

Abstract
We examined the fate of neutrophils following transmigration through an endothelial monolayer cultured on “Transwell” membrane filters. Treatment of human umbilical vein endothelial cells (HUVEC) with increasing doses of tumor necrosis factor-α increased the efficiency of transmigration and markedly reduced apoptosis among the transmigrated neutrophils in a dose-dependent manner. Apoptosis was also inhibited after transmigration of neutrophils through HUVEC stimulated with interleukin (IL)-1β but not so effectively after chemotaxis through unstimulated HUVEC driven by IL-8 added below the filter. Inhibition of β2-integrin binding after transmigration or coating the lower chamber with a nonadhesive polymer (polyhydroxyl-ethyl-methacrylate) abrogated neutrophil survival. Although integrin engagement during migration itself was not essential to inhibit apoptosis, activation of neutrophils through CXC chemokine receptors was necessary. Quite brief exposure to the HUVEC (30–120 min) was effective in reducing subsequent apoptosis, although if coincubation with the HUVEC were prolonged, neutrophil apoptosis was reduced further. Neutralization of granulocyte macrophage-colony stimulating factor inhibited this additional effect. Thus, a complex interplay between migration- and activation-dependent signals and adhesive interaction in tissue may combine to effectively prolong the survival of neutrophils recruited during inflammation.
Keywords: apoptosis, leukocyte trafficking, endothelial cells, adhesion molecules
INTRODUCTION
Peripheral blood neutrophils (PBN) circulate through the vascular system with a lifespan of 6–12 h, after which, they die by apoptosis (programmed cell death) and are removed by macrophages of the reticuloendothelial system [1, 2]. In vitro, neutrophils typically undergo spontaneous apoptosis over a period of 18–36 h, but under inflammatory conditions, neutrophil lifespan is thought to be prolonged, presumably allowing sustained effector function. This concept is based partly on findings that PBN from patients with systemic inflammation [3, 4] or from mice exposed to inflammatory agents [5, 6] underwent apoptosis relatively slowly in vitro. Functionally, however, the state of the neutrophils, which have undergone adhesion and migration through the endothelium, may be more important. In this context, neutrophils from inflammatory exudates from humans, rats, or mice typically showed evidence of protection against apoptosis ex vivo [5, 7, 8]. However, the mechanisms prolonging lifespan of the neutrophils derived from the vascular compartment or from tissues may not be the same.
During recruitment, circulating neutrophils are first captured by selectin receptors expressed by cytokine-activated endothelial cells (EC) and then engage chemokines or lipidderived chemotaxins on the endothelial surface [9]. These agents induce stable adhesion through activation of β2-integrins, which bind to intercellular adhesion molecule 1 (ICAM-1), and also stimulate motility, which enables migration over and through the endothelial monolayer. Neutrophils must then engage matrix proteins, probably using β1- as well as β2-integrins, and penetrate the basement membrane before chemotaxis to the inflammatory locus [10]. Although several factors associated with recruitment (soluble inflammatory mediators, adhesive interactions, chemotaxis, and transendothelial migration) might prolong the lifespan of a neutrophil, their relative importance remains hard to define.
Interleukin-8 (IL-8) and granulocyte macrophage-colony stimulating factor (GM-CSF) can be released by EC and tended to prolong neutrophil survival [11, 12], as did the cytokine interferon-β (IFN-β) [13]. It is perhaps surprising that adhesion of neutrophils to matrix proteins, such as fibronectin or laminin, tended to increase the rate of apoptosis compared with neutrophils adherent to plastic [14] or nonadherent cells [15]. Conversely, binding of neutrophils to the plasma protein fibrinogen, through the neutrophil αmβ2-integrin, (CD11b/CD18), delayed apoptosis [16]. This integrin was also implicated in the prolonged survival of neutrophils adhered to the surface of IL-1-treated EC [17]. Spontaneous apoptosis of neutrophils was delayed by engagement of CD11b or CD18 with clustering or activating antibodies or with surface-immobilized, purified ICAM-1 [8, 14]. Conversely, cross-linking or activation of CD11b/CD18 increased apoptosis in the presence of tumor necrosis factor (TNF) [14, 18]. Clearly, the mechanisms regulating apoptosis in the complex milieu of the migrating neutrophil are hard to deduce from the above.
Studies of apoptosis of neutrophils actually undergoing transendothelial migration have been rare. Coxon et al. [6] found that neutrophils, which had migrated through monolayers of EC that had been cultured on filters and treated with TNF and IL-1, exhibited a lower level of apoptosis than control neutrophils after 6 h. Protection was attributable to a transferable factor in the medium, most probably, GM-CSF released by the human umbilical vein EC (HUVEC). However, the migrated neutrophils were incubated on a nonadherent surface, and the delay in apoptosis was short-lived. In parallel studies of GM-CSF-deficient mice treated with a combination of TNF and IL-1 [6], prolonged survival of PBN ex vivo was attributable to GM-CSF, but the low level of apoptosis seen among migrated neutrophils in the cerebrospinal fluid was not. Thus, neutrophils migrating into tissue presumably receive signals other than those from soluble mediators. Chemotaxis across endothelial monolayers in response to the bacterial peptide formyl-Met-Leu-Phe (fMLP; rather than cytokine-induced transendothelial migration) has been shown to give a weak protection against apoptosis [6] or to accelerate it [8, 18]. In contrast, chemotaxis through epithelial cell lines toward fMLP or IL-8 caused a reduction in rate of neutrophil apoptosis [19, 20].
In summary, antiapoptotic effects have been attributed to soluble mediators, adhesive interactions, and migratory responses, but these effects can vary depending on the milieu in which the neutrophils are placed. During transendothelial migration, such responses will be integrated in a manner hard to tease apart by studying these factors in isolation. Therefore, we investigated the survival of neutrophils in a model where they migrated through an endothelial monolayer stimulated with varying levels of cytokines and were allowed to migrate away from the endothelial monolayer and re-establish adhesion. We modified adhesive interactions during and after transendothelial migration and revealed a cytokine dose-dependent and adhesion-dependent signal that inhibited apoptosis. Although leaving neutrophils in coincubation with the EC could also prolong survival through soluble mediators, this effect was clearly separate from the adhesion/migration-dependent effects and may be less important when neutrophils migrate away from endothelium into tissue.
MATERIALS AND METHODS
Isolation of human neutrophils
Venous blood from healthy individuals was collected in EDTA tubes (Sarstedt, Leicester, UK), and neutrophils were isolated using two-step histopaque density gradients as described previously [21]. The neutrophils were washed twice and resuspended in Medium-199 (M199; Gibco Invitrogen Compounds, Paisley, UK) containing 0.15% bovine serum albumin (BSA; 1/50 of 7.5% w/v sterile-filtered Fraction V solution, tissue-cultured and endotoxin-tested to be not more than 13 EU/ml, Sigma-Aldrich, Poole, UK).
Isolation and culture of EC
HUVEC were isolated from umbilical cords using collagenase as described previously [22] and cultured in M199 supplemented with 20% fetal calf serum (FCS), 10 ng/ml epithelial growth factor, 35 μg/ml gentamycin, 1 μg/ml hydrocortisone (all from Sigma-Aldrich), and 2.5 μg/ml amphotericin B (Gibco Invitrogen Compounds). Primary HUVEC were dissociated using trypsin/EDTA (Sigma-Aldrich), seeded onto uncoated, low-density, 3.0 μm pore polycarbonate Transwell filters, which were placed in matching 24-well plates (BD PharMingen, Oxford, UK), and cultured for 4 days. Each experiment used first-passage cells from a single, different cord. Immediately before assays, the HUVEC were stimulated for 4 h with 0–100 U/ml TNF-α or 50 pg/ml IL-1β (both from Sigma-Aldrich). In some experiments, 10 ng/ml IL-8 (R&D Systems, Abingdon, UK) was added to the lower chamber.
