

Abstract
Protein enzymes establish intricate networks of interactions to bind and position substrates and catalytic groups within active sites, enabling stabilization of the chemical transition state. Crystal structures of several RNA enzymes also suggest extensive interaction networks, despite RNA's structural limitations, but there is little information on the functional and energetic properties of these inferred networks. We used double mutant cycles and pre-steady state kinetic analyses to probe the putative interaction between the exocyclic amino group of the guanosine nucleophile and the N7 atom of residue G264 of the Tetrahymena group I ribozyme. As expected, the results supported the presence of this interaction, but remarkably, the energetic penalty for introducing a CH group at the 7-position of residue G264 accumulates as the reaction proceeds towards the chemical transition state to a total of 6.2 kcal/mol. Functional tests of neighboring interactions revealed that the presence of the CH group compromises multiple contacts within the interaction network that encompass the reactive elements, apparently forcing the nucleophile to bind and attack from an altered, suboptimal orientation. The energetic consequences of this indirect disruption of neighboring interactions as the reaction proceeds demonstrate that linkage between binding interactions and catalysis hinges critically on the precise structural integrity of a network of interacting groups.
Introduction
The discovery of the ability of RNA to act as an enzyme has led to a significant interest in understanding the catalytic strategies used by RNA enzymes, or ribozymes, and in comparing these strategies to those used by the more extensively investigated protein enzymes.1-3 Studies of several ribozymes have shown that, like protein enzymes, ribozymes bind and position substrates within active sites and use specific interactions to stabilize the reaction's transition state.1-6 Further, structural studies have provided evidence of extensive networks of interactions within ribozyme actives sites, suggesting that ribozymes may be able to use these networks for catalysis.1 Specifically, these networks might aid the precise positioning of substrates and of the functional groups needed for catalysis.
However, structural inspection alone is insufficient to define the role of these RNA interaction networks in catalysis, especially considering that RNA has limited ability to pack and is known to adopt alternative structures. Further, mutations that remove interactions observed in RNA structures can have small or negligible energetic effects, presumably due to formation of near isoenergetic alternative interactions.3,7-9 To test the role of networks in RNA catalysis, determination of the energetic consequences of perturbation of both single interactions and multiple interactions within these networks is needed. We have accomplished this for a network adjoining the active site of the Tetrahymena group I ribozyme.
The Tetrahymena group I ribozyme has served as a powerful system for uncovering the properties of catalytic RNA. This ribozyme catalyzes a site-specific attack of a guanosine molecule (G) on an oligonucleotide substrate (S), as shown in Figure 1 (for a review of the reaction, see ref. 4). In this reaction the oligonucleotide substrate S binds in two steps (Figure 1a). First, S binds to the ribozyme by base-pairing interactions,10 forming the ‘open complex’ denoted by the subscript ‘o’ in Figure 1a. Subsequently, the duplex formed by S and the ribozyme docks in the ribozyme's core to form the ‘closed complex’ denoted by the subscript ‘c’ in Figure 1a.11,12 The closed complex involves tertiary contacts between the substrate-ribozyme duplex (referred to as the P1 helix) and the ribozyme's core.
Figure 1. A model for the reaction catalyzed by the Tetrahymena group I ribozyme.
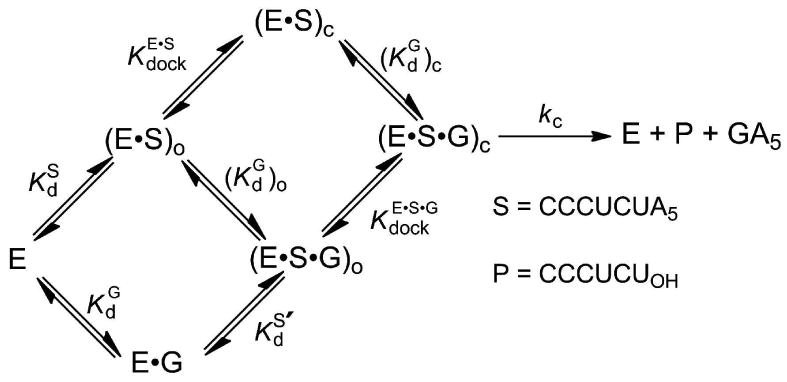
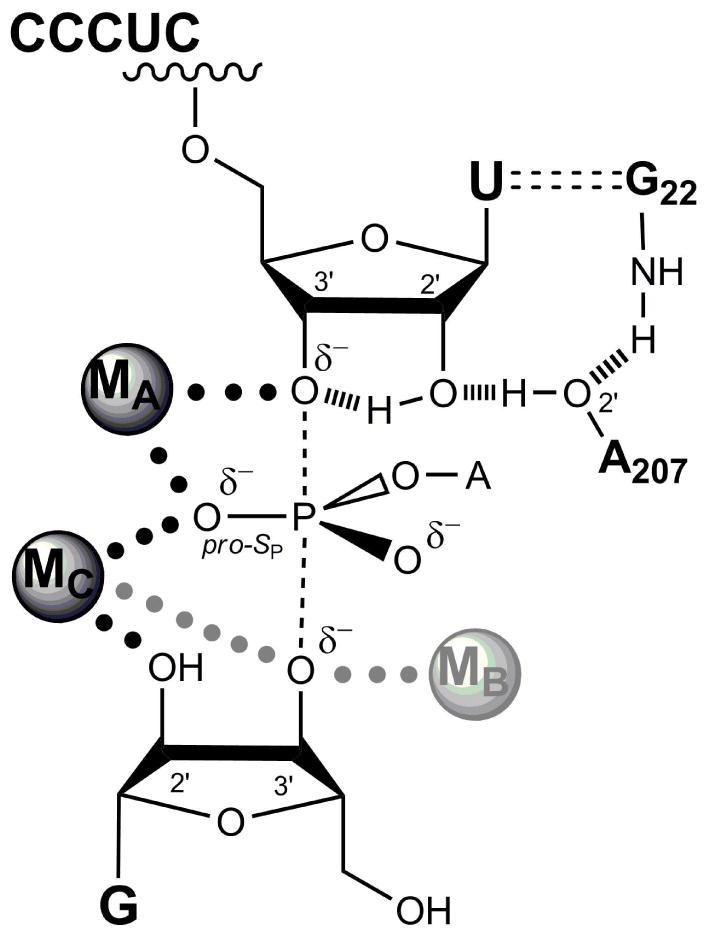
(a) Individual steps of the reaction. The subscript ‘o’ represents the open complex, while the subscript ‘c’ represents the closed complex. (b) Model for the transition state of the reaction. Hashed lines represent hydrogen bonds, dotted lines represent metal ion interaction, and dashed lines represent the bonds broken and formed in the reaction. MA, MB, and MC represent the three distinct metal ions implicated from functional data. MB (ligher shading) is not observed in the crystal structures of group I ribozymes, and it has been proposed, based on spatial proximity, that MC forms an additional interaction with the 3′-oxygen of the nucleophilic G in the transition state (gray dotted line).
