

Abstract
Flaviviruses are one of the most clinically important pathogens and their infection rates are increasing steadily. The phenylthiazole ring system has provided a template for the design and synthesis of antiviral agents that inhibit the flaviviruses by targeting their E-protein. Unfortunately, there is a correlation between phenylthiazole antiflaviviral activity and the presence of the reactive and therefore potentially toxic mono- or dibromomethyl moieties at thiazole-C4. Adding a linear hydrophobic tail para to the phenyl ring led to a new class of phenylthiazole antiflaviviral compounds that lack the toxic dibromomethyl moiety. This led to development of a drug-like phenylthiazole 12 that had high antiflaviviral selectivity (TI = 147).
Keywords: envelope protein, flaviviruses, phenylthiazoles, dengue virus, yellow fever virus
1. Introduction
Flavivirus is a genus of the positive-sense ssRNA family Flaviviridae, which includes many clinically important species such as dengue, Japanese encephalitis and West Nile viruses.
More than 50 million cases of dengue viral infection are reported per year in more than 80 countries in which the mosquito Aedes aegypti is endemic.1 Approximately 909,000 clinical cases of dengue viral infection were reported in 2008 in North, Central, and South America. Of these cases, there were 306 reported deaths as the consequence of the more severe illnesses dengue hemorrhagic fever and dengue shock syndrome, and the number is increasing steadily.2 In the United States, after decades of absence, the dengue virus is again emerging, causing an epidemic in Hawaii in 2001.3 The features of flavivirus infection range from an asymptomatic state to the severe hemorrhagic disorders that include the classical typical clinical manifestations (fever) and atypical symptoms that involve encephalitis, myocarditis, hepatitis and cholecystitis.4 Currently there are limited licensed flaviviral vaccines, but there are no human vaccines for the vast majority of flaviviruses including dengue viruses, nor effective therapy for treatment of the clinical cases.5
There are many flaviviral proteins that have been targeted for drug discovery such as helicase,6,7 methyl transferase,8,9 and serine protease.10,11 In addition, the viral RNA is also reported to be a target for some antiviral agents.12 Among the flaviviral targets, E-protein plays a crucial role at the first step in viral infection, since it contains the fusogenic loop.13 Structural comparisons of E protein in the immature and mature virus stages suggest that the E protein undergoes substantial conformational and translational changes through the virus replication cycle, thereby causing the native homodimer to change into a fusogenic homotrimer.13 Moreover, crystallization of a dengue virus type 2 E protein (Figure 1) in the presence and the absence of β-OG14 showed an orientation change between domains I and II and paved the road for structure-based design of antiviral agents that could occupy the β-OG pocket. Since the β-OG-containing crystal structure revealed conformational changes relative to the unoccupied protein, it is believed that the β-OG pocket is an ideal target for designing new antiflaviviral agents.
Figure 1.
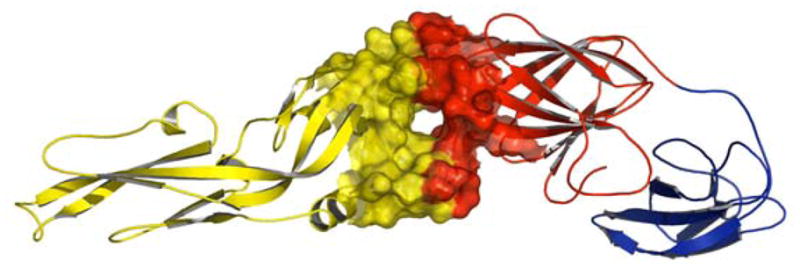
Dengue viral 2 E protein, Domain I: red; domain II: yellow; domain III: blue. The β-OG binding pocket is located between domains I and II.18
Starting from the hit phenylthiazole 1 that was obtained by virtual screening,15 followed by extensive structural optimization, compound 2 was developed with a notably more selective and potent antiflaviviral activity (Figure 2).16 These results have encouraged further structural optimization studies to search for more potent antiviral agents based on the phenylthiazole scaffold.17 Compound 2 has a high TI, but it had two main drawbacks. First, it is a simple methyl ester derivative with a short plasma half-life and its corresponding free acid was shown to lack any antiflaviviral activity.18 Second, it contains the dibromomethyl moiety that is expected to be vulnerable to endogenous nucleophiles and consequently a high in vivo toxicity could be expected. In the next step in this project,18 the focus was shifted to finding metabolically stable bioisosteres of the methyl ester that retained antiviral potency, combined with possibly less toxic dibromomethyl replacements. In those studies the SARs of the thiazole-C4 and -C5 substituents were defined and the pharmacophore model shown in Figure 2 was built.18 So far, several metabolically stable 4-chlorophenylthiazoles have been derived from this model with TI’s up to 256. In this article, the structure-activity relationships (SARs) at the thiazole-C2 position have been investigated in order to enable the rational design of more effective and less cytotoxic antiviral compounds.
Figure 2.

2. Result and Discussion
2.1. Chemistry
Treatment of methyl α-chloroacetoacetate (3) with the appropriate thioamide derivatives 4a-p in absolute ethanol afforded, in each case, the corresponding methyl ester derivatives 5a-p (Scheme 1). Bromination of intermediates 5a-k, utilizing NBS and UV light as a free radical initiator, gave the desired dibromomethyl derivatives 6a-k, usually in moderate to good yields (Scheme 1). The presence of the methine proton was confirmed by 1H NMR spectra which revealed, in each case, a singlet at about δ 7.8 ppm. In addition, the dibromomethyl carbons of the products were responsible for signals in the 13C NMR spectra that appeared between 30.9–31.7 ppm.
Scheme 1.

a
aReagents and conditions: (a) ethanol, heat to reflux, 12–24 h, 49–92% (b) NBS, UV irradiation, heat to reflux for 24 h, CCl4, 27–93%.
In order to synthesize methyl 4-pentylphenylthiazole-5-carboxylate 5q and its propyl analogue 5r, the thioamides 4q,r were prepared from the corresponding amides 7a,b using Lawesson’s reagent in dry THF (Scheme 2). These two thioamides 4q,r were treated with methyl α-chloroacetoacetate (3) as described for the synthesis of the other methyl thiazole esters 5a-p in Scheme 1.
Scheme 2.

a
aReagents and conditions: (a) Lawesson’s reagent, dry THF, 23 °C, 1 h, 55–57%; (b) methyl α-chloroacetoacetate, absolute ethanol, heat to reflux for 24 h, 51–55%.
Hydrolysis of methyl ester 5m afforded the corresponding free acid 8, which was converted into the acid chloride 9 by heating with thionyl chloride (Scheme 3). Treatment of acid chloride 9 with sodium methanethiolate afforded the corresponding thioester 10 as depicted in Scheme 3.
Scheme 3.

a
aReagents and conditions: (a) i, NaOH, methanol:H2O (3:5), heat to reflux for 2 h, ii, HCl, 100%; (b) SOCl2, heat to reflux for 2 h, 95%; (c) sodium methanethiolate, dry CH2Cl2, 23 °C, 30 min, 81%.
Thiazole methylketone derivative 11 was prepared in moderate yield by heating thioamide 4m with 3-chloropentane-2,4-dione in absolute ethanol (Scheme 4). The methyl ketone 11 was gently heated with aminoguanidine hydrochloride in the presence lithium chloride as a catalyst to afford hydrazinecarboximidamide derivative 12 (Scheme 4).
Scheme 4.

a
aReagents and conditions: (a) Ethanol, 3-chloro-2,4-pentanedione, heating for reflux, 24 h, 68%; (b) aminoguanidine hydrochloride, absolute ethanol, LiCl, heat to reflux for 24 h, 46%.
2.2. Biological Results
All of the thiazole derivatives have been evaluated in a yellow fever virus luciferase cellular assay. Compounds that showed sufficient inhibitory activity (over 50%) on viral replication at a concentration of 50 μM were considered to be active and were tested to determine their antiviral EC50 values for inhibition of viral replication, as well as their GI50 values for inhibition of growth of uninfected cells.
