

Pyruvate ferredoxin oxidoreductase (PFOR)1 is a thiamine pyrophosphate (TPP)-dependent enzyme that catalyzes the anaerobic oxidation of pyruvate (with CoA) to acetyl-CoA, CO2, and two electrons that are transferred to ferredoxin (eq 1).2 In this paper, the PFOR electron-transfer mechanism is examined. Early steps in the catalytic cycle follow the Breslow3 mechanism of TPP activation. After deprotonation, the carbanion at C-2 of TPP reacts with pyruvate to form 2-lactyl-TPP, which decarboxylates to generate CO2 and the resonant enamine/carbanion intermediate, hydroxyethylidene-TPP (HE-TPP). The fate of this intermediate differs among the TPP-dependent enzymes. In PFOR, oxidation of the intermediate is linked to reduction of an external electron acceptor, for example, the clostridial ferredoxin, which contains two [4Fe–4S] clusters.
| (1) |
After formation of HE-TPP, the next step in the PFOR mechanism is one-electron transfer from HE-TPP to one of three [4Fe–4S]2+ clusters, which generates a paramagnetic [4Fe–4S]1+ cluster and an HE-TPP radical.4 This paramagnetic intermediate forms at a rate constant of 140 s−1 at 10 °C (~3000 s−1 at the growth temperature of 55 °C).4 On the basis of the PFOR structure,5 cluster A, located only 9.5 Å from the thiazole sulfur of HE-TPP, is one of three [4Fe–4S] clusters that are separated by 10–12 Å (Figure 1).6 Since biological electron transfer is facile whenever the donor and acceptor are within ~15 Å, clusters A (proximal), B (medial), and C (distal, ~4 Å from the surface) are ideally positioned to sequentially transfer electrons to the surface and the external ferredoxin. Cluster A is logically the first to be reduced, since clusters B and C are >20 Å from the radical center on HE-TPP.
Figure 1.

HE-TPP radical and the Fe–S clusters (A, B, and C) in PFOR. The protein surface near cluster C is shown as a gray shell. From the PDB, accession 1KEK.7
The goal of the work described here is to better understand the paramagnetic intermediate with regard to which Fe–S cluster is reduced.4,8 A subsequent electron transfer from the HE-TPP radical leaves two reduced [4Fe–4S]1+ clusters. The rate of this redox reaction and, hence, the stability of the HE-TPP radical depends strongly on the presence of CoA. In its absence, the half-life of the HE-TPP radical at 10 °C is ~11.5 min; however, in the presence of CoA, it decreases to about 5 ms.4
The ground state of [4Fe–4S]2+ clusters is diamagnetic (S = 0), whereas the HE-TPP radical and the [4Fe–4S]1+ cluster have S = ½. This intermediate paramagnetic state is generated by reacting PFOR with pyruvate for 30 s and freeze quenching the reaction mixture in liquid nitrogen before the onset of radical decay.9 This simultaneous trapping makes it possible to determine the distance between the radical and the [4Fe–4S]1+ cluster by measuring their magnetic dipole interaction by pulsed electron paramagnetic resonance (EPR) techniques. In this work, two such techniques were used, electron–electron double resonance (ELDOR)10 and relaxation-induced dipolar modulation enhancement (RIDME).11
In the pulsed ELDOR experiment, the primary electron spin–echo (ESE) signal of the [4Fe–4S]1+ cluster was observed, while the pumping microwave pulse was in resonance with the HE-TPP radical. The Fourier transform (FT) spectrum of the time-domain ELDOR trace exhibits a prominent peak at 1.5 MHz and a smaller feature at 2.7 MHz (trace 1 in Figure 2). Although this spectral shape is reasonably close to that expected in a situation of complete orientational disorder, some orientational selectivity caused by the g-anisotropy of the [4Fe–4S]1+ center cannot be excluded. To remove the ambiguity, a RIDME experiment was performed.11 In this experiment, we observed a refocused stimulated ESE signal of the HE-TPP radical, while the longitudinal relaxation of the [4Fe–4S]1+ center (instead of pumping at a different microwave frequency) served the purpose of modifying the local magnetic field for the radical spin. Since the [4Fe–4S]1+ center at any orientation is subject to longitudinal relaxation,12 there is no orientational selectivity in the RIDME experiment.
Figure 2.
