation of MCF7 cells after treatment with a range of concentrations of the chemical or sample of interest, using E2 as a positive control. Using this assay, a variety of compounds were found to be estrogenic including alkylphenols, phthalates, some PCB congeners and hydroxylated PCBs. These compounds were further shown to bind ER and activate transcription of pS2, an estrogen responsive gene, in MCF7 cells (151). Proliferation assays like the E-SCREEN can be highly sensitive and results may be relevant given the system utilizes cells of human origin which express endogenous coactivators that may mediate estrogenic potency.
5.4 Metabolic assays
As mentioned previously, Tier 1 assays for identifying EDCs include two in vitro assays to detect metabolic inhibitors. A competitive binding assay aims to detect chemicals that will directly interfere with the conversion of androgens to estrogens by competing for the catalytic activity of aromatase and a steroidogenesis assay utilizing the adrenocarcinoma cell line H295R aims to detect chemicals that can interfere with estrogen or androgen production by measuring hormone production after exposure to suspected EDCs (135). H295R cells provide a tool to measure both steroid production and expression levels of steroidogenesis enzymes. Quantitative RT-PCR may be used to measure changes in the expression of enzymes required for normal estrogen synthesis (152). Human placental JEG-3 cells also express relatively high levels of aromatase and may be useful for identifying inhibitors of estrogen synthesis, but the cells are vulnerable to toxicity of compounds making high-throughput screening more difficult (153).
6. Conclusions and future perspectives
The impacts of EDCs on ER signaling are complex and often occur through multiple direct and indirect mechanisms making it difficult to predict the endpoints of EDC toxicity in cultured cells and animals. Though ERs are relatively promiscuous receptors, insight from crystal structures is useful for defining characteristic features of estrogenic ligands that act through direct interaction with ER LBDs. Such chemicals may be detected through screening assays aimed at measuring ER binding and dimerization such as fluorescence polarization and FRET/BRET. Current guidelines for detecting chemicals that interact with ER through binding do not discriminate between receptor subtypes despite the fact that many EDCs such as phytoestrogens display selectivity for ERβ. Tier 1 assays required by the U.S. Environmental Protection Agency also neglect transcriptional activation of ERβ. EDCs may also act indirectly through other signaling pathways such as AhR or through modification of normal estrogen synthesis. Cross-talk between AhR and ER is highly complex and dependent on cell context, availability of cofactors, and promoter context which makes it difficult to develop universal, effective in vitro assays for identifying EDCs that impact ER signaling through stimulation of AhR; such indirect EDCs may be detected more effectively using in vivo models. Finally, EDCs that impact ER signaling indirectly through modification of estrogen synthesis may be identified using assays currently required by the U.S. Environmental Protection Agency’s Tier 1 protocols.
Despite multiple approaches to identify EDCs using in vitro and cell-based assays, it is difficult to predict the toxic endpoints induced by EDCs in vivo. Though a chemical may not display estrogenic effects at low concentrations in highly controlled experiments, estrogenic effects may be elicited in the background of broad chemical exposure that occurs in our environment. EDCs may have additive or synergistic effects when combined (see reference (46)) and such complicated effects are compounded by individual differences in uptake, metabolism, and excretion. Tissue specific effects of EDCs are also difficult to predict using in vitro and cell-based assays, which underscores the importance of examining the effects of EDCs in vivo with animal models. Though in vitro and cell-based assays cannot replace in vivo models, approaches to predict tissue specific effects using in vitro techniques are developing. A recent study utilized hydrogen/deuterium exchange (HDX) mass spectrometry to predict the tissue selectivity of pharmaceutical SERMs used for the treatment of breast cancer (154). HDX mass spectrometry measures the rate of hydrogen and deuterium exchange at amide groups and variations in the exchange kinetics are a function of hydrogen bonding. Exchange rates are higher in regions where the ligand is bound to the receptor and in regions in which the receptor undergoes conformational changes. Based on the exchange kinetics, ligands could be classified based on tissue selectivity in most cases (154). Such an approach provides insight into the dynamic structural effects of ligands interacting with ERs and may prove useful for predicting tissue specificity of EDCs. In vitro and cell-based assays measuring specific mechanisms of ER signaling such as ligand binding, dimerization, DNA binding, and transcriptional activation are important initial screens for identifying EDCs. As our understanding of estrogen signaling mechanisms expands and technology advances, in vitro and cell-based assays will become more powerful tools for early identification of EDCs.
Acknowledgments
This publication was made possible by grant number T32 ES007015 from the National Institute of Environmental Health Sciences (NIEHS), NIH. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIEHS, NIH. W.X. is supported by NIH grants R01CA125387, R03MH089442, Markos Family Breast Cancer Research Grant and Shaw Scientist Award from Greater Milwaukee Foundation.
Footnotes