Guanosine can bind to the free enzyme, the open complex, or the closed complex (Figure 1a). The affinities of G for the free enzyme and the open complex are the same,13 and this and other observations14 suggest that the environment around the guanosine nucleophile does not change in these two forms of the ribozyme. In contrast, G binds the closed complex ∼5-fold more tightly than the open complex, indicating energetic coupling between G binding and docking of the P1 helix.13 After G is bound and S is docked, the chemical reaction takes place. The reaction involves deprotonation of the 3′-hydroxyl group of guanosine, which then attacks the phosphoryl group between the U and A residues of S. This chemical transformation is assisted by metal ions and other interactions (Figure 1b).4
In a seminal paper probing structure-activity relationships in an RNA enzyme, Bass and Cech defined specific atoms or groups on the guanosine nucleophile important for the group I self-splicing reaction.15 A subsequent specificity switch experiment by Michel and co-workers identified the location of a specific guanosine-binding site, provided evidence for a hydrogen bond between N1 of guanosine and O6 of G264, and suggested the presence of another hydrogen bond between the exocyclic amino group of guanosine and N7 of G26416 (Figure 2a). Although other models have been proposed,17,18 these two specific interactions are strongly supported by the proximity of these two groups in all of the structural models derived from group I ribozymes crystallographic data (Figure 2b-d, red and black dashed lines19-24).
Figure 2. The environment around the exocyclic amino group of G in the group I ribozyme.


The guanosine nucleophile is in green, residue G264 in red, residue A261 in blue. Dashed lines represent putative hydrogen bonds. The hydrogen bonds in black and blue are supported by functional data; the putative hydrogen bond represented in red is the interaction studied herein. (a) Two-dimensional chemical representation of the guanosine-binding site. (b) Superposition of the models derived from the Tetrahymena group I ribozyme crystals (PDB ID 1X8W), which contain four molecules. (c) Superposition of the models derived from the Twort group I ribozyme crystals (PDB entries 1Y0Q and 2RKJ). (d) Superposition of the models derived from the Azoarcus group I ribozyme crystals (PDB entries 1ZZN, 3BO2 and 3BO3). Models were superimposed using the guanosine nucleophile. Residues are numbered using the corresponding numbers from the Tetrahymena ribozyme.
Here, we have used functional tests, carried out with chemically modified ribozymes and substrates, to investigate the putative interaction between the exocyclic amino group of guanosine and N7 of G264 and the properties of the surrounding environment. Remarkably, perturbation of this interaction has a large energetic effect that accumulates along the reaction trajectory to a value far greater than expected from disruption of a single hydrogen bond. Our results provide powerful functional evidence for the existence of a tight, energetically linked network of interactions surrounding the active site that contributes to the ribozyme's catalytic efficiency and specificity.
Experimental Section
Materials
AUCG and AUCI were purchased from Dharmacon Inc. (Lafayette, CO) or synthesized by the PAN Facility (Stanford, CA). AUCG2′-NH2 was synthesized at the University of Chicago, using the phosphoramidite of G2′-NH2 as previously described.25 7-Deazaguanosine phosphoramidite was purchased from ChemGenes (Wilmington, MA). Oligonucleotides corresponding to nucleotides 260-274 of the ribozyme, containing a 5′-phosphoryl group, a guanosine or 7-deazaguanosine residue at position 264, and a 2′-OH or 2′-H group at position 261 were synthesized by the PAN Facility (Stanford, CA). Oligonucleotides were purified by reverse-phase HPLC as previously described.26 Wild type and variant ribozymes were constructed semi-synthetically using a single-step three-piece ligation,27 with a modified protocol previously described.26
General Kinetic Methods
All cleavage reactions were single turnover, with ribozyme in excess of radiolabeled oligonucleotide substrate (*S), which was always present in trace quantities (<100 pM). 5′-32P-End-labeling of the oligonucleotide substrates for kinetic experiments was performed by standard methods. The oligonucleotide substrates used in this work are CCCUCdUAAAAA (referred to as -1d,rSA5) and d(CCCUC)Ud(AAAAA) (referred to as -1r,dSA5). These substrates contain mixed ribose and deoxyribose residues, with deoxyribose residues indicated by a ‘d’, and allow the reactions to be monitored from different ground state E•S complexes (see below). Reactions were carried out at 30 °C in 45 mM NaHEPES/5 mM NaMOPS, pH 8.1, and 50 mM MgCl2, as previously described.26
Kinetic parameters for the WT and A261H ribozymes are from previous work.26 To obtain the kinetic parameters for the G264deaza and the G264deaza/A261H ribozymes, the rate constant for reaction of *S was determined as a function of AUCG or AUCI concentration for the different ribozymes. For the G264deaza ribozyme, reactions with up to 4 mM AUCG or with up to 1 mM AUCI were followed. For the G264deaza/A261H ribozyme, up to 300 μM AUCG was used. The ribozyme concentration was 50 nM, and native gels analysis28,29 confirmed that >95% of *S was bound to the ribozymes.
For the G264deaza ribozyme, the observed rate constant (kobs) for cleavage of *S was plotted as a function of AUCG (or AUCI) concentration and fit to Equation 1 to obtain kc, and (kcat /KM)AUCX, where AUCX represents AUCG or AUCI.
| (1) |
For the G264deaza/A261H ribozyme, reactions were performed under subsaturating concentrations of AUCG (0-200 μM), and values of (kcat / KM)AUCG were by fit to Equation 2
| (2) |
To determine the kinetic parameters for the closed complex we used the -1d,rSA5 substrate (CCCUCdUAAAAA). The deoxyribose residue at position -1 ensures that the chemical step is rate-limiting and that the observed (or ) equals [or ].13 To determine the kinetic parameters for the open complex, we used the oligonucleotide substrate -1r,dSA5 [d(CCCUC)Ud(AAAAA)], which favors the open complex because of the absence of specific 2′-OH groups involved in the tertiary interactions that stabilize the closed complex; reaction of this substrate has the chemical step rate-limiting, so that the observed rate constant reflects all reaction steps from the open complex ground state to the chemical transition state.30
Results
To test the importance of the putative hydrogen bond between the exocyclic amino group of guanosine and the N7 atom of residue G264 (Figure 2, red dashed line) in the reaction catalyzed by the Tetrahymena group I ribozyme, we used double-mutant cycles.31 Specifically, we determined the energetic penalty from removing the exocyclic amino group of the G nucleophile in the context of the wild-type (WT) ribozyme and a ribozyme modified at residue G264 so that it lacks the ability to accept a hydrogen bond at the 7 position. A similar energetic penalty for the WT and modified ribozymes would suggest no functional communication between the guanosine nucleophile and N7 of residue G264, and thus no contact; in contrast, a reduced effect from removing the exocyclic amino group of G in the context of the modified ribozyme would suggest functional communication and, given the proximity observed structurally, a direct contact.
