

Abstract
Objective
Rheumatoid arthritis (RA) is classically thought of as a Th1, T lymphocyte–driven disease of the adaptive immune system. However, cells of the innate immune system, including neutrophils, are prevalent within the diseased joint, and accumulate in large numbers. This study was undertaken to determine whether cells of the rheumatoid stromal microenvironment could establish an inflammatory environment in which endothelial cells are conditioned in a disease-specific manner to support neutrophil recruitment.
Methods
Human umbilical vein endothelial cells (ECs) and fibroblasts isolated from the synovium or skin of RA patients were established in coculture on opposite sides of porous transwell filters. After 24 hours of EC conditioning, the membranes were incorporated into a parallel-plate, flow-based adhesion assay and levels of neutrophil adhesion to ECs were measured.
Results
ECs cocultured with synovial, but not skin, fibroblasts could recruit neutrophils in a manner that was dependent on the number of fibroblasts. Antibody blockade of P-selectin or E-selectin reduced neutrophil adhesion, and an antibody against CD18 (the β2 integrin) abolished adhesion. Blockade of CXCR2, but not CXCR1, also greatly inhibited neutrophil recruitment. Interleukin-6 (IL-6) was detectable in coculture supernatants, and both IL-6 and neutrophil adhesion were reduced in a dose-dependent manner by hydrocortisone added to cocultures. Antibody blockade of IL-6 also effectively abolished neutrophil adhesion.
Conclusion
Synovial fibroblasts from the rheumatoid joint play an important role in regulating the recruitment of inflammatory leukocytes during active disease. This process may depend on a previously unsuspected route of IL-6–mediated crosstalk between fibroblasts and endothelial cells.
The recruitment of blood-borne leukocytes by endothelial cells (ECs) lining the blood vessels, typically postcapillary venules, is an early step in the inflammatory response (1). It is a process that is down-regulated in acute or resolving inflammation, but appears to continue in a disease-regulated manner in chronic inflammatory diseases (2). For example, in rheumatoid arthritis (RA), lymphocytes (B and T), monocytes, and neutrophils are recruited over protracted periods into the diseased joint. This dysregulation of the inflammatory response is probably related to the imposition of a disease-specific, continuously activated phenotype on the ECs of the joint vasculature (3). The mechanisms that regulate long-term EC activation and leukocyte accumulation in diseased tissue are poorly understood, but it is probable that part of the process is supported by cytokines and chemokines released by stromal cells of the affected tissue.
A number of studies have demonstrated the ability of stromal cells to alter the local microenvironment in a manner that contributes to disease. For example, in a model of atherosclerosis, smooth muscle cells could sensitize cocultured ECs in a manner dependent on transforming growth factor β1 so that the adhesion of neutrophils greatly increased in response to EC activation by tumor necrosis factor α (TNFα) (4,5). Fibroblasts and corneal stromal cells (keratocytes) alter the local microenvironment by inhibiting the release of the antiinflammatory cytokines interleukin-4 (IL-4) and IL-10 from activated T cells in a mechanism that enhances protective immune reactions in the anterior segment of the eye (6). Of particular relevance, there is growing evidence that synovial fibroblasts contribute to inflammatory processes that underpin RA and assist in the destruction of bone and cartilage by, for example, changing phenotypes so that they generate inflammatory cytokines and matrix metalloproteinases (7-14). An important alteration in the rheumatoid microenvironment is the high level of expression of IL-6 (15), IL-1, and TNFα (16). However, the source of these agents in the complex cellular environment of the arthritic joint remains unknown.
In clinical practice, various treatments target the production of proinflammatory cytokines. Such therapies include systemic administration of hydrocortisone or its analogs, which have been commonly used for more than 50 years. However, while short-term relief of symptoms is apparent, the long-term effects of this treatment are detrimental and the mechanism(s) of action are unclear. They may include down-regulation of proinflammatory cytokines and promotion of the synthesis of antiinflammatory cytokines and the inhibition of leukocyte adhesion (17-20). Other treatments directly target proinflammatory cytokines. Thus, anti-TNFα therapy has shown good efficacy in the reduction of symptoms associated with active rheumatoid disease, and some studies suggest that blocking the function of IL-1 protects bone and cartilage (21). However, it is still unclear whether reducing the bioactivity of these cytokines reduces EC activation and thereby the recruitment of inflammatory leukocytes. Additionally, 30% of patients treated with anti-TNFα do not respond to treatment (22), strongly implying that there are other mechanisms that promote chronic inflammation within the rheumatoid environment.
Because the spatial and temporal complexity of the rheumatoid environment makes it extremely difficult to examine the detail of the inflammatory processes that occur in vivo, we developed an in vitro coculture system that reconstitutes aspects of the rheumatoid stromal microenvironment and allows us to investigate the regulation of inflammation in this environment. This allowed us to test the hypothesis that synovial fibroblasts are imprinted with a proinflammatory phenotype that can promote the recruitment of leukocytes by activating cocultured ECs. Here we show that a previously unsuspected process of crosstalk involving IL-6 signaling occurs between synovial fibroblasts and vascular ECs, which results in up-regulation of adhesion molecules and chemokines that support neutrophil recruitment.
MATERIALS AND METHODS
Neutrophil preparation
Blood from healthy adult volunteers was collected and placed into EDTA (1.6 mg/ml) in accordance with local ethical guidelines and with the approval of the South Birmingham Local Research Ethics Committee. Neutrophils were separated using 2-step density gradients of Histopaque 1119 and 1077 (all reagents were obtained from Sigma, Poole, UK, unless otherwise stated), as previously described (1,2). Neutrophils were >95% pure based on volume distribution, which was determined using a Coulter Multisizer II (Beckman Coulter, Fullerton, CA).