Abbreviations: 3-MC, 3-methylcholanthrene; AF, activation function; AhR, aryl hydrocarbon receptor; ARNT, aryl hydrocarbon receptor nuclear translocator; bHLH, basic-helix-loop-helix; BPA, bisphenol A; BRET, bioluminescence resonance energy transfer; CYP, cytochrome P450-dependent monooxygenase; DBD, DNA binding domain; DDT, 1,1,1-trichloro-2,2-bis(p-chlorophenyl)ethane; DES, diethylstilbestrol; DPN, diarylpropionitrile; E2, 17β-estradiol; EDC, endocrine disrupting chemical; ER, estrogen receptor; ERE, estrogen response element; FRET, fluorescence resonance energy transfer; FSH, follicle stimulating hormone; HAH, halogenated aromatic hydrocarbon; HDX, hydrogen/deuterium exchange; HPTE, 2,2-bis(p-hydroxyphenyl)-1,1,1-trichloroethane; LH; leutinizing hormone; LBD, ligand binding domain; NR, nuclear receptor; PAH, polycyclic aromatic hydrocarbon; PAS, PER-ARNT-SIM; RBA, relative binding affinity; SERM, selective estrogen receptor modulator; TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin
References
- 1.Diamanti-Kandarakis E, Bourguignon JP, Giudice LC, Hauser R, Prins GS, Soto AM, Zoeller RT, Gore AC. Endocrine-disrupting chemicals: An Endocrine Society scientific statement. Endocr Rev. 2009;30:293–342. doi: 10.1210/er.2009-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kuiper GG, Carlsson B, Grandien K, Enmark E, Haggblad J, Nilsson S, Gustafsson JA. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology. 1997;138:863–870. doi: 10.1210/endo.138.3.4979. [DOI] [PubMed] [Google Scholar]
- 3.Kuiper GG, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson JA. Cloning of a novel receptor expressed in rat prostate and ovary. Proc Natl Acad Sci U S A. 1996;93:5925–5930. doi: 10.1073/pnas.93.12.5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Enmark E, Pelto-Huikko M, Grandien K, Lagercrantz S, Lagercrantz J, Fried G, Nordenskjold M, Gustafsson JA. Human estrogen receptor β-gene structure, chromosomal localization, and expression pattern. J Clin Endocrinol Metab. 1997;82:4258–4265. doi: 10.1210/jcem.82.12.4470. [DOI] [PubMed] [Google Scholar]
- 5.Taylor AH, Al-Azzawi F. Immunolocalisation of oestrogen receptor beta in human tissues. J Mol Endocrinol. 2000;24:145–155. doi: 10.1677/jme.0.0240145. [DOI] [PubMed] [Google Scholar]
- 6.Hewitt SC, Harrell JC, Korach KS. Lessons in estrogen biology from knockout and transgenic animals. Annu Rev Physiol. 2005;67:285–308. doi: 10.1146/annurev.physiol.67.040403.115914. [DOI] [PubMed] [Google Scholar]
- 7.Harris HA. Estrogen receptor-beta: Recent lessons from in vivo studies. Mol Endocrinol. 2007;21:1–13. doi: 10.1210/me.2005-0459. [DOI] [PubMed] [Google Scholar]
- 8.Paruthiyil S, Parmar H, Kerekatte V, Cunha GR, Firestone GL, Leitman DC. Estrogen receptor β inhibits human breast cancer cell proliferation and tumor formation by causing a G2 cell cycle arrest. Cancer Res. 2004;64:423–428. doi: 10.1158/0008-5472.can-03-2446. [DOI] [PubMed] [Google Scholar]
- 9.Murphy LC, Peng B, Lewis A, Davie JR, Leygue E, Kemp A, Ung K, Vendetti M, Shiu R. Inducible upregulation of oestrogen receptor-β1 affects oestrogen and tamoxifen responsiveness in MCF7 human breast cancer cells. J Mol Endocrinol. 2005;34:553–566. doi: 10.1677/jme.1.01688. [DOI] [PubMed] [Google Scholar]
- 10.Ström A, Hartman J, Foster JS, Kietz S, Wimalasena J, Gustafsson JA. Estrogen receptor β inhibits 17β-estradiol-stimulated proliferation of the breast cancer cell line T47D. Proc Natl Acad Sci U S A. 2004;101:1566–1571. doi: 10.1073/pnas.0308319100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sotoca AM, van den Berg H, Vervoort J, van der Saag P, Strom A, Gustafsson JA, Rietjens I, Murk AJ. Influence of cellular ERalpha/ERbeta ratio on the ERalpha-agonist induced proliferation of human T47D breast cancer cells. Toxicol Sci. 2008;105:303–311. doi: 10.1093/toxsci/kfn141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shanle EK, Xu W. Selectively targeting estrogen receptors for cancer treatment. Advanced Drug Delivery Reviews. doi: 10.1016/j.addr.2010.08.001. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, Tujague M, Strom A, Treuter E, Warner M, Gustafsson JA. Estrogen receptors: How do they signal and what are their targets. Physiol Rev. 2007;87:905–931. doi: 10.1152/physrev.00026.2006. [DOI] [PubMed] [Google Scholar]
- 14.Cowley SM, Parker MG. A comparison of transcriptional activation by ERα and ERβ. J Steroid Biochem Mol Biol. 1999;69:165–175. doi: 10.1016/s0960-0760(99)00055-2. [DOI] [PubMed] [Google Scholar]
- 15.Zwart W, de Leeuw R, Rondaij M, Neefjes J, Mancini MA, Michalides R. The hinge region of the human estrogen receptor determines functional synergy between AF-1 and AF-2 in the quantitative response to estradiol and tamoxifen. J Cell Sci. 2010;123:1253–1261. doi: 10.1242/jcs.061135. [DOI] [PubMed] [Google Scholar]
- 16.Lin CY, Vega VB, Thomsen JS, Zhang T, Kong SL, Xie M, Chiu KP, Lipovich L, Barnett DH, Stossi F, Yeo A, George J, Kuznetsov VA, Lee YK, Charn TH, Palanisamy N, Miller LD, Cheung E, Katzenellenbogen BS, Ruan Y, Bourque G, Wei CL, Liu ET. Whole-Genome Cartography of Estrogen Receptor α Binding Sites. PLoS Genet. 2007;3:e87. doi: 10.1371/journal.pgen.0030087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, Brodsky AS, Keeton EK, Fertuck KC, Hall GF, Wang Q, Bekiranov S, Sementchenko V, Fox EA, Silver PA, Gingeras TR, Liu XS, Brown M. Genome-wide analysis of estrogen receptor binding sites. Nat Genet. 2006;38:1289. doi: 10.1038/ng1901. [DOI] [PubMed] [Google Scholar]
- 18.Liu Y, Gao H, Marstrand TT, Ström A, Valen E, Sandelin A, Gustafsson J-Ãk, Dahlman-Wright K. The genome landscape of ERα- and ERβ-binding DNA regions. Proc Natl Acad Sci U S A. 2008;105:2604–2609. doi: 10.1073/pnas.0712085105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao C, Gao H, Liu Y, Papoutsi Z, Jaffrey S, Gustafsson J-Ãk, Dahlman-Wright K. Genome-Wide Mapping of Estrogen Receptor-β Binding Regions Reveals Extensive Cross-Talk with Transcription Factor Activator Protein-1. Cancer Res. 2010;70:5174–5183. doi: 10.1158/0008-5472.CAN-09-4407. [DOI] [PubMed] [Google Scholar]