To ablate interactions with the exocyclic amino group of G we used inosine (I) as the nucleophile, and to replace the N7 at position G264 of the Tetrahymena group I ribozyme with a group no longer capable of acting as hydrogen-bond acceptor we constructed a semi-synthetic ribozyme variant containing a CH group at the 7-position of residue 264 (see Experimental Section), referred to herein as the G264deaza ribozyme. To serve as a control for potential effects of the semi-synthetic procedure, we constructed a semi-synthetic variant of the WT ribozyme. This construct gave kinetic behavior identical to the transcribed WT ribozyme (data not shown), as found in other ribozyme constructs ligated by similar procedures.26,32-36 Because of the low solubility of G relative to its dissociation constant,13,37 we used a G analog, AUCG, instead of G; similarly we used AUCI instead of inosine. These analogs exhibit tighter binding without altering the reaction mechanism.38,39
Testing the putative interaction between the exocyclic amino group of the nucleophile and the N7 atom of residue G264
To determine the energetic consequence of removing the exocyclic amino group of AUCG in the context of the WT and the G264deaza ribozymes, we measured the second-order rate constant, , for reactions of the two ribozymes, where the superscript AUCX denotes AUCG or AUCI. (kcat / KM) is determined by all reaction steps from free enzyme and substrate up to and including the first irreversible step. Because the chemical step is the first irreversible step under all of the conditions used in this work,30 the value of (kcat / KM) is affected by this step and all steps that precede it, which are S binding, AUCG (or AUCI) binding, and S docking (Figure 1a). We monitored the reactions under conditions in which the oligonucleotide substrate (S) is already bound to the ribozyme in the so-called open complex [(E•S)o, Figure 1a], because the open complex gives a defined state that involves only base pairing of S with the ribozyme that is not expected to be affected by modifications in the guanosine binding site.10,40,41 To favor the open complex, we used the oligonucleotide substrate -1r,dSA5 (see General kinetic methods), in which specific 2′-OH groups that stabilize the docked state are replaced by 2′-H groups.
The interaction tested is represented by the red dashed line in Figure 3 and is enclosed by a black rectangle. The other interactions made by the G nucleophile and shown in Figure 3 have been established by previous functional data15,16,26,36 and are consistent with X-ray crystal structures.19-24 Figure 3 also summarizes the overall effects of the atomic perturbations on catalysis, showing the values for each of the four combinations of ribozyme and nucleophile, with these values converted into free energy differences relative to the wt ribozyme reaction with AUCG (upper left in figure). Previous results have shown that AUCI reacts ∼230-fold slower than AUCG in the WT ribozyme [Figure 3, and ], corresponding to an energetic penalty of 3.3 kcal/mol .26,36 Based on the interactions observed in the crystal structures, 20-22,24 this energy would correspond to the loss of two hydrogen bonds that are formed between the exocyclic amino group of G and the ribozyme, modulated by any contributions from favorable or unfavorable changes in solvation and steric effects upon complex formation. In contrast, we found that AUCI reacts 1200-fold faster than AUCG in the G264deaza ribozyme [Figure 3, and ], corresponding to an energetic gain of 4.2 kcal/mol . The observed coupling in the double-mutant cycle suggests that the interaction is indeed formed, as suggested by the structural data. Furthermore, the coupling energy (ΔΔΔGint) of 7.5 kcal/mol is remarkably large. We therefore investigated the origin of this coupling.
Figure 3. Testing the proposed contact between the exocyclic amino group of AUCG and the N7 atom of residue G264.
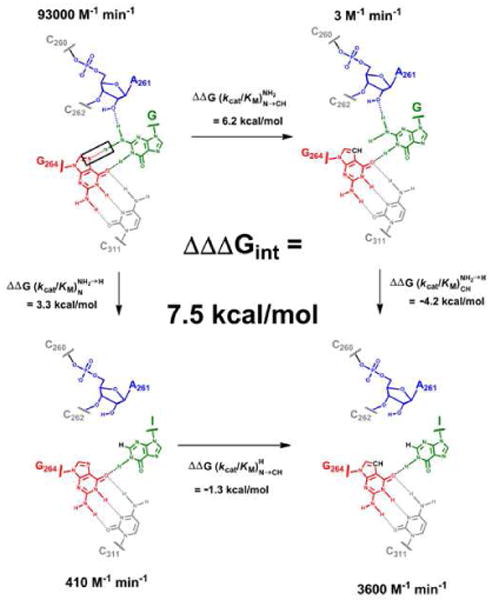
The contact tested is highlighted by a black rectangle, and corresponds to the red dashed line in Figure 2. Numbers with units of M-1 min-1 represent the second-order rate constant for reactions of AUCG (top) or AUCI (bottom) with the WT (left) or G264deaza (right) ribozymes. Vertical arrows: differences in reactivity between AUCG and AUCI for the WT ribozyme (left) and G264deaza ribozyme (right). Horizontal arrows: differences in reactivity between the WT and G264deaza ribozymes when AUCG (top) or AUCI (bottom) is used as the nucleophile. Values of ΔΔG are calculated from the relationship ΔΔG = RT ln[ratio (kcat/KM)], and were rounded to a single decimal figure to take into account the experimental errors. ΔΔΔGint is the difference between ΔΔG values on opposite sides of the cycle.