Isolation and culture of fibroblasts and ECs
Matched synovium and skin tissue was obtained during total knee arthroplasty from consenting patients who fulfilled the American College of Rheumatology (formerly, the American Rheumatism Association) criteria for RA (23). Fibroblasts were isolated by morselization of tissues, followed by dissociation in 5mM EDTA for 2 hours. Dissociated tissue was washed and transferred to culture flasks in RPMI 1640 medium supplemented with 20% fetal calf serum (FCS), 1% nonessential amino acids, 1% (100 mM) sodium pyruvate, 2 mM glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin. Once cells had attached, excess cells and tissue were removed. Medium was changed every 4 days until near-confluence, at which point cells were subcultured. Fibroblast phenotypes were confirmed by positive staining for prolyl-4-hydroxylase and fibronectin (Figure 1), and negative staining for contaminating cell populations using CD14, CD68, CD31, von Willebrand factor, and cytokeratin. Fibroblasts thus obtained from patient tissue biopsy samples were used between passages 3 and 10 for coculture experiments.
Figure 1.

Confirmation of fibroblast phenotype. Fibroblasts from 4 patients with rheumatoid arthritis (RA) were isolated from synovium (RASY) or skin (RADF) obtained at total knee arthroplasty. Cells were stained for the fibroblast marker fibronectin.
Human umbilical vein ECs were enzymatically isolated and cultured in medium (Medium 199; Invitrogen, Paisley, UK) containing 28 μg/ml gentamicin, 20% FCS, 1 μg/ml hydrocortisone, and 10 ng/ml epidermal growth factor, as previously described (24). ECs were grown to confluence in 25-cm2 culture flasks (BD Falcon, Oxford, UK) precoated with 1% gelatin solution (Sigma).
Preparation of coculture inserts
ECs and fibroblasts were cocultured on opposite sides of porous polyethylene terepthalate inserts (Becton Dickinson, Franklin Lakes, NJ) (Figure 2A). The membranes on the inserts had an effective culture area of 0.3 cm2, a pore size of 0.4 μm, and a pore density of 1 × 108/cm2. Both fibroblasts and ECs were dispersed in 0.02% EDTA–2.5 mg/ml−1 trypsin. The cells were washed with RPMI 1640 and centrifuged for 5 minutes at 400g followed by resuspension in fibroblast culture medium. After being counted with a hemocytometer, between 104 and 105 fibroblasts were added to the inside of the inserts and cultured for 24 hours. ECs were added to the opposite side of the inserts containing fibroblasts, or to empty inserts as controls, as previously described (4,5), and at a concentration that allowed a confluent monolayer after the cells were attached to and spread on the membrane. ECs were conditioned by coculture for 24 hours prior to flow-based adhesion assay. In some experiments, reagents that inhibited EC-activating agents were incorporated at the initiation of coculture.
Figure 2.

Assembly of coculture inserts. A, A porous polyethylene terepthalate culture insert on which cocultures were established by growing fibroblast and endothelial cells (ECs) on opposite sides of the porous membrane. B, The parallel-plate, flow-based adhesion assay assembled with coculture in situ. C, The components of the assay included an upper Perspex plate (i) that had a machined recess to accept a glass coverslip (ii), which formed the upper surface of the flow channel. A silicone gasket (iii) defined the depth of the flow channel (150 μm). The lower acetal plastic plate (iv) formed the lower surface of the flow channel and had a recess into which the culture insert fit so that the ECs also formed part of the lower surface of the flow channel. The insert (v) was held in place by a Perspex plate (vi) and a smaller silicone gasket (vii). Inlet and outlet conduits (viii) allowed the flow of leukocyte suspension or wash buffer through the flow channel. The plates were held together firmly by locating screws (ix and x).
Parallel-plate flow-based adhesion assay
Flow-based adhesion experiments were conducted in a parallel-plate assay adapted from one that was previously described (4,5). The system was composed of an upper Perspex plate (Figure 2B) that contained a recess to accept a glass coverslip which formed the upper wall of the flow channel, and through which microscopic observations were made. The lower plate, which formed the bottom of the flow channel, was separated from the upper plate by a noncompressible silicone gasket that defined the depth of the flow channel (150 μm). Inlet and outlet conduits allowed the flow of medium that contained neutrophils. The lower plate was engineered so that inserts with cocultures could be incorporated into the flow channel. When the insert was in situ, the EC monolayer formed the bottom of the flow channel, where the insert protruded through the lower plate. The parallel plates and gasket were sealed with hand-tightened screws so that medium could pass through the flow channel unhindered. The insert was held firmly in the lower plate by a small sealing plate and gasket, which were held in place with 4 locating screws.
The parallel-plate apparatus was placed on the stage of an upright fluorescence microscope (BX61 using an LCPlanFL 20× objective lens, numerical aperture of 0.4; Olympus, Melville, NY), and silicone tubing connected the inlet conduit to a microelectronic switching valve that regulated the choice between neutrophil suspension and wash buffer. The outlet conduit was connected to a Harvard syringe pump that drew medium through the plate and across the ECs at a wall shear stress of 0.1 Pa. The system was maintained at 37°C using a thermostatically controlled Perspex surround, which enclosed the microscope stage, sample holder, and parallel-plate system. Prior to measurement, neutrophils suspended in phosphate buffered saline containing 0.1% bovine serum albumin were fluorescently labeled for 15 minutes with 1 μg/ml of the dye bisbenzimide. Neutrophils were perfused at a concentration of 106/ml for 3 minutes, followed by addition of cell-free buffer to remove nonadherent cells. After 2 minutes of wash buffer perfusion, video records were made of 10 microscopic fields of view selected at random along the centerline of the insert. Video records were digitized using analysis software (Image Pro Plus; Media Cybernetics, Silver Spring, MD), adherent cells were counted, and data were expressed as cell number/mm2/106 perfused.