Initially, the para chlorine atom present on the C2-thiazole phenyl ring was removed to investigate its biological effect (compound 6a). Interestingly, the resulting unsubstituted phenyl analogue 6a revealed very weak antiflaviviral inhibitory activity (Table 1). This initial result emphasized the biological importance of the phenyl ring substitution. Next, the chlorine atom was replaced with fluorine, bromine, and a methoxy group. The bromine-containing derivative 6d showed a similar EC50 value to the lead compound 2 (Table 1), while its fluorinated 6f and its methoxy-containing derivative 6h revealed much weaker antiflaviviral activity (Table 1). As a preliminary conclusion, both the size and the electronegativity of the substituent may be considered to be important factors for the antiviral properties. To test this tentative conclusion, a more bulky trifluoromethyl-containing analogue 6i was synthesized and tested. Compound 6i showed a significantly higher EC50 value (Table 1). The effect of changing the halogen position was investigated next. Ortho and meta halogen-containing analogues 6b, 6c, 6e, and 6g were prepared. None of the ortho-substituted analogues showed any antiflaviviral activity, and only the meta-substituted derivative 6c revealed weak activity characterized by inhibition of viral replication occurring at the cytotoxic concentration (Table 1). Therefore, it was hypothesized that para substitution confers biological activity while meta substitution is less effective. Next, the disubstituted compounds 6j and 6k were synthesized. Both compounds produced less than 50% inhibition of viral replication at 50 μM in the preliminary screening test (Table 1). The non-brominated naphthyl derivative 5k was the only compound that does not contain a dibromomethyl moiety that had antiflaviviral activity (Table 1). This observation is an advancement because one of the original aims for optimizing the thiazole-C4 position was to find a more chemically stable alternative to the dibromomethyl moiety, since the presence of reactive bromides increases the risk of toxicity.18–21 Although a great deal of effort was expended to meet this goal, none of the dibromo replacements led to better antiviral activity than the brominated lead compound 2, with the exception of the monobromo analogue, which is expected to be more susceptible to endogenous nucleophilic substitution because of less steric hindrance and consequently more toxicity may be expected in vivo.18
Table 1.
Antiviral Activities and Cytotoxicities of Compounds vs. Yellow Fever Virus.a
| Comp. | % Inhibitiona | GI50b (μM) | EC50c (μM) | TI |
|---|---|---|---|---|
| 2 | 99.6 | 222.5 ± 35.0 | 2.83 ± 1.0 | 78 |
| 5a | < 50 | NDd | NDd | |
| 5b | < 50 | NDd | NDd | |
| 5c | < 50 | NDd | NDd | |
| 5d | < 50 | NDd | NDd | |
| 5e | < 50 | NDd | NDd | |
| 5f | < 50 | NDd | NDd | |
| 5g | < 50 | NDd | NDd | |
| 5h | < 50 | NDd | NDd | |
| 5i | −1672.6 | NDd | NDd | |
| 5j | −8.2 | NDd | NDd | |
| 5k | 78.4 | 433.5 ± 23.3 | 35.7 ± 22.7 | 12 |
| 5l | < 50 | NDd | NDd | |
| 5m | 91.9 | 63.1 ± 6.4 | 2.8 ± 0.8 | 22 |
| 5n | −222.5 | NDd | NDd | |
| 5o | −23.8 | NDd | NDd | |
| 5p | −6.5 | NDd | NDd | |
| 5q | 98.6 | 41.5 ± 7.6 | 38.9 ± 8.2 | 1.1 |
| 5r | 83.2 | 499.0 ± 0.9 | 34.3 ± 3.9 | 12 |
| 6a | < 50 | NDd | NDd | |
| 6b | < 50 | NDd | NDd | |
| 6c | 93 | 26.5 ± 1.5 | 26 | 1 |
| 6d | 81.9 | 407.5 ± 78.3 | 3.3 ± 2.1 | 121 |
| 6e | < 50 | NDd | NDd | |
| 6f | < 50 | NDd | NDd | |
| 6g | < 50 | NDd | NDd | |
| 6h | < 50 | NDd | NDd | |
| 6i | 91.0 | 261.7 ± 27.9 | 10.6 ± 3.1 | 26 |
| 6j | 42.2 | NDd | NDd | |
| 6k | 26.0 | NDd | NDd | |
| 8 | 83.2 | 365.7 ± 63.2 | 202.9 ± 14.7 | |
| 10 | 94.1 | 46.5 ± 5.6 | 45.6 ± 5.6 | 1 |
| 11 | 99.8 | 59.4 ± 4.2 | 10.1 ± 3.0 | 6 |
| 12 | 99.2 | 352.8 ± 28.8 | 2.4 ± 0.3 | 147 |
Measured as a reduction in luciferase activity of BHK cells infected with YF-IRES-Luc at 50 μM in comparison to the control.
The GI50 is the concentration of the compound causing a 50% growth inhibition of uninfected BHK cells.
The EC50 is the concentration of the compound resulting in a 50% inhibition in virus production.
ND indicates that the value was not determined; compounds that produced inhibitory activity of less than 50% in the preliminary screening test were considered to be inactive and their exact EC50 and GI50 values were not determined.
The moderate activity of compound 5k can be rationalized from the observation of the calculated position of the phenyl moiety of the phenylthiazoles. The phenyl moiety was calculated to be imbedded among hydrophobic residues (Val130, Phe193, Leu207, Leu191, Phe279 and Ile270) regardless of the position and the orientation of the top parts of the molecule (Figure 3). Therefore, a set of phenylthiazoles that carries different hydrophobic substitutions on the para position of the phenyl ring with different spatial characteristics was built (compounds 5l-5p). Among this new set of compounds 5l-5p, compound 5m showed a very interesting result (Table 1). It was the first phenylthiazole derivative that lacks the mono- or dibromomethyl moiety and has an equivalent EC50 value to the lead compound 2; however, its therapeutic index (TI) was still lower than the lead compound 2.
Figure 3.

The phenyl moiety of the highest-ranked binding poses of the lead compound 2 surrounded by hydrophobic residues (bottom view) (PDB ID: 1OKE). The stereoview is programmed for wall-eyed (relaxed) viewing.
Since the branched compound 5l and the more bulky derivatives 5o,p were found to be inactive (Table 1), it was hypothesized that the hydrophobic pocket of the flaviviral E-protein might prefer linear hydrophobic residues. Therefore, the optimal length of the alkyl chain was studied. One carbon longer and shorter homologues 5q and 5r of compound 5m were synthesized. The pentyl and propyl derivatives 5q,r showed 12 to 14 times higher EC50 values than the butyl derivative 5m (Table 1), which means that a length of four carbons is optimal.
Compound 5m has an EC50 value in the low micromolar range and it lacks the dibromomethyl moiety. Its TI is lower than the lead compound 2 and it contains a metabolically vulnerable methyl ester that is expected to be hydrolyzed in human plasma to the corresponding inactive free acid 8 (Table 1). To improve the metabolic stability as well as to increase the TI of 5m, the SAR analysis of substituents at the thiazole-5 position that was obtained from the previous study18 was applied to the methyl ester moiety. That investigation improved the metabolic stability of the lead compound 2 from 1.4 h to more than a day as measured in rat plasma and it also improved the TI up to 4 times.18 Accordingly, the corresponding thioester and methyl ketone analogues of 5m were synthesized. Although the corresponding methyl thioester and methyl ketone congeners of the lead compound 2 were previously found to be more active than the lead compound 2,18 the same chemical modifications on 5m produced two less active compounds 10 and 11 (Table 1).
The previously determined SAR model (Figure 2) contains one more feature, a hydrophilically favorable region of the receptor, that was assumed to exist around the ester moiety of the ligand (Figure 2).18 Also, active compounds that carried a polar amide moiety were calculated to form hydrogen bonds with E-protein polar residues Ser274 and Gln271 that are close to the solvent-accessible region. In addition, the hydrazinecarboximidamide moiety at the methyl ester position was previously proposed to be tightly bound to three polar residues in the solvent-accessible region (Gln200, Asp203, and Gln271).15 A hydrazinecarboximidamide moiety was chosen as a replacement for the methyl ester of 5m because it could possibly interact favorably with the polar residues on the surface of the E-protein. The hydrazinecarboximidamide derivative 12 was synthesized and it displayed a TI of 147 (Table 1). The TI of compound 12 is improved in comparison with its corresponding methyl ester 5m (Table 1) due to a decrease in cytotoxicity. That may be because introducing a hydrazinecarboximidamido moiety increases water solubility and that may hinder the ability of the cation of 12 to penetrate biological membranes. Compound 12 was subjected to hydrolytic stability analysis utilizing lyophilized rat plasma. The half-life of compound 12 was found to be 8.12 h. This value is around 6 times higher than the lead compound 2, which had a half-life of 1.41 h18 under the same experimental conditions. Therefore, compound 12 provided the desired increased metabolic stability, along with improved antiviral potency, lower cytotoxicity, and an improved TI. In order to confirm that the improved TI of compound 12 resulted from inhibition of viral replication and was not an artifact resulting from inhibition of luciferase, an inhibition assay was performed on cells transfected with luciferase-pcDNA3 (an optimized mammalian expression vector expressing the fire-fly luciferase gene). No reduction of luciferase activity was observed (data not shown).