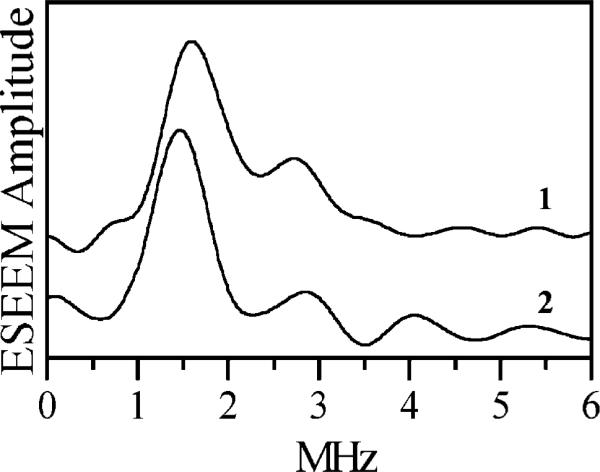
Cosine FTs of the dipolar ESE modulation (ESEEM) obtained in pulsed ELDOR (trace 1) and RIDME (trace 2) experiments. Trace 2 is obtained as the difference between the traces obtained at the temperatures of 13 and 4.2 K.13
The RIDME experiment was performed at 13 and 4.2 K. At 13 K, the cluster relaxes much more rapidly than at 4.2 K. The difference between the two stimulated ESE spectra gave the RIDME spectrum shown by trace 2 in Figure 2. The only reliable line observed in this spectrum is located at 1.48 MHz, close to the low-frequency feature in the ELDOR spectrum. The smaller features mostly originate from noise. By assigning 1.48 MHz to the dipolar coupling constant, Do, and using a point dipole approximation, we estimated the effective distance between the radical and the reduced FeS cluster to be Reff ≈ 33 Å. Numerical simulations taking into account the finite time integration window give a corrected value of Reff to be 31.8 ± 0.2 Å.
To assign this Reff to one of the FeS centers, it is necessary to take into account the distribution of unpaired spin in the HE-TPP radical and in the [4Fe–4S]1+ cluster. The detailed analysis (see Supporting Information) used the coordinates from the crystal structure and spin distribution in both paramagnetic centers from electronic structure calculations. The dipole–dipole tensor was calculated for each cluster using six possible geometries of the oxidized and reduced Fe's. Protonated and neutral forms of the HE-TPP radical were used with each geometry, thus requiring a total of 36 tensors. Tensors calculated for cluster B were all close to the experimentally determined dipolar interaction, whereas those for cluster A were far too large and those for cluster C were too small.
The pulsed EPR results indicate that the HE-TPP radical is magnetically coupled to cluster B. However, this cluster is much too far from HE-TPP to accommodate an electron-transfer rate greater than 100 s-1. Thus, the electron from HE-TPP must have been transferred from cluster A to cluster B. The pyruvate/acetyl-CoA half-cell reaction is at –520 mV, and the [4Fe–4S] centers in the D. africanus PFOR exhibit midpoint redox potentials of –540, –515, and –390 mV,14 although these potentials have not been assigned to specific clusters.
Rapid removal of the electron from cluster A to either of the other Fe–S clusters in the protein is important because it returns cluster A to the state of an oxidant. This scenario would facilitate the next electron-transfer reaction. However, in both PFOR4 and PO,15 the HE-TPP radical is relatively stable unless their co-substrates, CoA (PFOR) or phosphate (PO), are present. The rate enhancement by the co-substrate in each of the two systems indicates a similar mechanism for enhancing the reactivity of the radical in PFOR and PO. It has been proposed that rate enhancement could result from a kinetic coupling mechanism in which formation of an adduct between CoA (or phosphate) and the HE-TPP radical is linked to the electron-transfer reaction.4,15 For PFOR, the kinetic coupling mechanism would generate an anion radical adduct between CoA and the HE-TPP radical, which is estimated to have a negative redox potential of about –0.76 V (vs NHE).15 This potential is significantly more negative than that for the HE-TPP radical/acetyl-TPP couple, which has been estimated to be –487 mV.15 This enhanced reducing power would provide a strong driving force for reduction of cluster A (E0 = –540 mV). Therefore, this kinetic coupling mechanism would allow the protein to generate a stable radical that only undergoes electron transfer when the co-substrate is present.
Rapid electron transfer from the HE-TPP center is important so that reduced ferredoxin (PFOR) or NADH (PDH) can act as a low potential electron donor in energy metabolism. So essential is pyruvate oxidation to the anaerobic metabolism of some microbes and amitochondriate eukaryotes that it is the target of metronidazole and tinidazole, which are common treatments against infections by Helicobacter pylori and the protozoan parasites, Giardia and Trichomonas vaginalis.16 The low potential generated by the pyruvate/acetyl-CoA redox couple leads to the formation of nitro radicals by these pharmaceuticals.17
Supplementary Material
Acknowledgment
This work was supported by NIH Grants GM 39451 (S.W.R.) and GM 35752 (G.H.R.) and by an NIH Predoctoral Training Grant T32 GM08293 (S.O.M.). We are grateful to Dr. Russell LoBrutto and Dr. Arnold Raitsimring for helpful discussions, and thank Dr. L. Noodleman for suggestions on calculating spin projection factors for the S = ½ [4Fe–4S]1+ cluster.
Footnotes
Supporting Information Available: ESE field sweeps, experimental RIDME traces, inversion recovery traces for the reduced FeS cluster, simulated dipolar spectra, calculation of the dipolar tensors. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Abbreviations: PFOR, Pyruvate ferredoxin oxidoreductase; PDH, pyruvate dehydrogenase; PO, pyruvate oxidase; PDC, pyruvate decarboxylase; TPP, thiamine pyrophosphate; HE-TPP, hydroxyethylthiamine pyrophosphate; ELDOR, pulsed electron–electron double resonance; RIDME, relaxation-induced dipolar modulation enhancement; ESE, electron spin–echo; CoA, Coenzyme A.