Measurement of neutrophil adhesion and migration and subsequent apoptosis
The medium was removed from HUVEC, and 700 μl M199 + BSA was added to the lower chamber of the plate, and 200 μl neutrophils (2×106 cells/ml) were added to the upper chamber. The neutrophils were allowed to settle, adhere to HUVEC, and migrate at 37°C in a CO2 incubator. Migration was stopped at the chosen time by transferring the filter into a fresh well, leaving the transmigrated cells in the original, lower chamber. Medium containing nonadherent neutrophils was removed from the upper chamber and transferred to another well. The filter was washed twice, and cells in the washes were pooled with the upper chamber sample and taken to represent nonadherent neutrophils. These nonadherent cells and the transmigrated cells washed from the lower chamber were counted using a Coulter Multisizer II (Coulter Electronics Ltd., Essex, UK). The number of adherent cells remaining in the filter could be calculated by subtracting nonadherent and transmigrated cells from the known number of cells added. All counts were expressed as a percentage of the number added.
For analysis of apoptosis, nonadherent or transmigrated neutrophils were incubated at 37°C for up to 72 h, where time zero was the time of original addition of the neutrophils to the HUVEC. Samples of cells were centrifuged onto glass slides for 3 min at 10 g in a Shandon Cytospin II (Scientific Instruments, South Trentham, UK) and stained with Diff Quik. The level of apoptosis was determined by counting at least 100 cells using a light microscope. Apoptotic neutrophils were defined as cells containing one or more darkly stained, condensed nuclear fragments [13, 23]. Alternatively, we assessed the reduction in mitochondrial transmembrane potential as an early and irreversible step in apoptosis, reported by reduced incorporation of the lipophilic fluorochrome 3′3′-dihexyloxacarbocyanine iodide (DiOC6) [24]. Neutrophils were incubated with 40 nM DiOC6 (Molecular Probes, Cambridge, UK) for 30 min at 37°C, centrifuged at 800 g for 4 min, and resuspended in ice-cold phosphate-buffered saline containing 16% FCS. Samples were analyzed using a Coulter XL flow cytometer (Beckman Coulter, High Wycombe, UK), and data were expressed as a percentage of DiOC6 low cells, by comparison with freshly isolated, control neutrophils.
Treatments with antibodies and inhibitory agents
The following monoclonal antibodies (mAb) were used at 10 μg/ml: mAb 13 (β1-integrin/CD29 functional blocker, gift of Professor Martin J. Humphries, School of Biological Sciences, University of Manchester, UK); R6.5E (β2-integrin/CD18 functional blocker), KIM249 (αM-integrin/CD11b functional blocker), and 1G11 (anti-vascular cell adhesion molecule 1 (VCAM-1)/CD106 functional blocker, all gifts of Dr. Tony Shock, Celltech R&D, Slough UK); and SZ21 (β3-integrin/CD61 functional blocker, Immunotech, Marseille, France). Antibodies against CXC chemokine receptor 1 (CXCR1) or CXCR2 (Clones 501 and 19, Biosource International Inc., Camarillo, CA) were used at 2 μg/ml, according to the manufacturer’s recommendation. In recent, flow-based studies, we have found these antibodies to inhibit neutrophil migration through TNF-treated HUVEC and also to increase the percentage of the captured cells undergoing rolling [25, 26]. This indicates that they inhibit migration through blockade of activation rather than by mimicking the natural CXC chemokine agonists. Although these agonists can inhibit transmigration when added exogenously, they also have the opposite effect of converting rolling to stationary adhesion [27]. Neutralizing antibody against GM-CSF (Clone 3209, R&D Systems) was used at 2 μg/ml, which is four times the manufacturer’s stated neutralization dose for 500 pg/ml GM-CSF, a concentration well above that detected in supernatants from endothelial cultures and found to provide an antiapoptotic effect [6].
As all the antibodies were of immunoglobulin G1 isotype, those that had no functional effects (see Results) acted effectively as relevant isotype-matched controls for those that modified migration or apoptosis. In addition, antibody against VCAM-1 was included as an EC-binding control for any antibody, which was used to treat neutrophils but might also bind to EC (e.g., antibody against β1-integrin).
CT7010 is a low molecular weight, nonpeptide inhibitor of β2-integrin function (gift of Dr. Tony Shock, Celltech R&D), synthesized as a reference compound by Celltech R&D, and based on a compound patented by Genentech Inc. (San Francisco, CA; Patent No. WO99/49856) [28]. It was used at 1 μM, after verification of its antiadhesive action in independent studies of neutrophil migration through HUVEC in a flow-based assay (N. Thin Luu, G. B. Nash, unpublished observations). The inhibitor of phosphatidylinositol-3 kinase (PI-3K) LY294002 was used at 10 μM [29]. The specific inhibitor of Src kinase was PD0173952 (10 μM, gift from Pfizer Ltd., Ann Arbor, MI) [30].
Neutrophils were incubated with inhibitor or mAb for 30 min at room temperature before the assay, and agents were present throughout subsequent incubations in the upper chamber. HUVEC were treated with mAb against VCAM-1 for the last 30 min of the 4-h treatment with TNF, and mAb was washed out prior to the addition of neutrophils to the upper chamber. In some experiments, the antibodies, including the neutralizing antibody against GM-CSF, or inhibitors were added to the lower chamber of the Transwell at the time of neutrophil addition to the upper chamber. They were present throughout the assay, including any subsequent incubation of the transmigrated neutrophils in the lower chamber.
Before some experiments, lower chambers of the tissue-culture plate were made nonadhesive by coating with 50 mg/ml polyhydroxyl-ethyl-methacrylate (polyHEMA; Sigma-Aldrich) for 48 h at 37°C, as described [15, 31]. Independent tests with neutrophils stimulated with 10 ng/ml IL-8 and allowed to settle in polyHEMA-coated or uncoated wells showed that coating reduced adhesion by 99 ± 0.5% (mean±sem from three experiments).
Statistical analysis
Variation between multiple treatments was evaluated using ANOVA, followed by comparison of treatments with controls by the Dunnett test when indicated. Comparisons of individual treatments with controls were done by paired t-test.
RESULTS
Neutrophil migration through TNF-stimulated HUVEC and subsequent apoptosis
When neutrophils were added to TNF-stimulated HUVEC for 2 h, transmigration increased in a TNF dose-dependent manner (Fig. 1A). It is interesting that the transmigrated neutrophils were strongly protected against apoptosis over the next 22 h (compared with controls incubated in empty wells), and the protection increased the higher the dose of TNF used to pretreat the HUVEC (Fig. 1B). The protection evidenced by maintenance of normal nuclear morphology (Fig. 1D) was also evident if mitochondrial membrane integrity was assessed by measuring fluorescence intensity of DiOC6 staining (Fig. 1C). There was a significant, TNF dose-dependent effect of transmigration on fluorescence intensity. A higher proportion of transmigrated neutrophils showed loss of DiOC6 fluorescence rather than nuclear condensation. This was presumably because the DiOC6 fluorescence reflects an early stage in apoptosis (changes in mitochondrial membrane potential), and nuclear morphology reflects an end stage. Thus, the strong protection against nuclear changes seen in transmigrated cells at 24 h, but lesser protection against changes in membrane potential, suggests that some of the cells were in the early stages of apoptosis (i.e., migration caused a delay in apoptosis over 24 h rather than total protection). To investigate this point further, apoptosis was assessed at intervals up to 72 h (Fig. 2). It was evident that apoptosis was essentially complete for all samples by 72 h and that apoptosis was delayed (or slowed) more effectively, i.e., the higher the dose of TNF that was used to pretreat the HUVEC. For instance, protection at 48 h was only evident for the highest dose of TNF (Fig. 2).