- 20.Secreto FJ, Monroe DG, Dutta S, Ingle JN, Spelsberg TC. Estrogen receptor alpha/beta isoforms, but not betacx, modulate unique patterns of gene expression and cell proliferation in Hs578T cells. J Cell Biochem. 2007;101:1125–1147. doi: 10.1002/jcb.21205. [DOI] [PubMed] [Google Scholar]
- 21.Monroe D, Barbara JG, Steven AJ, Riggs BL, Sundeep K, Thomas CS. Estrogen receptor isoform-specific regulation of endogenous gene expression in human osteoblastic cell lines expressing either ERalpha or ERbeta. J Cell Biochem. 2003;90:315–326. doi: 10.1002/jcb.10633. [DOI] [PubMed] [Google Scholar]
- 22.Pettersson K, Grandien K, Kuiper GG, Gustafsson JA. Mouse estrogen receptor beta forms estrogen response element-binding heterodimers with estrogen receptor alpha. Mol Endocrinol. 1997;11:1486–1496. doi: 10.1210/mend.11.10.9989. [DOI] [PubMed] [Google Scholar]
- 23.Powell E, Xu W. Intermolecular interactions identify ligand-selective activity of estrogen receptor α/β dimers. Proc Natl Acad Sci U S A. 2008;105:19012–19017. doi: 10.1073/pnas.0807274105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kushner PJ, Agard DA, Greene GL, Scanlan TS, Shiau AK, Uht RM, Webb P. Estrogen receptor pathways to AP-1. J Steroid Biochem Mol Biol. 2000;74:311–317. doi: 10.1016/s0960-0760(00)00108-4. [DOI] [PubMed] [Google Scholar]
- 25.Saville B, Wormke M, Wang F, Nguyen T, Enmark E, Kuiper G, Gustafsson JA, Safe S. Ligand-, cell-, and estrogen receptor subtype (α/β)-dependent activation at GC-rich (Sp1) promoter elements. J Biol Chem. 2000;275:5379–5387. doi: 10.1074/jbc.275.8.5379. [DOI] [PubMed] [Google Scholar]
- 26.Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engstrom O, Ohman L, Greene GL, Gustafsson JA, Carlquist M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389:753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 27.Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 28.Pike AC, Brzozowski AM, Hubbard RE, Bonn T, Thorsell AG, Engstrom O, Ljunggren J, Gustafsson J, Carlquist M. Structure of the ligand-binding domain of oestrogen receptor beta in the presence of a partial agonist and a full antagonist. EMBO J. 1999;18:4608–4618. doi: 10.1093/emboj/18.17.4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lonard DM, O’Malley BW. Nuclear receptor coregulators: Judges, juries, and executioners of cellular regulation. Mol Cell. 2007;27:691–700. doi: 10.1016/j.molcel.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 30.Bjornstrom L, Sjoberg M. Mechanisms of estrogen receptor signaling: Convergence of genomic and nongenomic actions on target genes. Mol Endocrinol. 2005;19:833–842. doi: 10.1210/me.2004-0486. [DOI] [PubMed] [Google Scholar]
- 31.Filardo EJ, Quinn JA, Bland KI, Frackelton AR., Jr Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol. 2000;14:1649–1660. doi: 10.1210/mend.14.10.0532. [DOI] [PubMed] [Google Scholar]
- 32.Migliaccio A, Di Domenico M, Castoria G, de Falco A, Bontempo P, Nola E, Auricchio F. Tyrosine kinase/p21ras/MAP-kinase pathway activation by estradiol-receptor complex in MCF-7 cells. EMBO J. 1996;15:1292–1300. [PMC free article] [PubMed] [Google Scholar]
- 33.Chen Z, Yuhanna IS, Galcheva-Gargova Z, Karas RH, Mendelsohn ME, Shaul PW. Estrogen receptor α mediates the nongenomic activation of endothelial nitric oxide synthase by estrogen. J Clin Invest. 1999;103:401–406. doi: 10.1172/JCI5347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Improta-Brears T, Whorton AR, Codazzi F, York JD, Meyer T, McDonnell DP. Estrogen-induced activation of mitogen-activated protein kinase requires mobilization of intracellular calcium. Proc Natl Acad Sci U S A. 1999;96:4686–4691. doi: 10.1073/pnas.96.8.4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aronica SM, Kraus WL, Katzenellenbogen BS. Estrogen action via the cAMP signaling pathway: stimulation of adenylate cyclase and cAMP-regulated gene transcription. Proc Natl Acad Sci U S A. 1994;91:8517–8521. doi: 10.1073/pnas.91.18.8517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nadal A, Ropero AB, Laribi O, Maillet M, Fuentes E, Soria B. Nongenomic actions of estrogens and xenoestrogens by binding at a plasma membrane receptor unrelated to estrogen receptor α and estrogen receptor β. Proc Natl Acad Sci U S A. 2000;97:11603–11608. doi: 10.1073/pnas.97.21.11603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Safe S, Kim K. Non-classical genomic estrogen receptor (ER)/specificity protein and ER/activating protein-1 signaling pathways. J Mol Endocrinol. 2008;41:263–275. doi: 10.1677/JME-08-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu F, Khan S, Wu Q, Barhoumi R, Burghardt R, Safe S. Ligand structure-dependent activation of estrogen receptor [alpha]/Sp by estrogens and xenoestrogens. J Steroid Biochem Mol Biol. 2008;110:104–115. doi: 10.1016/j.jsbmb.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chadwick CC, Chippari S, Matelan E, Borges-Marcucci L, Eckert AM, Keith JC, Albert LM, Leathurby Y, Harris HA, Bhat RA, Ashwell M, Trybulski E, Winneker RC, Adelman SJ, Steffan RJ, Harnish DC. Identification of pathway-selective estrogen receptor ligands that inhibit NFκB transcriptional activity. Proc Natl Acad Sci U S A. 2005;102:2543–2548. doi: 10.1073/pnas.0405841102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kalaitzidis D, Gilmore TD. Transcription factor cross-talk: the estrogen receptor and NF-[kappa]B. Trends Endocrinol Metab. 2005;16:46–52. doi: 10.1016/j.tem.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 41.Swaby R, Sharma C, Jordan V. SERMs for the treatment and prevention of breast cancer. Rev Endocr and Metab Disord. 2007;8:229–239. doi: 10.1007/s11154-007-9034-4. [DOI] [PubMed] [Google Scholar]
- 42.Shang Y, Brown M. Molecular determinants for the tissue specificity of SERMs. Science. 2002;295:2465–2468. doi: 10.1126/science.1068537. [DOI] [PubMed] [Google Scholar]