Measurement of soluble IL-6
Soluble IL-6 in supernatants from cultured or cocultured cells was measured using a commercially available sandwich enzyme-linked immunosorbent assay according to the manufacturer’s instructions (R&D Systems, Abingdon, UK). Data were expressed in pg/ml as determined using a calibration curve constructed using recombinant IL-6.
Cell treatments
In some experiments, we included hydrocortisone (2.5 × 10−9−2.5 × 10−3M) or neutralizing antibodies against IL-6 (50 μg/ml−1; IgG1) (clone 6708; R&D Systems), TNFα (50 μg/ml−1; IgG1) (clone 1825.12; R&D Systems), or IL-1β (50 μg/ml−1; IgG1) (clone 8516.311; R&D Systems) when cocultures were established. In experiments to identify the molecules that directly supported neutrophil adhesion and activation, we treated cocultured ECs with rabbit polyclonal antibodies against P-selectin (a gift of Michael Berndt, Baker Medical Research Institute, Melbourne, Australia), E-selectin (1 μg/ml, F[ab’]2 of clone ENA2; Bradshaw Biologicals, Chepshet, UK), or a nonimmune control antibody (25 μg/ml−1, rabbit polyclonal antibody; Dako, High Wycombe, UK) prior to neutrophil perfusion. Alternatively, neutrophils were treated with antibodies against CD18 (β2 integrin) (10 μg/ml−1; IgG1) (clone R6.5E; Genentech, South San Francisco, CA) or CXCR1 or CXCR2 (both 2 μg/ml−1; IgG1) (clones 501 and 19, respectively; BioSource International, Camarillo, CA) prior to perfusion. In some experiments, function-neutralizing antibodies against CXC chemokines (all IgG1 and all used at 10 μg/ml) (clone 33160.111 against epithelial neutrophil–activating peptide 78 [ENA-78], clone 31716.11 against growth-related oncogene α [GROα], clone 6217.111 against IL-8; R&D Systems) were used prior to adhesion assay.
Statistical analysis
Statistical comparisons of individual treatments were performed using Student’s paired 2-tailed t-test. Effects of hydrocortisone dosage were tested using analysis of covariance followed by Student’s paired t-test, where appropriate. Correlations between IL-6 levels in coculture medium and the levels of neutrophil adhesion supported by cocultured ECs were calculated using Pearson’s correlation test assuming a Gaussian distribution. P values less than or equal to 0.05 (2-sided) were considered significant.
RESULTS
Adhesion of flowing neutrophils supported by ECs cocultured with synovial fibroblasts
When ECs were cultured for 24 hours with fibroblasts from the rheumatoid synovium and then incorporated into the parallel-plate flow adhesion assay, the ECs were able to support the adhesion of flowing neutrophils (Figure 3). Importantly, the levels of neutrophil adhesion to ECs cocultured with skin fibroblasts explanted from the same patients were negligible (Figure 3). The levels of adhesion supported by ECs cocultured with different rheumatoid fibroblast explants varied (Figure 3A), although neutrophils were recruited by ECs cocultured with all the synovial fibroblast explants tested (Figures 3A and B). We also examined the effects of varying the number of fibroblasts in coculture with ECs. Fibroblasts at concentrations between 1 × 104/insert and 1 × 105/insert induced a graded increase of neutrophil recruitment to cocultured ECs (Figure 3C). Importantly, skin fibroblasts did not show an ability to support neutrophil adhesion at any density of seeding.
Figure 3.

Adhesion of flowing neutrophils to endothelial cells (ECs) cocultured with fibroblasts. A, A comparison of synovial (RASY) and skin (RADF) fibroblast explants from 5 different patients with rheumatoid arthritis (RA). B, The mean ± SEM response of ECs cultured with fibroblasts from the 5 donors. C, Effects of varying the number of fibroblasts in coculture with ECs. Values are the mean ± SEM in the 5 donors.
Necessity of selectin and β2 integrin receptors and CXC chemokine ENA-78 for neutrophil adhesion
To identify the receptors involved in the initial capture and adhesion of neutrophils, antibodies against endothelial and neutrophil receptors were used at the time of flow assay. The blockade of E-selectin or P-selectin on ECs reduced the levels of neutrophil adhesion to synovial fibroblast/EC cocultures, with the P-selectin blockade more efficient than the blockade of E-selectin (Figure 4A). The effect of combining both antibodies resulted in a further decrease in adhesion, indicating that both E-selectin and P-selectin contributed to the adhesion of neutrophils from flow. Interestingly, the inclusion of a β2 integrin–blocking antibody completely inhibited stable neutrophil adhesion (Figure 4A), which suggests that in the absence of integrin-mediated interactions, neutrophil interactions with the EC monolayer via selectin receptors were transient, and prolonged rolling adhesion was not established.
Figure 4.

Levels of neutrophil adhesion achieved with antibody blockade. A, Effects of anti–E-selectin, anti–P-selectin, a combination of these antibodies, a control antibody, or blockade of CD18 (β2 integrin). B, Neutrophil adhesion levels with the use of anti-CXCR1, anti-CXCR2, or a combination of these antibodies. C, Effects of anti–epithelial neutrophil–activating peptide 78 (ENA-78), growth-related oncogene α (GROα), or interleukin-8 (IL-8) (all CXC chemokines). Values are the mean and SEM from at least 4 experiments using different endothelial cells and fibroblast explants. RASY = fibroblasts isolated from synovium of rheumatoid arthritis patients. * = P < 0.05 versus untreated control, by Student’s paired t-test.