The antiflaviviral activity of compound 12 can be rationalized from molecular modeling (Figure 4). The hydrazinecarboximidamido moiety of the ligand is calculated to hydrogen bond to the Glu49 carboxylate. High flexibility was observed for the hydrazinecarboximidamido moiety in lower ranked binding poses where different hydrogen bonding possibilities have been observed between the hydrazinecarboximidamide and other polar amino acid residues such as Lys47 and Glu126. The hydrophobic n-butylphenyl tail is modeled within the hydrophobic core of the β-OG pocket as shown in Figure 4.
Figure 4.

Hypothetical model of compound 12 in the dengue viral 2 E protein β-OG binding pocket (PDB ID: 1OKE). The stereoview is programmed for wall-eyed (relaxed) viewing.
3. Conclusion
Optimizing the thiazole-C2 position of the phenylthiazole scaffold led to the first non-brominated phenylthiazole 5k to show antiflaviviral activity. Further structural optimization documented a significant impact of substituents present on the para position of the phenyl ring on antiflaviviral activity. With this in mind, the 2-naphthyl moiety of 5k was optimized to the n-butylphenyl analogue 5m. Compound 5m was the first non-brominated phenylthiazole derivative that had a EC50 value that was equivalent to the lead compound 2. To improve the metabolic stability and antiviral selectivity of 5m, the SAR analysis of the thiazole-5 position that was obtained from the previous study18 was applied to the methyl ester moiety of 5m and that furnished a drug-like compound 12 with an excellent TI value and higher plasma stability character. Lastly, a new comprehensive SAR model has been established that includes the thiazole-C2, -C4, and C5 positions (Figure 5).
Figure 5.

New SAR model of phenylthiazoles as antiflaviviral agents.
4. Experimental Section
4.1. General
Melting points were determined in capillary tubes using a Mel-Temp apparatus and are not corrected. 1H NMR spectra were run at 300 MHz and 13C spectra were determined at 75.46 MHz in deuterated chloroform (CDCl3) or dimethyl sulfoxide (DMSO-d6). Chemical shifts are given in parts per million (ppm) on the delta (δ) scale. Chemical shifts were calibrated relative to those of the solvents. Mass spectra were recorded at 70 eV. All reactions were conducted under argon or nitrogen atmosphere, unless otherwise specified. Compounds 5i,19 5l,20 5o and 5p21 were previously reported.
4.2. Preparation of Thioamides
General Procedure
Amides 7 (1 mmol) and Lawesson’s reagent (490 mg, 1.2 mmol) were added to dry THF (15 mL). The reaction mixture was stirred at room temperature for 1 h. The solvent was evaporated under reduced pressure and the residue was partitioned between aq NaHCO3 (25 mL) and ethyl acetate (25 mL). The organic solvent was separated and dried over anhydrous MgSO4. The crude product was further purified by silica gel flash chromatography, using hexane-ethyl acetate (4:1), to yield the corresponding thioamides as yellow solids (42–60%). Non-commercially available amides 4-n-pentylbenzamide (7a),22 and n-propylbenzamide (7b)23 were prepared as previously reported.
4.2.1. 4-Pentylbenzothioamide (4q)
Yellow solid (57%): mp 56 °C. 1H NMR (CDCl3) δ 8.24 (brs, 1 H), 7.75 (d, J = 8.1 Hz, 2 H), 7.50 (brs, 1 H), 7.15 (d, J = 8.1 Hz, 1 H), 2.59 (t, J = 7.5 Hz, 2 H), 1.58 (m, J = 7.2 Hz, 2 H), 1.29 (m, 4 H), 0.88 (t, J = 7.2 Hz, 3 H); 13C NMR (CDCl3) δ 202.05, 147.80, 136.18, 128.42, 127.11, 35.71, 31.36, 30.75, 22.46, 14.00; CIMS m/z (rel intensity) 208 (MH+, 100); HRMS (CI), m/z 208.1158 MH+, calcd for C12H18NS 208.1160.
4.2.2. 4-Propylbenzothioamide (4r)
Yellow solid (55%): mp 57 °C. 1H NMR (CDCl3) δ 7.88 (brs, 1 H), 7.77 (d, J = 8.4 Hz, 2 H), 7.24 (brs, 1 H), 7.23 (d, J = 8.4 Hz, 1 H), 2.54 (t, J = 7.5 Hz, 2 H), 1.57 (m, J = 7.5 Hz, 2 H), 0.86 (t, J = 7.5 Hz, 3 H); 13C NMR (CDCl3) δ 202.49, 147.51, 136.35, 128.52, 126.93, 37.77, 24.18, 13.68; ESIMS m/z (rel intensity) 180 (MH+, 100); HRMS (ESI), m/z 180.0841 MH+, calcd for C10H14NS 180.0841.
4.3. Preparation of Methyl Thiazole-5-carboxylates 5a-r
General Procedure
Appropriate thiobenzamides or thioacetamides (1 mmol) and α-chloroacetoacetate 3 (150 mg, 1.2 mmol) were added to absolute ethanol (15 mL). The reaction mixture was heated at reflux for 24 h. After removal of solvent under reduced pressure, the residue was purified by silica gel chromatography using hexanes-ethyl acetate (7:3) to provide the desired compounds.
4.3.1. Methyl 4-Methyl-2-phenylthiazole-5-carboxylate (5a)
White solid (120 mg, 70%): mp 110–112 °C. 1H NMR (CDCl3) δ 7.94 (m, 2 H), 7.44 (m, 3 H), 3.87 (s, 3 H), 2.77 (s, 3 H); 13C NMR (CDCl3) δ 169.9, 162.5, 161.2, 132.7, 130.9, 128.9 × 2, 126.7 × 2, 121.2, 52.0, 17.4; IR (KBr) 2925, 2851, 1717, 1266, 1095, 764 cm−1; ESI-MS m/z (rel intensity) 233.97 (MH+, 100). Anal. Calcd for C12H9Br2NO2S: C, 36.85; H, 2.32; N, 3.58. Found: C, 37.23; H, 2.14; N, 3.51.
4.3.2. Methyl 2-(2-Chlorophenyl)-4-methylthiazole-5-carboxylate (5b)
White solid (60 mg, 73%): mp 146–148 °C. 1H NMR (CDCl3) δ 8.32 (m, 1 H), 7.49 (m, 1 H), 7.38 (m, 2 H), 3.90 (s, 3 H), 2.80 (s, 3 H); 13C NMR (CDCl3) δ 131.0, 130.8, 130.6, 127.0, 52.0, 17.3; IR (KBr) 1714, 1266, 1100, 763 cm−1; ESIMS m/z (rel intensity) 267.87 (MH+, 100). Anal. Calcd for C12H10ClNO2S: C, 53.83; H, 3.76; N, 5.23. Found: C, 53.48; H, 3.59; N, 5.08.
4.3.3. Methyl 2-(3-Bromophenyl)-4-methylthiazole-5-carboxylate (5c)
White solid (65 mg, 70%): mp 96–98 °C. 1H NMR (CDCl3) δ 8.14 (s, 1 H), 7.86 (d, J = 7.5 Hz, 1 H), 7.58 (d, J = 7.5, 1 H), 7.32 (dd, J = 7.5, 7.5 Hz, 1 H), 3.90 (s, 3 H), 2.78 (s, 3 H); 13C NMR (CDCl3) δ 133.7, 130.4, 129.4, 125.3, 52.2, 17.3; IR (KBr) 2990, 2848, 1713, 1518, 1424, 1257, 1098, 779 cm−1; ESI-MS m/z (rel intensity) 311.88 (MH+, 100). Anal. Calcd for C12H10BrNO2S: C, 46.17; H, 3.23; N, 4.49. Found: C, 46.07; H, 3.10; N, 4.39.