Fig. 1.
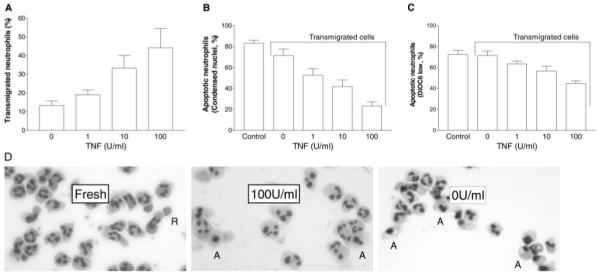
Percentage of neutrophils undergoing transmigration and subsequent apoptosis after incubation with HUVEC on Transwell filters. HUVEC were treated with different concentrations of TNF for 4 h and washed free of TNF, and then neutrophils were added for 2 h before removal of the filter. (A) Percentage of added neutrophils migrating into the lower chamber of the Transwell. (B) Percentage of transmigrated neutrophils undergoing apoptosis after 24 h, judged by nuclear morphology. Control = Freshly isolated neutrophils incubated for 24 h in matching plates. (C) Percentage of transmigrated neutrophils showing decreased mitochondrial transmembrane potential (i.e., low level of fluorescence of DiOC6). (D) Photomicrographs of stained neutrophils used to assess apoptosis from nuclear morphology: Fresh = Freshly isolated cells; 100 U/ml = neutrophils 24 h after transmigration through HUVEC treated with 100 U/ml TNF; 0 U/ml = neutrophils 24 h after transmigration through untreated HUVEC. A = Examples of apoptotic cells; R = red blood cell found in the fresh isolate. Data are the mean ± sem from seven to 10 independent experiments. ANOVA showed a significant effect of TNF concentration on migration and on apoptosis assessed by either method (P<0.01 in each case).
Fig. 2.
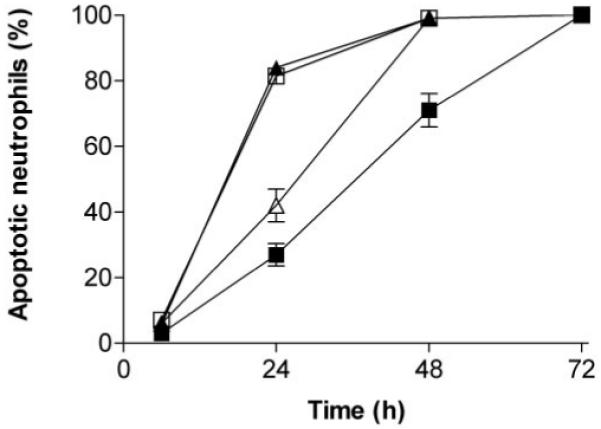
Time course of apoptosis of neutrophils, which have transmigrated through HUVEC. Comparisons were made between freshly isolated neutrophils (▲) and neutrophils that had migrated for 2 h through unstimulated HUVEC (□) or through HUVEC that had been treated with 1 U/ml TNF (△) or 100 U/ml TNF (■). Data are the mean ± sem from three of more independent experiments, except at 72 h, where n = 2. ANOVA showed significant effects of time and treatment on apoptosis (P<0.01 in each case).
HUVEC that had been stimulated with IL-1β (50 pg/ml) supported neutrophil transmigration at a similar level to TNF (33±5% of added neutrophils; mean±sem, n=4). Again, neutrophil apoptosis was reduced markedly 22 h following transmigration through IL-1β-stimulated HUVEC (percentage of cells with abnormal nuclear morphology=9±2% compared with 80±3% for cells migrated through unstimulated HUVEC; mean±sem, n=4; P<0.01 by paired t-test). In contrast, in the same experiments, migration through unstimulated HUVEC in response to IL-8 (10 ng/ml) added to the lower chamber had only a modest effect on apoptosis, which did not reach statistical significance (percentage of cells with abnormal nuclear morphology=60±10%; mean±sem, n=4). The IL-8 did induce effective neutrophil chemotaxis (41±5% of added neutrophils migrated; mean±sem, n=4).
To test the specificity of the effect of transmigration on apoptosis further, we compared survival of freshly isolated neutrophils, nonadherent neutrophils, which had settled on HUVEC for 2 h, and transmigrated cells, all cultured for 24 h in medium conditioned by 2 h contact with the HUVEC (Table 1). There were no significant differences between the different types of neutrophils when suspended in medium from unstimulated HUVEC, although there was a trend toward lesser apoptosis for the cells that had transmigrated through the unstimulated HUVEC. Nor was apoptosis significantly reduced for any cells compared with freshly isolated cells in nonconditioned medium (Table 1). Neutrophils suspended in medium from HUVEC which had been treated with 100 U/ml TNF, did tend to show increased survival compared with fresh cells in non-conditioned medium, although this effect was only statistically significant for the cells that had transmigrated through the stimulated HUVEC, which showed the lowest level of apoptosis. Thus, contact with the apical surface of HUVEC, and any soluble factor(s) released by TNF-treated HUVEC over the 2-h assay, were not the major contributors to survival of transmigrated cells. It is interesting that in the medium from HUVEC, which had been treated with 100 U/ml TNF, there was a trend for the freshly isolated cells to survive better than the nonadherent cells in the same medium. This suggests that the improved survival of the transmigrated cells may have been partly a result of a subpopulation of efficient migrators being inherently more resistant to apoptosis, and nonmigrators were less resistant than the original, “fresh” cells. However, this is not consistent with the finding that when a progressively greater proportion of neutrophils migrated through HUVEC with an increasing dose of TNF (i.e., the process became less selective), the survival of the migrated cells increased. Thus, overall, the data indicate that migration through cytokine-stimulated HUVEC provides a specific, dose-dependent survival signal to the migrating neutrophils— greater, e.g., than that generated during neutrophil chemotaxis.
TABLE 1.
Apoptosis of Freshly Isolated, Nonadherent, or Transmigrated Neutrophils in Fresh or Endothelial-Conditioned Media
| Neutrophils | Apoptosis (% of cells) |
|
|---|---|---|
| A. Fresh medium | Freshly isolated | 79 ± 6 |
| B. Medium conditioned by HUVEC treated with 0 U/ml TNF |
Nonadherent | 80 ± 9 |
| Freshly isolated | 74 ± 8 | |
| Transmigrated | 72 ± 4 | |
| C. Medium conditioned by HUVEC treated with 100 U/ml TNF |
Nonadherent | 67 ± 7 |
| Freshly isolated | 53 ± 8 | |
| Transmigrated | 29 ± 4* |
Neutrophils were added to HUVEC for 2 h after any TNF had been removed. Nonadherent neutrophils from the upper chamber were removed and cultured for 24 h in the conditioned medium. In addition, freshly isolated and transmigrated (lower chamber) neutrophils were cultured overnight in the conditioned medium. Freshly isolated neutrophils were also cultured in fresh medium for 24 h. Data are mean ± SEM from four experiments. ANOVA showed a significant effect overall of different treatments (P<0.01).
P<0.01 compared with the freshly isolated neutrophils cultured in fresh medium by Dunnett test.