A physical model for the extremely large coupling energy
Site-directed mutagenesis that removes one hydrogen-bond partner in neutral hydrogen bonded complexes typically gives energetic effects of 0.5-1.5 kcal/mol.42,43 How then does the G264 N7 hydrogen bond to the exocyclic amino group of the G nucleophile give an energetic effect of 6.2 kcal/mol and a coupling energy of 7.5 kcal/mol (Figure 3)? Replacement of the nitrogen atom at the 7-position of residue G264 with a CH group introduces changes beyond the loss of the hydrogen-bond acceptor properties at the 7-position: the pKa of the purine N1 atom is altered,44 stacking properties differ,45,46 a larger volume is occupied by the CH group of 7-deazaguanosine compared to the N7 atom of guanosine, and a hydrophobic group replaces a polar group. Because the nucleophilic G molecule does not interact directly with the N1 of G264 and does not stack with this residue, differences in the pKa and in the stacking properties of residue 264 would be predicted to affect reactivity of AUCG and AUCI to the same extent and therefore would be unlikely to have large differential energetic effects and thus give little or no coupling energy. However, steric and polar differences could give differential effects and contribute to the observed coupling energy, as observed occasionally in other RNA systems.47
Simple consideration of van der Waals radii shows that replacement of the nitrogen atom at the 7-position of residue G264 with a CH group could introduce a steric or a polar clash between the hydrogen atom of the CH group and a hydrogen atom of the exocyclic amino group of the AUCG nucleophile (Supplemental Figure 1). Such unfavorable interactions may result in weaker binding and, subsequent to binding, altered positioning of the AUCG nucleophile relative to the transferred phosphoryl group or the catalytic metal ions depicted in Figure 1b. This model predicts that AUCI, in which the exocyclic amino group is replaced by a smaller, apolar H atom, would relieve the unfavorable interaction and thus would react faster than AUCG in the G264deaza ribozyme. In agreement with this prediction, as noted above, AUCI reacts 1200-fold faster (corresponding to 4.2 kcal/mol) than AUCG with the G264deaza ribozyme (Figure 3, 3600 M-1 min-1 and 3 M-1 min-1, respectively). An additional factor that could lead to increased reactivity of AUCI in the G264deaza ribozyme (relative to the WT ribozyme) would be easier desolvation of the 7-CH group of the G264deaza ribozyme than the N7 of the WT ribozyme.
Unfavorable steric or polar interactions might simply eliminate a single hydrogen bond. Alternatively, they might cause the loss of multiple interactions. For example, the interaction made by the other hydrogen atom of the exocyclic amino group of AUCG with the 2′-OH group of residue A261 of the WT ribozyme36 might be broken to relieve a steric clash. To distinguish between these models, we measured the reactivity of a ribozyme containing both a CH group at the 7-position of G264 and a 2′-deoxy substitution at position A261 (i.e., the G264deaza/A261H ribozyme). If the interaction between the exocyclic amino group of AUCG and the 2′-OH group of residue A261 were not formed when the N7 atom of G264 is replaced by a CH group, the G264deaza/A261H and the G264deaza ribozymes would react with the same second-order rate constant (Figure 4, red arrow). In contrast, if the interaction were still formed, the second-order rate constant of the G264deaza/A261H ribozyme would be expected to drop by 180-fold (3.3 kcal/mol), which corresponds to the energetic penalty observed for the A261 substitution alone (Figure 4, blue arrow).36 In agreement with the former model, we found that the reaction of the G264deaza/A261H ribozyme is within two-fold of that for the G264deaza ribozyme, corresponding to only 0.5 kcal/mol of destabilization from the second substitution rather than the 3.3 kcal/mol predicted for independent contributions (Figure 4). This result strongly suggests that the introduction of 7-deazaguanosine at position 264 leads to rearrangements that result in the weakening or loss of the interaction between the exocyclic amino group of AUCG and the 2′-OH group of residue A261 in the transition state.
Figure 4. Decrease in reactivity of single and double modified ribozymes compared to the WT ribozyme.
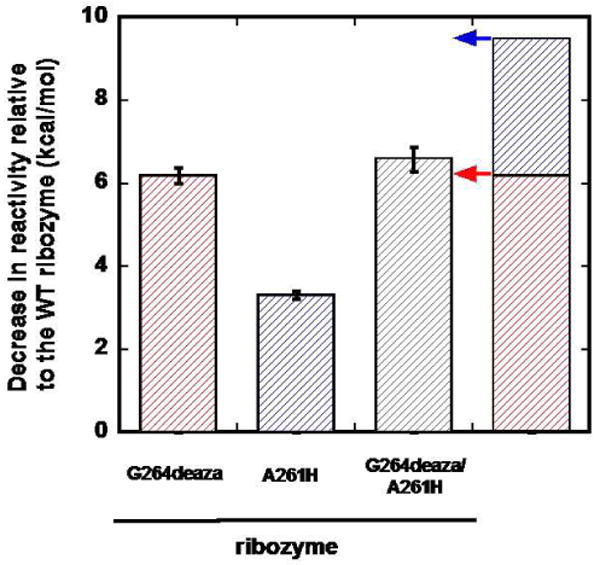
The three bars on the left bars represent the measured energetic penalty on the second-order rate constant from introducing a 7-deazaguanosine at position 264 (G264deaza, red), a 2′-H group at position 261 (A261H, blue), and the two groups simultaneously (G264deaza/A261H, gray), relative to the WT ribozyme. The stacked bar on the right represents the sum of the energetic penalties of the G264deaza and the A261H ribozymes. The arrows next to this bar represent the energetic penalty expected if the G264deaza and the A261H effects were fully coupled (red arrow) or fully independent (blue arrow). As described in the text, there is only a 0.5 kcal/mol difference between energetic penalty corresponding to the red arrow and the measured value for the G264deaza/A261H ribozyme.
The results presented above imply that the introduction of a CH group at the 7-position of residue 264 leads to structural rearrangements that alter the position of the AUCG nucleophile relative to other groups. The ribozyme appears to be unable to rearrange locally to accommodate the structural perturbation and maintain transition state stabilization, and the results therefore suggest a substantial rigidity in and around the guanosine-binding site. To measure this energetic penalty along the reaction pathway, we monitored the coupling energy for the interaction between the exocyclic amino group of AUCG and the N7 atom of residue G264 in AUCG binding, S docking, and the chemical step, as described below.
Tightening of the network of interactions along the reaction pathway
We first determined the energetic penalty of removing the exocyclic amino group of G on binding of the nucleophile to the open complexes [(E·S)o] of the WT and G264deaza ribozymes. As noted above, binding to the open complex is expected to be identical to binding to the free enzyme13 but is more convenient to measure. AUCG binding to the open complex of the G264deaza ribozyme is destabilized by 1.5 kcal/mol, relative to binding of the same nucleophile to the WT ribozyme (Figure 5a). This energetic penalty is about half of the energetic penalty arising from the use of AUCI as the nucleophile in reactions of the WT ribozyme (Figure 5a and ref. 36). Because AUCI lacks two hydrogen bonds with the ribozyme, compared to AUCG, the result above suggests, most simply, that in the open complex of the G264deaza ribozyme AUCG forms only one hydrogen bond with the ribozyme (the one between the exocyclic amino group of the G nucleophile and an unknown group).26 Consistent with this model, there is an additional penalty from removing the exocyclic amino group of AUCG for the G264deaza ribozyme (0.9 kcal/mol, Figure 5a). AUCI binds with the same affinities to the open complexes of the WT and the G264deaza ribozymes, as expected if no gross perturbations are introduced in the G264deaza ribozyme.