- 43.Wu F, Safe S. Differential activation of wild-type estrogen receptor [alpha] and C-terminal deletion mutants by estrogens, antiestrogens and xenoestrogens in breast cancer cells. J Steroid Biochem Mol Biol. 2007;103:1–9. doi: 10.1016/j.jsbmb.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 44.Kuiper GG, Lemmen JG, Carlsson B, Corton JC, Safe SH, van der Saag PT, van der Burg B, Gustafsson JA. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor β. Endocrinology. 1998;139:4252–4263. doi: 10.1210/endo.139.10.6216. [DOI] [PubMed] [Google Scholar]
- 45.Minutolo F, Macchia M, Katzenellenbogen BS, Katzenellenbogen JA. Estrogen receptor beta ligands: Recent advances and biomedical applications. Med Res Rev. 2009 doi: 10.1002/med.20186. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 46.Kortenkamp A. Ten Years of Mixing Cocktails: A Review of Combination Effects of Endocrine-Disrupting Chemicals. Environ Health Perspect. 2007:115. doi: 10.1289/ehp.9357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Swedenborg E, Ruegg J, Makela S, Pongratz I. Endocrine disruptive chemicals: mechanisms of action and involvement in metabolic disorders. J Mol Endocrinol. 2009;43:1–10. doi: 10.1677/JME-08-0132. [DOI] [PubMed] [Google Scholar]
- 48.le Maire A, Bourguet W, Balaguer P. A structural view of nuclear hormone receptor: endocrine disruptor interactions. Cell Mol Life Sci. 2010;67:1219. doi: 10.1007/s00018-009-0249-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Herbst AL, Cole P, Colton T, Robboy SJ, Scully RE. Age-incidence and risk of diethylstilbestrol-related clear cell adenocarcinoma of the vagina and cervix. Am J Obstet Gynecol. 1977;128:43–50. doi: 10.1016/0002-9378(77)90293-9. [DOI] [PubMed] [Google Scholar]
- 50.Giusti RM, Iwamoto K, Hatch EE. Diethylstilbestrol Revisited: A Review of the Long-Term Health Effects. Ann Intern Med. 1995;122:778–788. doi: 10.7326/0003-4819-122-10-199505150-00008. [DOI] [PubMed] [Google Scholar]
- 51.Newbold RR, Padilla-Banks E, Jefferson WN. Adverse Effects of the Model Environmental Estrogen Diethylstilbestrol Are Transmitted to Subsequent Generations. Endocrinology. 2006;147:s11–17. doi: 10.1210/en.2005-1164. [DOI] [PubMed] [Google Scholar]
- 52.Bredfeldt TG, Greathouse KL, Safe SH, Hung MC, Bedford MT, Walker CL. Xenoestrogen-Induced Regulation of EZH2 and Histone Methylation via Estrogen Receptor Signaling to PI3K/AKT. Mol Endocrinol. 2010;24:993–1006. doi: 10.1210/me.2009-0438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tiemann U. In vivo and in vitro effects of the organochlorine pesticides DDT, TCPM, methoxychlor, and lindane on the female reproductive tract of mammals: A review. Reprod Toxicol. 2008;25:316. doi: 10.1016/j.reprotox.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 54.Turusov V, Rakitsky V, Tomatis L. Dichlorodiphenyltrichloroethane (DDT): Ubiquity, Persistence, and Risks. Environ Health Perspect. 2002:110. doi: 10.1289/ehp.02110125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.U.S. Environmental Protection Agency. Methoxychlor Reregistration Eligibility Decision (RED) EPA Publication No. EPA 738-R-04–010. Jun 30, 2004. [Google Scholar]
- 56.Cummings AM. Methoxychlor as a Model for Environmental Estrogens. Crit Rev Toxicol. 1997;27:367–379. doi: 10.3109/10408449709089899. [DOI] [PubMed] [Google Scholar]
- 57.Gaido KW, Leonard LS, Maness SC, Hall JM, McDonnell DP, Saville B, Safe S. Differential Interaction of the Methoxychlor Metabolite 2,2-Bis-(p-Hydroxyphenyl)-1,1,1-Trichloroethane with Estrogen Receptors {alpha} and {beta} Endocrinology. 1999;140:5746–5753. doi: 10.1210/endo.140.12.7191. [DOI] [PubMed] [Google Scholar]
- 58.Gaido KW, Maness SC, McDonnell DP, Dehal SS, Kupfer D, Safe S. Interaction of Methoxychlor and Related Compounds with Estrogen Receptor α and β, and Androgen Receptor: Structure-Activity Studies. Mol Pharmacol. 2000;58:852–858. [PubMed] [Google Scholar]
- 59.Hewitt SC, Korach KS. Estrogenic Activity of Bisphenol A and 2,2-bis(p-hydroxyphenyl)-1,1,1-trichloroethane (HPTE) Demonstrated in Mouse Uterine Gene Profiles. Environ Health Perspect. 2010 doi: 10.1289/ehp.1002347. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Calafat AM, Ye X, Wong LY, Reidy JA, Needham LL. Exposure of the U.S. population to bisphenol A and 4-tertiary-octylphenol: 2003–2004. Environ Health Perspect. 2007;116:39–44. doi: 10.1289/ehp.10753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vandenberg LN, Maffini MV, Sonnenschein C, Rubin BS, Soto AM. Bisphenol-A and the great divide: A review of controversies in the field of endocrine disruption. Endocr Rev. 2009;30:75–95. doi: 10.1210/er.2008-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Paris F, Balaguer P, Térouanne B, Servant N, Lacoste C, Cravedi JP, Nicolas JC, Sultan C. Phenylphenols, biphenols, bisphenol-A and 4-tert-octylphenol exhibit [alpha] and [beta] estrogen activities and antiandrogen activity in reporter cell lines. Mol Cell Endocrin. 2002;193:43–49. doi: 10.1016/s0303-7207(02)00094-1. [DOI] [PubMed] [Google Scholar]
- 63.Markey CM, Michaelson CL, Veson EC, Sonnenschein C, Soto AM. The Mouse Uterotrophic Assay: A Reevaluation of Its Validity in Assessing the Estrogenicity of Bisphenol A. Environ Health Perspect. 2001;109:55. doi: 10.1289/ehp.0110955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Watson CS, Bulayeva NN, Wozniak AL, Finnerty CC. Signaling from the membrane via membrane estrogen receptor-[alpha]: Estrogens, xenoestrogens, and phytoestrogens. Steroids. 2005;70:364–371. doi: 10.1016/j.steroids.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 65.Thomas P, Dong J. Binding and activation of the seven-transmembrane estrogen receptor GPR30 by environmental estrogens: A potential novel mechanism of endocrine disruption. J Steroid Biochem Mol Biol. 2006;102:175–179. doi: 10.1016/j.jsbmb.2006.09.017. [DOI] [PubMed] [Google Scholar]
- 66.Li X, Zhang S, Safe S. Activation of kinase pathways in MCF-7 cells by 17[beta]-estradiol and structurally diverse estrogenic compounds. J Steroid Biochem Mol Biol. 2006;98:122–132. doi: 10.1016/j.jsbmb.2005.08.018. [DOI] [PubMed] [Google Scholar]
