

Abstract
Pathological increases in cell death in the liver as well as in peripheral tissues has emerged as an important mechanism involved in the development and progression of nonalcoholic fatty liver disease (NAFLD). An increase in hepatocyte cell death by apoptosis is typically present in patients with NAFLD and in experimental models of steatohepatitis, while an increase in adipocyte cell death in visceral adipose tissue may be an important mechanism triggering insulin resistance and hepatic steatosis. The two fundamental pathways of apoptosis, the extrinsic (death receptor-mediated) and intrinsic (organelle-initiated) pathways, are both involved. This article summarizes the current knowledge related to the distinct molecular and biochemical pathways of cell death involved in NAFLD pathogenesis. In particular, it will highlight the efforts for the development of both novel diagnostic and therapeutic strategies based on this knowledge.
Keywords: apoptosis, caspase inhibitor, cell death, cytokeratin 18, fibrosis, nonalcoholic fatty liver disease, nonalcoholic steatohepatitis
Nonalcoholic fatty liver disease (NAFLD) is the most common form of chronic liver disease in both children and adults in the USA [1,2]. NAFLD is a clinicopathological syndrome including a wide spectrum of liver damage, ranging from hepatic steatosis, to nonalcoholic steatohepatitis (NASH), to cirrhosis [3]. NASH represents a more severe form of NAFLD, characterized by steatosis along with inflammation and hepatocellular damage in patients with no history of significant alcohol consumption [4]. Over the last decade there has been a growing understanding of the role of cell death and the specific molecular pathways that trigger apoptosis in the pathogenesis of NASH and disease progression, which has allowed the targeting of these pathways in the development of both novel therapeutic and diagnostic strategies for this disease.
Molecular mechanisms of apoptosis
Apoptosis is a highly organized and genetically controlled form of cell death. During apoptosis, cells are fragmented into small membrane-bound apoptotic bodies that contain cleaved DNA and proteolytic fragments (Figure 1). The apoptotic bodies are subsequently cleared by phagocytosis. Apoptosis may be executed by two fundamental pathways (Figure 2) [5,6]: the extrinsic pathway is mediated by death receptors on the cellular surface and the intrinsic pathway is organelle-based.
Figure 1.
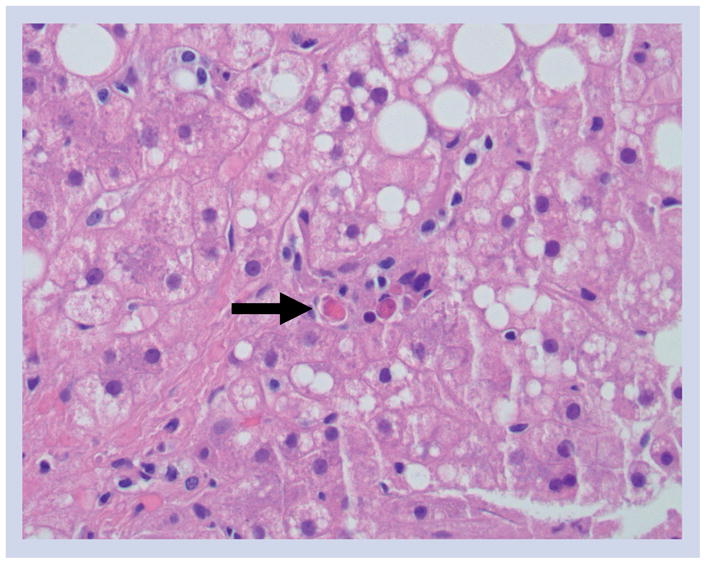
Hematoxylin and eosin-stained liver tissue showing apoptotic hepatocytes (arrow) in a patient with nonalcoholic steatohepatitis (magnification ×200).
Figure 2. Apoptotic pathways in nonalcoholic fatty liver disease.

Apoptosis occurs via two molecular pathways, extrinsic and intrinsic. The extrinsic pathway involves death ligand-induced activation of receptors, resulting in the formation of the death complex. The intrinsic pathway is activated by cellular stress and mitochondrial dysfunction. ER stress can induce apoptosis through JNK and CHOP, which activate the proapoptotic protein Bax, leading to mitochondrial dysfunction. Both apoptotic pathways activate intracellular proteases, the caspases, that cleave cell components in an organized way. The engulfment of the resulting apoptotic bodies by Kupffer cells promotes hepatic fibrosis and inflammation through the activation of stellate cells and the release of cytokines.
CHOP: C/EBP homologous protein; Cyto c: Cytochrome c; ER: Endoplasmic reticulum; FasL: Fas ligand; PERK: PKR-like ER kinase; TRAIL: TNF-related apoptosis-inducing ligand.
The extrinsic pathway is initiated by death receptors, including Fas, TNF receptor (R)1 and TNF-related apoptosis-inducing ligand (TRAIL) receptors. When engaged by their natural ligands, these receptors trigger intracellular cascades that activate death-inducing proteolytic enzymes, especially caspases.
In the intrinsic pathway, apoptosis can be initiated by several intracellular organelles. In fact, mitochondrial dysfunction, lysosomal permeabilization, endoplasmic reticulum (ER) stress and nuclear DNA damage can all trigger apoptosis (Table 1). In liver cells, mitochondrial dysfunction plays a critical role by amplifying the apoptotic signal and integrating both pathways into a final common pathway. Mitochondrial dysfunction results in the release of several proapoptotic proteins into the cytosol, including cytochrome c, which then forms an activation complex with apoptotic-protein activation factor-1 (Apaf-1) and caspase 9, known as the apoptosome. This complex activates the downstream effector caspases 3, 6 and 7, which execute the final apoptotic changes [7]. The mitochondrial events are regulated by the Bcl-2 family of proteins, which comprises at least 20 members with various degree of homology within four conserved regions (BH1–4 domains) [8,9]. The family can be divided into three subclasses: the first includes the antiapoptotic proteins Bcl-2, Bcl-xL and Mcl-1, which localize predominantly at the mitochondria. The second and third subclasses include only proapoptotic proteins: the multi-domain Bax-like proteins, such as Bax and Bak, and the BH3-only proteins, such as Bid, Bad, Bim, Noxa and PUMA. Bax resides in the cytoplasm in healthy cells; however, following an apoptotic stimulus, it can integrate into the outer mitochondrial membrane, causing membrane permeabilization [10,11]. The BH3-only protein Bid provides crosstalk between the extrinsic and intrinsic pathways. Indeed, activated caspase 8 (by death receptor engagement) cleaves the cytosolic protein Bid to a truncated active fragment, tBid, which translocates to mitochondria and induces cytochrome c release [12].
Table 1.
Key molecules involved in apoptosis during nonalcoholic steatohepatitis development.
| Pathway | Mediator | Role |
|---|---|---|
| Extrinsic pathway | Death ligands (FasL, TNF-α and TRAIL) | Formation of death complexes and activation of initiator caspases |
|
| ||
|
Intrinsic pathway
| ||
| Mitochondrial | Bcl-2, Bcl-xL and Mcl-1 | Antiapoptotic |
| Bax and Bak | Proapoptotic | |
| Bid, Bad, Bim, Noxa and PUMA | Proapoptotic (Bid mediates the crosstalk between the extrinsic and intrinsic pathways) | |
|
| ||
| Lysosomal | Cathepsin B | Lysosomal protease that leads to mitochondrial dysfunction |
|
| ||
| Endoplasmic reticulum | PERK and IRE-1α | Induce CHOP and JNK, which activate Bax |
CHOP: C/EBP homologous protein; FasL: Fas ligand; IRE-1α: Inositol-requiring enzyme 1α;
PUMA: p53-upregulated modulator of apoptosis; TRAIL: TNF-related apoptosis-inducing ligand.
Lysosomes are also an important source of proapoptotic signaling. Selective lysosomal permeabilization appears to augment death receptor-mediated apoptosis. Cathepsin B, a lysosomal protease, is released from lysosomes during TNF-α signaling, which leads to mitochondrial dysfunction and the release of cytochrome c [13,14]. TRAIL cytotoxicity is associated with lysosomal permeabilization and the redistribution of cathepsin B from lysosomes into the cytosol [15,16]. Lysosomal extracts can also cleave Bid into its active form [17].
Another important organelle in which proapoptotic signals may also originate is the ER, which plays a central role in the synthesis, folding and trafficking of proteins [18]. ER stress, initiated by protein overload or misfolding, leads to an adaptive response known as the unfolded protein response (UPR), which dampen the stress [19]. If the cell fails to adapt, alarm pathways are activated, including JNK, which results in apoptosis [20].