4.3.4. Methyl 2-(4-Bromophenyl)-4-methylthiazole-5-carboxylate (5d)
White solid (70 mg, 75%): mp 136–138 °C. 1H NMR (CDCl3) δ 7.82 (s, J = 8.5 Hz, 2 H), 7.58 (d, J = 8.5 Hz, 2 H), 3.89 (s, 3 H), 2.77 (s, 3 H); 13C NMR (CDCl3) δ 133.0, 130.4, 128.0, 52.2, 17.3; IR (KBr) 2918, 2848, 1716, 1519, 1431, 1277, 1096, 821 cm−1; ESI-MS m/z (rel intensity) 312.05 (MH+, 100). Anal. Calcd for C12H10BrNO2S: C, 46.17; H, 3.23; N, 4.49. Found: C, 45.78; H, 3.10; N, 4.50.
4.3.5. Methyl 2-(2-Bromophenyl)-4-methylthiazole-5-carboxylate (5e)
White solid (70 mg, 75%): mp 126–128 °C. 1H NMR (CDCl3) δ 8.14 (d, J = 6.6 Hz, 1 H), 7.70 (d, J = 6.6 Hz, 1 H), 7.40 (dd, J = 6.6, 6.6 Hz, 1 H), 7.29 (dd, J = 6.6, 6.6 Hz, 1 H). 3.90 (s, 3 H), 2.80 (s, 3 H); 13C NMR (CDCl3) δ 166.5, 162.6, 159.7, 134.1, 133.3, 131.5, 131.1, 127.5, 122.8, 121.6, 52.1, 17.2; IR (KBr) 2920, 2850, 1716, 1527, 1265, 1101, 761 cm−1; ESI-MS m/z (rel intensity) 312.01 (MH+, 100). Anal. Calcd for C12H10BrNO2S: C, 46.17; H, 3.23; N, 4.49. Found: C, 46.07; H, 3.11; N, 4.40.
4.3.6. Methyl 2-(4-Fluorophenyl)-4-methylthiazole-5-carboxylate (5f)
White solid (52 mg, 70%): mp 90–92 °C. 1H NMR (CDCl3) δ 7.95 (m, 2 H), 7.14 (m, 2 H), 3.89 (s, 3 H), 2.77 (s, 3 H); 13C NMR (CDCl3) δ 169.8, 162.7, 161.8, 161.1, 128.7 × 2, 125.6, 120.2, 116.2, 115.9, 52.1, 17.4; IR (KBr) 2958, 2924, 2850, 1726, 1521, 1439, 1264, 1091, 834, 756 cm−1; ESIMS m/z (rel intensity) 252 (MH+, 100). Anal. Calcd for C12H10FNO2S: C, 57.36; H, 4.01; N, 5.57. Found: C, 56.64; H, 4.10; N, 5.70.
4.3.7. Methyl 2-(2-Fluorophenyl)-4-methylthiazole-5-carboxylate (5g)
White solid (60 mg, 80%): mp 100–102 °C. 1H NMR (CDCl3) δ 8.34 (t, J = 9.0 Hz, 1 H), 7.43 (m, 1 H), 7.19 (m, 2 H); 13C NMR (CDCl3) δ 165.13, 160.99, 161.05, 159.15, 132.02, 131.91, 128.88, 124.57, 116.26, 52.04, 17.29; ESIMS m/z (rel intensity) 252 (MH+, 100); HRMS (ESI), m/z 252.0492 MH+, calcd for C12H11FNO2S 252.0489; HPLC purity 95.00%
4.3.8. Methyl 2-(4-Methoxyphenyl)-4-methylthiazole-5-carboxylate (5h)
White solid (62 mg, 79%): mp 118–120 °C. 1H NMR (CDCl3) δ 7.90 (d, J = 8.7 Hz, 2 H), 6.93 (d, J = 8.5 Hz, 2 H), 3.87 (s, 3 H), 3.85 (s, 3 H), 2.75 (s, 3 H); 13C NMR (CDCl3) δ 169.8, 162.7, 161.8, 161.1, 128.3 × 2, 125.6, 120.2, 114.2 × 2, 55.3, 51.9, 17.4; IR (KBr) 2994, 2949, 2840, 1715, 1521, 1437, 1271, 1258, 1096, 823, 758 cm−1; ESIMS m/z (rel intensity) 263.94 (MH+, 100). Anal. Calcd for C13H13NO3S: C, 59.30; H, 4.98; N, 5.32. Found: C, 59.12; H, 4.92; N, 5.21.
4.3.9. Methyl 2-(3,4-Dichlorophenyl)-4-methylthiazole-5-carboxylate (5j)
White solid (88%): mp 68 °C. 1H NMR (CDCl3) δ 8.05 (d, J = 1.2 Hz, 1 H), 7.73 (dd, J = 1.5, 8.1 Hz, 1 H), 7.47 (dd, J = 1.5, 8.0 Hz, 1 H), 3.88 (s, 3 H), 2.75 (s, 3 H); 13C NMR (CDCl3) δ 166.88, 162.28, 161.36, 135.05, 133.47, 132.60, 130.95, 128.27, 125.69, 122.14, 52.25, 17.41; ESIMS m/z (rel intensity) 304/302 (MH+, 40/56); HRMS (ESI), m/z 301.9806 MH+, calcd for C12H10Cl2NO2S 301.9804; HPLC purity 95.05%.
4.3.10. Methyl 4-Methyl-2-(naphthalen-2-yl)thiazole-5-carboxylate (5k)
White solid (49%): mp 58 °C. 1H NMR (CDCl3) δ 8.43 (s, 1 H), 7.97–7.80 (m, 4 H), 7.52 (dt, J = 1.5, 4.8, 7.5 Hz, 2 H), 3.88 (s, 3 H), 2.81 (s, 3 H); 13C NMR (CDCl3) δ 169.97, 162.60, 161.34, 134.47, 133.03, 130.10, 128.80, 127.80, 127.43, 126.88, 126.56, 123.73, 121.31, 52.12, 17.55; ESIMS m/z (rel intensity) 284 (MH+, 100); HRMS (ESI), m/z 284.0748 MH+, calcd for C16H14NO2S 284.0745; HPLC purity 95.30%.
4.3.11. Methyl 2-(4-Butylphenyl)-4-methylthiazole-5-carboxylate (5m)
Colorless viscous oil (92%). 1H NMR (CDCl3) δ 7.85 (d, J = 8.0 Hz, 2 H), 7.22 (d, J = 8.4 Hz, 2 H), 3.85 (s, 3 H), 2.75 (s, 3 H), 2.64 (t, J = 7.8 Hz, 3 H), 1.58 (m, J = 7.4 Hz, 2 H), 1.34 (m, J = 7.4 Hz, 2 H), 0.91 (t, J = 7.4 Hz, 3 H); 13C NMR (CDCl3) δ 170.21, 162.67, 161.21, 146.43, 130.40, 129.02, 126.71, 120.70, 52.01, 35.52, 33.26, 22.26, 17.48, 13.87; ESIMS m/z (rel intensity) 290 (MH+, 98); HRMS (ESI), m/z 290.1215 MH+, calcd for C16H20NO2S 290.1209; HPLC purity 98.05%.
4.3.12. Methyl 2-(4-Iodophenyl)-4-methylthiazole-5-carboxylate (5n)
White solid (86%): mp 148–149 °C. 1H NMR (CDCl3) δ 7.74 (d, J = 8.4 Hz, 2 H), 7.64 (d, J = 8.4 Hz, 2 H), 3.87 (s, 3 H), 2.75 (s, 3 H); 13C NMR (CDCl3) δ 168.67, 162.43, 161.32, 138.14, 132.25, 128.10, 121.57, 97.52, 52.21, 17.48; ESIMS m/z (rel intensity) 360 (MH+, 100); HRMS (ESI), m/z 359.9555 MH+, calcd for C12H11INO2S 359.9550; HPLC purity 97.35%.
4.3.13. Methyl 4-Methyl-2-(4-pentylphenyl)thiazole-5-carboxylate (5q)
Colorless oil (166 mg, 55%). 1H NMR (CDCl3) δ 7.82 (d, J = 8.4 Hz, 2 H), 7.20 (d, J = 8.4 Hz, 2 H), 3.83 (s, 3 H), 2.74 (s, 3 H), 2.59 (t, J = 7.5 Hz, 2 H), 1.59 (m, J = 7.2 Hz, 2 H), 1.30 (m, 4 H), 0.87 (t, J = 7.5 Hz, 3 H); 13C NMR (CDCl3) δ 170.16, 162.61, 146.41, 130.37, 128.99, 126.68, 120.67, 51.96, 35.78, 31.38, 30.79, 22.46, 17.44, 13.95; ESIMS m/z (rel intensity) 304 (MH+, 100); HRMS (ESI), m/z 304.1368 MH+, calcd for C17H22NO2S 304.1366; HPLC purity 95.10%.