- 67.Routledge EJ, White R, Parker MG, Sumpter JP. Differential effects of xenoestrogens on coactivator recruitment by estrogen receptor (ER) α and ERβ. J Biol Chem. 2000;275:35986–35993. doi: 10.1074/jbc.M006777200. [DOI] [PubMed] [Google Scholar]
- 68.White R, Jobling S, Hoare SA, Sumpter JP, Parker MG. Environmentally persistent alkylphenolic compounds are estrogenic. Endocrinology. 1994;135:175–182. doi: 10.1210/endo.135.1.8013351. [DOI] [PubMed] [Google Scholar]
- 69.Terasaka S, Inoue A, Tanji M, Kiyama R. Expression profiling of estrogen-responsive genes in breast cancer cells treated with alkylphenols, chlorinated phenols, parabens, or bis- and benzoylphenols for evaluation of estrogenic activity. Toxicol Lett. 2006;163:130. doi: 10.1016/j.toxlet.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 70.Kochukov MY, Jeng YJ, Watson CS. Alkylphenol Xenoestrogens with Varying Carbon Chain Lengths Differentially and Potently Activate Signaling and Functional Responses in GH3/B6/F10 Somatomammotropes. Environ Health Perspect. 2009;117:723–730. doi: 10.1289/ehp.0800182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kuhnle GGC, Dell’Aquila C, Aspinall SM, Runswick SA, Mulligan AA, Bingham SA. Phytoestrogen content of beverages, nuts, seeds, and oils. J Agric Food Chem. 2008;56:7311–7315. doi: 10.1021/jf801534g. [DOI] [PubMed] [Google Scholar]
- 72.Mense SM, Hei TK, Ganju RK, Bhat HK. Phytoestrogens and Breast Cancer Prevention: Possible Mechanisms of Action. Environ Health Perspect. 2008;116:426. doi: 10.1289/ehp.10538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Strom SS, Yamamura Y, Duphorne CM, Spitz MR, Babaian RJ, Pillow PC, Hursting SD. Phytoestrogen intake and prostate cancer: A case-control study using a new database. Nutr Cancer. 1999;33:20–25. doi: 10.1080/01635589909514743. [DOI] [PubMed] [Google Scholar]
- 74.de Kleijn MJJ, van der Schouw YT, Wilson PWF, Adlercreutz H, Mazur W, Grobbee DE, Jacques PF. Intake of Dietary Phytoestrogens Is Low in Postmenopausal Women in the United States: The Framingham Study1–4. J Nutr. 2001;131:1826–1832. doi: 10.1093/jn/131.6.1826. [DOI] [PubMed] [Google Scholar]
- 75.Cassidy A, Brown JE, Hawdon A, Faughnan MS, King LJ, Millward J, Zimmer-Nechemias L, Wolfe B, Setchell KDR. Factors Affecting the Bioavailability of Soy Isoflavones in Humans after Ingestion of Physiologically Relevant Levels from Different Soy Foods. J Nutr. 2006;136:45–51. doi: 10.1093/jn/136.1.45. [DOI] [PubMed] [Google Scholar]
- 76.Kumar NB, Cantor A, Allen K, Riccardi D, Cox CE. The specific role of isoflavones on estrogen metabolism in premenopausal women. Cancer. 2002;94:1166. doi: 10.1002/cncr.10320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cao Y, Calafat AM, Doerge DR, Umbach DM, Bernbaum JC, Twaddle NC, Ye X, Rogan WJ. Isoflavones in urine, saliva, and blood of infants: data from a pilot study on the estrogenic activity of soy formula. J Expos Sci Environ Epidemiol. 2008;19:223–234. doi: 10.1038/jes.2008.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Penza M, Montani C, Romani A, Vignolini P, Ciana P, Maggi A, Pampaloni B, Caimi L, Di Lorenzo D. Genistein Accumulates in Body Depots and Is Mobilized during Fasting, Reaching Estrogenic Levels in Serum that Counter the Hormonal Actions of Estradiol and Organochlorines. Toxicol Sci. 2007;97:299–307. doi: 10.1093/toxsci/kfm036. [DOI] [PubMed] [Google Scholar]
- 79.Mersereau JE, Levy N, Staub RE, Baggett S, Zogric T, Chow S, Ricke WA, Tagliaferri M, Cohen I, Bjeldanes LF, Leitman DC. Liquiritigenin is a plant-derived highly selective estrogen receptor beta agonist. Mol Cell Endocrinol. 2008;283:49–57. doi: 10.1016/j.mce.2007.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Escande A, Pillon A, Servant N, Cravedi JP, Larrea F, Muhn P, Nicolas JC, Cavaillès V, Balaguer P. Evaluation of ligand selectivity using reporter cell lines stably expressing estrogen receptor alpha or beta. Biochem Pharmacol. 2006;71:1459. doi: 10.1016/j.bcp.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 81.Manas ES, Xu ZB, Unwalla RJ, Somers WS. Understanding the selectivity of genistein for human estrogen receptor-[beta] using X-ray crystallography and computational methods. Structure. 2004;12:2197–2207. doi: 10.1016/j.str.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 82.An J, Tzagarakis-Foster C, Scharschmidt TC, Lomri N, Leitman DC. Estrogen receptor β-selective transcriptional activity and recruitment of coregulators by phytoestrogens. J Biol Chem. 2001;276:17808–17814. doi: 10.1074/jbc.M100953200. [DOI] [PubMed] [Google Scholar]
- 83.Nguyen LP, Bradfield CA. The search for endogenous activators of the aryl hydrocarbon receptor. Chem Res Toxicol. 2007;21:102. doi: 10.1021/tx7001965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Beischlag T, Morales JL, Hollingshead B, Perdew G. The aryl hydrocarbon receptor complex and the control of gene expression. Crit Rev Eukaryot Gene Expr. 2008;18:207–250. doi: 10.1615/critreveukargeneexpr.v18.i3.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Safe S, Wormke M. Inhibitory aryl hydrocarbon receptor α estrogen receptor β cross-talk and mechanisms of action. Chemical Research in Toxicology. 2003;16:807. doi: 10.1021/tx034036r. [DOI] [PubMed] [Google Scholar]
- 86.Burbach KM, Poland A, Bradfield CA. Cloning of the Ah-receptor cDNA reveals a distinctive ligand-activated transcription factor. Proc Natl Acad Sci U S A. 1992;89:8185–8189. doi: 10.1073/pnas.89.17.8185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Perdew GH. Association of the Ah receptor with the 90-kDa heat shock protein. J Biol Chem. 1988;263:13802–13805. [PubMed] [Google Scholar]
- 88.Wilhelmsson A, Cuthill S, Denis M, Wikström AC, Gustafsson J, Poellinger L. The specific DNA binding activity of the dioxin receptor is modulated by the 90 kd heat shock protein. EMBO J. 1990;9:69–76. doi: 10.1002/j.1460-2075.1990.tb08081.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kazlauskas A, Poellinger L, Pongratz I. Evidence That the Co-chaperone p23 Regulates Ligand Responsiveness of the Dioxin (Aryl Hydrocarbon) Receptor. J Biol Chem. 1999;274:13519–13524. doi: 10.1074/jbc.274.19.13519. [DOI] [PubMed] [Google Scholar]