The role of adhesive interactions in enhanced survival of transmigrated neutrophils
We inhibited binding to various adhesion molecules, which could potentially deliver a survival signal. First, CT7010 (a low molecular weight, nonpeptide inhibitor of β2-integrin function [28]) or mAb against β1-, β2-, or β3-integrins were incubated with neutrophils and were present in the upper chamber during the 2-h adhesion and migration assay. Neutrophil migration through HUVEC was reduced in the presence of CT7010 or mAb against β2-integrins but not by the other antibodies (Fig. 3A). However, none of the agents significantly modified the level of apoptosis among the transmigrated neutrophils (Fig. 3B). This suggests that engagement of these receptors prior to transmigration was not necessary for survival or that the small number of cells that did undergo transmigration after treatment with antibody against β2-integrins still had some remaining, unblocked CD18 so that an adhesive signal was still operative. The results also indicate that binding of the antibodies themselves was not proapoptotic. As β2-integrins may have become up-regulated upon neutrophil transmigration [32], antibody against β2-integrins or CT7010 was added to the lower chamber prior to the addition of the neutrophils to the upper chamber. The neutrophils were allowed to transmigrate for 2 h through the TNF-treated HUVEC into the lower chamber and were incubated in the medium in the presence of the inhibitor or antibodies for a further 22 h. Under these conditions, there was a significant increase in the percentage of transmigrated neutrophils undergoing apoptosis when β2-integrins were inhibited in the lower chamber, which was not observed when β1-integrins were blocked (Fig. 4).
Fig. 3.
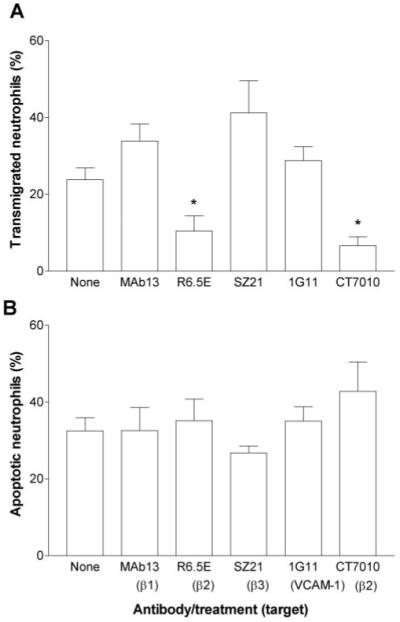
Effects of function-blocking mAb or agents against β1-, β2-, or β3-integrins or mAb against VCAM-1 on (A) transmigration of neutrophils or (B) apoptosis of transmigrated cells. HUVEC were treated with 100 U/ml TNF for 4 h and washed free of TNF, and then neutrophils were added for 2 h before removal of the filter. Antibodies against integrins (mAb13=anti-β1; R6.5E=anti-β2; SZ21=anti-β3) or CT7010 were added to the neutrophils, or mAb against VCAM-1 (1G11) were added to HUVEC for 30 min prior to addition of neutrophils to the HUVEC. Data are the mean ± sem from four experiments. ANOVA showed a significant effect of treatment on transmigration (P<0.05). *, P < 0.05, compared with untreated control (None) by Dunnett test.
Fig. 4.
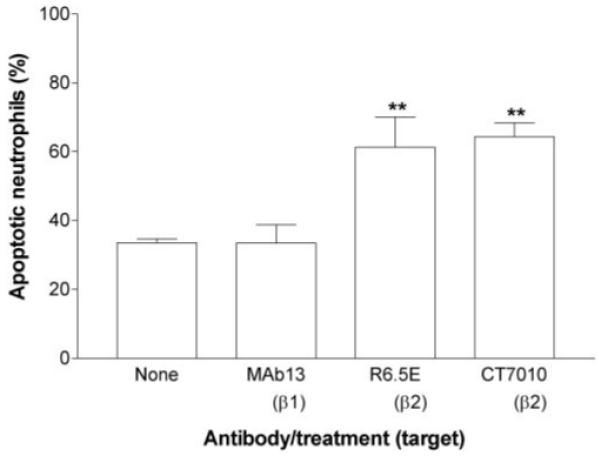
Effects of adding function-blocking antibodies or agents against β1- or β2-integrins to the lower chamber on apoptosis of transmigrated cells. HUVEC were treated with 100 U/ml TNF for 4 h and washed free of TNF, and then neutrophils were added for 2 h before removal of the filter. Antibodies or CT7010 were added to the lower chamber immediately before addition of neutrophils. Data are the mean ± sem from four independent experiments. ANOVA showed a significant effect of treatment on apoptosis (P<0.01). **, P < 0.01, compared with untreated control (None) by Dunnett test.
These results suggested that β2-integrin engagement was important for the survival of transmigrated neutrophils, presumably by supporting adhesion to the bottom of the chamber. The only protein in the medium added to this chamber was albumin, which would tend to coat the surface. Albumin-coated surfaces are known to support neutrophil adhesion through the β2-integrin CD11b/CD18 [33], and so, we carried out a series of experiments where antibody KIM249 against CD11b was added to the lower chamber. In this case, after 24 h, apoptosis of neutrophils, which had transmigrated through HUVEC stimulated with 100 U/ml TNF, was increased to 71 ± 5.3% compared with 33 ± 1.2% for untreated controls (mean±sem, n=5, P<0.05 by paired t-test). Taking this further, we precoated the lower chamber of the Transwell with the nonadhesive substrate polyHEMA [15, 31]. With this coating, neutrophil apoptosis was increased to 80 ± 5.7% compared with 30 ± 3.5% for uncoated wells (mean±sem, n=4, P<0.01 by paired t-test).
The results thus far suggest that an increasing signal for transmigration at higher doses of TNF occurred along with an increasing signal for survival, which induced and required adhesion for its protective function. CXC chemokine(s) presented by the TNF-treated HUVEC are able to induce transmigration [27]. To test their role here, we used function-blocking antibodies against the neutrophil chemokine receptors CXCR1 and CXCR2. We found that migration of neutrophils through TNF-treated HUVEC was partially reduced by each of these antibodies and more efficiently reduced when both receptors on neutrophils were blocked (Fig. 5A). It is interesting that the neutrophils that did transmigrate received lesser protection against apoptosis, and apoptosis was greatest when both receptors were blocked (Fig. 5B). These results suggest that the chemokine-activating signal(s) transduced through CXCR during transmigration were responsible for the increased adhesion-dependent survival of neutrophils in this model.
Fig. 5.
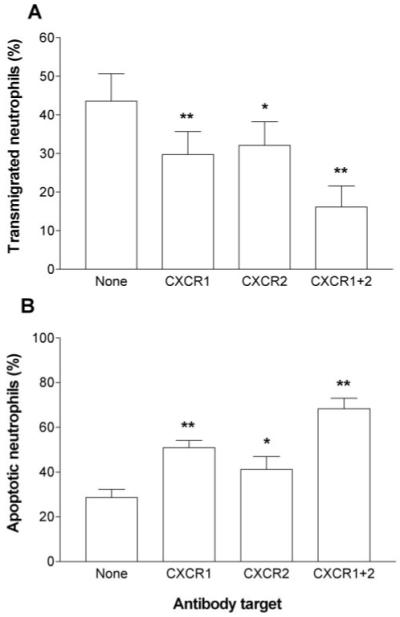
Effects of function-blocking antibodies against CXCR1 and/or CXCR2 on (A) transmigration of neutrophils or (B) apoptosis of transmigrated cells. HUVEC were treated with 100 U/ml TNF for 4 h and washed free of TNF, and then neutrophils were added for 2 h before removal of the filter. Antibodies against CXCR1, CXCR2, or both were added to the neutrophils for 30 min prior to addition of neutrophils to the HUVEC. Data are the mean ± sem of three to seven experiments. ANOVA showed a significant effect of treatment on transmigration and on apoptosis (P<0.01 in each case). *, P < 0.05; **, P < 0.01, compared with untreated control (None) by Dunnett test.