Hepatocyte apoptosis in chronic liver injury
Apoptosis has evolved as an important, if not critical, mechanism contributing to the progression of many human liver diseases [21–26]. The ensuing responses of cell repair, inf lammation, regeneration and fibrosis may all be triggered by apoptosis of adjacent cells [23]. Of these processes, hepatic fibrosis has the potential to be the most deleterious, as progressive fibrosis may result in cirrhosis and end-stage liver disease [27,28]. Experimental studies suggest that uncontrolled hepatocyte apoptosis may be a central mechanism, triggering liver fibrogenesis and fibrosis [29]. For instance, attenuation of hepatocyte apoptosis also reduces fibrogenesis in animal models of cholestasis [30–32], while hepatocyte-specific genetic disruption of the antiapoptotic member of the Bcl-2 family Bcl-xL results in hepatocyte apoptosis and liver fibrotic responses [33]. This latter model is highly illustrative because it directly demonstrates that hepatocyte apoptosis is profibrogenic. Engulfment of apoptotic bodies by hepatic stellate cells (HSCs) stimulates the fibrogenic activity of these cells and this may be, at least in part, through the induction of the activation of reduced nicotinamide adenine dinucleotide phosphate oxidase 2 (NOX2), the phagocytic NADPH oxidase, in HSCs [34,35]. Recent data also demonstrated that DNA from apoptotic hepatocytes acts as an important mediator of HSC activation and differentiation [36]. Activated stellate cells also secrete matrix metalloproteinases (MMPs), which play a significant role in hepatic fibrogenesis and can mediate the release of cell-bound TNF-α into the circulation, which in turn can induce and activate other MMPs in a feed-forward damage response [37]. In fact, inhibition of MMPs reduced liver injury, hepatocyte apoptosis and fibrosis in two mice models of liver disease [38,39].
Hepatocyte apoptosis in NAFLD
Lipotoxicity as a key trigger of apoptosis
During NAFLD development, while the majority of lipids are stored in the form of triglycerides (TGs), several other lipid metabolites, such as free fatty acids (FFAs), cholesterol, sphingolipids and phospholipids, may also accumulate. Considerable data now indicate that TG accumulation is not harmful to hepatocytes per se and, in fact, may represent a protective mechanism against lipotoxicity [40,41]. Studies from our laboratory, as well as others, have demonstrated that saturated fatty acids (SFAs), as well as free cholesterol, are key mediators of lipotoxicity by triggering specific signaling pathways resulting in apoptotic cell death [42,43]. FFAs cross the hepatocyte membrane passively and actively via fatty acid transport proteins (FATPs). Recently, Bechmann et al. demonstrated that the FATP CD36/fatty acid translocase was upregulated in patients with NASH, in association with serum FFA and mediators of apoptosis [44]. The role and relevance of FATPs in the pathogenesis of NASH deserves further elucidation in future studies.
The extrinsic pathway
Fas & Fas ligand
Fas is a glycosylated protein that is widely expressed in the liver [45] and its activation by Fas ligand (FasL) leads to receptor trimerization and the formation of the death-inducing signaling complex (DISC) [46]. The DISC contains a Fas-associated protein with death domain (FADD). FADD is required for the recruitment and activation of caspase 8, which can activate executioner caspases (caspases 3, 6 and 7), either directly (type 1 pathway) or indirectly via mitochondrial involvement (type 2 pathway) as in hepatocytes [47,48].
We reported that Fas protein expression is increased in liver samples from NASH patients [49]. By performing semiquantitative analyses we demonstrated that Fas immunostaining was significantly higher in NASH patients compared with patients with simple steatosis and normal controls. In a subsequent study we demonstrated an increased sensitivity to Fas-mediated hepatocyte apoptosis in a dietary model of NAFLD induced by feeding mice with a high carbohydrate diet, which recapitulates many of the cardinal features of human NAFLD, including obesity, insulin resistance, hyperleptinemia, elevated serum FFA and hepatic steatosis [50]. Moreover, exposure of human liver cells to FFA resulted in upregulation of Fas expression and increased sensitivity to Fas-mediated apoptosis. The precise mechanism by which FFA promotes Fas generation remains unknown and will require further investigation. A recent report demonstrated that the hepatocyte growth factor receptor Met associates directly with Fas in normal liver tissues, preventing Fas activation [51]. Fas sequestration by Met is abrogated in both human and experimental NAFLD, resulting in increased Fas–FasL complex formation and apoptosis.
TNF-R1 & TNF-α
Tumor necrosis factor α is a pleiotropic proinflammatory cytokine produced mainly by activated macrophages. TNF-α can signal through two different receptors: TNF-R1 and TNF-R2 [52]. TNF-R1, the best studied of these receptors, can induce both proapoptotic and prosurvival signaling. Upon stimulation by TNF-α, TNF-R1 engages TNF receptor-associated protein with death domain (TRADD), receptor interacting protein 1 (RIP1) and TNF receptor-associated factor 2 (TRAF2) to form the so-called complex I [53]. This complex internalizes and binds to FADD, resulting in caspase 8 activation and cell death. TNF-α/TNF-R1 can also induce JNK activation, which can contribute to cell death.
In normal liver TNF-R1 expression is low, but TNF-R1 expression increases in various liver diseases. An increase in both TNF-α and TNF-R1 mRNA levels in the liver of NASH patients has been reported [54]. This increase of TNF-α mRNA was higher in patients with a more advanced stage of fibrosis. Consistent with these results, another group reported an increased protein expression of TNF-R1 in liver specimens from NASH patients [55]. In addition, Hui and colleagues demonstrated an increase in serum levels of TNF-α in NASH patients [56]. Experimental studies have also provided strong evidence for a role of the TNF-α–TNF-R1 system in the pathogenesis of NAFLD. Li et al. demonstrated that treatment with antibodies to TNF-α attenuated liver injury in a mice model of NAFLD [57]. In addition, we demonstrated that TNF-R1 knockout mice are protected against diet-induced steatosis and liver injury [58]. We also demonstrated that lysosomal destabilization by FFA promotes hepatic lipotoxicity by releasing cathepsin B into the cytosol and by stimulating TNF-α expression via a NF-κB-dependent pathway [58]. TNF-α may also further promote lysosomal destabilization, resulting in a feed-forward, self-perpetuating pathway, further accentuating liver injury.
An intriguing study by Mari and colleagues found that free cholesterol loading sensitized hepatocytes to TNF-α and Fas-induced apoptosis and steatohepatitis development [59]. In rats fed a hypercholesterolemic diet, TNF-α induced the release of cytochrome c, caspase 3 activation and reactive oxygen species (ROS) generation. Mitochondrial glutathione was found to be selectively depleted. The authors proposed a critical role for mitochondrial free cholesterol loading in precipitating steatohepatitis by sensitizing hepatocytes to TNF-α and Fas through mitochondrial glutathione depletion.
TRAIL-R2/DR5 & TRAIL
Of the five known TRAIL receptors, only TRAIL-R1/DR4 and TRAIL-R2/DR5 contain a death domain and can initiate apoptosis [60]. The signaling pathways for TRAIL largely overlap with Fas signaling, with the formation of DISC and activation of caspase 8.
TRAIL is known to induce apoptosis in a variety of transformed cells while sparing normal cells [61]. A recent study by Malhi and colleagues demonstrated that FFA sensitized normal hepatocytes to TRAIL-mediated apoptosis [62]. Oleic acid treatment led to upregulation of TRAIL-R2/DR5 but not TRAIL-R1/DR4 and this upregulation was JNK dependent. Furthermore, enhanced DR5 expression was observed in liver biopsy samples from patients with NASH [55].
The intrinsic pathway
Mitochondria
Impaired mitochondrial function is a central abnormality responsible for the progression from hepatic steatosis to steatohepatitis. Studies in experimental models of NAFLD, as well as in humans with NAFLD, have demonstrated that liver cells have both structural and functional mitochondrial abnormalities [63,64]. Structural abnormalities include mitochondrial enlargement and development of crystalline inclusions, whereas functional mitochondrial abnormalities are characterized by enhanced production of ROS, accumulation of lipid peroxides and release of cytochrome c into the cytoplasm [63,65].
However, the molecular mechanisms responsible for the mitochondrial dysfunction remained poorly understood until recently. We recently demonstrated that incubation of human and murine hepatocytes with FFA results in a dose- and saturation- dependent mitochondrial dysfunction [58]. The SFA palmitate, at concentrations that mimic the levels of this FFA present in the circulation of humans with the metabolic syndrome, induced significant mitochondrial membrane permeabilization and increased ROS production. Recent studies from the Gregory J Gores laboratory have implicated different proteins from the Bcl-2 family in FFA-induced hepatocyte apoptosis. They demonstrated that SFAs induce JNK-dependent hepatocyte lipoapoptosis by activating the proapoptotic proteins Bim and Bax, which trigger the mitochondrial apoptotic pathway [66]. Hepatocyte apoptosis was abrogated by siRNA-targeted knockdown of Bim. Bim mRNA expression is regulated by the transcription factor FoxO3a. In a subsequent study, they observed activation of FoxO3a, its nuclear translocation and binding to the Bim promoter following treatment of hepatocytes with FFA [67]. Furthermore, siRNA-targeted knockdown of FoxO3a abrogated the increase in Bim and cell death by saturated FFA. The same group demonstrated that the SFA palmitate, but not the monounsaturated fatty acid oleate, induced a JNK1-dependent PUMA expression in primary murine hepatocytes, which led to Bax activation and apoptosis by palmitate [68]. Their data demonstrated that PUMA expression was upregulated and JNK was activated in patients with NASH compared with patients with hepatic steatosis. Finally, a recent study revealed that the antiapoptotic protein Mcl-1 was rapidly degraded in liver cells in response to the SFAs palmitate and stearate by a proteasome-dependent pathway [69]. The inhibition of Mcl-1 degradation attenuated hepatocyte apoptosis by SFAs.