4.3.14. Methyl 4-Methyl-2-(4-propylphenyl)thiazole-5-carboxylate (5r)
White solid (140 mg, 51%): mp 41 °C. 1H NMR (CDCl3) δ 7.84 (d, J = 8.4 Hz, 2 H), 7.22 (d, J = 8.4 Hz, 2 H), 3.85 (s, 3 H), 2.75 (s, 3 H), 2.60 (t, J = 7.5 Hz, 2 H), 1.64 (m, J = 7.5 Hz, 2 H), 0.93 (t, J = 7.2 Hz, 3 H); 13C NMR (CDCl3) δ 170.22, 162.68, 161.20, 146.20, 130.43, 129.08, 126.70, 120.72, 52.01, 37.86, 24.23, 17.47, 13.69; ESIMS m/z (rel intensity) 276 (MH+, 100); HRMS (ESI), m/z 276.1058 MH+, calcd for C15H18NO2S 276.1053; HPLC purity 95.20%.
4.4. Preparation of 6a-k. General Procedure
The ester 5 (0.5 mmol) and NBS (531 mg, 3 mmol) were added to CCl4 (10 mL). The reaction mixture was heated at reflux for 6 h, during which time it was irradiated by an ultraviolet sunlamp (GE, 215 W). After removal of solvent under reduced pressure, the residue was purified by silica gel column chromatography (ethyl acetate-hexanes 1:4) to provide the desired compounds.
4.4.1. Methyl 4-(Dibromomethyl)-2-phenylthiazole-5-carboxylate (6a)
White solid (117 mg, 60%): mp 176–178 °C. 1H NMR (CDCl3) δ 8.03 (m, 2 H), 7.71 (s, 1 H), 7.50 (m, 3 H), 3.94 (s, 3 H); 13C NMR (CDCl3) δ 171.5, 161.0, 158.8, 132.1, 131.7, 129.0 × 2, 127.0 × 2, 116.9, 52.8, 31.2; IR (KBr) 3057, 2947, 2918, 2849, 1716, 1521, 1434, 1283, 1092, 719, 629 cm−1; ESI-MS m/z (rel intensity) 389.69 (MH+, 100). Anal. Calcd for C12H9Br2NO2S: C, 36.85; H, 2.32; N, 3.58. Found, C, 37.23; H, 2.14; N, 3.51.
4.4.2. Methyl 2-(2-Chlorophenyl)-4-(dibromomethyl)thiazole-5-carboxylate (6b)
White solid (133 mg, 63%): mp 154–156 °C. 1H NMR (CDCl3) δ 8.53 (m, 1 H), 7.72 (s, 1 H), 7.50 (m, 1 H), 7.43 (m, 2 H), 3.96 (s, 3 H); 13C NMR (CDCl3) δ 166.5, 161.2, 157.3, 132.3, 131.7, 131.4, 130.6, 130.5, 127.3, 121.0, 52.8, 31.3; IR (KBr) 3045, 2956, 2848, 1706, 1520, 1433, 1283, 1099, 756, 632 cm−1; ESI-MS m/z (rel intensity) 423.49 (MH+, 100). Anal. Calcd for C12H9Br2NO2S: C, 36.85; H, 2.32; N, 3.58. Found: C, 37.23; H, 2.14; N, 3.51
4.4.3. Methyl 2-(3-Bromophenyl)-4-(dibromomethyl)thiazole-5-carboxylate (6c)
White solid (140 mg, 60%): mp 111–113 °C. 1H NMR (CDCl3) δ 8.21 (s, 1 H), 7.92 (dd, J = 10.8, 0.6 Hz, 1 H), 7.69 (s, 1 H), 7.63 (dd, J = 10.8, 0.6 Hz, 1 H), 7.35 (m, 1 H), 3.95 (s, 3 H); 13C NMR (CDCl3) δ 169.5, 160.8, 158.8, 134.5, 133.8, 130.5, 129.7, 125.6, 123.2, 120.3, 52.9, 30.9; IR (KBr) 3221, 1705, 1427, 1316, 1176, 814, 640 cm−1; ESI-MS m/z (rel intensity) 467.65 (MH+, 100); HPLC purity 100%.
4.4.4. Methyl 2-(4-Bromophenyl)-4-(dibromomethyl)thiazole-5-carboxylate (6d)
White solid (140 mg, 60%): mp 120–122 °C. 1H NMR (CDCl3) δ 7.93 (d, J = 8.4 Hz, 2 H), 7.70 (s, 1 H), 7.61 (d, J = 8.4 Hz, 2 H), 3.95 (s, 3 H); 13C NMR (CDCl3) δ 170.1, 160.8, 158.9, 132.3 × 2, 131.0, 128.4, 126.3, 120.0, 52.9, 31.0; IR (KBr) 3046, 1712, 1447, 1289, 1093, 829, 622 cm−1; ESI-MS m/z (rel intensity) 467.48 (MH+, 100); HPLC purity 96.00%.
4.4.5. Methyl 2-(2-Bromophenyl)-4-(dibromomethyl)thiazole-5-carboxylate (6e)
White solid (128 mg, 63%): mp 159–161 °C. 1H NMR (CDCl3) δ 8.25 (d, J = 8.1 Hz, 1 H), 7.63 (s, 1 H), 7.72 (d, J = 8.1 Hz, 1 H), 7.52 (m, 2 H), 3.82 (s, 3 H); 13C NMR (CDCl3) δ 164.74, 162.74, 159.80, 133.03, 132.10, 131.87, 131.56, 131.04, 128.75, 123.26, 53.29, 31.4; ESIMS m/z (rel intensity) 468/470/472/474 (MH+, 23/91/100/26); HRMS (ESI), m/z 467.7902 MH+, calcd for C12H9Br3NO2S 467.7899; HPLC purity 95.86%.
4.4.6. Methyl 4-(Dibromomethyl)-2-(4-fluorophenyl)thiazole-5-carboxylate (6f)
White solid (128 mg, 63%): mp 160–162 °C. 1H NMR (CDCl3) δ 8.05 (m, 2 H), 7.69 (s, 1 H), 7.17 (m, 2 H), 3.94 (s, 3 H); 13C NMR (CDCl3) δ 170.27, 166.53, 160.99, 158.89, 129.34, 128.58, 119.84, 116.49, 52.94, 31.16; IR (KBr) 3063, 2960, 1717, 1287, 842, 631 cm−1; ESI-MS m/z (rel intensity) 407.73 (MH+, 100); HPLC purity 96.87%.
4.4.7. Methyl 4-(Dibromomethyl)-2-(2-fluorophenyl)thiazole-5-carboxylate (6g)
White solid (142 mg, 70%): mp 132–134 °C. 1H NMR (CDCl3) δ 8.49 (ddd, J = 7.5, 7.5, 1.8 Hz, 1 H), 7.73 (s, 1 H), 7.49 (m, 1 H), 7.32 (m, 1 H), 7.23 (m, 1 H), 3.95 (s, 3 H); 13C NMR (CDCl3) δ 163.6, 161.4, 161.1, 159.4, 157.7, 132.8, 129.5, 124.8, 120.7, 116.1, 52.8, 31.3; IR (KBr) 1707, 1587, 1516, 1455, 1291, 1100, 633 cm−1; ESIMS m/z (rel intensity) 407.72 (MH+, 100); HPLC purity 96.81%.
4.4.8. Methyl 4-(Dibromomethyl)-2-(4-methoxyphenyl)thiazole-5-carboxylate (6h)
White solid (127 mg, 60%): mp 104–106 °C. 1H NMR (CDCl3) δ 7.97 (m, 2 H), 7.69 (s, 1 H), 6.96 (m, 2 H), 3.92 (s, 3 H), 3.87 (s, 3 H); 13C NMR (CDCl3) δ 171.4, 162.4, 161.2, 158.7, 128.7 × 2, 128.6, 125.0, 114.3 × 2, 55.4, 52.7, 31.4; IR (KBr) 2919, 2850, 1701, 1422, 1321, 1167, 814, 640 cm−1; ESIMS m/z (rel intensity) 263.94 (MH+, 100). Anal. Calcd for C13H11Br2NO3S): C, 37.08; H, 2.63; N, 3.33. Found: C, 37.05; H, 2.59; N, 3.24.