Figure 5. Testing individual reaction steps for the proposed contact between the exocyclic amino group of AUCG and the N7 atom of residue G264.
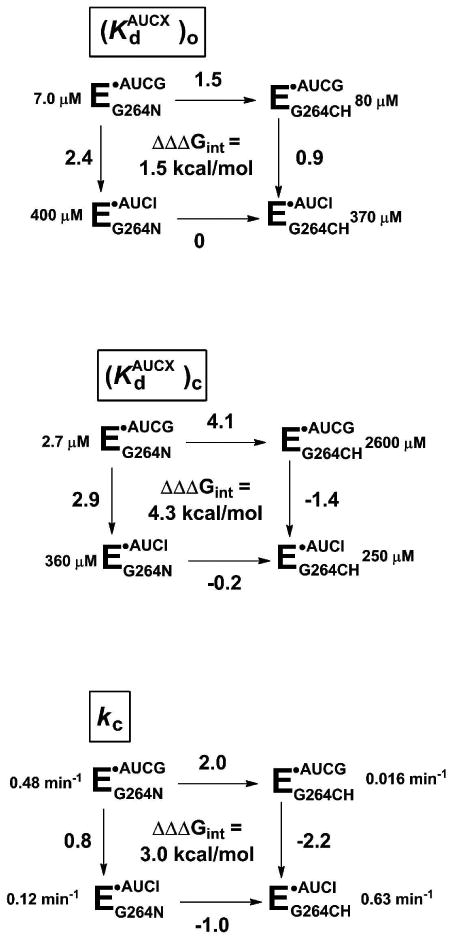
Reactions for the WT (EG264N) or G264deaza (EG264CH) ribozymes with AUCG or AUCI. AUCX represents either AUCG or AUCI. The numbers next to each arrow represent the functional effect (ΔΔG in kcal/mol) of either replacing the N7 of residue G264 with a CH group (horizontal arrows) or ablating the exocyclic amine of AUCG, by use of AUCI as the nucleophile (vertical arrows). Values of ΔΔΔGint are calculated by subtracting the value on the right from the value on the left or, equivalently, the value on the bottom from the value on the top. The individual reaction steps (defined in Figure 2a) are as follows: (a) nucleophile binding to the open complex ; (b) nucleophile binding to the closed complex ; and (c) the chemical step (kc). ΔΔG values were rounded to a single decimal place to take in account the experimental errors. The sum of the coupling energies on the individual reaction steps matches the overall effect reported in Figure 3 (see Supporting Information).
Quantitative analysis of all of the results for this reaction step, shown in Figure 5a, reveals a coupling energy of 1.5 kcal/mol (Figure 5a, ΔΔΔGint), much smaller than the overall coupling energy of 7.5 kcal/mol (Figure 3). This result suggests that the (E·S·AUCG)o complex of the G264deaza ribozyme can readily accommodate via rearrangements and/or solvent reorganization the perturbation introduced by the CH group at the 7-position of residue 264, while maintaining other interactions formed between the guanosine nucleophile and the ribozyme. Thus, the environment around the guanosine nucleophile is relatively ‘loose’ or flexible in the open complex (Figure 6).
Figure 6. Schematic model of the differences between the guanosine-binding site of the WT ribozyme and that of the G264deaza ribozyme.

Dashed lines represent interactions between the guanosine and the ribozyme; pink curved lines represent the unfavorable interaction between the 7-deaza group of the G264deaza ribozyme and the exocyclic amino group of the nucleophilic G. In the open complex there is sufficient flexibility to maintain the other interaction of the guanosine exocyclic amine in the G264deaza ribozyme, but the tighter and more extensive network of interactions in the closed complex prevents reorientation of the other hydrogen bond acceptor and thus leads to the loss of both hydrogen bonds in the G264deaza ribozyme.
We next monitored the energetic penalty from removal of the exocyclic amino group of G on the binding affinity of the nucleophile for the closed complexes [(E·S)c] of the WT and G264deaza ribozymes. In this case, AUCG binding to the closed complex of the G264deaza ribozyme is destabilized by 4.1 kcal/mol, relative to binding of the same nucleophile to the WT ribozyme (Figure 5b), consistent with the loss of the additional contact between the nucleophile and the 2′-OH of A261 in the G264deaza ribozyme (Figure 4). Surprisingly, this value is even larger than the 2.9 kcal/mol measured for the loss of the two hydrogen bonds made by the exocyclic amino group of AUCG (left vertical arrow in Figure 5b)15,26,36,48 and suggests that more than two interactions are lost in the closed complex of the G264deaza ribozyme. We further investigate the reason for such large energetic effect below (see “Introduction of 7-deazaguanosine at position 264 perturbs the environment around MC” below).
Unlike the open complex, the closed complex of the G264deaza ribozyme does not seem to be able to accommodate the unfavorable interaction arising from the N7 substitution at position 264. This inability suggests that in the closed complex the nucleophile binding energy derives from a tighter or more extensive network of interactions within the ribozyme's scaffold compared to the open complex (Figure 6). When the unfavorable interaction between the exocyclic amino group of AUCG and the CH group of the G264deaza ribozyme is removed, by using AUCI, the nucleophile binds tighter than AUCG, suggesting that the lack of the exocyclic amino group, which eliminates the unfavorable interaction, allows AUCI to maintain its position within the active site without disrupting the alignment of groups in the surrounding network of interactions.
The transition state for the reaction with AUCG is destabilized by 2.0 kcal/mol in the G264deaza ribozyme, relative to that in the WT ribozyme (Figure 5c). Thus, there is an additional penalty, not present in the closed complex, that arises as the closed complex proceeds to the transition state of the reaction. In the transition state for the chemical step, AUCI is again favored (by 2.2 kcal/mol) relative to AUCG in the G264deaza ribozyme, indicating that ablating the exocyclic amino group relieves an energetic penalty. These results reveal a large coupling energy between the exocyclic amine and G264 N7 in the transition state (Figure 5c, 3.0 kcal/mol). Further, in this case AUCI reacts within the G264deaza ribozyme as fast as AUCG reacts within the WT ribozyme (0.63 and 0.48 min-1, Figure 5c). This result provides further support for the existence of a tight network of interactions: once the unfavorable interactions are removed, other interactions surrounding the nucleophile appear to be sufficient to orient it for optimal catalysis. It is also possible that apolar interactions between the hydrophobic CH at position 264 and the hydrogen of AUCI might help alignment of the nucleophile in the chemical step.