- 90.Knoblauch R, Garabedian MJ. Role for Hsp90-Associated Cochaperone p23 in Estrogen Receptor Signal Transduction. Mol Cell Biol. 1999;19:3748–3759. doi: 10.1128/mcb.19.5.3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Reyes H, Reisz-Porszasz S, Hankinson O. Identification of the Ah Receptor Nuclear Translocator Protein (Arnt) as a Component of the DNA Binding Form of the Ah Receptor. Science. 1992;256:1193–1195. doi: 10.1126/science.256.5060.1193. [DOI] [PubMed] [Google Scholar]
- 92.Hankinson O. Role of coactivators in transcriptional activation by the aryl hydrocarbon receptor. Arch Biochem Biophys. 2005;433:379. doi: 10.1016/j.abb.2004.09.031. [DOI] [PubMed] [Google Scholar]
- 93.Whitlock JP. Induction of cytochrome P4501A1. Annu Rev Pharmacol Toxicol. 2003;39:103. doi: 10.1146/annurev.pharmtox.39.1.103. [DOI] [PubMed] [Google Scholar]
- 94.Ohtake F, Takeyama K-i, Matsumoto T, Kitagawa H, Yamamoto Y, Nohara K, Tohyama C, Krust A, Mimura J, Chambon P, Yanagisawa J, Fujii-Kuriyama Y, Kato S. Modulation of oestrogen receptor signalling by association with the activated dioxin receptor. Nature. 2003;423:545. doi: 10.1038/nature01606. [DOI] [PubMed] [Google Scholar]
- 95.Shipley JM, Waxman DJ. Aryl hydrocarbon receptor-independent activation of estrogen receptor-dependent transcription by 3-methycholanthrene. Toxicol Appl Pharmacol. 2006;213:87. doi: 10.1016/j.taap.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 96.Safe S, Astroff B, Harris M, Zacharewski T, Dickerson R, Romkes M, Biegel L. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) and related compounds as antioestrogens: Characterization and mechanism of action. Pharmacol Toxicol. 1991;69:400–409. doi: 10.1111/j.1600-0773.1991.tb01321.x. [DOI] [PubMed] [Google Scholar]
- 97.Astroff B, Rowlands C, Dickerson R, Safe S. 2,3,7,8-Tetrachlorodibenzo-p-dioxin inhibition of 17 beta-estradiol-induced increases in rat uterine epidermal growth factor receptor binding activity and gene expression. Mol Cell Endocrinol. 1990;72:247–252. doi: 10.1016/0303-7207(90)90149-3. [DOI] [PubMed] [Google Scholar]
- 98.Zacharewski TR, Bondy KL, McDonell P, Wu ZF. Antiestrogenic effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin on 17β-estradiol-induced pS2 expression. Cancer Res. 1994;54:2707–2713. [PubMed] [Google Scholar]
- 99.DuSell CD, Nelson ER, Wittmann BM, Fretz JA, Kazmin D, Thomas RS, Pike JW, McDonnell DP. Regulation of aryl hydrocarbon receptor function by selective estrogen receptor modulators. Mol Endocrinol. 2010;24:33–46. doi: 10.1210/me.2009-0339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Amakura Y, Tsutsumi T, Nakamura M, Kitagawa H, Fujino J, Sasaki K, Toyoda M, Yoshida T, Maitani T. Activation of the aryl hydrocarbon receptor by some vegetable constituents determined using in vitro reporter gene assay. Biol Pharm Bull. 2003;26:532–539. doi: 10.1248/bpb.26.532. [DOI] [PubMed] [Google Scholar]
- 101.Zhang S, Qin C, Safe S. Flavonoids as aryl hydrocarbon receptor agonists/antagonists: Effects of structure and cell context. Environ Health Perspect. 2003;111:1877–1882. doi: 10.1289/ehp.6322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Murray IA, Morales JL, Flaveny CA, DiNatale BC, Chiaro C, Gowdahalli K, Amin S, Perdew GH. Evidence for Ligand-Mediated Selective Modulation of Aryl Hydrocarbon Receptor Activity. Mol Pharmacol. 2010;77:247–254. doi: 10.1124/mol.109.061788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Swedenborg E, Rüegg J, Hillenweck A, Rehnmark S, Faulds MH, Zalko D, Pongratz I, Pettersson K. 3-Methylcholanthrene Displays Dual Effects on Estrogen Receptor (ER) α and ERβ Signaling in a Cell-Type Specific Fashion. Mol Pharmacol. 2008;73:575–586. doi: 10.1124/mol.107.036384. [DOI] [PubMed] [Google Scholar]
- 104.Abdelrahim M, Smith R, Safe S. Aryl Hydrocarbon Receptor Gene Silencing with Small Inhibitory RNA Differentially Modulates Ah-Responsiveness in MCF-7 and HepG2 Cancer Cells. Mol Pharmacol. 2003;63:1373–1381. doi: 10.1124/mol.63.6.1373. [DOI] [PubMed] [Google Scholar]
- 105.Spink DC, Hayes CL, Young NR, Christou M, Sutter TR, Jefcoate CR, Gierthy JF. The effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on estrogen metabolism in MCF-7 breast cancer cells: Evidence for induction of a novel 17[beta]-estradiol 4-hydroxylase. J Steroid Biochem Mol Biol. 1994;51:251–258. doi: 10.1016/0960-0760(94)90037-x. [DOI] [PubMed] [Google Scholar]
- 106.Spink DC, Lincoln DW, Dickerman HW, Gierthy JF. 2,3,7,8-Tetrachlorodibenzo-p-dioxin causes an extensive alteration of 17 beta-estradiol metabolism in MCF-7 breast tumor cells. Proc Natl Acad Sci U S A. 1990;87:6917–6921. doi: 10.1073/pnas.87.17.6917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Krishnan V, Porter W, Santostefano M, Wang X, Safe S. Molecular mechanism of inhibition of estrogen-induced cathepsin D gene expression by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in MCF-7 cells. Mol Cell Biol. 1995;15:6710–6719. doi: 10.1128/mcb.15.12.6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ahmed S, Valen E, Sandelin A, Matthews J. Dioxin increases the interaction between aryl hydrocarbon receptor and estrogen receptor alpha at human promoters. Toxicol Sci. 2009;111:254–266. doi: 10.1093/toxsci/kfp144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Duan R, Porter W, Samudio I, Vyhlidal C, Kladde M, Safe S. Transcriptional Activation of c-fos Protooncogene by 17{beta}-Estradiol: Mechanism of Aryl Hydrocarbon Receptor-Mediated Inhibition. Mol Endocrinol. 1999;13:1511–1521. doi: 10.1210/mend.13.9.0338. [DOI] [PubMed] [Google Scholar]