4.4.9. Methyl 4-(Dibromomethyl)-2-(4-(trifluoromethyl)phenyl)thiazole-5-carboxylate (6i)
White solid (67 mg, 87%): mp 81–82 °C. 1H NMR (CDCl3) δ 8.16 (d, J = 8.4 Hz, 2 H), 8.75 (d, J = 8.4 Hz, 2 H), 7,70 (s, 1 H), 3.96 (s, 3 H); 13C NMR (CDCl3) δ 169.48, 160.81, 159.13, 135.19, 131.01, 127.59, 127.24, 126.37, 125.94, 53.25, 31.12; CIMS m/z (rel intensity) 462/460/458 (MH+, 57/100/50); HRMS (ESI), m/z 457.8670 MH+, calcd for C13H9Br2F3NO2S 456.8667.
4.4.10. Methyl 4-(Dibromomethyl)-2-(3,4-dichlorophenyl)thiazole-5-carboxylate (6j)
White solid (849 mg, 93%): mp 144–145 °C. 1H NMR (CDCl3) δ 8.16 (d, J = 2.1 Hz, 1 H), 7.83 (dd, J = 2.1, 8.4 Hz, 1 H), 7.68 (s, 1 H), 7.55 (d, J = 8.4 Hz, 1 H), 3.95 (s, 3 H); 13C NMR (CDCl3) δ 168.63, 161.53, 160.76, 159.05, 136.00, 133.72, 131.90, 131.12, 128.66, 126.04, 120.58, 53.08, 30.83; APCIMS m/z (rel intensity) 464/462/460/458 (MH+, 21/80/100/36); HRMS (CI), m/z 457.8019 MH+, calcd for C12H8Br2Cl2NO2S 457.9014; HPLC purity 97.09%.
4.4.11. Methyl 4-(Dibromomethyl)-2-(naphthalen-2-yl)thiazole-5-carboxylate (6k)
Off-white solid (224 mg, 27%): mp > 360 °C. 1H NMR (CDCl3) δ 8.57 (s, 1 H), 8.08 (d, J = 1.8 Hz, 1 H), 7.94–7.80 (m, 3 H), 7.75 (s, 1 H), 7.59–7.56 (m, 2 H), 3.96 (s, 3 H); 13C NMR (CDCl3) δ 171.65, 161.12, 159.01, 134.82, 132.96, 129.50, 129.01, 128.96, 127.87, 127.26, 127.08, 123.79, 119.84, 52.93, 31.36; CIMS m/z (rel intensity) 443/441/439 (MH+, 50/100/64); HRMS (CI), m/z 439.8958 MH+, calcd for C16H12Br2NO2S 439.8955; HPLC purity 95.09%.
4.5. 2-(4-Butylphenyl)-4-methylthiazole-5-carboxylic Acid (8)
NaOH (200 mg, 5 mmol) was added to a solution of methyl ester 5m (290 mg, 1 mmol) in methanol (6 mL) and water (10 mL). The reaction mixture was heated under reflux for 2 h, and then allowed to cool to room temperature. The reaction mixture was filtered and the pH of the liquid phase was adjusted to 2 with hydrochloride acid. The solid was filtered and dried to provide the corresponding carboxylic acid as a white solid (100%): mp 147–148 °C. 1H NMR (DMSO-d6) δ 7.87 (d, J = 8.1 Hz, 2 H), 7.33 (d, J = 8.1 Hz, 2 H), 2.65 (s, 3 H), 2.62 (t, J = 7.2 Hz, 3 H), 1.60 (m, J = 7.2 Hz, 2 H), 1.36 (m, J = 7.2 Hz, 2 H), 0.86 (t, J = 7.2 Hz, 3 H); 13C NMR (DMSO-d6) δ 170.21, 163.89, 160.50, 148.44, 130.17, 127.40, 35.57, 33.69, 22.64, 18.04, 14.69; ESIMS m/z (rel intensity) 276 (MH+, 36), 178 (100); HRMS (ESI), m/z 276.1063 MH+, calcd for C15H8NO2S 276.1058.
4.6. 2-(4-Butylphenyl)-4-methylthiazole-5-carbonyl Chloride (9)
The carboxylic acid 8 (275 mg, 1 mmol) was heated under reflux with thionyl chloride (6 mL) for 2 h. The solvent was evaporated under reduced pressure. The brown residue was collected and purified by silica gel flash chromatography, using hexane-ethyl acetate (7:3), to yield the corresponding acid chloride as viscous colorless oil (95%). 1H NMR (CDCl3) δ 7.88 (d, J = 8.4 Hz, 2 H), 7.26 (d, J = 8.4 Hz, 2 H), 2.72 (s, 3 H), 2.66 (t, J = 7.8 Hz, 3 H), 1.61 (m, J = 7.5 Hz, 2 H), 1.36 (m, J = 7.5 Hz, 2 H), 0.94 (t, J = 7.5 Hz, 3 H); 13C NMR (CDCl3) δ 174.11, 164.12, 158.19, 147.79, 129.74, 129.25, 126.97, 126.82, 35.63, 33.21, 22.29, 18.51, 13.89; CIMS m/z (rel intensity) 295/293 (MH+, 36/100); HRMS (CI), m/z 294.0725 MH+, calcd for C15H17ClNOS 294.0719.
4.7. S-Methyl 2-(4-Butylphenyl)-4-methylthiazole-5-carbothioate (10)
The acid chloride 9 (58 mg, 0.2 mmol) was stirred with sodium methanthiol (12 mg, 1.7 mmol) in dry dichloromethane for 30 min. The solvent was evaporated under reduced pressure and the solid residue was partitioned between EtOAc (10 mL) and water (10 mL). The organic layer was separated, dried and evaporated. The yellow precipitate was further purified by crystallization from MeOH to afford the title compound as a yellow solid (81%): mp 35 °C. 1H NMR (CDCl3) δ 7.85 (d, J = 8.1 Hz, 2 H), 7.24 (d, J = 8.1 Hz, 2 H), 2.76 (s, 3 H), 2.63 (t, J = 7.6 Hz, 3 H), 1.60 (m, J = 7.5 Hz, 2 H), 1.35 (m, J = 7.5 Hz, 2 H), 0.92 (t, J = 7.5 Hz, 3 H); 13C NMR (CDCl3) δ 183.58, 169.68, 158.13, 146.71, 130.20, 129.43, 129.08, 126.85, 35.56, 33.27, 22.29, 18.34, 13.89, 12.58; ESIMS m/z (rel intensity) 306 (MH+, 100); HRMS (ESI), m/z 306.0990 MH+, calcd for C16H20NOS2 306.0986; HPLC purity 95.01%.
4.8. 1-(2-(4-Butylphenyl)-4-methylthiazol-5-yl)ethanone (11)
4-n-Butylthiobenzamide (4m, 450 mg, 2.3 mmol) and 3-chloropentane-2,4-dione (0.32 mL, 2.8 mmol) were added to absolute ethanol (10 mL). The reaction mixture was heated at reflux for 24 h. After evaporation of solvent under reduced pressure, the brown residue was collected and purified by silica gel flash chromatography, using hexane-ethyl acetate (9:1), to yield the desired compound as a yellowish oil (432 mg, 68%). 1H NMR (CDCl3) δ 7.75 (d, J = 8.7 Hz, 2 H), 7.13 (d, J = 8.7 Hz, 2 H), 2.65 (s, 3 H), 2.53 (t, J = 5.2 Hz, 2 H), 2.40 (s, 3 H), 1.52 (m, J = 5.2 Hz, 2 H), 1.29 (m, J = 5.2 Hz, 2 H), 0.85 (t, J = 5.0 Hz, 3 H); 13C NMR (CDCl3) δ 190.10, 169.35, 159.26, 146.47, 130.62, 130.18, 128.95, 126.68, 35.47, 33.17, 30.53, 22.26, 18.33, 13.85; ESIMS m/z (rel intensity) 274 (MH+, 100); HRMS (ESI), m/z 274.1262 MH+, calcd for C16H20NOS 274.1260; HPLC purity 99.38%.