Lysosomes
Accumulating evidence suggests a central role of lysosomes in both necrotic and apoptotic cell death. In both instances, lysosomal permeabilization appears to be an early event that precedes other features characteristic of the cell death processes [70]. Lysosomal permeabilization results in the release of lysosomal proteases into the cytosol. The lysosomal cysteine proteases, also called cathepsins, represent the largest group of proteolytic enzymes in the lysosomes, with cathepsin B, D and L being the most abundant [70]. All three cathepsins are active at the neutral pH of the cytosol [71]. Cathepsin B, which is one of the most stable proteases at physiologic pH, has been demonstrated to be essential in different models of liver injury, including bile acid-induced hepatocyte apoptosis, TNF-α-induced liver damage, and ischemia– reperfusion injury in steatotic livers [14,72–76]. The release of lysosomal enzymes may set off a cascade of events culminating in cell death. A possible mechanism is the direct cleavage and activation of caspases. Indeed, cathepsin B may activate caspase 2 in vitro, while cathepsin L may indirectly activate procaspase 3. However, a growing body of evidence suggests that lysosomal proteases induce cell death by acting directly or indirectly on mitochondria to induce mitochondrial dysfunction [76,77].
Reactive oxygen species generated following mitochondrial damage, and other possible factors of mitochondria origin, could also feedback to the lysosome, resulting in further lysosomal breakdown and cell damage [77]. We initially reported that cathepsin B is released into the cytosol in response to FFA in vitro, and that the redistribution of cathepsin B into the cytoplasm is present in human liver tissue from patients with NAFLD [78]. More recently we extended these observations by demonstrating that during FFA treatment of liver cells, lysosomal permeabilization and release of cathepsin B into the cytosol is an early event that occurs hours prior to mitochondrial depolarization and cytochrome c redistribution into the cytosol [79]. More importantly, cathepsin B silencing by siRNA, or chemical inhibition by two different selective pharmacological inhibitors, significantly prevented FFA-induced mitochondrial dysfunction, proving cathepsin B is essential for FFA-induced mitochondrial dysfunction. Supporting these results are the in vivo studies demonstrating that cathepsin B knockout mice are protected against diet-induced mitochondrial dysfunction, increased oxidative stress and liver damage. Taken together, these data strongly suggest a central role for the lysosomal–mitochondrial axis in NAFLD progression.
Endoplasmic reticulum
The potential role of ER stress responses in NAFLD development has received a great deal of interest recently. The ER is a subcellular organelle where the vast majority of secreted and membrane proteins are folded. A variety of cellular perturbations can lead to the accumulation of unfolded proteins [80]. These proteins tend to form aggregates that activate a compensatory response, termed the UPR. The UPR is a coordinated response that includes cell cycle arrest, transient attenuation of global protein synthesis, induction of ER-localized chaperone proteins and folding catalysts, and activation of ER-associated protein degradation. Failure of the UPR to re-establish ER homeostasis can lead to apoptotic cell death. Three different ER transmembrane signaling proteins sense the accumulation of unfolded protein and induce ER stress responses. These proteins are PKR-like ER kinase (PERK), inositol-requiring enzyme (IRE)-1α and activating transcription factor 6 (ATF6). PERK can induce the transcription factor C/EBP homologous protein (CHOP) and IRE-1α activates the JNK pathway [81,82]. Both JNK and CHOP can activate the proapoptotic protein Bax, leading to mitochondrial dysfunction.
Overaccumulation of SFA may result in ER stress and apoptosis [83,84]. Incubation of a rat hepatoma cell line with SFA resulted in a significant increase in ER stress response genes, including CHOP, GADD34 and GRP78, followed by an increase in apoptotic cell death, which was mitochondrial-dependent. In vivo studies using dietary models of NAFLD that result in different compositions of hepatic FFA support this concept [85]. Feeding male Wistar rats either a high saturated fat diet or a high sucrose diet, but not a high polyunsaturated fat diet, resulted in a significant increase in hepatic SFA, as well as increased ER stress, caspase-3 activation and liver injury.
Puri and colleagues studied the potential role of ER stress in human NAFLD [86]. They demonstrated a variable degree of UPR activation in liver biopsies from patients with NAFLD and NASH, compared with subjects with the metabolic syndrome and normal liver histology. Human NASH was specifically associated with activation of JNK and failure to generate splicing X box protein-1 (sXBP-1), a key transcription factor involved in the ER stress response. Pagliassotti’s laboratory have recently demonstrated that exposure of hepatocytes to palmitate and stearate reduced ER luminal calcium stores and increased markers of ER stress, changes that preceded liver cell death [87]. Their data suggested that redistribution of ER luminal calcium contributes to SFA-mediated ER stress. Interestingly, co-incubation with oleate prevented the reduction in calcium stores and the induction of ER stress markers. More recently, Cazanave et al. demonstrated that palmitate can mediate ER stress to induce CHOP and phosphorylated- c-Jun containing activator protein (AP)-1 complex. This can lead to upregulation of PUMA expression, with subsequent activation of Bax, mitochondrial dysfunction and, eventually, apoptosis [88].
Adipocyte apoptosis & hepatic steatosis
Abnormal adipose tissue metabolism has been identified as a critical mechanistic link between obesity and NAFLD [89,90]. The increased release of FFA from adipose tissue in a state of insulin resistance characteristic of obesity represents the main source of FFA during the development of hepatic steatosis in NAFLD [91]. In addition, a state of low-grade chronic inflammation characterized by macrophage infiltration of adipose tissues, plays a crucial role in the development of insulin resistance [92–94]. We have recently identified adipocyte apoptosis as a key initial event that contributes to macrophage infiltration into adipose tissue, insulin resistance and hepatic steatosis in obese mice and humans [95]. We found that both the extrinsic and intrinsic pathways were activated in adipose tissue from obese animals. Furthermore, inhibition of adipocyte apoptosis in Bid knockout mice protected against macrophage infiltration of adipose tissue and hepatic steatosis. A recent study demonstrated that selectively deleting Fas from adipose tissue (adipose-specific Fas-knockout mice) protected from high-fat diet-induced insulin resistance and liver steatosis [96]. In addition, Fas and FasL were found to be upregulated in the adipose tissue of obese patients and even more so in obese diabetic patients, which suggests a role for Fas in obesity-induced insulin resistance.
Development of NASH diagnostics by targeting apoptotic pathways
Cytokeratin 18 fragment levels & other hepatocyte apoptotic markers as noninvasive biomarkers for NASH
The effector caspases (mainly caspase 3) cleave different substrates inside the cell – including cytokeratin 18 (CK18), the major intermediate filament protein in the liver – resulting in the characteristic morphological alterations noted in apoptotic cells. Caspase 3 cleaves CK18 into an N-terminal and a C-terminal at a typical cleavage sequence at Asp 237, and the C-terminal is further cleaved at a second site at Asp 398 [97,98]. These fragments, but not full-length CK18, can be detected using the monoclonal antibody M30 both in the liver and blood (Figure 3). By using a specific immunoELISA assay, our group demonstrated that CK18 fragments were strikingly increased in the serum of patients with NASH compared with patients with simple steatosis and those with normal liver biopsies [99]. Several groups have assessed the accuracy of CK18 fragments for predicting the presence of NASH, including one recent study in children [100–106]. The results were encouraging, with an area under the receiver operating characteristic (ROC) curve between 0.71 and 0.93 (Table 2). The utility of CK18 fragments has been validated in a multicenter study that demonstrated that for every 50 U/l increase in the plasma level of CK18, the likelihood of having NASH as opposed to simple steatosis increased by 74% (odds ratio: 1.74; 95% CI: 1.31–2.31) [107]. The CK18 test has several features that fulfill the requirements for an ideal biomarker for NAFLD, including that the test is simple, easy to measure and is reproducible. This test identifies the presence of NASH and the risk of associated fibrosis, and allows the monitoring of disease progression over time. More recently, we have demonstrated that serum levels of soluble Fas and soluble FasL were significantly higher in patients with biopsy-proven NASH compared with patients with simple steatosis (Figure 3) [108]. We generated a prediction model to diagnose NASH, which included CK18, soluble Fas and age, with an almost perfect area under the ROC (0.953). A recent Chinese study found that soluble TRAIL concentrations were significantly higher in the sera of patients with NAFLD than those of controls [109]. However, no histological data were available to compare these levels between patients with NASH and simple steatosis.
Figure 3. Cytokeratin 18 and soluble Fas levels as noninvasive markers for nonalcoholic steatohepatitis.

Caspase 3-generated CK18 fragments are produced during hepatocyte apoptosis and can be detected using a monoclonal antibody (M30), both in the liver and in blood. The Fas–Fas ligand complex is a key part of the extrinsic pathway in the hepatocyte apoptotic cascade, and soluble Fas can be measured in the serum using ELISA.