The integrin-mediated adhesion of neutrophils to ECs typically requires activation of the neutrophils. We have previously reported that in a flow-based model using TNFα-activated ECs, activation through the neutrophil chemokine receptor CXCR2 was required for β2 integrin-mediated adhesion (25). Here, the use of an anti-CXCR2 antibody completely inhibited neutrophil adhesion to ECs cocultured with synovial fibroblasts, while an anti-CXCR1 antibody had no consistent effect on neutrophil behavior (Figure 4B). When function-neutralizing antibodies against CXC chemokines were incorporated into the coculture system, neutrophil adhesion was effectively inhibited by an anti–ENA-78 antibody but not by anti-GROα or anti–IL-8 antibodies (Figure 4C), which strongly indicates that a single chemokine (ENA-78), which operated through CXCR2, was responsible for the activation of neutrophils in this system.
Inhibition of EC responses to synovial fibroblasts by hydrocortisone or inhibition of IL-6 activity
The addition of hydrocortisone to the coculture medium over a broad range of concentrations inhibited neutrophil adhesion to ECs cocultured with synovial fibroblasts in a dose-dependent manner (Figure 5A). Because hydrocortisone has been reported to reduce the expression of IL-6 by fibroblasts (26), we measured the concentration of this cytokine in our coculture medium in the presence of different concentrations of hydrocortisone. Hydrocortisone reduced the levels of IL-6 detectable in the coculture medium in a dose-dependent manner (Figure 5B). Furthermore, there was a significant correlation between the levels of IL-6 in the coculture medium and the levels of neutrophil adhesion to ECs cocultured with synovial fibroblasts (Figure 5C), indicating that IL-6 activity in the coculture system might be important in the process of EC activation. To test this hypothesis, at the initiation of coculture we added a function-neutralizing anti–IL-6 antibody. This completely inhibited neutrophil adhesion to the ECs cocultured with synovial fibroblasts (Figure 6). Interestingly, isotype-matched, function-neutralizing antibodies against TNFα or IL-1β had no consistent effect on neutrophil adhesion in the coculture system.
Figure 5.

Role of hydrocortisone and interleukin-6 (IL-6) activity in neutrophil adhesion. A and B, Effects of hydrocortisone on A, the adhesion of neutrophils to ECs cocultured with RASY and RADF, and B, the levels of IL-6 released into the medium by cocultured cells. Values are the mean and SEM from at least 4 experiments using different ECs and fibroblast explants. C, Correlation between levels of IL-6 released into coculture medium and levels of neutrophil adhesion to ECs cocultured with fibroblasts. See Figure 3 for other definitions.
Figure 6.
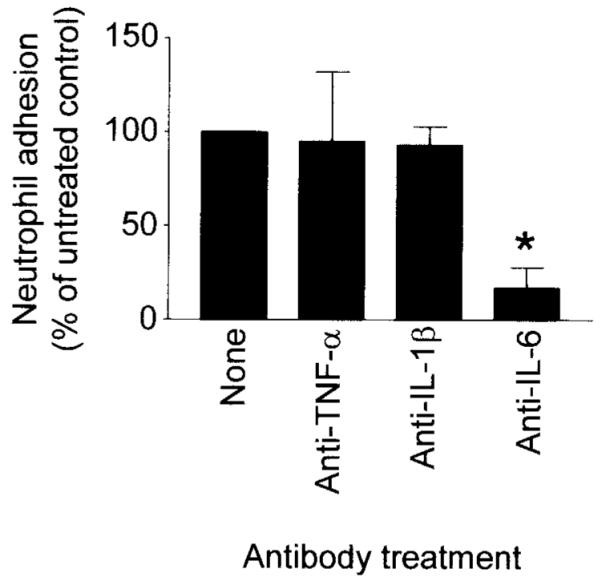
Effect of antibody blockade of the biologic function of interleukin-6 (IL-6), tumor necrosis factor α (TNFα), or IL-1β on neutrophil adhesion. Values are the mean and SEM levels of neutrophil adhesion. * = P < 0.05, by Pearson’s correlation test assuming a Gaussian distribution.
DISCUSSION
The concept that inflammation is regulated by cells of the tissue stroma is well established, although the key regulatory components are usually considered to be highly specialized cells, such as tissue macrophages and mast cells, that have differentiated from leukocytic precursors. Recently it has become clear that cells such as fibroblasts, which were thought to be relatively inert structural components of the tissue, actually play important roles in regulating tissue homeostasis in health and disease. These cells may play a particularly important role in chronic inflammatory diseases (including RA), in which they undergo a disease-specific transition in phenotype, often show hyperplastic responses to the disease environment (leading to degradation of the normal tissue architecture), and help perpetuate disease-associated pathology by promoting inflammation (7-15).
In the current study, we investigated the ability of fibroblasts derived from different tissue sites to regulate the recruitment of flowing neutrophils to cocultured ECs. We tested the hypothesis that fibroblasts derived from the rheumatic joint would have a proinflammatory phenotype conferred on them by residence in chronically inflamed tissue. Using a unique multicellular model of the synovial microenvironment, we identified a previously undescribed interaction between rheumatoid fibroblasts and ECs that promotes the recruitment of flowing neutrophils. The process of cellular crosstalk that supported EC activation depended on IL-6 signaling. Importantly, the ability to promote neutrophil recruitment was restricted to fibroblasts derived from the rheumatoid environment, because fibroblasts from the skin of the same patients were unable to activate ECs.