The simplest explanation for the large effect on the chemical step is that the unfavorable interaction between AUCG and the CH group of 7-deazaguanosine at position 264 alters the positioning of the nucleophile and that the network of interactions surrounding the reactive elements makes it energetically costly to accommodate the modification without the loss of native interactions. The 2′- and 3′-oxygen atoms of the guanosine nucleophile are involved in catalytic interactions (Figure 1b), so it is possible that an alteration in guanosine positioning would affect interactions with these atoms. To test this possibility, we determined whether the contact between the 2′-oxygen and the catalytic metal ion MC is altered in the G264deaza ribozyme.
Introduction of 7-deazaguanosine at position 264 perturbs the MC environment
Previous results with the WT and several modified ribozymes have shown that 2′-aminoguanosine reacts slower than guanosine and that metal ions such as Mn2+ and Cd2+ stimulate the reactivity of 2′-aminoguanosine, relative to that of guanosine.49,50 Because Mn2+ and Cd2+ interact better than Mg2+ with nitrogen atoms, these and other observations suggested that a metal ion interacts with the 2′-group of the guanosine nucleophile. Increasing Mn2+ concentration results in increased Mn2+-occupancy of site C (Figure 1b), which allows a more favorable interaction with the amino group of 2′-aminoguanosine in the transition state compared to Mg2+.
We have used the above information to test whether the contact between the 2′-oxygen and MC is suboptimal for function in the G264deaza ribozyme. Specifically, we determined the effect of addition of Mn2+ ions on the reactions of AUCG and AUCG2′-NH2, a guanosine analog containing a 2′-amino group instead of a 2′-hydroxyl, with the WT and G264deaza ribozymes. If the interaction between MC and the 2′-moiety of the nucleophile were perturbed in the G264deaza ribozyme, a lessened deleterious effect upon introduction of the 2′-amino group on the nucleophile in the G264deaza ribozyme compared to the wt ribozyme would be predicted. Further, if there were altered positioning of MC and of the 2′-moiety of the nucleophilic G in the G264deaza ribozyme, then Mn2+ might not be able to rescue the reactivity of AUCG2′-NH2. As described below, both predictions were met.
When only Mg2+ is present, AUCG2′-NH2 reacts with the WT (E•S)o complex ∼20-fold slower than AUCG. In contrast, AUCG2′-NH2 reacts with the G264deaza (E•S)o complex within three-fold of the AUCG rate (Figure 7a). Further, addition of 5 mM Mn2+ stimulates the reactivity of AUCG2′-NH2 with the (ES)o complex of the WT ribozyme, relative to that of AUCG, such that now the reaction rates are within two-fold (Figure 7b). This corresponds to a 12-fold greater stimulation of AUCG2′-NH2 than AUCG; in contrast, this stimulation is only two-fold in the G264deaza ribozyme (Figure 7c). The ∼5-fold Mn2+ stimulation for reactions of both AUCG and AUCG2′-NH2 with the G264deaza ribozyme (Figure 7c and Supplemental Figure 4) is consistent with the effect from a distinct metal ion, referred to as MD, which stabilizes the closed complex.51 The simplest model to explain these results is that the interaction between MC and the 2′-moiety of the nucleophile is absent or substantially altered in the G264deaza ribozyme. Such a perturbed interaction may be responsible for the 30-fold slower chemical step for the reaction of the G264deaza ribozyme with AUCG relative to the WT ribozyme (Figure 5c).
Figure 7. Observed rate constant for reactions of AUCG and AUCG2′-NH2 with the WT and G264deaza ribozymes.



Reactions were carried out with 0.6 μM nucleophile for the WT ribozyme and with 6 μM nucleophile for the G264deaza ribozyme, starting from the (E•S)o complex in both cases. Reactions of AUCG are represented by the black bars; reactions of AUCG2′-NH2 are represented by gray bars. (a) Rate constants for reactions in 50 mM Mg2+. (b) Rate constants for reactions in 50 mM Mg2+ and 5 mM Mn2+. (c) Ratio between rate constants in panel (b) and in panel (a). Rate constants for reactions at intermediate Mn2+ concentrations are given in Supplemental Figure S4.
Discussion
Since the original discovery by Buchner that fermentation can occur without intact cells,52 many enzymes have been identified. For many classes of enzymes, researchers have established reasonable reaction mechanisms based on the chemical properties of individual active site functional groups in isolation.53,54 However, active site residues are not present in enzymes as disjoined entities, and structural inspections of enzymes almost inevitably reveal intricate networks of interactions surrounding active sites. These networks of interactions surrounding the active sites can be important for function. For example, trypsin was converted to chymotrypsin by changing surface loops that do not interact with the substrate;55 statistical and mutational analysis of the PDZ domain family uncovered networks of interactions that allow energetic coupling between distal residues;56 16 of the 17 mutations needed to switch the specificity of an aminotransferase from aspartate to valine involve residues located outside the active site;57 and the activity of a catalytic antibody was increased 100-fold without any change in the residues directly contacting the substrate.58 Guided by structural data, chemical approaches such as specific replacements of atoms involved in these putative networks, in conjunction with careful thermodynamic and kinetic analysis of the resulting functional effects, can be used to understand the origin of energetic coupling at an atomic level.
RNA enzymes are excellent systems for these types of studies, because of their amenability to single-atom substitution59 and the highly developed kinetic and thermodynamic frameworks available to facilitate interpretation. For the group I intron in particular, functional and structural studies have uncovered the groups directly involved in catalysis and the architecture of the substrates' binding sites.4 In this ribozyme, previous functional16 and structural19-21,24 data predicted an interaction between the N7 of residue G264 and the exocyclic amino group of the nucleophilic guanosine. Our results, based on the double-mutant cycle shown in Figure 3, not surprisingly, provide experimental evidence for this interaction. However, the additional functional and structural consequences arising from the introduction of a CH group at the 7-position of residue 264 has revealed remarkable unforeseen properties of an RNA active site -strikingly large energetic effects and loss of interactions in addition to the one monitored.
We have shown that the introduction of a CH group at the 7-position of residue 264 affects binding of guanosine to the open complex of the ribozyme by 1.5 kcal/mol, an energetic effect often seen for the loss of a single hydrogen bond.42,43 This energetic effect is depicted in the free-energy profile of Figure 8 by the difference in energy between the (E·S·G)o complexes of the wild-type (black line) and the G264deaza (red line) ribozymes. This result suggests that, despite the differences in size and polarity between the native nitrogen atom and the introduced CH group, the remaining interactions between the nucleophilic guanosine and the ribozyme are the same in the WT and G264deaza ribozymes. These interactions likely include the hydrogen bond between the N1 of G and the O6 of G26416 (shown in magenta in Figure 9, top) and a hydrogen bond between the exocyclic amino group of G and an unknown partner (not shown in Figure 9).60 Thus, the guanosine nucleophile communicates within a relatively small, loose network of residues, highlighted in magenta in the top panel of Figure 9.