CK18: Cytokeratin 18.
Table 2.
Accuracy of cytokeratin 18 fragments in predicting the presence of nonalcoholic steatohepatitis.
| Study (year) | Population | Patients (n) | AUC | Sensitivity (%) | Specificity (%) | Ref. |
|---|---|---|---|---|---|---|
| Wieckowska et al. (2006) | Single-center clinic patients | 44 | 0.93 | 86 | 99.9 | [99] |
| Yilmaz et al. (2007) | Single-center clinic patients | 83 | 0.83 | 62 | 100 | [104] |
| Diab et al. (2008) | Single-center bariatric surgery patients | 99 | 0.88 | 77 | 100 | [101] |
| Malik et al. (2009) | Single-center clinic patients | 95 | 0.80 | – | – | [103] |
| Younossi et al. (2008) | Single-center morbidly obese patients | 101 | 0.71 | 70 | 84 | [105] |
| Feldstein et al. (2009) | Multicenter clinic patients | 139 | 0.83 | 75 | 81 | [107] |
| Fitzpatrick et al. (2010) | Pediatric patients | 45 | 0.85 | 84 | 88 | [102] |
| Musso et al. (2010) | Single-center clinic patients | 125 | 0.83 | 78 | 88 | [106] |
| Anty et al. (2010) | Morbidly obese patients, the Nice Model including CK18, ALT and the metabolic syndrome | 310 (training) | 0.88 | 84 | 86 | [100] |
| 154 (validation) | 0.84 | – | – |
ALT: Alanine transaminase; AUC: Area under the curve; CK18: Cytokeratin 18.
Treatment of NASH by targeting apoptotic pathways
Given the potential severe complications of NASH, including cirrhosis, liver failure and death, it is imperative that medical therapies targeted at halting the progression of NASH and improving steatosis and inflammation be developed. It is clear from the literature that weight loss alone is not an adequate longterm solution for the treatment of NAFLD. Thus, developing a pharmacological therapy to improve NASH is the next step. To date, there are no US FDA-approved pharmacological therapies to treat NASH. Several studies have been performed to evaluate potential therapies, with the most promising medications including insulin sensitizers and antioxidants. Given the key role of apoptosis in the progression of NAFLD, as outlined in this article, it is a logical progression to target key players in apoptosis for potential medical therapies for NASH (Table 3).
Table 3.
Treatment of nonalcoholic steatohepatitis targeting apoptotic pathways.
| Therapeutic agent | Pathway | Target | Trial | Ref. |
|---|---|---|---|---|
| R-3020 | Intrinsic | Cathepsin B inhibitor | Preclinical | [31,42] |
| VBY-376 | Intrinsic | Cathepsin B inhibitor | Preclinical and Phase I | [110] |
| Pentoxifylline | Extrinsic | TNF-α inhibitor | Phase II RCT | [114] |
| YLGA peptides | Extrinsic | Fas | Preclinical | [51] |
| VX-166 | Effector caspases | Pan-caspase inhibitor | Preclinical | [118,119] |
| IDN-6556 | Effector caspases | Pan-caspase inhibitor | Multicenter, double-blind, placebo-controlled, dose-ranging | [120] |
| GS-9450 | Effector caspases | Inhibitor of caspases 8, 9 and 1 | Phase II RCT | [201] |
| PF-03491390 (formerly IDN-6556) | Effector caspases | Pan-caspase inhibitor | Preclinical | [122] |
RCT: Randomized controlled trial; YLGA: Tyr Leu Gly Ala.
Targeting of mediators of the intrinsic pathway
There are several mitochondrial-, lysosomal- and ER stressrelated pathways that could potentially be targeted to attenuate apoptosis. It may be possible to decrease lysosomal effects by targeting the action of cathepsin B. We have already described the proteolytic effects of cathepsin B, including the activation of cell signaling pathways ultimately leading to cell death. In a preclinical study using a mouse model of the effects of NASH, administration of a cathepsin B inhibitor (R-3020) to mice fed a high-carbohydrate diet led to decreased lysosomal permeabilization and decreased steatosis and liver injury [37]. Both genetic and pharmacologic inhibition of cathepsin B decreased hepatocyte apoptosis and histologic evidence of liver injury in a mouse model of cholestasis [31]. More recently, the reversible cathepsin B inhibitor VBY-376 demonstrated potent activity in a mouse model of liver fibrosis. Furthermore, a Phase I study to evaluate the safety and pharmacokinetics of VBY-376 in humans was conducted and no serious adverse events were observed [110]. Wu et al. used 18β-glycyrrhetinic acid, a biologically active metabolite of licorice root extract, and demonstrated that it prevented FFA-induced lipid accumulation and hepatocyte apoptosis both in vitro in a human liver cell line and in vivo in a rat NAFLD model [111]. The proposed mechanism involved stabilizing the lysosomal membrane, inhibiting cathepsin B expression and enzyme activity, and reducing mitochondrial cytochrome c release.
Targeting of mediators of the extrinsic pathway
Mediators of the extrinsic pathway are also potential therapeutic targets for patients with NASH. Blocking the activation of death receptors, such as TNF-R, TRAIL and FADD, and their associated signaling cascades may lead to decreased inflammation in the presence of steatosis. There has been active research on components of the TNF-α and TNF-R cascade, including the evaluation of several TNF-α inhibitors as potential therapies for NASH. Pentoxifylline, a weak TNF-α inhibitor, has been studied in several open-label trials [112,113]. Preliminary results did demonstrate improvement in transaminases and liver histology; however, significant side effects were documented as well, including severe nausea and other gastrointestinal symptoms. More recently, a double-blind, randomized, placebo-controlled trial demonstrated the efficacy of pentoxifylline in improving alanine transaminase (ALT) and histological features of NASH, including hepatocyte apoptosis, without significant side effects [114]. Etanercept, a TNF-α receptor compound that reduces TNF-α bioactivity, may also be effective in the treatment of NASH. However, caution must be taken as a new randomized placebo-controlled trial has demonstrated that etanercept was associated with a significantly higher mortality rate in patients with moderate-to-severe alcoholic hepatitis [115]. Future therapies may also target interrupting the Fas–FasL interaction and formation of the DISC. Zou et al. performed studies utilizing Tyr–Leu–Gly–Ala (YLGA) peptides, short strands of peptides corresponding to a portion of FasL that bind readily to Fas. YLGA tetramers were administered to ob/ob mice daily for up to 4 weeks. Mice injected with YLGA peptides had significant reductions in hepatic apoptosis, hepatic inflammation and ALT levels compared with control animals [51].
Targeting effector caspases
Given the key role that caspases play in the intracellular cascade leading to apoptosis, several groups have developed caspase inhibitors as a means to decrease apoptosis. The majority of inhibitors that have been developed are pan-caspase inhibitors. The first pancaspase inhibitor to enter clinical trials was IDN-6556 and it has been studied extensively [116]. When administered to bile-duct ligated (BDL) mice as a model for hepatocyte apoptosis and liver fibrosis, apoptosis, bile infarcts, chemokine activation and serum ALT levels were reduced [30]. Similarly to the other caspase inhibitors described in this section, IDN-6556 not only inhibits proapoptotic caspases but also proinflammatory capsases such as caspase 1, which has been involved in the processing and activation of various proteins with functions in inflammation and tissue repair during tissue damage. Thus, part of the effects of these drugs may be related to inhibition of this alternative pathway [117].
The pan-caspase inhibitor VX-166 was used in a mouse model of NASH (genetically obese male db/db mice fed the methionine– choline-deficient [MCD] diet). VX-166 improved liver fibrosis; however, it failed to improve ALT levels or the net liver injury, as assessed by the NAFLD activity score [118]. The mechanism for decreased fibrosis was thought to be related to the fact that phagocytosis of apoptotic hepatocytes activates HSCs and VX-166 inhibited HSC activation by apoptotic bodies. More recently, Anstee et al. evaluated VX-166 in mice models of steatosis (high-fat diet) and steatohepatitis (db/db mice on MCD diet) and demonstrated that VX-166 did not reduce steatosis but reduced histological inflammation, apoptosis, ALT levels and oxidative stress, particularly in the MCD model [119].