- 110.Gillesby BE, Stanostefano M, Porter W, Safe S, Wu ZF, Zacharewski TR. Identification of a motif within the 5′ regulatory region of pS2 which is responsible for AP-1 binding and TCDD-mediated suppression. Biochemistry (Mosc) 1997;36:6080–6089. doi: 10.1021/bi962131b. [DOI] [PubMed] [Google Scholar]
- 111.Porter W, Wang F, Duan R, Qin C, Castro-Rivera E, Kim K, Safe S. Transcriptional activation of heat shock protein 27 gene expression by 17beta-estradiol and modulation by antiestrogens and aryl hydrocarbon receptor agonists. J Mol Endocrinol. 2001;26:31–42. doi: 10.1677/jme.0.0260031. [DOI] [PubMed] [Google Scholar]
- 112.Kumar MB, Tarpey RW, Perdew GH. Differential recruitment of coactivator RIP140 by Ah and estrogen receptors: Absence of a role for LXXLL motifs. J Biol Chem. 1999;274:22155–22164. doi: 10.1074/jbc.274.32.22155. [DOI] [PubMed] [Google Scholar]
- 113.Beischlag TV, Wang S, Rose DW, Torchia J, Reisz-Porszasz S, Muhammad K, Nelson WE, Probst MR, Rosenfeld MG, Hankinson O. Recruitment of the NCoA/SRC-1/p160 Family of Transcriptional Coactivators by the Aryl Hydrocarbon Receptor/Aryl Hydrocarbon Receptor Nuclear Translocator Complex. Mol Cell Biol. 2002;22:4319–4333. doi: 10.1128/MCB.22.12.4319-4333.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Brunnberg S, Pettersson K, Rydin E, Matthews J, Hanberg A, Pongratz I. The basic helix–loop–helix–PAS protein ARNT functions as a potent coactivator of estrogen receptor-dependent transcription. Proc Natl Acad Sci U S A. 2003;100:6517–6522. doi: 10.1073/pnas.1136688100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ruegg J, Swedenborg E, Wahlstrom D, Escande A, Balaguer P, Pettersson K, Pongratz I. The transcription factor aryl hydrocarbon receptor nuclear translocator functions as an estrogen receptor {beta}-selective coactivator, and its recruitment to alternative pathways mediates antiestrogenic effects of dioxin. Mol Endocrinol. 2008;22:304–316. doi: 10.1210/me.2007-0128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sanderson JT. The Steroid Hormone Biosynthesis Pathway as a Target for Endocrine-Disrupting Chemicals. Toxicol Sci. 2006;94:3–21. doi: 10.1093/toxsci/kfl051. [DOI] [PubMed] [Google Scholar]
- 117.Miller WL. Molecular Biology of Steroid Hormone Synthesis. Endocr Rev. 1988;9:295–318. doi: 10.1210/edrv-9-3-295. [DOI] [PubMed] [Google Scholar]
- 118.Ghayee H, Auchus R. Basic concepts and recent developments in human steroid hormone biosynthesis. Rev Endocr and Metab Disord. 2007;8:289–300. doi: 10.1007/s11154-007-9052-2. [DOI] [PubMed] [Google Scholar]
- 119.Nebert DW, Russell DW. Clinical importance of the cytochromes P450. The Lancet. 2002;360:1155. doi: 10.1016/S0140-6736(02)11203-7. [DOI] [PubMed] [Google Scholar]
- 120.Tsuchiya Y, Nakajima M, Yokoi T. Cytochrome P450-mediated metabolism of estrogens and its regulation in human. Cancer Letters. 2005;227:115. doi: 10.1016/j.canlet.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 121.Pinter A, Torok G, Borzsonyi M, Surjan A, Csik M, Kelecsenyi Z, Kocsis Z. Long-term carcinogenicity bioassay of the herbicide atrazine in F344 rats. Neoplasma. 1990;37:533–544. [PubMed] [Google Scholar]
- 122.Wetzel LT, Luempert LG, III, Breckenridge CB, Tisdel MO, Stevens JT, Thakur AK, Extrom PJ, Eldridge JC. Chronic effects of atrazine on estrus and mammary tumor formation in female Sprague-Dawley and Fischer 344 rats. J Toxicol Environ Health. 1994;43:169–182. doi: 10.1080/15287399409531913. [DOI] [PubMed] [Google Scholar]
- 123.Sanderson JT, Boerma J, Lansbergen GWA, van den Berg M. Induction and inhibition of aromatase (CYP19) activity by various classes of pesticides in H295R human adrenocortical carcinoma cells. Toxicol Appl Pharmacol. 2002;182:44–54. doi: 10.1006/taap.2002.9420. [DOI] [PubMed] [Google Scholar]
- 124.Laville N, Balaguer P, Brion F, Hinfray N, Casellas C, Porcher JM, Aït-Aïssa S. Modulation of aromatase activity and mRNA by various selected pesticides in the human choriocarcinoma JEG-3 cell line. Toxicology. 2006;228:98–108. doi: 10.1016/j.tox.2006.08.021. [DOI] [PubMed] [Google Scholar]
- 125.Xu X, Duncan AM, Merz BE, Kurzer MS. Effects of soy isoflavones on estrogen and phytoestrogen metabolism in premenopausal women. Cancer Epidemiol Biomarkers Prev. 1998;7:1101–1108. [PubMed] [Google Scholar]
- 126.Moon YJ, Wang X, Morris ME. Dietary flavonoids: Effects on xenobiotic and carcinogen metabolism. Toxicol In Vitro. 2006;20:187–210. doi: 10.1016/j.tiv.2005.06.048. [DOI] [PubMed] [Google Scholar]
- 127.Kao Y, Zhou C, Sherman M, Laughton C, Chen S. Molecular basis of the inhibition of human aromatase (estrogen synthetase) by flavone and isoflavone phytoestrogens: A site-directed mutagenesis study. Environ Health Perspect. 1998;106:85–92. doi: 10.1289/ehp.9810685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.van Meeuwen JA, Nijmeijer S, Mutarapat T, Ruchirawat S, de Jong PC, Piersma AH, van den Berg M. Aromatase inhibition by synthetic lactones and flavonoids in human placental microsomes and breast fibroblasts -- A comparative study. Toxicol Appl Pharmacol. 2008;228:269–276. doi: 10.1016/j.taap.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 129.Hayes CL, Spink DC, Spink BC, Cao JQ, Walker NJ, Sutter TR. 17 beta-estradiol hydroxylation catalyzed by human cytochrome P450 1B1. Proc Natl Acad Sci U S A. 1996;93:9776–9781. doi: 10.1073/pnas.93.18.9776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hammond DK, Zhu BT, Wang MY, Ricci MJ, Liehr JG. Cytochrome P450 metabolism of estradiol in hamster liver and kidney. Toxicol Appl Pharmacol. 1997;145:54. doi: 10.1006/taap.1997.8167. [DOI] [PubMed] [Google Scholar]
- 131.Belous AR, Hachey DL, Dawling S, Roodi N, Parl FF. Cytochrome P450 1B1-mediated estrogen metabolism results in estrogen-deoxyribonucleoside adduct formation. Cancer Res. 2007;67:812–817. doi: 10.1158/0008-5472.CAN-06-2133. [DOI] [PubMed] [Google Scholar]