The adhesion receptors that support the prolonged recruitment of neutrophils into the rheumatoid synovium in vivo remain to be definitively described. However, during active disease, elevated levels of soluble E-selectin and intercellular adhesion molecule 1 (ICAM-1) in the serum have been reported, with the implication that these receptors are shed from the inflamed vasculature of the rheumatoid pannus (27). Paeolog and collegues reported that treatment of patients with anti-TNFα antibody (cA2) reduced the circulating levels of these receptors in a manner that correlated with clinical benefit (28). They proposed that the observed decrease reflected diminished activation of ECs in the synovial microvasculature, which might lead to reduced migration of leukocytes (28). The expression of ICAM-1, E-selectin, and P-selectin has also been demonstrated immunohistochemically in human synovial tissues (29,30). Additionally, in a number of rodent models of resolving arthritis, the use of antibodies targeting E-selectin or P-selectin reduced leukocyte (including neutrophil) recruitment into affected joints (31-33). Taken together, these observations strongly implicate both E-selectin and P-selectin in the recruitment of neutrophils in RA and might also indicate that TNFα plays an important role in the activation of the synovial microvasculature in RA.
In our in vitro model of the RA synovium, we identified roles for both E-selectin and P-selectin in neutrophil recruitment. Interestingly, however, we were able to exclude a role for TNFα in the process of crosstalk between RA fibroblasts and ECs. The identification of a novel route of cellular crosstalk that results in EC activation independent of TNFα signaling has implications for targeted treatment of RA. Approximately 30% of patients do not respond to treatment with anti-TNFα antibodies and it is tempting to speculate that in these individuals, alternative routes of EC activation, such as that demonstrated here, may support the prolonged recruitment of inflammatory leukocytes.
In the current study, we identified a critical role for IL-6 in the regulation of neutrophil adhesion to cocultured ECs. IL-6 was first identified and cloned as the 26-kd B cell differentiation factor (34). However, more recent evidence shows that IL-6 is a pleiotropic agent, which plays an important role in hematopoiesis, the acute-phase response, and in many inflammatory diseases, including RA (35). In RA, IL-6 is required to activate autoreactive T cells, induce the production of rheumatoid factor and other autoreactive antibodies, and promote the acute-phase response (35). In murine models of antigen-induced monarthritis (36), collagen-induced arthritis (37), and spontaneous T cell–mediated chronic autoimmune arthritis (38), knockout of the IL-6 gene significantly reduced joint swelling, synovial infiltration of leukocytes, and bone and cartilage erosion. These effects were reversible in some models upon the infusion of a soluble IL-6 receptor (sIL-6R)/IL-6 fusion protein (36). Moreover, clinical trials in RA patients using humanized anti–IL-6R antibody showed a dose-related reduction in inflammation (39,40).
Thus, IL-6 appears to play a critical role in the processes that typify human and animal arthritic disease. Cognate IL-6 receptors are restricted to hepatocytes, megakaryocytes, leukocytes, and fibroblasts, but have not been reported on ECs (26). However, there is also an sIL-6R, which is proteolytically shed by various leukocyte subsets upon activation (41-44), as well as an alternatively spliced product of the IL-6R gene reported to be secreted exclusively in the inflamed synovium of RA patients. Trans-signaling by sIL-6R requires the formation of a heterodimeric complex of IL-6/sIL-6R and the nonligand binding signal transducer gp130 (45). Signaling via this complex has been shown to promote EC expression of E-selectin, IL-8, ICAM-1, and vascular cellular adhesion molecule 1 (VCAM-1) (37).
In our model of the RA synovium, we did not detect sIL-6R when we screened coculture supernatants (data not shown). Thus, although IL-6 was essential for promoting neutrophil adhesion in our coculture system, it is probable that it did not directly activate ECs. Rather, we propose that IL-6 stimulates fibroblasts, which are known to express IL-6R (46) and which, in the presence of accessory signals derived from the coculture environment, generate an EC-activating agent(s) that is responsible for promoting neutrophil recruitment. Interestingly, we could find no role for TNFα or IL-1β in our system, nor were these agents released into coculture supernatants (data not shown), indicating that the agent(s) responsible for EC activation may be one that is not commonly associated with inflammation in RA.
Hydrocortisone is a potent antiinflammatory steroid that has been used in the treatment of RA for the past 50 years. However, there is still much debate about the mechanisms of this agent in RA. The inhibitory actions of glucocorticoid steroids on the leukocyte adhesion cascade have been previously described. For example, Cronstein et al (47) showed that dexamethasone caused a dose-dependent decrease in neutrophil adhesion to lipopolysaccharide-stimulated ECs in a static assay, and that this was due to a decrease in both E-selectin and ICAM-1 messenger RNA (mRNA) and protein. Dexamethasone also reduced E-selectin mRNA expression in IL-1β- but not TNFα-stimulated ECs in their system (47), although the TNFα- or IL-1β–induced surface expression of E-selectin and VCAM-1 was decreased in another series of experiments (48).
Our own flow-based studies on the regulation of neutrophil adhesion to TNFα-stimulated ECs show that hydrocortisone is ineffective on ECs stimulated with high concentrations of TNFα (100 units/ml) but efficiently inhibits neutrophil adhesion at lower TNFα concentrations (1 unit/ml) (Nash GB, Rainger GE: unpublished observations). Additionally, neutrophil recruitment into inflamed human conjunctiva and P-selectin expression in this tissue were decreased by hydrocortisone treatment (49). Thus, hydrocortisone can have direct effects on ECs and the expression of molecules that support the adhesion of leukocytes.