4.9. 2-(1-(2-(4-Butylphenyl)-4-methylthiazol-5-yl)ethylidene)hydrazinecarboximidamide (12)
The thiazole derivative 11 (230 mg, 0.83 mmol) was dissolved in absolute ethanol (10 mL), and aminoguanidine hydrochloride (110 mg, 1 mmol) and a catalytic amount of LiCl (5 mg) were added. The reaction mixture was heated at reflux for 24 h. The solvent was evaporated under reduced pressure. The crude product was purified by crystallization from 70% methanol, then recrystallized from methanol to afford the desired compound as a off-white solid (78 mg, 46%): mp 230–231 °C. 1H NMR (DMSO-d6) δ 11.49 (brs, 1 H), 7.80 (d, J = 7.8 Hz, 2 H) 7.76 (brs, 3 H), 7.31 (d, J = 7.8 Hz, 2 H), 2.61 (t, J = 7.2 Hz, 2 H), 2.59 (s, 3 H), 2.42 (s, 3 H), 1.56 (m, J = 6.9 Hz, 2 H), 1.30 (m, J = 6.9 Hz, 2 H), 0.89 (t, J = 7.5 Hz, 3 H); 13C NMR (DMSO-d6) δ 170.23, 161.05, 157.50, 152.36, 150.52, 135.40, 135.33, 134.34, 131.14, 39.80, 37.97, 26.92, 23.36, 23.30, 18.93; ESIMS m/z (rel intensity) 330 (MH+, 100); HRMS (ESI), m/z 330.1751 MH+, calcd for C17H24N5S; HPLC purity 95.43%.
4.10. Bioassay Methods
4.10.1. BHK Cells
BHK-15 cells obtained from the American Type Culture Collection (ATCC, Rockville, MD) were maintained in MEM (Invitrogen, Carlsbad, CA) containing 10% FBS. Cells were grown in incubators at 37 °C in the presence of 5% CO2.
4.10.2. YFV-IRES-Luc
A fire-fly luciferase reporter gene was inserted into pYF23, a derivative of pACNR which is the full-length cDNA clone of YFV 17D, to construct YFV-IRES-Luc, a luciferase-reporting full-length virus. To facilitate this construction, an NsiI restriction site was introduced at the beginning of the 3′NTR immediately following the UGA termination codon of NS5 in pYF23 using standard overlapping PCR mutagenesis. To construct YFV-IRES-Luc, an IRES-FF.Luc (EMCV IRES-fire fly luciferase) cassette was amplified by PCR from YFRP-IRES-Luc, a YFV replicon, and inserted into the NsiI restriction site.24
4.10.3. Generation of YFV-IRES-Luc Virus
In vitro transcribed YFV-IRES-Luc RNA was transfected into BHK-15 cells using Lipofectamine (Invitrogen, Carlsbad, CA). At 4 days post-transfection, the resulting YFV-IRES-Luc virus was harvested and the titer of the virus determined by a standard plaque assay. The infectivity of the virus could be assayed directly as a measure of the luciferase amounts produced in infected cells over a period of time.
4.10.4. Inhibition of YFV-IRES-Luc Virus Growth
BHK cells were plated in a 96-well plate and grown at 37 ºC. At confluency, cells were infected with YF-IRES-Luc virus at a multiplicity of infection (MOI) of 0.1. A low MOI was utilized to ensure that fewer cells were infected so that the spread of released virus could be monitored. Cells were then overlaid with culture media containing serial dilutions of compounds at concentrations below the GI50 values. Controls included uninfected cells, infected cells, and DMSO-treated infected cells. Cells were incubated at 37 °C, 5% CO2 for ~36 h, lysed using 50 μL of cell culture lysis buffer (Promega Inc., Madison, WI), and 10 μL of cell extracts placed into a 96-well opaque plate. Luciferase activity was determined from the luminescence generated with fire-fly luciferase substrate (Promega Inc., Madison, WI). Luminescense was measured in a 96-well-plate luminometer, LMax II (Molecular Devices, Sunnyvale, CA). A reduction in luciferase activity indicates inhibition of YFV-IRES-Luc virus growth. The luciferase luminescence as a function of compound concentration was analyzed by non-linear regression analysis using GraphPadPrizm to estimate the EC50 of each compound. The EC50 was defined as the concentration of the compound to cause 50% reduction of luciferase activity in infected cells as compared to the DMSO-treated cells.
4.10.5. Cell Viability Assay
BHK cells were plated in a 96-well plate and grown at 37 ºC. At confluency, cells were overlaid with culture media containing serial dilutions of compounds (compound stocks were generated by dissolving compounds in DMSO). Untreated and DMSO-treated cells served as positive controls. Cells were then incubated at 37 °C, 5% CO2 for ~36 h. At ~36 h post-treatment, media on cells was replaced with fresh media to remove the compounds. Then 10 μL of XTT-substrate from the Quick Cell Proliferation Kit (Biovision Inc., CA) was added to each well. Cells were incubated at 37 °C for a further 2 h. Plates were then removed and OD450 measured using a 96-well plate reader (Molecular Devices, Sunnyvale, CA). The OD450 value for cells treated with a compound was compared to that obtained from cells treated with 1% DMSO and the GI50 for each compound was calculated.
4.10.6. Luciferase-pcDNA3
A fire-fly luciferase gene was inserted into a pcDNA3 backbone to construct a mammalian expression vector of fire-fly luciferase (Addgene plasmid 18964). Briefly, the fire-fly luciferase gene was excised from pGL3-Basic Vector (Promega Inc., Madison, WI), and inserted in pcDNA3(Invitrogen, Carlsbad, CA) by using the HindIII and XbaI restriction sites.25
4.10.7. Direct Luciferase Inhibition Assay
BHK cells were plated in a 96-well plate and grown at 37 °C. At confluency, the cells were visually inspected for uniform growth and the Luciferase-pcDNA3 plasmid was introduced into the cells using Lipofectamine (Invitrogen, Carlsbad, CA) transfection reagent according to the manufacturer’s instructions. At 3 h post infection, the complexes were removed and replaced with culture media containing serial dilutions of compound 12 (diluted in DMSO), above and below the EC50 value. Controls included uninfected cells, infected cells and DMSO-treated infected cells. Cells were then incubated at 37 °C, 5% CO2 for 20 h, lysed using 50 μL of cell culture lysis buffer (Promega, Inc., Madison, WI), and 10 μL of cell extracts were placed into a 96-well opaque plate. Luciferase activity was determined from the luminescence generated with 50 μL fire-fly luciferase substrate (Promega Inc., Madison, WI). An LMax II 96-well-plate luminometer was used to measure luminescence (Molecular Devices, Sunnyvale, CA). The luciferase readings for cells overlaid with Compound 12 were compared to those obtained from cells overlaid with 1% DMSO using GraphPadPrizm. A cell viability assay was also performed as detailed in section 4.10.5.
4.11. Molecular Modeling
The energy-optimized compounds were docked into the β-OG binding domain in the E-protein of the dengue virus after removal of the n-octyl-β-D-glucoside (β-OG). The parameters were set as the default values for GOLD. The maximum distance between hydrogen bond donors and acceptors for hydrogen bonding was set to 3.5 Å. After docking, the first pose conformations of compounds of interest were merged into the ligand-free protein. The new ligand–protein complex was subsequently subjected to energy minimization using the Amber force field with Amber charges. During the energy minimization, the structure of the compounds of interest and only chain A of the viral E protein were allowed to move. Chain B was kept frozen. The energy minimization was performed using the Powell method with a 0.05 kcal/(mol Å) energy gradient convergence criterion and a distance dependent dielectric function.
4.12. In Vitro Hydrolytic Stability Assay Utilizing Rat Plasma
Compound 12 was tested for its hydrolytic stability in solutions of reconstituted rat plasma. Compounds 12 (10 μmol) and 7 μmol of 4-bromopyrazole as an internal standard were dissolved in DMSO (1.0 mL). This solution was filtered through a 0.45 μM filter (Millex-HN). Lyophilized rat plasma (1.0 mL) (LOT# 048K7420, Sigma Chemical Co., St. Louis, Mo) was reconstituted with water of HPLC (1.0 mL). The plasma solution was incubated at 37 °C for 15 min and was then diluted with 0.01 M saline (0.250 mL) to afford an 80% plasma solution. The plasma solution was incubated again at 37 °C for an additional 5 min. An aliquot of the compound 12 in DMSO (100 μL) as added to the rat plasma (0.75 mL) and the mixture was incubated at 37 °C throughout the course of the experiment. Aliquots (10 μL) of the compound-plasma mixture were collected at various time intervals and diluted with methanol (90 μL) to precipitate any proteins present. The aliquots were mixed and centrifuged at 10,000 rpm for 5–10 min to pellet the precipitated proteins. After centrifugation, the supernatants (20 μL) of the aliquots were analyzed by HPLC to determine the residual amount of tested compounds present in the sample. The aliquot supernatants were analyzed using a Waters binary HPLC system (Model 1525, 10 μL injection loop) and a Waters dual wavelength absorbance UV detector (Model 2487) set for 254 nM. Data were collected and processed using the Breeze software (version 3.3) on a Dell Optiplex GX280 personal computer. The mobile phase consisted of 85:15 (v/v) methanol/water and the Sunrise® HPLC column (4.6 mm × 150 mm) was packed with C18 Silica from Waters. The column was maintained at room temperature during the analyses. The half-life of 12 was calculated from regression curves fitted to plots of the compound concentration versus time.