The use of caspase inhibitors in human liver disease is being explored. A multicenter, placebo-controlled trial used IDN-6556 in 105 patients with chronic liver diseases, mainly chronic hepatitis C (n = 80), and few NASH patients (n = 5) [120]. IDN-6556, given for 14 days, significantly lowered aminotransferase activity in both hepatitis C virus and NASH patients and appeared to be well tolerated. In a follow-up, larger, double-blind, randomized, placebo-controlled, parallel-dose study using the same compound, now renamed as PF-03491390, 204 chronic hepatitis C patients were treated with placebo or this compound orally, twice daily for up to 12 weeks. They demonstrated that PF-03491390 significantly reduced serum aspartate transaminase (AST) and ALT levels and was well tolerated over the study period [121]. More recently, the PF-03491390 compound was found to also decrease liver injury, as well as fibrosis in a murine model of NASH [122]. Another caspase inhibitor, GS-9450, was evaluated in a Phase II randomized, double- blind, placebo-controlled study in adult patients with NASH, which was recently completed [201]. In this study, patients (n = 124, principally male, mean age 45 years, with BMI >30 kg/m2) with biopsy-proven NASH were randomized to receive 1, 5, 10 or 40 mg GS-9450 or placebo once daily for 4 weeks [123]. After 4 weeks on treatment, patients in the 40-mg treatment group experienced the greatest reduction in ALT and AST levels. At week 4, linear regression of ALT versus GS-9450 dose was highly significant (p < 0.0001), with 35% achieving ALT levels within the normal range (7–56 U/l) compared with 0% at study baseline, and 48% achieving normal levels of AST (5–40 U/l) compared with 20% at baseline. Placebo treatment showed no meaningful change for ALT or AST. In addition, the on-treatment measure of CK18 fragments declined in the 10- and 40-mg dose groups (median baseline and week 4 values were 540 and 445 U/l in the 10-mg group and 562 and 386 U/l in the 40-mg group, respectively), although no dose-related response was seen. Unfortunately, another study evaluating this caspase inhibitor in patient with chronic hepatitis C was subsequently stopped due to safety concerns. Thus, the future clinical development of this particular compound is uncertain.
Expert commentary
Nonalcoholic fatty liver disease is threatening to become a major public-health problem in the USA. In this article, we examined the role of hepatocyte apoptosis in NAFLD progression from the benign form of simple steatosis to the more aggressive form of NASH. We provided evidence that both apoptotic pathways are activated in humans with NASH, as well as animal models of NASH. FasL, TNF-α and TRAIL activate the extrinsic pathways, whereas mitochondrial dysfunction, lysosomal permeabilization and ER stress activate the intrinsic pathway. These findings open a new and exciting era in our understanding of why some patients progress to NASH rapidly and may give us the chance to stop this process by inhibiting specific molecular pathways involved in hepatocyte apoptosis.
Five-year view
Over the last decade, our understanding of the pathogenesis of NAFLD has increased exponentially. Hepatocyte apoptosis is at the center of the mechanisms that are responsible for disease progression to NASH. More recently, the role of adipocyte apoptosis in metabolic disorders, including insulin resistance and hepatic steatosis, is being elucidated. The uncovering of apoptotic mechanisms in NAFLD has led to major breakthroughs in terms of the noninvasive diagnosis of NASH. New therapeutic agents that can modulate hepatocyte apoptosis are being explored and can provide attractive options to treat patients with steatohepatitis.
Key issues
Apoptosis is a highly organized form of cell death that contributes to liver injury and fibrosis in many human liver diseases.
Hepatocyte apoptosis is a key mechanism for disease progression in patients with nonalcoholic fatty liver disease (NAFLD), the most common chronic liver disease in the USA.
Both the extrinsic pathway (mediated by death receptors) and the intrinsic pathway (organelle-based) of apoptosis are activated in human NAFLD.
Saturated free fatty acids may induce hepatocyte apoptosis through death receptors (Fas, TNF receptor 1 and TNF-related apoptosisinducing ligand [TRAIL]-R2/DR5), the lysosomal-mitochondrial pathway and endoplasmic reticulum stress.
Recently, a role for adipocyte apoptosis in insulin resistance and hepatic steatosis has been suggested. Both pathways of apoptosis were activated in adipocytes from obese animals and the inhibition of adipocyte apoptosis resulted in the resolution of hepatic steatosis in these animals.
Markers of hepatocyte apoptosis in the serum (cytokeratin 18 and soluble Fas) can be used as noninvasive markers to diagnose nonalcoholic steatohepatitis, the most severe form of NAFLD.
Modulating hepatocyte apoptosis has great implications for developing new therapeutic agents for nonalcoholic steatohepatitis. This can be achieved by targeting mediators of the extrinsic and intrinsic pathways and by inhibiting the effector caspases.
Footnotes
Financial & competing interests disclosure
This work was supported by NIH grants DK076852 and DK082451 to Ariel E Feldstein. Ariel E Feldstein is named as co-inventor on pending patents filed by the Cleveland Clinic that refer to the use of biomarkers in fatty liver disorders. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
- 1.Angulo P. Nonalcoholic fatty liver disease. N Engl J Med. 2002;346:1221–1231. doi: 10.1056/NEJMra011775. [DOI] [PubMed] [Google Scholar]
- 2.Wieckowska A, Feldstein AE. Nonalcoholic fatty liver disease in the pediatric population: a review. Curr Opin Pediatr. 2005;17:636–641. doi: 10.1097/01.mop.0000172816.79637.c5. [DOI] [PubMed] [Google Scholar]
- 3.Matteoni CA, Younossi ZM, Gramlich T, Boparai N, Liu YC, McCullough AJ. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology. 1999;116:1413–1419. doi: 10.1016/s0016-5085(99)70506-8. [DOI] [PubMed] [Google Scholar]
- 4.Harrison SA, Neuschwander-Tetri BA. Nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Clin Liver Dis. 2004;8:861–879. ix. doi: 10.1016/j.cld.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 5.Delhalle S, Duvoix A, Schnekenburger M, Morceau F, Dicato M, Diederich M. An introduction to the molecular mechanisms of apoptosis. Ann NY Acad Sci. 2003;1010:1–8. doi: 10.1196/annals.1299.001. [DOI] [PubMed] [Google Scholar]
- 6.Yuan J, Horvitz HR. A first insight into the molecular mechanisms of apoptosis. Cell. 2004;116:S53–S56. doi: 10.1016/s0092-8674(04)00028-5. [DOI] [PubMed] [Google Scholar]
- 7.Riedl SJ, Shi Y. Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol. 2004;5:897–907. doi: 10.1038/nrm1496. [DOI] [PubMed] [Google Scholar]
- 8.Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2:647–656. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- 9.Cory S, Huang DC, Adams JM. The Bcl-2 family: roles in cell survival and oncogenesis. Oncogene. 2003;22:8590–8607. doi: 10.1038/sj.onc.1207102. [DOI] [PubMed] [Google Scholar]
- 10.Antonsson B, Montessuit S, Sanchez B, Martinou JC. Bax is present as a high molecular weight oligomer/complex in the mitochondrial membrane of apoptotic cells. J Biol Chem. 2001;276:11615–11623. doi: 10.1074/jbc.M010810200. [DOI] [PubMed] [Google Scholar]
- 11.Nechushtan A, Smith CL, Lamensdorf I, Yoon SH, Youle RJ. Bax and Bak coalesce into novel mitochondria-associated clusters during apoptosis. J Cell Biol. 2001;153:1265–1276. doi: 10.1083/jcb.153.6.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- 13.Foghsgaard L, Wissing D, Mauch D, et al. Cathepsin B acts as a dominant execution protease in tumor cell apoptosis induced by tumor necrosis factor. J Cell Biol. 2001;153:999–1010. doi: 10.1083/jcb.153.5.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guicciardi ME, Deussing J, Miyoshi H, et al. Cathepsin B contributes to TNF-α-mediated hepatocyte apoptosis by promoting mitochondrial release of cytochrome c. J Clin Invest. 2000;106:1127–1137. doi: 10.1172/JCI9914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guicciardi ME, Bronk SF, Werneburg NW, Gores GJ. cFLIPL prevents TRAIL-induced apoptosis of hepatocellular carcinoma cells by inhibiting the lysosomal pathway of apoptosis. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1337–G1346. doi: 10.1152/ajpgi.00497.2006. [DOI] [PubMed] [Google Scholar]