In the current study we found that the addition of hydrocortisone to the coculture medium reduced neutrophil adhesion dose-dependently and in a manner that correlated closely with a reduction in IL-6 release, an observation that is consistent with the reported ability of synthetic glucocorticoids to inhibit IL-6 expression at the mRNA and protein levels in synovial fibroblasts (26). This observation led us to directly test the hypothesis that neutralizing IL-6 would inhibit the adhesion of neutrophils to cocultured ECs. Thus, although hydrocortisone may directly affect EC expression of adhesion receptors that support neutrophil adhesion, it is also likely that effects on the phenotype of cocultured fibroblasts play an important role in moderating the inflammatory pathways operating in this model.
In conclusion, an important advantage of using in vitro models that recapitulate aspects of the stromal microenvironment is the ability to interfere directly in the crosstalk between specific cell types, a difficult process in the complex cellular environment of the diseased tissue. Using simple inhibitor strategies (e.g., the addition of a function-neutralizing anti–IL-6 antibody), we have been able to demonstrate a previously unsuspected route of fibroblast-EC interaction, which may support the ongoing process of leukocyte recruitment into the rheumatoid microenvironment. Thus, our system may faithfully mimic important in vivo processes involved in leukocyte accumulation within the synovium, and might provide a useful screen for new antiinflammation therapies in RA.
Acknowledgments
Ms Smith’s work was supported by the Biotechnology and Biological Sciences Research Council (grant 02/A4/C/08602). Dr. Lally’s work was supported by the Arthritis Research Campaign (grant B0694). Dr. Rainger’s work was supported by a British Heart Foundation Non-Clinical Senior Lectureship (BS/97001).
REFERENCES
- 1.Dunon D, Piali L, Imhof BA. To stick or not to stick: the new leukocyte homing paradigm. Curr Opin Cell Biol. 1996;8:714–23. doi: 10.1016/s0955-0674(96)80114-1. review. [DOI] [PubMed] [Google Scholar]
- 2.Wong SH, Lord JM. Factors underlying chronic inflammation in rheumatoid arthritis. Arch Immunol Ther Exp (Warsz) 2004;52:379–88. review. [PubMed] [Google Scholar]
- 3.Middleton J, Americh L, Gayon R, Julien D, Aguilar L, Amalric F, et al. Endothelial cell phenotypes in the rheumatoid synovium: activated, angiogenic, apoptotic and leaky. Arthritis Res Ther. 2004;6:60–72. doi: 10.1186/ar1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rainger GE, Nash GB. Cellular pathology of atherosclerosis: smooth muscle cells prime cocultured endothelial cells for enhanced leukocyte adhesion. Circ Res. 2001;88:615–22. doi: 10.1161/01.res.88.6.615. [DOI] [PubMed] [Google Scholar]
- 5.Rainger GE, Stone P, Morland CM, Nash GB. A novel system for investigating the ability of smooth muscle cells and fibroblasts to regulate adhesion of flowing leukocytes to endothelial cells. J Immunol Methods. 2001;255:73–82. doi: 10.1016/s0022-1759(01)00427-6. [DOI] [PubMed] [Google Scholar]
- 6.Holan V, Vitova A, Pindjakova J, Krulova M, Zajicova A, Filipec M. Corneal stromal cells selectively inhibit production of anti–inflammatory cytokines by activated T cells. Clin Exp Immunol. 2004;136:200–6. doi: 10.1111/j.1365-2249.2004.02457.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kontoyiannis D, Kollias G. Fibroblast biology: synovial fibroblasts in rheumatoid arthritis: leading role or chorus line? Arthritis Res. 2000;2:342–3. doi: 10.1186/ar109. review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pap T, Muller-Ladner U, Gay RE, Gay S. Fibroblast biology: role of synovial fibroblasts in the pathogenesis of rheumatoid arthritis. Arthritis Res. 2000;2:361–7. doi: 10.1186/ar113. review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ritchlin C. Fibroblast biology: effector signals released by the synovial fibroblast in arthritis. Arthritis Res. 2000;2:356–60. doi: 10.1186/ar112. review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Konttinen YT, Li TF, Hukkanen M, Ma J, Xu JW, Virtanen I. Fibroblast biology: signals targeting the synovial fibroblast in arthritis. Arthritis Res. 2000;2:348–55. doi: 10.1186/ar111. review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Edwards JC. Fibroblast biology: development and differentiation of synovial fibroblasts in arthritis. Arthritis Res. 2000;2:344–7. doi: 10.1186/ar110. review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perlman H, Bradley K, Liu H, Cole S, Shamiyeh E, Smith RC, et al. IL-6 and matrix metalloproteinase-1 are regulated by the cyclin-dependent kinase inhibitor p21 in synovial fibroblasts. J Immunol. 2003;170:838–45. doi: 10.4049/jimmunol.170.2.838. [DOI] [PubMed] [Google Scholar]