Acknowledgments
This work was sponsored by the NIH/NIAID Regional Center of Excellence for Biodefense and Emerging Infectious Diseases Research (RCE) Program. Support is gratefully acknowledged from the Region V 3Great Lakes2 RCE (NIH award 1-U54-AI-057153) and NIAID (P01AI055672). This research was also supported by a fellowship to A.S.M. from the Egyptian government. Figure 1 and part of Figure 2 are reproduced from reference 18 and from Mayboub, A. S. et al. J. Med. Chem. 2011, 54, 1704 and appear here with permission from the American Chemical Society.
Abbreviations
- BHK
baby hamster kidney cells
- β-OG
n-octyl-β-D-glucoside
- EMCV
encephalomyocarditis virus
- E-protein
Envelop-protein
- FBS
fetal bovine serum
- Luc
luciferase
- MEM
minimal essential medium
- NBS
N-bromosuccinimide
- NCS
N-chlorosuccinimide
- PCR
polymerase chain reaction
- SAR
structure-activity relationship
- ssRNS
single-stranded RNA
- TI
therapeutic index
- YFV
yellow fever virus
- IRES
internal ribosome entry site
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Munoz-Jordan JL, Sanchez-Burgos GG, Laurent-Rolle M, Garcia-Sastre A. Proc Natl Acad Sci USA. 2003;100:14333–14338. doi: 10.1073/pnas.2335168100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pan American Health Organization website. Number of Reported Cases of Dengue and Dengue Hemorrhagic Fever in the Americans, by country: figures for 2008 (to week noted by each country) [Accessed October 6, 2010]; http://www.paho.org/English/AD/DPC/CD/dengue-cases-2008.pdf.
- 3.Morens DM, Fauci AS. J Am Med Assoc. 2008;299:214–216. [Google Scholar]
- 4.a) Gulati S, Maheshwari A. Trop Med Int Health. 2007;12:1087–1095. doi: 10.1111/j.1365-3156.2007.01891.x. [DOI] [PubMed] [Google Scholar]; b) Varatharaj A. Neurol India. 2010;58:585–591. doi: 10.4103/0028-3886.68655. [DOI] [PubMed] [Google Scholar]; c) Sejvar JJ, Haddad MB, Tierney BC. J Am Med Assoc. 2003;290:511–515. doi: 10.1001/jama.290.4.511. [DOI] [PubMed] [Google Scholar]
- 5.Sampath A, Padmanabhan R. Antiviral Res. 2009;81:6–15. doi: 10.1016/j.antiviral.2008.08.004.and references sited within; Petersen LR, Marfin AA, Gubler DJ. J Am Med Assoc. 2003;290:524–528. doi: 10.1001/jama.290.4.524.
- 6.Borowski P, Lang M, Haag A, Schmitz H, Choe J, Chen HM, Hosmane RS. Antimicrob Agents Chemother. 2002;46:1231–1239. doi: 10.1128/AAC.46.5.1231-1239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang N, Ming Chen H, Koch V, Schmitz H, Minczuk M, Stepien P, Fattom AI, Naso RB, Kalicharran K, Borowski P, Hosmane RS. J Med Chem. 2003;46:4776–4789. doi: 10.1021/jm030277k. [DOI] [PubMed] [Google Scholar]
- 8.Luzhkov VB, Selisko B, Nordqvist A, Peyrane F, Decroly E, Alvarez K, Karlen A, Canard B, Qvist JA. Bioorg Med Chem. 2007;15:7795–7802. doi: 10.1016/j.bmc.2007.08.049. [DOI] [PubMed] [Google Scholar]
- 9.Fabrega C, Hausmann S, Shen V, Shuman S, Lima C. DMol Cell. 2004;13:77–89. doi: 10.1016/s1097-2765(03)00522-7. [DOI] [PubMed] [Google Scholar]
- 10.Mueller NH, Pattabiraman N, Ansarah-Sobrinho C, Viswanathan P, Pierson TC, Padmanabhan R. Antimicrob Agents Chemother. 2008;52:3385–3393. doi: 10.1128/AAC.01508-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mueller NH, Yon C, Ganesh VK, Padmanabhan R. Int J Biochem Cell Biol. 2007;39:606–614. doi: 10.1016/j.biocel.2006.10.025. [DOI] [PubMed] [Google Scholar]
- 12.Puig-Basagoiti F, Tilgner M, Forshey BM, Philpott SM, Espina NG, Wentworth DE, Goebel SJ, Masters PS, Falgout B, Ren P, Ferguson DM, Shi P. Antimicrob Agents Chemother. 2006;50:1320–1329. doi: 10.1128/AAC.50.4.1320-1329.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Zhang Y, Zhang W, Ogata S, Clements D, Strauss JH, Baker TS, Kuhn RJ, Rossmann MG. Structure. 2004;12:1607–1618. doi: 10.1016/j.str.2004.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang W, Chipman PR, Corver J, Johnson PR, Zhang Y, Mukhopadhyay S, Baker TS, Strauss JH, Rossmann MG, Kuhn RJ. Nat Struct Biol. 2003;10:907–912. doi: 10.1038/nsb990. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Perera Rk, Kuhn RJ. Curr Opin Microbiol. 2008;11:369–377. doi: 10.1016/j.mib.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Modis Y, Ogata S, Clements D, Harrison SC. Proc Natl Acad Sci USA. 2003;100:6986–6991. doi: 10.1073/pnas.0832193100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou Z, Khaliq M, Suk J, Patkar C, Li L, Kuhn RJ, Post CB. Chem Biol. 2008;3:765–775. doi: 10.1021/cb800176t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ze L, Khaliq M, Zhou Z, Post CB, Kuhn RJ. J Med Chem. 2008;51:4660–4671. doi: 10.1021/jm800412d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.a) Wang Q, Patel SJ, Vangrevelinghe E, Xu HY, Rao R, Jaber D, Schul W, Gu F, Heudi O, Ma NL, Poh MK, Phong WY, Keller TH, Jacoby E, Vasudevan SG. Antimicrob Agents Chemother. 2009;53:1823–1831. doi: 10.1128/AAC.01148-08. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Poh MK, Yip A, Zhang S, Priestle JP, Ma NL, Smit JM, Wilschut J, Shi P, Wenk MR, Schul W. Antiviral Res. 2008;84:260–266. doi: 10.1016/j.antiviral.2009.09.011. [DOI] [PubMed] [Google Scholar]
- 18.Mayhoub AS, Khaliq M, Kuhn RJ, Cushman M. J Med Chem. 2011;54:1704–1714. doi: 10.1021/jm1013538. [DOI] [PubMed] [Google Scholar]
- 19.DePierre JW. Handb Environ Chem. 2003;3:205–251. Pr. R. [Google Scholar]
- 20.Yang RS, Witt KL, Alden CJ, Cockerham LG. Rev Environ Contam Toxicol. 1995;142:65–85. doi: 10.1007/978-1-4612-4252-9_3. [DOI] [PubMed] [Google Scholar]
- 21.Miller DP, Haggard HW. J Ind Hyg Toxicol. 1943;25:423–433. [Google Scholar]
- 22.Oates LJ, Jackson RFW, Block MH. Org Biomol Chem. 2003;1:140–144. doi: 10.1039/b208632h. [DOI] [PubMed] [Google Scholar]
- 23.Willard ML, Maresh C. J Am Chem Soc. 1940;62:1253–1257. [Google Scholar]
- 24.Jones CT, Patkar CG, Kuhn RJ. Virology. 2005;331:247–259. doi: 10.1016/j.virol.2004.10.034. [DOI] [PubMed] [Google Scholar]
- 25.Safran M, Kim WY, O’Connell F, Flippin L, Günzler V, Horner JW, Depinho RA, Kaelin WG. Proc Natl Acad Sci USA. 2006;103:105–110. doi: 10.1073/pnas.0509459103. [DOI] [PMC free article] [PubMed] [Google Scholar]