FIGURE 8.

Free energy profiles for the wild-type ribozyme (black line) and the G264deaza ribozyme (red line) obtained from the data in Figure 5. Free energy differences were obtained from the rate and equilibrium constants using the standard conversions: and ΔG = -RT ln Keq. The profiles are qualitative but the noted values of ΔΔG are quantitative.
Figure 9. The network of interactions around the nucleophilic guanosine tightens and becomes more extensive in the transition state of the reaction.

The nucleophilic guanosine is shown in green; residues of the oligonucleotide substrate are in orange; ribozyme residues interacting with the C-2 and U-3 residues of the oligonucleotide substrate in the docked conformation are in dark yellow; other residues are in gray. MA is in yellow and MC is in cyan. The bond formed in the transition state is indicated by a black dashed line. Residues for which there is evidence for energetic or functional communication with the guanosine nucleophile in the WT ribozyme are shown in magenta; interactions involved in the communication networks are shown by magenta dashed lines; specific atoms involved in this network are also in magenta. Other atoms known to be important for function are shown in red (oxygen) or in blue (nitrogen). This Figure was generated using Pymol (Schrödinger, LLC) and the 3BO3 Azoarcus structural model. Residues are numbered using the corresponding numbers from the Tetrahymena group I ribozyme.
When the oligonucleotide substrate docks in the ribozyme's active site, forming tertiary interactions with the ribozyme's backbone and contacting the catalytic metal ions,4,22 the nucleophilic guanosine forms additional interactions with the 2′-OH group of residue A261 (via its exocyclic amino group)26 and with the catalytic metal ion MC.22,49 This establishes additional connectivity between the guanosine nucleophile and parts of the active site, highlighted in magenta in the middle panel of Figure 9. Thus, the guanosine nucleophile communicates with a larger network of interactions in the closed complex than in the open complex (cf. Figure 9, top and middle part), which includes the bonds shown in magenta in middle panel of Figure 9. An oxygen atom of the phosphoryl group connecting residues A261 and C262 is a ligand of MC21,22,24,34 and is thus part of this network, and the bases of A261 and C262 stack above and below the guanine base of the bound guanosine.20-22,24 Further, the base of A306 stacks over A261,20,21 and its phosphoryl oxygen atoms ligand both MC and MA.22,24,35 Such a network might be important for selective recognition of guanosine for catalysis and may help precisely position the reactive groups for the chemical step.
The energetic penalty of 4.2 kcal/mol on AUCG binding to the closed complex of the ribozyme, arising from the introduction of a CH group at the 7-position of residue 264, is larger than the value typically observed from the loss of a single hydrogen bond (0.5-1.5 kcal/mol).42,43 This energetic penalty is significantly larger than the penalty measured in the open complex, as shown in Figure 8. The results shown above (see Figures 4 and 7) strongly suggest that the hydrogen bond with residue A261 and the contact with MC are perturbed in the G264deaza ribozyme. The inability of the guanosine nucleophile to rearrange to accommodate the atomic change in the G264deaza ribozyme without disruption of distal interactions strongly implies the presence of a tight, rigid network of interactions around the guanosine nucleophile (Figure 9, residues in magenta in the middle panel).
If the precise positioning of the nucleophilic guanosine were not important for the reaction, the chemical step for the G264deaza ribozyme would be unaffected by the deaza substitution. In contrast, the ∼30-fold slower reactivity in the chemical step and the lack of rescue of AUCG2′-NH2 by Mn2+ strongly suggests that the nucleophilic guanosine does not rearrange within the active site to form the most favorable contact with MC, and likely sits in a suboptimal position for attack on the transferred phosphoryl group. This is an additional penalty of ∼2.0 kcal/mol, which is not present in the closed complex and brings the total destabilization given by the deaza substitution at position G264 to 6.2 kcal/mol (Figure 8). Our results suggest that such large energetic penalty is given by the difficulty to rearrange the tight network of interactions that impart catalysis once even a remote aspect of that network is perturbed (shown in magenta in the bottom panel of Figure 9).
In contrast to the evidence for tight coupling from the substitutions studied herein, substitution of sulfur atoms for active site oxygen ligands of metal ions and of Cd2+ for Mg2+, which both introduce groups larger than the native ones, have smaller effects on reactivity.35 Perhaps the multiple rotatable bonds around the phosphoryl groups ease relaxation at these sites, whereas the multiple reinforcing hydrogen bonding, stacking, and covalent linkages associated with guanosine binding leave less room for rearrangement. In addition, hydrogen bonding interactions require precise directionality, whereas interactions with metal ions may be more permissive.
Like protein enzymes, ribozymes use binding interactions to position reactive groups, substrates and catalytic residues.3,5,6,15 We have used chemical modifications and functional tests to show that in the Tetrahymena ribozyme at least some of these binding interactions are used to establish a tight network of interactions around the active site. Ultimately, these networks may be used by ribozymes, as well as protein enzymes, to limit dynamics and provide the precise positioning of the substrates and the catalytic residues that appear to contribute substantially to the remarkable catalytic power of enzymes.61 Such networks, by connecting distal regions of an RNA to active centers, also introduce the potential for highly responsive, long range conformational coupling, as occurs in RNA-based machinery such as the ribosome and the spliceosome.
Supplementary Material
Acknowledgments
This work was supported by a grant from the NIH (GM 49243) to D.H. We thank members of the Herschlag lab for helpful comments on the manuscript.