To test possible mechanisms by which adhesion-dependent signals might prolong survival, we treated neutrophils with inhibitors of Src kinases (PD0173952) or PI-3K (LY294002), which have been implicated in signaling through ligated β2-integrins [34]. Neither agent had a significant effect on neutrophil transmigration through HUVEC treated with 100 U/ml TNF when neutrophils were treated before assay or when the agent was added to the lower well (Table 2). However, although PD0173952 had no significant effect on subsequent apoptosis of the transmigrated cells, LY294002 significantly increased apoptosis, whether added to neutrophils in advance or only to the lower well (Table 2).
TABLE 2.
Effects of Inhibitors of Src Kinase (PD0173952) or PI-3K (LY294002) on Transmigration of Neutrophils through HUVEC, which Had Been Treated with 100 U/ml TNF or Apoptosis of the Transmigrated Cells
| Treatment | Transmigrated neutrophils (%) |
Apoptotic neutrophils (%) |
|---|---|---|
| A. Inhibition of Src kinase | ||
| Control | 44 ±5 | 41 ± 5 |
| PD added to upper well | 40 ±5 | 34 ± 10 |
| PD added to lower well | 44 ±6 | 33 ± 7 |
| B. Inhibition of PI-3K | ||
| Control | 39 ±8 | 27 ± 3 |
| LY294002 added to upper well | 31 ±4 | 51 ± 5* |
| LY294002 added to lower well | 34 ±4 | 46 ± 6* |
Data are mean± SEM from four or five experiments. Neutrophils were treated with inhibitors before being added to the upper well, or inhibitors were added to the lower well only.
P < 0.05 compared with the untreated controls by paired t-test.
Ability of soluble mediators to prolong survival of transmigrated neutrophils
In light of the data in Table 1, suggesting that some contribution to survival might come from released substances, we carried out studies to vary the duration of coincubation of neutrophils with HUVEC and hence, the time over which soluble mediators were released. Neutrophils were added to Transwell filters containing unstimulated or TNF-stimulated HUVEC for 0.5, 2, or 24 h before removal of the Transwell filter and assessment of apoptosis among the migrated cells at 24 h. Neutrophil transmigration through unstimulated or TNF-stimulated HUVEC increased with time (Fig. 6A), and there were several trends in apoptosis. First, neutrophils that migrated through TNF-treated monolayers within 30 min underwent markedly reduced apoptosis compared with those migrating through unstimulated HUVEC (Fig. 6B). Second, although apoptosis of neutrophils that had transmigrated through TNF-treated HUVEC tended to decrease the longer the coculture, this effect was not strong (Fig. 6B). In contrast, apoptosis of neutrophils that had migrated through unstimulated HUVEC did decrease strongly, the longer the coincubation. Finally, neutrophils which had transmigrated through TNF-treated HUVEC, underwent a lesser degree of apoptosis than those that had migrated through unstimulated HUVEC at every time-point. These results indicated that progressive release of soluble factor(s) over a 24-h period enhanced survival of neutrophils. Although this effect could improve survival of neutrophils that had migrated through TNF-treated HUVEC, it was not great compared with the effect of transmigration itself.
Fig. 6.
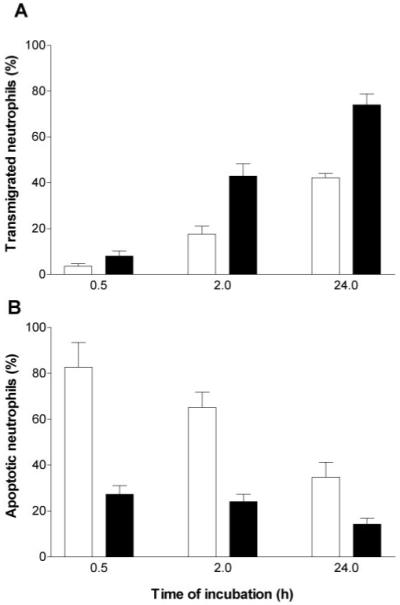
Percentage of neutrophils undergoing transmigration and subsequent apoptosis after incubation with HUVEC on Transwell filters for different times. HUVEC were unstimulated (open bars) or treated with 100 U/ml TNF (solid bars) for 4 h and washed free of TNF, and then neutrophils were added for 0.5, 2, or 24 h before removal of the filter. (A) Percentage of added neutrophils migrating into the lower chamber of the Transwell. (B) Percentage of transmigrated neutrophils undergoing apoptosis after 24 h, judged by nuclear morphology. Data are the mean ± sem from four to eight independent experiments. ANOVA showed a significant effect of TNF concentration and of time of coincubation on migration and on apoptosis (P<0.01 in each case).
To test the possible role of GM-CSF [6], a neutralizing antibody was added to the lower chamber prior to the addition of the neutrophils to the upper chamber for 2 h or 24 h. Neutralization of GM-CSF did not have any consistent effect on the percentage of neutrophils migrating through unstimulated or TNF-treated HUVEC (data not shown). In addition, it had little effect on the subsequent apoptosis of neutrophils that had migrated through either type of HUVEC after 2 h (Fig. 7). Neutralization did increase apoptosis of neutrophils that had migrated and been coincubated with TNF-treated or unstimulated HUVEC for 24 h (Fig. 7). Nevertheless, after neutralization, a survival advantage was retained by neutrophils that had migrated through TNF-treated HUVEC. Conversely, increased survival of neutrophils that had transmigrated through unstimulated HUVEC was largely attributable to the effects of GM-CSF (Fig. 7). We also carried out limited studies with double the antibody concentration (4 μg/ml) in the 24-h coincubations but found no extra increase in apoptosis (e.g., 81% of transmigrated neutrophils underwent apoptosis after incubation with unstimulated HUVEC, and 53% after incubation with TNF-treated HUVEC; means from two experiments in the presence of antibody). As these values are nearly identical to those shown in Figure 7, we conclude that adequate neutralizing antibody was present in those studies. It seems therefore that soluble mediators had little effect on the apoptosis of neutrophils which were exposed to HUVEC for up to 2 h, but had greater effect in prolonged coincubations.
Fig. 7.
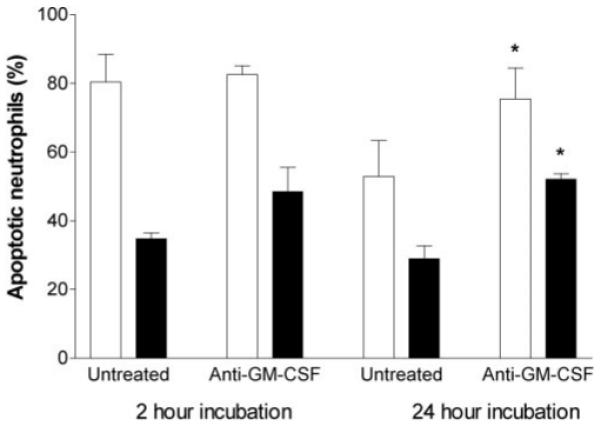
Effect of neutralizing mAb against GM-CSF on apoptosis of neutrophils, which had transmigrated through HUVEC for different periods. HUVEC were unstimulated (open bars) or treated with 100 U/ml TNF (solid bars) for 4 h and washed free of TNF, and then neutrophils were added and incubated for 2 or 24 h before removal of the filter. Apoptosis was evaluated after a total of 24 h incubation. Antibody was added to the lower chamber prior to the addition of the neutrophils. Data are the mean ± sem from four independent experiments. ANOVA showed a significant effect of TNF dose (P<0.01) but not antibody on apoptosis for 2-h incubations and a significant effect of TNF dose and antibody on apoptosis for 24-h incubations (P<0.01 in each case). *, P < 0.05, compared with control without antibody at the same time of incubation by Dunnett test.