- 13.Dasu MR, Barrow RE, Spies M, Herndon DN. Matrix metalloproteinase expression in cytokine stimulated human dermal fibroblasts. Burns. 2003;29:527–31. doi: 10.1016/s0305-4179(03)00154-2. [DOI] [PubMed] [Google Scholar]
- 14.Feldmann M, Brennan FM, Maini RN. Role of cytokines in rhuematoid arthritis. Annu Rev Immunol. 1996;14:397–440. doi: 10.1146/annurev.immunol.14.1.397. review. [DOI] [PubMed] [Google Scholar]
- 15.Ohshima S, Saeki Y, Mima T, Sasai M, Nishioka K, Nomura S, et al. Interleukin 6 plays a key role in the development of antigen-induced arthritis. Proc Natl Acad Sci USA. 1998;95:8222–6. doi: 10.1073/pnas.95.14.8222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Danis VA, Franic GM, Rathjen DA, Laurent RM, Brooks PM. Circulating cytokine levels in patients with rheumatoid arthritis: results of a double blind trial with sulphasalazine. Ann Rheum Dis. 1992;51:946–50. doi: 10.1136/ard.51.8.946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pipitone N, Sinha M, Theodoridis E, Goulding N, Hall M, Lanchbury J, et al. The glucocorticoid inhibition of LFA-1 and CD2 expression by human mononuclear cells is reversed by IL-2, IL-7 and IL-15. Eur J Immunol. 2001;31:2135–42. doi: 10.1002/1521-4141(200107)31:7<2135::aid-immu2135>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 18.Pitzalis C, Pipitone N, Perreti M. Regulation of leukocyte-endothelial interactions by glucocorticoids. Ann N Y Acad Sci. 2002;966:108–18. doi: 10.1111/j.1749-6632.2002.tb04208.x. review. [DOI] [PubMed] [Google Scholar]
- 19.Filep JG, Delalandre A, Payette Y, Foldes-Filep E. Glucocorticoid receptor regulates expression of L-selectin and CD11/CD18 on human neutrophils. Circulation. 1997;96:295–301. doi: 10.1161/01.cir.96.1.295. [DOI] [PubMed] [Google Scholar]
- 20.Getting SJ, Flower RJ, Perretti M. Inhibition of neutrophil and monocyte recruitment by endogenous and exogenous lipocortin 1. Br J Pharmacol. 1997;120:1075–82. doi: 10.1038/sj.bjp.0701029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abramson SB, Amin A. Blocking the effects of IL-1 in rheumatoid arthritis protects bone and cartilage. Rheumatology (Oxford) 2002;41:972–80. doi: 10.1093/rheumatology/41.9.972. review. [DOI] [PubMed] [Google Scholar]
- 22.Su C, Lichtenstein GR. Are there predictors of Remicade treatment success or failure? Adv Drug Deliv Rev. 2005;57:237–45. doi: 10.1016/j.addr.2004.08.006. review. [DOI] [PubMed] [Google Scholar]
- 23.Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–24. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 24.Cooke BM, Usami S, Perry I, Nash GB. A simplified method for culture of endothelial cells and analysis of adhesion of blood cells under conditions of flow. Microvasc Res. 1993;45:33–45. doi: 10.1006/mvre.1993.1004. [DOI] [PubMed] [Google Scholar]
- 25.Luu NT, Rainger GE, Nash GB. Differential ability of exogenous chemotactic agents to disrupt transendothelial migration of flowing neutrophils. J Immunol. 2000;164:5961–9. doi: 10.4049/jimmunol.164.11.5961. [DOI] [PubMed] [Google Scholar]
- 26.Miyazawa K, Mori A, Miyata H, Akahane M, Ajisawa Y, Okudaira H. Regulation of interleukin-1β-induced interleukin-6 gene expression in human fibroblast-like synoviocytes by p38 mitogen-activated protein kinase. J Biol Chem. 1998;273:24832–8. doi: 10.1074/jbc.273.38.24832. [DOI] [PubMed] [Google Scholar]
- 27.Littler AJ, Buckley CD, Wordsworth P, Collins I, Martinson J, Simmons DL. A distinct profile of six soluble adhesion molecules (ICAM-1, ICAM-3, VCAM-1, E-selectin, L-selectin and P-selectin) in rheumatoid arthritis. Br J Rheumatol. 1997;36:164–9. doi: 10.1093/rheumatology/36.2.164. [DOI] [PubMed] [Google Scholar]
- 28.Paeolog EM, Hunt M, Elliott MJ, Feldmann M, Maini RN, Woody JN. Deactivation of vascular endothelium by monoclonal anti–tumour necrosis factor α antibody in rheumatoid arthritis. Arthritis Rheum. 1996;39:1082–91. doi: 10.1002/art.1780390703. [DOI] [PubMed] [Google Scholar]
- 29.Koch AE, Burrows JC, Haines GK, Carlos TM, Harlan JM, Leibovich SJ. Immunolocalization of endothelial and leukocyte adhesion molecules in human rheumatoid and osteoarthritic synovial tissues. Lab Invest. 1991;64:313–20. [PubMed] [Google Scholar]
- 30.Johnson BA, Haines GK, Harlow LA, Koch AE. Adhesion molecule expression in human synovial tissue. Arthritis Rheum. 1993;36:137–46. doi: 10.1002/art.1780360203. [DOI] [PubMed] [Google Scholar]
- 31.Issekutz AC, Mu JY, Liu G, Melrose J, Berg EL. E-selectin, but not P-selectin, is required for the development of adjuvant-induced arthritis in the rat. Arthritis Rheum. 2001;44:1428–37. doi: 10.1002/1529-0131(200106)44:6<1428::AID-ART238>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 32.Verdrengh M, Erlandsson-Harris H, Tarkowski A. Role of selectins in experimental Staphylococcus aureus-induced arthritis. Eur J Immunol. 2000;30:1606–13. doi: 10.1002/1521-4141(200006)30:6<1606::AID-IMMU1606>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 33.Walter UM, Issekutz AC. The role of E- and P-selectin in neutrophil and monocyte migration in adjuvant-induced arthritis in the rat. Eur J Immunol. 1997;27:1498–505. doi: 10.1002/eji.1830270628. [DOI] [PubMed] [Google Scholar]