- 16.Kahraman A, Barreyro FJ, Bronk SF, et al. TRAIL mediates liver injury by the innate immune system in the bile duct-ligated mouse. Hepatology. 2008;47:1317–1330. doi: 10.1002/hep.22136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stoka V, Turk B, Schendel SL, Kim TH, et al. Lysosomal protease pathways to apoptosis. Cleavage of bid, not procaspases, is the most likely route. J Biol Chem. 2001;276:3149–3157. doi: 10.1074/jbc.M008944200. [DOI] [PubMed] [Google Scholar]
- 18.Lemasters JJ. Dying a thousand deaths: redundant pathways from different organelles to apoptosis and necrosis. Gastroenterology. 2005;129:351–360. doi: 10.1053/j.gastro.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 19.Liu CY, Kaufman RJ. The unfolded protein response. J Cell Sci. 2003;116:1861–1862. doi: 10.1242/jcs.00408. [DOI] [PubMed] [Google Scholar]
- 20.Rao RV, Ellerby HM, Bredesen DE. Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ. 2004;11:372–380. doi: 10.1038/sj.cdd.4401378. [DOI] [PubMed] [Google Scholar]
- 21.Feldstein AE, Gores GJ. Apoptosis in alcoholic and nonalcoholic steatohepatitis. Front Biosci. 2005;10:3093–3099. doi: 10.2741/1765. [DOI] [PubMed] [Google Scholar]
- 22.Green DR. Apoptotic pathways: ten minutes to dead. Cell. 2005;121:671–674. doi: 10.1016/j.cell.2005.05.019. [DOI] [PubMed] [Google Scholar]
- 23.Guicciardi ME, Gores GJ. Apoptosis: a mechanism of acute and chronic liver injury. Gut. 2005;54:1024–1033. doi: 10.1136/gut.2004.053850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malhi H, Gores GJ. Cellular and molecular mechanisms of liver injury. Gastroenterology. 2008;134:1641–1654. doi: 10.1053/j.gastro.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patel T, Steer CJ, Gores GJ. Apoptosis and the liver: a mechanism of disease, growth regulation, and carcinogenesis. Hepatology. 1999;30:811–815. doi: 10.1002/hep.510300334. [DOI] [PubMed] [Google Scholar]
- 26.Rust C, Gores GJ. Apoptosis and liver disease. Am J Med. 2000;108:567–574. doi: 10.1016/s0002-9343(00)00370-3. [DOI] [PubMed] [Google Scholar]
- 27.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Friedman SL. Liver fibrosis – from bench to bedside. J Hepatol. 2003;38(Suppl 1):S38–S53. doi: 10.1016/s0168-8278(02)00429-4. [DOI] [PubMed] [Google Scholar]
- 29.Canbay A, Friedman S, Gores GJ. Apoptosis: the nexus of liver injury and fibrosis. Hepatology. 2004;39:273–278. doi: 10.1002/hep.20051. [DOI] [PubMed] [Google Scholar]
- 30.Canbay A, Feldstein A, Baskin-Bey E, Bronk SF, Gores GJ. The caspase inhibitor IDN-6556 attenuates hepatic injury and fibrosis in the bile duct ligated mouse. J Pharmacol Exp Ther. 2004;308:1191–1196. doi: 10.1124/jpet.103.060129. [DOI] [PubMed] [Google Scholar]
- 31.Canbay A, Guicciardi ME, Higuchi H, et al. Cathepsin B inactivation attenuates hepatic injury and fibrosis during cholestasis. J Clin Invest. 2003;112:152–159. doi: 10.1172/JCI17740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Canbay A, Higuchi H, Bronk SF, Taniai M, Sebo TJ, Gores GJ. Fas enhances fibrogenesis in the bile duct ligated mouse: a link between apoptosis and fibrosis. Gastroenterology. 2002;123:1323–1330. doi: 10.1053/gast.2002.35953. [DOI] [PubMed] [Google Scholar]
- 33.Takehara T, Tatsumi T, Suzuki T, et al. Hepatocyte-specific disruption of Bcl-xL leads to continuous hepatocyte apoptosis and liver fibrotic responses. Gastroenterology. 2004;127:1189–1197. doi: 10.1053/j.gastro.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 34.Canbay A, Taimr P, Torok N, Higuchi H, Friedman S, Gores GJ. Apoptotic body engulfment by a human stellate cell line is profibrogenic. Lab Invest. 2003;83:655–663. doi: 10.1097/01.lab.0000069036.63405.5c. [DOI] [PubMed] [Google Scholar]
- 35.Jiang JX, Venugopal S, Serizawa N, et al. Reduced nicotinamide adenine dinucleotide phosphate oxidase 2 plays a key role in stellate cell activation and liver fibrogenesis in vivo. Gastroenterology. 139:1375–1384. doi: 10.1053/j.gastro.2010.05.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Watanabe A, Hashmi A, Gomes DA, et al. Apoptotic hepatocyte DNA inhibits hepatic stellate cell chemotaxis via Toll-like receptor 9. Hepatology. 2007;46:1509–1518. doi: 10.1002/hep.21867. [DOI] [PubMed] [Google Scholar]
- 37.Malhi H, Guicciardi ME, Gores GJ. Hepatocyte death: a clear and present danger. Physiol Rev. 2010;90:1165–1194. doi: 10.1152/physrev.00061.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kahraman A, Bronk SF, Cazanave S, et al. Matrix metalloproteinase inhibitor, CTS-1027, attenuates liver injury and fibrosis in the bile duct-ligated mouse. Hepatol Res. 2009;39:805–813. doi: 10.1111/j.1872-034X.2009.00541.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wielockx B, Lannoy K, Shapiro SD, et al. Inhibition of matrix metalloproteinases blocks lethal hepatitis and apoptosis induced by tumor necrosis factor and allows safe antitumor therapy. Nat Med. 2001;7:1202–1208. doi: 10.1038/nm1101-1202. [DOI] [PubMed] [Google Scholar]
- 40.McClain CJ, Barve S, Deaciuc I. Good fat/bad fat. Hepatology. 2007;45:1343–1346. doi: 10.1002/hep.21788. [DOI] [PubMed] [Google Scholar]
- 41.Yamaguchi K, Yang L, McCall S, et al. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology. 2007;45:1366–1374. doi: 10.1002/hep.21655. [DOI] [PubMed] [Google Scholar]
- 42.Nolan CJ, Larter CZ. Lipotoxicity: why do saturated fatty acids cause and monounsaturates protect against it? J Gastroenterol Hepatol. 2009;24:703–706. doi: 10.1111/j.1440-1746.2009.05823.x. [DOI] [PubMed] [Google Scholar]
- 43.Li ZZ, Berk M, McIntyre TM, Feldstein AE. Hepatic lipid partitioning and liver damage in nonalcoholic fatty liver disease: role of stearoyl-CoA desaturase. J Biol Chem. 2009;284:5637–5644. doi: 10.1074/jbc.M807616200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bechmann LP, Gieseler RK, Sowa JP, et al. Apoptosis is associated with CD36/fatty acid translocase upregulation in non-alcoholic steatohepatitis. Liver Int. 2010;30:850–859. doi: 10.1111/j.1478-3231.2010.02248.x. [DOI] [PubMed] [Google Scholar]
- 45.Akazawa Y, Gores GJ. Death receptor-mediated liver injury. Semin Liver Dis. 2007;27:327–338. doi: 10.1055/s-2007-991510. [DOI] [PubMed] [Google Scholar]
- 46.Peter ME, Krammer PH. The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ. 2003;10:26–35. doi: 10.1038/sj.cdd.4401186. [DOI] [PubMed] [Google Scholar]
- 47.Baskin-Bey ES, Gores GJ. Death by association: BH3 domain-only proteins and liver injury. Am J Physiol Gastrointest Liver Physiol. 2005;289:G987–G990. doi: 10.1152/ajpgi.00371.2005. [DOI] [PubMed] [Google Scholar]
- 48.Scaffidi C, Fulda S, Srinivasan A, et al. Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 1998;17:1675–1687. doi: 10.1093/emboj/17.6.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Feldstein AE, Canbay A, Angulo P, et al. Hepatocyte apoptosis and Fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology. 2003;125:437–443. doi: 10.1016/s0016-5085(03)00907-7. [DOI] [PubMed] [Google Scholar]
- 50.Feldstein AE, Canbay A, Guicciardi ME, Higuchi H, Bronk SF, Gores GJ. Diet associated hepatic steatosis sensitizes to Fas mediated liver injury in mice. J Hepatol. 2003;39:978–983. doi: 10.1016/s0168-8278(03)00460-4. [DOI] [PubMed] [Google Scholar]
- 51.Zou C, Ma J, Wang X, et al. Lack of Fas antagonism by Met in human fatty liver disease. Nat Med. 2007;13:1078–1085. doi: 10.1038/nm1625. [DOI] [PubMed] [Google Scholar]
- 52.Ding WX, Yin XM. Dissection of the multiple mechanisms of TNF-α-induced apoptosis in liver injury. J Cell Mol Med. 2004;8:445–454. doi: 10.1111/j.1582-4934.2004.tb00469.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–190. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- 54.Crespo J, Cayon A, Fernandez-Gil P, et al. Gene expression of tumor necrosis factor α and TNF-receptors, p55 and p75, in nonalcoholic steatohepatitis patients. Hepatology. 2001;34:1158–1163. doi: 10.1053/jhep.2001.29628. [DOI] [PubMed] [Google Scholar]