- 132.Bolton JL, Thatcher GRJ. Potential mechanisms of estrogen quinone carcinogenesis. Chem Res Toxicol. 2007;21:93–101. doi: 10.1021/tx700191p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.OECD. Report of the First Meeting of the OECD Endocrine Disrupter Testing and Assessment (EDTA) Working Group. OECD; Paris: 1998. [Google Scholar]
- 134.EDSTAC. Endocrine disruptor screening and testing advisory committee final report, 1998. U. S. Environmental Protection Agency; 1998. [Google Scholar]
- 135.EDSP. Endocrine Disruptor Screening Program (EDSP); Announcing the Availability of the Tier 1 Screening Battery and Related Test Guidelines. U S Environmental Protection Agency; 2009. pp. 54415–54422. [Google Scholar]
- 136.Stokes WS. Selecting appropriate animal models and experimental designs for endocrine disruptor research and testing studies. ILAR J. 2004;45:387–393. doi: 10.1093/ilar.45.4.387. [DOI] [PubMed] [Google Scholar]
- 137.Sonneveld E, Riteco JAC, Jansen HJ, Pieterse B, Brouwer A, Schoonen WG, van der Burg B. Comparison of In Vitro and In Vivo Screening Models for Androgenic and Estrogenic Activities. Toxicol Sci. 2006;89:173–187. doi: 10.1093/toxsci/kfj009. [DOI] [PubMed] [Google Scholar]
- 138.Bolger R, Wiese TE, Ervin K, Nestich S, Checovich W. Rapid screening of environmental chemicals for estrogen receptor binding capacity. Environ Health Perspect. 1998;106:551–557. doi: 10.1289/ehp.98106551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Tamrazi A, Carlson KE, Daniels JR, Hurth KM, Katzenellenbogen JA. Estrogen receptor dimerization: Ligand binding regulates dimer affinity and dimer dissociation rate. Mol Endocrinol. 2002;16:2706–2719. doi: 10.1210/me.2002-0250. [DOI] [PubMed] [Google Scholar]
- 140.Liu J, Knappenberger KS, Kack H, Andersson G, Nilsson E, Dartsch C, Scott CW. A homogeneous in vitro functional assay for estrogen receptors: Coactivator recruitment. Mol Endocrinol. 2003;17:346–355. doi: 10.1210/me.2002-0331. [DOI] [PubMed] [Google Scholar]
- 141.Weatherman RV, Chang CY, Clegg NJ, Carroll DC, Day RN, Baxter JD, McDonnell DP, Scanlan TS, Schaufele F. Ligand-selective interactions of ER detected in living cells by fluorescence resonance energy transfer. Mol Endocrinol. 2002;16:487–496. doi: 10.1210/mend.16.3.0813. [DOI] [PubMed] [Google Scholar]
- 142.Xu X, Soutto M, Xie Q, Servick S, Subramanian C, von Arnim AG, Johnson CH. Imaging protein interactions with bioluminescence resonance energy transfer (BRET) in plant and mammalian cells and tissues. Proc Natl Acad Sci U S A. 2007;104:10264–10269. doi: 10.1073/pnas.0701987104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Powell E, Huang S-X, Xu Y, Rajski SR, Wang Y, Peters N, Guo S, Eric Xu H, Hoffmann FM, Shen B, Xu W. Identification and characterization of a novel estrogenic ligand actinopolymorphol A. Biochem Pharmacol. 2010 doi: 10.1016/j.bcp.2010.06.030. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wilson VS, Bobseine K, Gray LE., Jr Development and characterization of a cell line that stably expresses an estrogen-responsive luciferase reporter for the detection of estrogen receptor agonist and antagonists. Toxicol Sci. 2004;81:69–77. doi: 10.1093/toxsci/kfh180. [DOI] [PubMed] [Google Scholar]
- 145.Balaguer P, Francois F, Comunale F, Fenet H, Boussioux A, Pons M, Nicolas J, Casellas C. Reporter cell lines to study the estrogenic effects of xenoestrogens. Sci Total Environ. 1999;233:47–56. doi: 10.1016/s0048-9697(99)00178-3. [DOI] [PubMed] [Google Scholar]
- 146.Lemaire G, Mnif W, Mauvais P, Balaguer P, Rahmani R. Activation of [alpha]-and [beta]-estrogen receptors by persistent pesticides in reporter cell lines. Life Sci. 2006;79:1160–1169. doi: 10.1016/j.lfs.2006.03.023. [DOI] [PubMed] [Google Scholar]
- 147.Barkhem T, Carlsson B, Nilsson Y, Enmark E, Gustafsson J-Ãk, Nilsson S. Differential response of estrogen receptor α and estrogen receptor β to partial estrogen agonists/antagonists. Mol Pharmacol. 1998;54:105–112. doi: 10.1124/mol.54.1.105. [DOI] [PubMed] [Google Scholar]
- 148.Li J, Li N, Ma M, Giesy JP, Wang Z. In vitro profiling of the endocrine disrupting potency of organochlorine pesticides. Toxicol Lett. 2008;183:65–71. doi: 10.1016/j.toxlet.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 149.Leusch F, de Jager C, Levi Y, Lim R, Puijker L, Sacher F, Tremblay L, Wilson V, HFC Comparison of five in vitro bioassays to measure estrogenic activity in environmental waters. Env Sci Tech. 2010;44:3853–3860. doi: 10.1021/es903899d. [DOI] [PubMed] [Google Scholar]
- 150.Silva E, Scholze M, Kortenkamp A. Activity of xenoestrogens at nanomolar concentrations in the E-Screen assay. Environ Health Perspect. 2007;115:91–97. doi: 10.1289/ehp.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Soto AM, Sonnenschein C, Chung KL, Fernandez MF, Olea N, Serrano FO. The E-SCREEN Assay as a tool to identify estrogens: An update on estrogenic environmental pollutants. Environ Health Perspect. 1995;103:113–122. doi: 10.1289/ehp.95103s7113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zhang X, Yu RMK, Jones PD, Lam GKW, Newsted JL, Gracia T, Hecker M, Hilscherova K, Sanderson JT, Wu RSS, Giesy JP. Quantitative RT-PCR methods for evaluating toxicant-induced effects on steroidogenesis using the H295R cell line. Env Sci Tech. 2005;39:2777–2785. doi: 10.1021/es048679k. [DOI] [PubMed] [Google Scholar]
- 153.Letcher RJ, van Holsteijn I, Drenth HJ, Norstrom RJ, Bergman Å, Safe S, Pieters R, van den Berg M. Cytotoxicity and aromatase (CYP19) activity modulation by organochlorines in human placental JEG-3 and JAR choriocarcinoma cells. Toxicol Appl Pharmacol. 1999;160:10–20. doi: 10.1006/taap.1999.8746. [DOI] [PubMed] [Google Scholar]
- 154.Dai SY, Chalmers MJ, Bruning J, Bramlett KS, Osborne HE, Montrose-Rafizadeh C, Barr RJ, Wang Y, Wang M, Burris TP, Dodge JA, Griffin PR. Prediction of the tissue-specificity of selective estrogen receptor modulators by using a single biochemical method. Proc Natl Acad Sci U S A. 2008;105:7171–7176. doi: 10.1073/pnas.0710802105. [DOI] [PMC free article] [PubMed] [Google Scholar]