- 34.Hirano T, Yasukawa K, Harada H, Taga T, Watanabe Y, Matsuda T, et al. Complementary DNA for a novel human interleukin (BSF-2) that induces B lymphocytes to produce immunoglobulin. Nature. 1986;324:73–6. doi: 10.1038/324073a0. [DOI] [PubMed] [Google Scholar]
- 35.Ishihara K, Hirano T. IL-6 in autoimmune disease and chronic inflammatory proliferative disease. Cytokine Growth Factor Rev. 2002;13:357–68. doi: 10.1016/s1359-6101(02)00027-8. review. [DOI] [PubMed] [Google Scholar]
- 36.Nowell MA, Richards PJ, Horiuchi S, Yamamoto N, Rose-John S, Topley N, et al. Soluble IL-6 receptor governs IL-6 activity in experimental arthritis: blockade of arthritis severity by soluble glycoprotein 130. J Immunol. 2003;171:3202–9. doi: 10.4049/jimmunol.171.6.3202. [DOI] [PubMed] [Google Scholar]
- 37.Modur V, Li Y, Zimmerman GA, Prescott SM, McIntyre TM. Retrograde inflammatory signaling from neutrophils to endothelial cells by soluble interleukin-6 receptor α. J Clin Invest. 1997;100:2752–6. doi: 10.1172/JCI119821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hata H, Sakaguchi N, Yoshitomi H, Iwakura Y, Sekikawa K, Azuma Y, et al. Distinct contribution of IL-6, TNF-α, IL-1, and IL-10 to T cell-mediated spontaneous autoimmune arthritis in mice. J Clin Invest. 2004;114:582–8. doi: 10.1172/JCI21795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Choy EH, Isenberg DA, Garrood T, Farrow S, Ioannou Y, Bird H, et al. Therapeutic benefit of blocking interleukin-6 activity with an anti–interleukin-6 receptor monoclonal antibody in rheumatoid arthritis: a randomized, double-blind, placebo-controlled, dose-escalation trial. Arthritis Rheum. 2002;46:3143–50. doi: 10.1002/art.10623. [DOI] [PubMed] [Google Scholar]
- 40.Nishimoto N, Yoshizaki K, Miyasaka N, Yamamoto K, Kawai S, Takeuchi T, et al. Treatment of rheumatoid arthritis with humanized anti–interleukin-6 receptor antibody: a multicenter, double-blind, placebo-controlled trial. Arthritis Rheum. 2004;50:1761–9. doi: 10.1002/art.20303. [DOI] [PubMed] [Google Scholar]
- 41.Mullberg J, Schooltink H, Stoyan T, Gunther M, Graeve L, Buse G, et al. The soluble interleukin-6 receptor is generated by shedding. Eur J Immunol. 1993;23:473–80. doi: 10.1002/eji.1830230226. [DOI] [PubMed] [Google Scholar]
- 42.Gallea-Robache S, Morand V, Millet S, Bruneau JM, Bhatnagar N, Chouaib S, et al. A metalloproteinase inhibitor blocks the shedding of soluble cytokine receptors and processing of transmembrane cytokine precursors in human monocytic cells. Cytokine. 1997;9:340–6. doi: 10.1006/cyto.1996.0174. [DOI] [PubMed] [Google Scholar]
- 43.Banning U, Bonig H, Pafferath B, Klein-Vehne A, Burdach S, Korholz D. Release of the soluble interleukin-6 receptor from human T-cells. Immunol Invest. 1998;27:47–55. doi: 10.3109/08820139809070889. [DOI] [PubMed] [Google Scholar]
- 44.Jones SA, Novick D, Horiuchi S, Yamamoto N, Szalai AJ, Fuller GM. C-reactive protein: a physiological activator of interleukin 6 receptor shedding. J Exp Med. 1999;189:599–604. doi: 10.1084/jem.189.3.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kishimoto T, Akira S, Taga T. IL-6 receptor and mechanism of signal transduction. Int J Immunopharmacol. 1992;14:431–8. doi: 10.1016/0192-0561(92)90173-i. review. [DOI] [PubMed] [Google Scholar]
- 46.Nishimoto N, Ito A, Ono M, Tagoh H, Matsumoto T, Tomita T, et al. IL-6 inhibits the proliferation of fibroblastic synovial cells from rheumatoid arthritis patients in the presence of soluble IL-6 receptor. Int Immunol. 2000;12:187–93. doi: 10.1093/intimm/12.2.187. [DOI] [PubMed] [Google Scholar]
- 47.Cronstein BN, Kimmel SC, Levin RI, Martiniuk F, Weissmann G. A mechanism for the antiinflammatory effects of corticosteroids: the glucocorticoid receptor regulates leukocyte adhesion to endothelial cells and expression of endothelial-leukocyte adhesion molecule 1 and intercellular adhesion molecule 1. Proc Natl Acad Sci USA. 1992;89:9991–5. doi: 10.1073/pnas.89.21.9991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aziz KE, Wakefield D. Modulation of endothelial cell expression of ICAM-1, E-selectin, and VCAM-1 by β-estradiol, progesterone, and dexamethasone. Cell Immunol. 1996;167:79–85. doi: 10.1006/cimm.1996.0010. [DOI] [PubMed] [Google Scholar]
- 49.Kirveskari J, Helinto M, Moilanen JA, Paavonen T, Tervo TM, Renkonen R. Hydrocortisone reduced in vivo, inflammation-induced slow rolling of leukocytes and their extravasation into human conjunctiva. Blood. 2002;100:2203–7. doi: 10.1182/blood-2002-04-1017. [DOI] [PubMed] [Google Scholar]