- 55.Ribeiro PS, Cortez-Pinto H, Sola S, et al. Hepatocyte apoptosis, expression of death receptors, and activation of NF-κB in the liver of nonalcoholic and alcoholic steatohepatitis patients. Am J Gastroenterol. 2004;99:1708–1717. doi: 10.1111/j.1572-0241.2004.40009.x. [DOI] [PubMed] [Google Scholar]
- 56.Hui JM, Hodge A, Farrell GC, Kench JG, Kriketos A, George J. Beyond insulin resistance in NASH: TNF-α or adiponectin? Hepatology. 2004;40:46–54. doi: 10.1002/hep.20280. [DOI] [PubMed] [Google Scholar]
- 57.Li Z, Yang S, Lin H, et al. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology. 2003;37:343–350. doi: 10.1053/jhep.2003.50048. [DOI] [PubMed] [Google Scholar]
- 58.Feldstein AE, Werneburg NW, Canbay A, et al. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-α expression via a lysosomal pathway. Hepatology. 2004;40:185–194. doi: 10.1002/hep.20283. [DOI] [PubMed] [Google Scholar]
- 59.Mari M, Caballero F, Colell A, et al. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006;4:185–198. doi: 10.1016/j.cmet.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 60.MacFarlane M. TRAIL-induced signalling and apoptosis. Toxicol Lett. 2003;139:89–97. doi: 10.1016/s0378-4274(02)00422-8. [DOI] [PubMed] [Google Scholar]
- 61.Sadarangani A, Kato S, Espinoza N, et al. TRAIL mediates apoptosis in cancerous but not normal primary cultured cells of the human reproductive tract. Apoptosis. 2007;12:73–85. doi: 10.1007/s10495-006-0492-z. [DOI] [PubMed] [Google Scholar]
- 62.Malhi H, Barreyro FJ, Isomoto H, Bronk SF, Gores GJ. Free fatty acids sensitise hepatocytes to TRAIL mediated cytotoxicity. Gut. 2007;56:1124–1131. doi: 10.1136/gut.2006.118059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Caldwell SH, Chang CY, Nakamoto RK, Krugner-Higby L. Mitochondria in nonalcoholic fatty liver disease. Clin Liver Dis. 2004;8:595–617. x. doi: 10.1016/j.cld.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 64.Fromenty B, Robin MA, Igoudjil A, Mansouri A, Pessayre D. The ins and outs of mitochondrial dysfunction in NASH. Diabetes Metab. 2004;30:121–138. doi: 10.1016/s1262-3636(07)70098-8. [DOI] [PubMed] [Google Scholar]
- 65.Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest. 2004;114:147–152. doi: 10.1172/JCI22422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Malhi H, Bronk SF, Werneburg NW, Gores GJ. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J Biol Chem. 2006;281:12093–12101. doi: 10.1074/jbc.M510660200. [DOI] [PubMed] [Google Scholar]
- 67.Barreyro FJ, Kobayashi S, Bronk SF, Werneburg NW, Malhi H, Gores GJ. Transcriptional regulation of Bim by FoxO3A mediates hepatocyte lipoapoptosis. J Biol Chem. 2007;282:27141–27154. doi: 10.1074/jbc.M704391200. [DOI] [PubMed] [Google Scholar]
- 68.Cazanave SC, Mott JL, Elmi NA, et al. JNK1-dependent PUMA expression contributes to hepatocyte lipoapoptosis. J Biol Chem. 2009;284:26591–26602. doi: 10.1074/jbc.M109.022491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Masuoka HC, Mott J, Bronk SF, et al. Mcl-1 degradation during hepatocyte lipoapoptosis. J Biol Chem. 2009;284:30039–30048. doi: 10.1074/jbc.M109.039545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Guicciardi ME, Leist M, Gores GJ. Lysosomes in cell death. Oncogene. 2004;23:2881–2890. doi: 10.1038/sj.onc.1207512. [DOI] [PubMed] [Google Scholar]
- 71.Turk B, Bieth JG, Bjork I, et al. Regulation of the activity of lysosomal cysteine proteinases by pH-induced inactivation and/or endogenous protein inhibitors, cystatins. Biol Chem Hoppe Seyler. 1995;376:225–230. doi: 10.1515/bchm3.1995.376.4.225. [DOI] [PubMed] [Google Scholar]
- 72.Baskin-Bey ES, Canbay A, Bronk SF, et al. Cathepsin B inactivation attenuates hepatocyte apoptosis and liver damage in steatotic livers after cold ischemia–warm reperfusion injury. Am J Physiol Gastrointest Liver Physiol. 2005;288:G396–G402. doi: 10.1152/ajpgi.00316.2004. [DOI] [PubMed] [Google Scholar]
- 73.Guicciardi ME, Miyoshi H, Bronk SF, Gores GJ. Cathepsin B knockout mice are resistant to tumor necrosis factor-alpha-mediated hepatocyte apoptosis and liver injury: implications for therapeutic applications. Am J Pathol. 2001;159:2045–2054. doi: 10.1016/s0002-9440(10)63056-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Roberts LR, Adjei PN, Gores GJ. Cathepsins as effector proteases in hepatocyte apoptosis. Cell Biochem Biophys. 1999;30:71–88. doi: 10.1007/BF02737885. [DOI] [PubMed] [Google Scholar]
- 75.Roberts LR, Kurosawa H, Bronk SF, et al. Cathepsin B contributes to bile salt-induced apoptosis of rat hepatocytes. Gastroenterology. 1997;113:1714–1726. doi: 10.1053/gast.1997.v113.pm9352877. [DOI] [PubMed] [Google Scholar]
- 76.Zhao M, Antunes F, Eaton JW, Brunk UT. Lysosomal enzymes promote mitochondrial oxidant production, cytochrome c release and apoptosis. Eur J Biochem. 2003;270:3778–3786. doi: 10.1046/j.1432-1033.2003.03765.x. [DOI] [PubMed] [Google Scholar]
- 77.Boya P, Andreau K, Poncet D, et al. Lysosomal membrane permeabilization induces cell death in a mitochondrion-dependent fashion. J Exp Med. 2003;197:1323–1334. doi: 10.1084/jem.20021952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Feldstein AE, Werneburg NW, Li Z, Bronk SF, Gores GJ. Bax inhibition protects against free fatty acid-induced lysosomal permeabilization. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1339–G1346. doi: 10.1152/ajpgi.00509.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li Z, Berk M, McIntyre TM, Gores GJ, Feldstein AE. The lysosomal–mitochondrial axis in free fatty acid-induced hepatic lipotoxicity. Hepatology. 2008;47:1495–1503. doi: 10.1002/hep.22183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mao C, Dong D, Little E, Luo S, Lee AS. Transgenic mouse model for monitoring endoplasmic reticulum stress in vivo. Nat Med. 2004;10:1013–1014. doi: 10.1038/nm1004-1013. author reply 1014. [DOI] [PubMed] [Google Scholar]
- 81.Marciniak SJ, Yun CY, Oyadomari S, et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18:3066–3077. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Urano F, Wang X, Bertolotti A, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 83.Wei Y, Wang D, Pagliassotti MJ. Saturated fatty acid-mediated endoplasmic reticulum stress and apoptosis are augmented by trans-10, cis-12-conjugated linoleic acid in liver cells. Mol Cell Biochem. 2007;303:105–113. doi: 10.1007/s11010-007-9461-2. [DOI] [PubMed] [Google Scholar]
- 84.Wei Y, Wang D, Topczewski F, Pagliassotti MJ. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am J Physiol Endocrinol Metab. 2006;291:E275–E281. doi: 10.1152/ajpendo.00644.2005. [DOI] [PubMed] [Google Scholar]
- 85.Wang D, Wei Y, Pagliassotti MJ. Saturated fatty acids promote endoplasmic reticulum stress and liver injury in rats with hepatic steatosis. Endocrinology. 2006;147:943–951. doi: 10.1210/en.2005-0570. [DOI] [PubMed] [Google Scholar]
- 86.Puri P, Mirshahi F, Cheung O, Natarajan R, Maher JW, Kellum JM, Sanyal AJ. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology. 2008;134:568–576. doi: 10.1053/j.gastro.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 87.Wei Y, Wang D, Gentile CL, Pagliassotti MJ. Reduced endoplasmic reticulum luminal calcium links saturated fatty acid-mediated endoplasmic reticulum stress and cell death in liver cells. Mol Cell Biochem. 2009;331:31–40. doi: 10.1007/s11010-009-0142-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cazanave SC, Elmi NA, Akazawa Y, Bronk SF, Mott JL, Gores GJ. CHOP and AP-1 cooperatively mediate PUMA expression during lipoapoptosis. Am J Physiol Gastrointest Liver Physiol. 2010;299:G236–G243. doi: 10.1152/ajpgi.00091.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jou J, Choi SS, Diehl AM. Mechanisms of disease progression in nonalcoholic fatty liver disease. Semin Liver Dis. 2008;28:370–379. doi: 10.1055/s-0028-1091981. [DOI] [PubMed] [Google Scholar]
- 90.Wree A, Kahraman A, Gerken G, Canbay A. Obesity affects the liver – the link between adipocytes and hepatocytes. Digestion. 2011;83:124–133. doi: 10.1159/000318741. [DOI] [PubMed] [Google Scholar]