Finally, although HUVEC were washed free of TNF before addition of neutrophils, we checked whether survival could have been influenced by any residual TNF. A wide range of concentrations of TNF was added to fresh neutrophils (increasing in decades from 0.001 to 100 U/ml), which were then cultured alone for 24 h. In three experiments, an average of 88% of untreated neutrophils underwent apoptosis, judged from nuclear morphology. Means for those treated with the six different doses of TNF ranged from 83% to 94% with no significant trends or effects of dose detectable. Thus, any residual TNF could not have been responsible for reduction in apoptosis of migrating neutrophils.
DISCUSSION
There is extensive literature about the signaling pathways and biochemical and environmental factors that regulate neutrophil apoptosis (e.g., refs. [35, 36]), but much less is known about the mechanisms by which apoptosis may be delayed during migration into tissue. Here, we found that when neutrophils transmigrated through endothelial monolayers, which had been stimulated with different concentrations of TNF, apoptosis was delayed in a dose-dependent manner. Although transmigration through IL-1-treated HUVEC had a similar effect, chemotaxis through resting HUVEC in response to a gradient of IL-8 was much less effective in delaying neutrophil apoptosis. Migration-induced survival required engagement of β2-integrins after transmigration, and apoptosis was accelerated if migrated neutrophils were incubated on a nonadhesive substrate. In prolonged coincubations with HUVEC, neutrophil survival was enhanced further through the action of soluble GM-CSF. However, this effect was distinct from the signal transduced through CXCRs during migration, which was apparent after short-term contact with EC. Thus, cytokine-stimulated EC can signal to migrating neutrophils to delay their apoptosis in a manner that is dependent on the degree of endothelial stimulation and on subsequent attachment to an adhesive substrate.
Coxon et al. [6] found that neutrophils, which had migrated through HUVEC that had been stimulated with a combination of IL-1 and TNF, exhibited a lower level of apoptosis than control neutrophils after 6 h. However, the migrated cells were incubated on a nonadherent surface, the protection was short-lived, and it was attributable to GM-CSF released by the HUVEC during the 1-h migration phase. Here, we observed low levels of apoptosis at 6 h under all conditions but obtained long-term suppression of apoptosis in transmigrated cells, which was lost when a nonadherent surface was used. Moreover, there was much less protection if neutrophils, which had been incubated on HUVEC for 2 h (but not transmigrated), were incubated in the conditioned medium for prolonged periods. Nevertheless, we could clearly demonstrate antiapoptotic effects of soluble mediator(s) on top of the migration-induced signal if HUVEC and neutrophils were cultured together for longer periods. By using a neutralizing antibody, we also identified the dominant agent in this effect as GM-CSF. A variety of soluble agents, including IL-8, TNF, IFN-β, and GM-CSF, can influence the rate of neutrophil apoptosis [6, 11, 13, 37]. However, it is uncertain how long endothelial-derived, soluble agents can be expected to continue to act on neutrophils in vivo after they have migrated away from the subendothelial space into tissue. For instance, Coxon et al. [6] found that in cytokine-treated mice, the prolonged survival of PBN was dependent on the presence of GM-CSF, but the survival of neutrophils that had migrated into the cerebrospinal fluid was not.
Other studies have tested effects of chemotaxis across the unstimulated cell line ECV304 (previously thought to be of endothelial in origin) in response to a gradient of fMLP [8, 18]. Apoptosis was actually accelerated by chemotaxis, although in one of the studies, it was slower compared with unmigrated controls if the neutrophils had been preactivated with lipopolysacharride [8]. We found that chemotaxis across unstimulated HUVEC toward IL-8 caused a modest effect on apoptosis, as did Coxon et al. [6] for chemotaxis to fMLP. Studies of chemotaxis toward fMLP across epithelial cell lines also showed protection against apoptosis [19, 20]. Although chemotactic stimulation of neutrophils may delay apoptosis, there does not seem to be a specific role for EC in this process. Recruitment and transmigration induced by endogenous, endothelial-derived receptors and agents mimic an inflammatory response in vivo more accurately than chemotaxis across unstimulated monolayers. In the former case, the EC play an active part in prolonging neutrophil survival, and our results indicate that presentation of CXC chemokines underlies this effect. Growth-regulating oncogene-α [CXC chemokine ligand 1 (CXCL1)], epithelial cell-derived, neutrophil-activating peptide-78 (CXCL5), granulocyte chemotactic protein-2 (CXCL6), neutrophil-activating protein 2 (CXCL7), and IL-8 (CXCL8) have been reported to be expressed by HUVEC and are able to transduce signals through CXCR1 and/or CXCR2 [38-40]. We have not previously been able to define the roles of the individual CXC chemokines in neutrophil transmigration through TNF-treated HUVEC, although it seems likely that increasing transmigration with an increasing dose of TNF is a result of increasing presentation of chemokines [27, 41]. Increasing presentation could also explain the TNF dose-dependent increase in survival of transmigrated neutrophils. As IL-8 added alone to unstimulated HUVEC did not induce comparable survival, it seems that the antiapoptotic effect of the chemokines operates more effectively when they are presented on the EC surface or when a combination of chemokines is present.
It is interesting that our results indicated that binding of β2-integrins after transmigration was necessary for the survival signal. The requirement for integrin engagement during the transendothelial migration was not so clear-cut. Antibody against β2-integrin or a small molecule inhibitor (CT7010) markedly reduced the level of transmigration, as expected from previous reports [41, 42]. However, the neutrophils that did transmigrate showed prolonged survival. Although this might mean that β2-engagement during this phase was not essential for survival, it is also possible that the residual migration arose from imperfect blockade of integrins, which still contributed to survival. During transmigration, de novo expression of β2-integrin CD11b/CD18 occurs, and this “unblocked” integrin could also have supported survival upon attachment to the lower chamber. In any case, when the same blocking agents were added to the lower chamber of the Transwell system, the neutrophils transmigrated, but their survival was reduced greatly. Coating the lower chamber with polyHEMA so that it was nonadherent or adding antibody to CD11b had similar effects. Thus, regardless of a contribution during transendothelial migration, adhesion after transmigration did seem essential for protection against apoptosis. Clustering of β2-integrins on neutrophils has been reported to induce activation and signaling through Src kinases and PI-3K (see ref. [34] for review). Moreover, PI-3K phosphorylation is associated with a number of antiapoptotic pathways [43]. Here, treatment with the Src kinase inhibitor had no effect on the survival of neutrophils post-transmigration, but there was an increase in apoptosis of transmigrated neutrophils in the presence of LY294002 (PI-3K inhibitor). These findings imply that β2-integrin engagement generates a Src kinase-independent signal, which acts, in part, at least, through PI-3K to increase survival.
The role of β2-integrins in neutrophil apoptosis has not always been clear-cut. Others found a prosurvival effect when neutrophils were incubated with IL-1-stimulated HUVEC for 24 h, which was reduced by blockade of β2-integrin or ICAM-1 [17]. However, all the neutrophils added to the monolayers were tested, not just the transmigrated cells. The effect of integrin inhibition could have arisen because transmigration itself was reduced, or because cells on the upper surface required adhesion to respond to soluble agents released in these prolonged incubations. Engagement of CD11b or CD18 with antibodies (with or without cross-linking) or with immobilized ICAM-1 could accelerate or delay apoptosis, depending on whether neutrophils were cotreated with TNF or not, respectively [8, 14, 18]. It is not obvious how relevant these results are to the migration process in vivo. Studies of inflammatory responses in mice deficient in CD11b indicated that low levels of apoptosis among neutrophils recruited to peritoneal fluid or cerebrospinal fluid did not require expression of CD11b [5, 6]. Ex vivo survival of neutrophils lacking CD11b/CD18 was also unimpaired, but that was for cells incubated on a nonadherent surface, and so integrin engagement and signaling presumably could not occur. Overall, it seems that in the “correct” milieu, β2-integrin engagement can promote survival, possibly when combined with migration-dependent signal(s).