- 2.Ragsdale SW. Chem. ReV. 2003;103:2333–2346. doi: 10.1021/cr020423e. [DOI] [PubMed] [Google Scholar]
- 3.Breslow RJ. Am. Chem. Soc. 1958;80:3719–3726. [Google Scholar]
- 4.Furdui C, Ragsdale SW. Biochemistry. 2002;41:9921–9937. doi: 10.1021/bi0257641. [DOI] [PubMed] [Google Scholar]
- 5.The high degree of sequence identity (59%) between the M. thermoacetica and the D. africanus PFORs, including high homology around the TPP active site and the three FeS clusters, indicates that their active sites and FeS clusters are located at the same positions.4
- 6.Chabriere E, Charon M-H, Volbeda A, Pieulle L, Hatchikian EC, Fontecilla-Camps J-C. Nat. Struct. Biol. 1999;6:182–190. doi: 10.1038/5870. [DOI] [PubMed] [Google Scholar]
- 7.Chabriere E, Vernede X, Guigliarelli B, Charon MH, Hatchikian EC, Fontecilla-Camps JC. Science. 2001;294:2559–2563. doi: 10.1126/science.1066198. [DOI] [PubMed] [Google Scholar]
- 8.Menon S, Ragsdale SW. Biochemistry. 1997;36:8484–8494. doi: 10.1021/bi970403k. [DOI] [PubMed] [Google Scholar]
- 9.M. thermoacetica was grown and PFOR was purified as described.8 PFOR was reacted with pyruvate (in the absence of CoA) in an EPR tube as described.4 The reaction was quenched by freezing the sample at 77 K in a dewar filled with liquid nitrogen. The spin intensities of HE-TPP radical (0.4 spins/mol) and reduced cluster (0.46 spins/mol) were determined by EPR spectroscopy as described.4
- 10.Milov AD, Salikhov KM, Shirov MD. Fiz. Tverd. Tela. 1981;23:975–982. [Google Scholar]
- 11.Kulik LV, Dzuba SA, Grigoryev IA, Tsvetkov YD. Chem. Phys. Lett. 2001;343:315. [Google Scholar]
- 12.a Bencini A, Gatteschi D. Electron Paramagnetic Resonance of Exchange Coupled Systems. Springer-Verlag; Berlin: 1990. [Google Scholar]; b Mukai K, Sogabe A. J. Chem. Phys. 1980;72:598–601. [Google Scholar]; c Rupp H, Rao KK, Hall DO, Cammack R. Biochim. Biophys. Acta. 1978;537:255–269. doi: 10.1016/0005-2795(78)90509-3. [DOI] [PubMed] [Google Scholar]; d Bouchev VF, Furdui CM, Menon S, Muthukumaran RB, Ragsdale SW, McCracken J. J. Am. Chem. Soc. 1999;121:3724–3729. [Google Scholar]
- 13.Experimental conditions for trace 1: observation microwave frequency, 9.3405 GHz; pumping microwave frequency, 9.4405 GHz; Bo = 337 mT; microwave pulses, 3 × 15 ns; temperature, 4.2 K. Experimental conditions for trace 2: microwave frequency, 9.4405 GHz; Bo = 337 mT; microwave pulses, 3 × 15 + 1 × 10 ns; time interval between the second and third pulses, 20 μs. The experiments were performed on the X/Ku-band pulsed EPR spectrometer at the University of Arizona.
- 14.Pieulle L, Guigliarelli B, Asso M, Dole F, Bernadac A, Hatchikian EC. Biochim. Biophys. Acta: Protein Struct. Mol. Enz. 1995;1250:49–59. doi: 10.1016/0167-4838(95)00029-t. [DOI] [PubMed] [Google Scholar]
- 15.Tittmann K, Wille G, Golbik R, Weidner A, Ghisla S, Hubner G. Biochemistry. 2005;44:13291–13303. doi: 10.1021/bi051058z. [DOI] [PubMed] [Google Scholar]
- 16.a Upcroft J, Upcroft P. Bioessays. 1998;20:256–263. doi: 10.1002/(SICI)1521-1878(199803)20:3<256::AID-BIES9>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]; b Hughes NJ, Chalk PA, Clayton CL, Kelly DJ. J. Bacteriol. 1995;177:3953–3959. doi: 10.1128/jb.177.14.3953-3959.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.a Moreno SN, Docampo R. Environ. Health Perspect. 1985;64:199–208. doi: 10.1289/ehp.8564199. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Yarlett N, Rowlands CC, Evans JC, Yarlett NC, Lloyd D. Mol. Biochem. Parasitol. 1987;24:255–261. doi: 10.1016/0166-6851(87)90157-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.