- 91.Alkhouri N, Dixon LJ, Feldstein AE. Lipotoxicity in nonalcoholic fatty liver disease: not all lipids are created equal. Expert Rev Gastroenterol Hepatol. 2009;3:445–451. doi: 10.1586/egh.09.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Neels JG, Olefsky JM. Inflamed fat: what starts the fire? J Clin Invest. 2006;116:33–35. doi: 10.1172/JCI27280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. 2005;115:1111–1119. doi: 10.1172/JCI25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Alkhouri N, Gornicka A, Berk MP, et al. Adipocyte apoptosis: a link between obesity, insulin resistance and hepatic steatosis. J Biol Chem. 2010;285(5):3428–3438. doi: 10.1074/jbc.M109.074252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wueest S, Rapold RA, Schumann DM, et al. Deletion of Fas in adipocytes relieves adipose tissue inflammation and hepatic manifestations of obesity in mice. J Clin Invest. 2010;120:191–202. doi: 10.1172/JCI38388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Caulin C, Salvesen GS, Oshima RG. Caspase cleavage of keratin 18 and reorganization of intermediate filaments during epithelial cell apoptosis. J Cell Biol. 1997;138:1379–1394. doi: 10.1083/jcb.138.6.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ku NO, Liao J, Omary MB. Apoptosis generates stable fragments of human type I keratins. J Biol Chem. 1997;272:33197–33203. doi: 10.1074/jbc.272.52.33197. [DOI] [PubMed] [Google Scholar]
- 99.Wieckowska A, Zein NN, Yerian LM, Lopez AR, McCullough AJ, Feldstein AE. In vivo assessment of liver cell apoptosis as a novel biomarker of disease severity in nonalcoholic fatty liver disease. Hepatology. 2006;44:27–33. doi: 10.1002/hep.21223. [DOI] [PubMed] [Google Scholar]
- 100.Anty R, Iannelli A, Patouraux S, et al. A new composite model including metabolic syndrome, alanine aminotransferase and cytokeratin-18 for the diagnosis of non-alcoholic steatohepatitis in morbidly obese patients. Aliment Pharmacol Ther. 2010;32:1315–1322. doi: 10.1111/j.1365-2036.2010.04480.x. [DOI] [PubMed] [Google Scholar]
- 101.Diab DL, Yerian L, Schauer P, et al. Cytokeratin 18 fragment levels as a noninvasive biomarker for nonalcoholic steatohepatitis in bariatric surgery patients. Clin Gastroenterol Hepatol. 2008;6:1249–1254. doi: 10.1016/j.cgh.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fitzpatrick E, Mitry RR, Quaglia A, Hussain MJ, DeBruyne R, Dhawan A. Serum levels of CK18 M30 and leptin are useful predictors of steatohepatitis and fibrosis in paediatric NAFLD. J Pediatr Gastroenterol Nutr. 2010;51:500–506. doi: 10.1097/MPG.0b013e3181e376be. [DOI] [PubMed] [Google Scholar]
- 103.Malik R, Chang M, Bhaskar K, et al. The clinical utility of biomarkers and the nonalcoholic steatohepatitis CRN liver biopsy scoring system in patients with nonalcoholic fatty liver disease. J Gastroenterol Hepatol. 2009;24:564–568. doi: 10.1111/j.1440-1746.2008.05731.x. [DOI] [PubMed] [Google Scholar]
- 104.Yilmaz Y, Dolar E, Ulukaya E, et al. Soluble forms of extracellular cytokeratin 18 may differentiate simple steatosis from nonalcoholic steatohepatitis. World J Gastroenterol. 2007;13:837–844. doi: 10.3748/wjg.v13.i6.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Younossi ZM, Jarrar M, Nugent C, et al. A novel diagnostic biomarker panel for obesity-related nonalcoholic steatohepatitis (NASH) Obes Surg. 2008;18:1430–1437. doi: 10.1007/s11695-008-9506-y. [DOI] [PubMed] [Google Scholar]
- 106.Musso G, Gambino R, Durazzo M, Cassader M. Noninvasive assessment of liver disease severity with liver fat score and CK-18 in NAFLD: prognostic value of liver fat equation goes beyond hepatic fat estimation. Hepatology. 2010;51:715–717. doi: 10.1002/hep.23255. [DOI] [PubMed] [Google Scholar]
- 107.Feldstein AE, Wieckowska A, Lopez AR, Liu YC, Zein NN, McCullough AJ. Cytokeratin-18 fragment levels as noninvasive biomarkers for nonalcoholic steatohepatitis: a multicenter validation study. Hepatology. 2009;50:1072–1078. doi: 10.1002/hep.23050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tamimi TA, Berk MP, Alkhouri N, et al. An apoptosis panel for nonalcoholic steatohepatitis diagnosis. Gastroenterology. 2009;136:A89. doi: 10.1016/j.jhep.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yan X, Xu L, Qi J, Liang X, et al. sTRAIL levels and TRAIL gene polymorphisms in Chinese patients with fatty liver disease. Immunogenetics. 2009;61:551–556. doi: 10.1007/s00251-009-0389-4. [DOI] [PubMed] [Google Scholar]
- 110.Holsinger L, Coakley D, Dener J, Green M, Booth R, Dalrymple S. Efficacy of a reversible cathepsin B inhibitor in a rodent model of liver fibrosis and human pharmacokinetic profile. Hepatology. 2010;52:1128A. [Google Scholar]
- 111.Wu X, Zhang L, Gurley E, et al. Prevention of free fatty acid-induced hepatic lipotoxicity by 18β-glycyrrhetinic acid through lysosomal and mitochondrial pathways. Hepatology. 2008;47:1905–1915. doi: 10.1002/hep.22239. [DOI] [PubMed] [Google Scholar]
- 112.Adams LA, Zein CO, Angulo P, Lindor KD. A pilot trial of pentoxifylline in nonalcoholic steatohepatitis. Am J Gastroenterol. 2004;99:2365–2368. doi: 10.1111/j.1572-0241.2004.40064.x. [DOI] [PubMed] [Google Scholar]
- 113.Satapathy SK, Sakhuja P, Malhotra V, Sharma BC, Sarin SK. Beneficial effects of pentoxifylline on hepatic steatosis, fibrosis and necroinflammation in patients with non-alcoholic steatohepatitis. J Gastroenterol Hepatol. 2007;22:634–638. doi: 10.1111/j.1440-1746.2006.04756.x. [DOI] [PubMed] [Google Scholar]
- 114.Zein CO, Gogate P, Yerian L, Kirwan J, McCullough A. Pentoxifylline improves nonalcoholic steatohepatitis: results of a double-blinded, randomized, placebo-controlled trial. Am J Gastroenterol. 2010;105:S114. doi: 10.1002/hep.24544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Boetticher NC, Peine CJ, Kwo P, et al. A randomized, double-blinded, placebo-controlled multicenter trial of etanercept in the treatment of alcoholic hepatitis. Gastroenterology. 2008;135:1953–1960. doi: 10.1053/j.gastro.2008.08.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Valentino KL, Gutierrez M, Sanchez R, Winship MJ, Shapiro DA. First clinical trial of a novel caspase inhibitor: anti-apoptotic caspase inhibitor, IDN-6556, improves liver enzymes. Int J Clin Pharmacol Ther. 2003;41:441–449. doi: 10.5414/cpp41441. [DOI] [PubMed] [Google Scholar]
- 117.Dixon L, Berk M, Thapaliya S, Feldstein A. Caspase 1-mediated regulation of fibrogenesis in diet-induced steatohepatitis. Hepatology. 2010;52:A351. doi: 10.1038/labinvest.2012.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Witek RP, Stone WC, Karaca FG, et al. Pan-caspase inhibitor VX-166 reduces fibrosis in an animal model of nonalcoholic steatohepatitis. Hepatology. 2009;50:1421–1430. doi: 10.1002/hep.23167. [DOI] [PubMed] [Google Scholar]
- 119.Anstee QM, Concas D, Kudo H, et al. Impact of pan-caspase inhibition in animal models of established steatosis and non-alcoholic steatohepatitis. J Hepatol. 2010;53:542–550. doi: 10.1016/j.jhep.2010.03.016. [DOI] [PubMed] [Google Scholar]
- 120.Pockros PJ, Schiff ER, Shiffman ML, et al. Oral IDN-6556, an antiapoptotic caspase inhibitor, may lower aminotransferase activity in patients with chronic hepatitis C. Hepatology. 2007;46:324–329. doi: 10.1002/hep.21664. [DOI] [PubMed] [Google Scholar]
- 121.Shiffman ML, Pockros P, McHutchison JG, Schiff ER, Morris M, Burgess G. Clinical trial: the efficacy and safety of oral PF-03491390, a pancaspase inhibitor – a randomized placebo-controlled study in patients with chronic hepatitis C. Aliment Pharmacol Ther. 2010;31:969–978. doi: 10.1111/j.1365-2036.2010.04264.x. [DOI] [PubMed] [Google Scholar]
- 122.Barreyro F, Holod S, Finocchietto P, et al. PF-03491390 Pan-caspase inhibitor decreases liver injury and fibrosis in a murine model of non-alcoholic steatohepatitis (NASH) J Hepatol. 2010;52:S303. doi: 10.1111/liv.12570. [DOI] [PubMed] [Google Scholar]
- 123.Ratziu V, Chojkier M, Sheikh M, et al. Safety, tolerability and preliminary activity of GS-9450, a selective caspase inhibitor, in patients with non-alcoholic steatohepatitis (NASH) J Hepatol. 2010;52:S38. [Google Scholar]
Website
- 201.Safety, Tolerability, Pharmacokinetics and Activity of GS-9450 in Adults With Non-Alcoholic Steatohepatitis (NASH) http://clinicaltrials.gov/ct2/show/NCT00740610.