The foregoing indicates that a linked sequence of responses prolong neutrophil survival during the physical process of migration through cytokine-stimulated endothelium. At an early stage, CXCR-mediated signaling acts as the major inducer of β2-integrin activation and migration. The TNF dose-dependent signals also promote prolonged survival, which becomes more effective with increasing stimulation of the EC. This response requires subsequent binding to substrate through β2-integrins (especially CD11b/CD18) for its effect to be felt. Activation of PI-3K (which is not required for transmigration itself) contributes to the integrin-mediated survival signal. Although soluble growth factors such as GM-CSF can clearly augment survival, they are not necessary in the context of the relatively brief contact between neutrophils and EC during migration and do not appear to require adhesion judging from previous studies [6]. In vivo, prolongation of survival may depend on different mediators in different compartments. In mice treated with cytokines, circulating neutrophils had increased survival ex vivo, which was reduced in mice lacking GM-CSF [6]. However, prolonged survival of migrated neutrophils from cerebrospinal fluid was not reduced in these mice. Within solid tissue, it may be that migration-induced, adhesive signals have the dominant effect on lifespan rather than soluble mediators. Thus, while progressing from the vascular compartment into sites of inflammation, neutrophils may benefit from a series of signals that prolongs their lifespan and their protective function.
ACKNOWLEDGMENTS
This work was supported by an MRC Ph.D. studentship awarded to H. M. M., a program grant from the British Heart Foundation (No. RG/2000011), and a British Heart Foundation Nonclinical Senior Lectureship Award to G. E. R. (BS/97001).
REFERENCES
- 1.Whyte M, Renshaw S, Lawson R, Bingle C. Apoptosis and the regulation of neutrophil lifespan. Biochem. Soc. Trans. 1999;27:802–807. doi: 10.1042/bst0270802. [DOI] [PubMed] [Google Scholar]
- 2.Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat. Rev. Immunol. 2002;2:965–975. doi: 10.1038/nri957. [DOI] [PubMed] [Google Scholar]
- 3.Jimenez MF, Watson RW, Parodo J, Evans D, Foster D, Steinberg M, Rotstein OD, Marshall JC. Dysregulated expression of neutrophil apoptosis in the systemic inflammatory response syndrome. Arch. Surg. 1997;132:1263–1269. doi: 10.1001/archsurg.1997.01430360009002. [DOI] [PubMed] [Google Scholar]
- 4.Ertel W, Keel M, Infanger M, Ungethum U, Steckholzer U, Trentz O. Circulating mediators in serum of injured patients with septic complications inhibit neutrophil apoptosis through up-regulation of protein-tyrosine phosphorylation. J. Trauma. 1998;44:767–775. doi: 10.1097/00005373-199805000-00005. [DOI] [PubMed] [Google Scholar]
- 5.Coxon A, Rieu P, Barkalow FJ, Askari S, Sharpe AH, von Andrian UH, Arnaout MA, Mayadas TN. A novel role for the β 2 integrin CD11b/CD18 in neutrophil apoptosis: a homeostatic mechanism in inflammation. Immunity. 1996;5:653–666. doi: 10.1016/s1074-7613(00)80278-2. [DOI] [PubMed] [Google Scholar]
- 6.Coxon A, Tang T, Mayadas TN. Cytokine-activated endothelial cells delay neutrophil apoptosis in vitro and in vivo. A role for granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 1999;190:923–934. doi: 10.1084/jem.190.7.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hotta K, Niwa M, Hara A, Ohno T, Wang X, Matsuno H, Kozawa O, Ito H, Kato K, Otsuka T, Matsui N, Uematsu T. The loss of susceptibility to apoptosis in exudated tissue neutrophils is associated with their nuclear factor-κ B activation. Eur. J. Pharmacol. 2001;433:17–27. doi: 10.1016/s0014-2999(01)01480-7. [DOI] [PubMed] [Google Scholar]
- 8.Watson RWG, Rotstein OD, Nathens AB, Parodo J, Marshall JC. Neutrophil apoptosis is modulated by endothelial transmigration and adhesion molecule engagement. J. Immunol. 1997;158:945–953. [PubMed] [Google Scholar]
- 9.Springer TA. Traffic signals on endothelium for lymphocyte recirculation and leukocyte emigration. Annu. Rev. Physiol. 1995;57:827–872. doi: 10.1146/annurev.ph.57.030195.004143. [DOI] [PubMed] [Google Scholar]
- 10.Yadav R, Larbi KY, Young RE, Nourshargh S. Migration of leukocytes through the vessel wall and beyond. Thromb. Haemost. 2003;90:598–606. doi: 10.1160/TH03-04-0220. [DOI] [PubMed] [Google Scholar]
- 11.Kettritz R, Gaido ML, Haller H, Luft FC, Jennette CJ, Falk RJ. Interleukin-8 delays spontaneous and tumor necrosis factor-α-mediated apoptosis of human neutrophils. Kidney Int. 1998;53:84–91. doi: 10.1046/j.1523-1755.1998.00741.x. [DOI] [PubMed] [Google Scholar]
- 12.Klein JB, Rane MJ, Scherzer JA, Coxon PY, Kettritz R, Mathiesen JM, Buridi A, McLeish KR. Granulocyte-macrophage colony-stimulating factor delays neutrophil constitutive apoptosis through phosphoinositol 3-kinase and extracellular signal-regulated kinase pathways. J. Immunol. 2000;164:4286–4291. doi: 10.4049/jimmunol.164.8.4286. [DOI] [PubMed] [Google Scholar]
- 13.Wang K, Scheel-Toellner D, Wong SH, Craddock R, Caamano J, Akbar AN, Salmon M, Lord JM. Inhibition of neutrophil apoptosis by type 1 IFN depends on cross-talk between phosphoinositol 3-kinase, protein kinase C-δ, and NF-κ B signaling pathways. J. Immunol. 2003;171:1035–1041. doi: 10.4049/jimmunol.171.2.1035. [DOI] [PubMed] [Google Scholar]
- 14.Whitlock BB, Gardai S, Fadok V, Bratton D, Henson PM. Differential roles for αMβ2 integrin clustering or activation in the control of apoptosis via regulation of Akt or ERk survival mechanisms. J. Cell Biol. 2000;151:1305–1320. doi: 10.1083/jcb.151.6.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kettritz R, Xu YX, Kerren T, Quass P, Klein JB, Luft FC, Haller H. Extracellular matrix regulates apoptosis in human neutrophils. Kidney Int. 1999;55:562–571. doi: 10.1046/j.1523-1755.1999.00280.x. [DOI] [PubMed] [Google Scholar]
- 16.Rubel C, Gomez S, Fernandez GC, Isturiz MA, Caamano J, Palermo MS. Fibrinogen-CD11b/CD18 interaction activates the NF-κB pathway and delays apoptosis in human neutrophils. Eur. J. Immunol. 2003;33:1429–1438. doi: 10.1002/eji.200323512. [DOI] [PubMed] [Google Scholar]
- 17.Ginis I, Faller DV. Protection from apoptosis in human neutrophils is determined by the surface of adhesion. Am. J. Physiol. 1997;272:C295–C309. doi: 10.1152/ajpcell.1997.272.1.C295. [DOI] [PubMed] [Google Scholar]
- 18.Walzog B, Jeblonski F, Zakrzewicz A, Gaehtgens P. β2 integrins (CD11/CD18) promote apoptosis of human neutrophils. FASEB J. 1997;11:1177–1186. doi: 10.1096/fasebj.11.13.9367353. [DOI] [PubMed] [Google Scholar]
- 19.Le’Negrate G, Rostagno P, Auberger P, Rossi B, Hofman P. Down regulation of caspases and fas ligand expression, and increased lifespan of neutrophils after transmigration across intestinal epithelium. Cell Death Differ. 2003;10:153–162. doi: 10.1038/sj.cdd.4401110. [DOI] [PubMed] [Google Scholar]
- 20.Hu M, Miller EJ, Lin X, Simms HH. Transmigration across a lung epithelial monolayer delays apoptosis of polymorphonuclear leukocytes. Surgery. 2004;135:87–98. doi: 10.1016/s0039-6060(03)00347-7. [DOI] [PubMed] [Google Scholar]
- 21.Buttrum SM, Hatton R, Nash GB. Selectin-mediated rolling of neutrophils on immobilized platelets. Blood. 1993;82:1165–1174. [PubMed] [Google Scholar]
- 22.Cooke BM, Usami S, Perry I, Nash GB. A simplified method for culture of endothelial cells and analysis of adhesion of blood cells under conditions of flow. Microvasc. Res. 1993;45:33–45. doi: 10.1006/mvre.1993.1004. [DOI] [PubMed] [Google Scholar]
- 23.Scheel-Toellner D, Wang K, Henriquez NV, Webb PR, Craddock R, Pilling D, Akbar AN, Salmon M, Lord JM. Cytokine-mediated inhibition of apoptosis in non-transformed T cells and neutrophils can be dissociated from protein kinase B activation. Eur. J. Immunol. 2002;32:486–493. doi: 10.1002/1521-4141(200202)32:2<486::AID-IMMU486>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 24.Zamzami N, Marchetti P, Castedo M, Zanin C, Vayssiere JL, Petit PX, Kroemer G. Reduction in mitochondrial potential constitutes an early irreversible step of programmed lymphocyte death in vivo. J. Exp. Med. 1995;181:1661–1672. doi: 10.1084/jem.181.5.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sheikh S, Rainger GE, Gale Z, Luu N-T, Rahman M, Nash GB. Differing mechanisms of leukocyte recruitment and sensitivity to conditioning by shear stress for endothelial cells treated with tumor necrosis factor-α or interleukin-1. Br. J. Pharmacol. 2005;145:1052–1061. doi: 10.1038/sj.bjp.0706281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Calderwood JW, Williams JM, Morgan MD, Nash GB, Savage CO. ANCA induces β2 integrin and CXC chemokine-dependent neutrophil-endothelial cell interactions that mimic those of highly cytokine-activated endothelium. J. Leukoc. Biol. 2005;77:33–43. doi: 10.1189/jlb.0104054. [DOI] [PubMed] [Google Scholar]
- 27.Luu NT, Rainger GE, Nash GB. Differential ability of exogenous chemotactic agents to disrupt transendothelial migration of flowing neutrophils. J. Immunol. 2000;164:5961–5969. doi: 10.4049/jimmunol.164.11.5961. [DOI] [PubMed] [Google Scholar]
- 28.Genentech Inc. Antagonists for Treatment of CD11/CD18 Adhesion Receptor-Mediated Disorders. Patent No. WO99/49856. 1999
- 29.Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002) J. Biol. Chem. 1994;269:5241–5248. [PubMed] [Google Scholar]
- 30.Marshall SJ, Senis YA, Auger JM, Feli R, Hofmann F, Salmon G, Peterson JT, Burslem F, Watson SP. GPIb-dependent platelet activation is dependent on Src kinases but not MAP kinase or cGMP-dependent kinase. Blood. 2004;103:2601–2609. doi: 10.1182/blood-2003-09-3319. [DOI] [PubMed] [Google Scholar]
- 31.Folkman J, Moscona A. Role of cell shape in growth control. Nature. 1978;273:345–349. doi: 10.1038/273345a0. [DOI] [PubMed] [Google Scholar]
- 32.Freyer DR, Morganroth ML, Todd RF., III Surface Mo1 (CD11b/CD18) glycoprotein is up-modulated by neutrophils recruited to sites of inflammation in vivo. Inflammation. 1989;13:495–505. doi: 10.1007/BF00916757. [DOI] [PubMed] [Google Scholar]
- 33.Hughes BJ, Hollers JC, Crockett-Torabi E, Smith CW. Recruitment of CD11b/CD18 to the neutrophil surface and adherence-dependent cell locomotion. J. Clin. Invest. 1992;90:1687–1696. doi: 10.1172/JCI116041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berton G, Lowell CA. Integrin signaling in neutrophils and macrophages. Cell. Signal. 1999;11:621–635. doi: 10.1016/s0898-6568(99)00003-0. [DOI] [PubMed] [Google Scholar]
- 35.Webb PR, Wang KQ, Scheel-Toellner D, Pongracz J, Salmon M, Lord JM. Regulation of neutrophil apoptosis: a role for protein kinase C and phosphatidylinositol-3-kinase. Apoptosis. 2000;5:451–458. doi: 10.1023/a:1009601220552. [DOI] [PubMed] [Google Scholar]
- 36.Simon HU. Neutrophil apoptosis pathways and their modifications in inflammation. Immunol. Rev. 2003;193:101–110. doi: 10.1034/j.1600-065x.2003.00038.x. [DOI] [PubMed] [Google Scholar]
- 37.Murray J, Barbara JSA, Dunkley SA, Lopez AF, Ostade XV, Condliffe AM, Dransfield I, Haslett C, Chilvers ER. Regulation of neutrophil apoptosis by tumor necrosis factor-α: requirement for TNFR55 and TNF75 for induction of apoptosis in vitro. Blood. 1997;90:2772–2783. [PubMed] [Google Scholar]
- 38.Murphy PM. Neutrophil receptors for interleukin-8 and related CXC chemokines. Semin. Hematol. 1997;34:311–318. [PubMed] [Google Scholar]
- 39.Beck GC, Yard BA, Breedijk AJ, van Ackern K, van der Woude FJ. Release of CXC-chemokines by human lung microvascular endothelial cells (LMVEC) compared with macrovascular umbilical vein endothelial cells. Clin. Exp. Immunol. 1999;118:298–303. doi: 10.1046/j.1365-2249.1999.01052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luster AD. Chemokines–chemotactic cytokines that mediate inflammation. N. Engl. J. Med. 1998;338:436–445. doi: 10.1056/NEJM199802123380706. [DOI] [PubMed] [Google Scholar]
- 41.Bahra P, Rainger GE, Wautier JL, Luu N-T, Nash GB. Each step during transendothelial migration of flowing neutrophils is regulated by the stimulatory concentration of tumor necrosis factor-α. Cell Adhes. Commun. 1998;6:491–501. doi: 10.3109/15419069809010797. [DOI] [PubMed] [Google Scholar]
- 42.Smith CW, Marlin SD, Rothlein R, Toman C, Anderson DC. Cooperative interactions of LFA-1 and Mac-1 with intercellular adhesion molecule-1 in facilitating adherence and transendothelial migration of human neutrophils in vitro. J. Clin. Invest. 1989;83:2008–2017. doi: 10.1172/JCI114111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lindemans CA, Coffer PJ. Regulation of granulocyte apoptosis by phosphatidylinositol 3-kinase. Biochem. Soc. Trans. 2004;32:480–484. doi: 10.1042/BST0320480. [DOI] [PubMed] [Google Scholar]