Footnotes
Supporting Information Available. Figures with the representation of possible clashes in the G264deaza ribozyme, analysis of the coupling energy on the individual reaction steps, plots of kobs as a function of [AUCG] in the closed complexes of the WT and G264deaza ribozyme, and plots of kobs as a function of [Mn2+] for reactions of the open complexes of the WT and G264deaza ribozymes with AUCG and AUCGNH2. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Hoogstraten CG, Sumita M. Biopolymers. 2007;87:317. doi: 10.1002/bip.20836. [DOI] [PubMed] [Google Scholar]
- 2.Lilley DM. Trends Biochem Sci. 2003;28:495. doi: 10.1016/S0968-0004(03)00191-9. [DOI] [PubMed] [Google Scholar]
- 3.Narlikar GJ, Herschlag D. Annu Rev Biochem. 1997;66:19. doi: 10.1146/annurev.biochem.66.1.19. [DOI] [PubMed] [Google Scholar]
- 4.Hougland JL, Piccirilli JA, Forconi M, Lee J, Herschlag D. In: The RNA World. 3rd. Gesteland RF, Cech TR, Atkins JF, editors. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 2006. p. 133. [Google Scholar]
- 5.Narlikar GJ, Gopalakrishnan V, McConnell TS, Usman N, Herschlag D. Proc Natl Acad Sci USA. 1995;92:3668. doi: 10.1073/pnas.92.9.3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hertel KJ, Peracchi A, Uhlenbeck OC, Herschlag D. Proc Natl Acad Sci USA. 1997;94:8497. doi: 10.1073/pnas.94.16.8497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sigler PB. Annu Rev Biophys Bioeng. 1975;4:477. doi: 10.1146/annurev.bb.04.060175.002401. [DOI] [PubMed] [Google Scholar]
- 8.Antonioli AH, Cochrane JC, Lipchock SV, Strobel SA. RNA. 2010;16:762. doi: 10.1261/rna.1883810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Santalucia J, Kierzek R, Turner DH. Science. 1992;256:217. doi: 10.1126/science.1373521. [DOI] [PubMed] [Google Scholar]
- 10.Herschlag D, Cech TR. Biochemistry. 1990;29:10159. doi: 10.1021/bi00496a003. [DOI] [PubMed] [Google Scholar]
- 11.Bevilacqua PC, Turner DH. Biochemistry. 1991;30:10632. doi: 10.1021/bi00108a005. [DOI] [PubMed] [Google Scholar]
- 12.Herschlag D. Biochemistry. 1992;31:1386. doi: 10.1021/bi00120a015. [DOI] [PubMed] [Google Scholar]
- 13.McConnell TS, Cech TR, Herschlag D. Proc Natl Acad Sci USA. 1993;90:8362. doi: 10.1073/pnas.90.18.8362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Latham JA, Cech TR. Science. 1989;245:276. doi: 10.1126/science.2501870. [DOI] [PubMed] [Google Scholar]
- 15.Bass BL, Cech TR. Nature. 1984;308:820. doi: 10.1038/308820a0. [DOI] [PubMed] [Google Scholar]
- 16.Michel F, Hanna M, Green R, Bartel DP, Szostak JW. Nature. 1989;342:391. doi: 10.1038/342391a0. [DOI] [PubMed] [Google Scholar]
- 17.Ortoleva-Donnelly L, Szewczak AA, Gutell RR, Strobel SA. RNA. 1998;4:498. doi: 10.1017/s1355838298980086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yarus M, Illangesekare M, Christian E. J Mol Biol. 1991;222:995. doi: 10.1016/0022-2836(91)90590-3. [DOI] [PubMed] [Google Scholar]
- 19.Adams PL, Stahley MR, Kosek AB, Wang J, Strobel SA. Nature. 2004;430:45. doi: 10.1038/nature02642. [DOI] [PubMed] [Google Scholar]
- 20.Golden BL, Kim H, Chase E. Nature Struct Mol Biol. 2005;12:82. doi: 10.1038/nsmb868. [DOI] [PubMed] [Google Scholar]
- 21.Guo F, Gooding AR, Cech TR. Mol Cell. 2005;16:351. doi: 10.1016/j.molcel.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 22.Lipchock SV, Strobel SA. Proc Natl Acad Sci USA. 2008;105:5699. doi: 10.1073/pnas.0712016105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paukstelis PJ, Chen JH, Chase E, Lambowitz AM, Golden BL. Nature. 2008;451:94. doi: 10.1038/nature06413. [DOI] [PubMed] [Google Scholar]
- 24.Stahley MR, Strobel SA. Science. 2005;309:1587. doi: 10.1126/science.1114994. [DOI] [PubMed] [Google Scholar]
- 25.Dai Q, Deb SK, Hougland JL, Piccirilli JA. Bioorg Med Chem. 2006;14:705. doi: 10.1016/j.bmc.2005.08.050. [DOI] [PubMed] [Google Scholar]
- 26.Forconi M, Sengupta RN, Piccirilli JA, Herschlag D. Biochemistry. 2010;49:2753. doi: 10.1021/bi902200n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moore MJ, Sharp PA. Science. 1992;256:992. doi: 10.1126/science.1589782. [DOI] [PubMed] [Google Scholar]
- 28.Mei R, Herschlag D. Biochemistry. 1996;35:5796. doi: 10.1021/bi9527653. [DOI] [PubMed] [Google Scholar]
- 29.Pyle AM, Mcswiggen JA, Cech TR. Proc Natl Acad Sci USA. 1990;87:8187. doi: 10.1073/pnas.87.21.8187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knitt DS, Herschlag D. Biochemistry. 1996;35:1560. doi: 10.1021/bi9521147. [DOI] [PubMed] [Google Scholar]
- 31.Horovitz A. Fold Des. 1996;1:R121. doi: 10.1016/S1359-0278(96)00056-9. [DOI] [PubMed] [Google Scholar]
- 32.Strobel SA, Ortoleva-Donnelly L. Chem Biol. 1999;6:153. doi: 10.1016/S1074-5521(99)89007-3. [DOI] [PubMed] [Google Scholar]
- 33.Szewczak AA, Kosek AB, Piccirilli JA, Strobel SA. Biochemistry. 2002;41:2516. doi: 10.1021/bi011973u. [DOI] [PubMed] [Google Scholar]
- 34.Hougland JL, Kravchuk AV, Herschlag D, Piccirilli JA. PLoS Biol. 2005;3:1536. doi: 10.1371/journal.pbio.0030277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Forconi M, Lee J, Lee JK, Piccirilli JA, Herschlag D. Biochemistry. 2008;47:6883. doi: 10.1021/bi800519a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Forconi M, Sengupta RN, Liu MC, Sartorelli AC, Piccirilli JA, Herschlag D. Angew Chem Int Ed. 2009;48:7171. doi: 10.1002/anie.200903006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pitha PM, Huang WM, Ts'o PO. Proc Natl Acad Sci USA. 1968;61:332. doi: 10.1073/pnas.61.1.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moran S, Kierzek R, Turner DH. Biochemistry. 1993;32:5247. doi: 10.1021/bi00070a037. [DOI] [PubMed] [Google Scholar]
- 39.Russell R, Herschlag D. RNA. 1999;5:158. doi: 10.1017/s1355838299981839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Narlikar GJ, Bartley LE, Koshla M, Herschlag D. Biochemistry. 1999;38:14192. doi: 10.1021/bi9914309. [DOI] [PubMed] [Google Scholar]
- 41.Shi X, Mollova ET, Pljevaljcic G, Millar DP, Herschlag D. J Am Chem Soc. 2009;131:9571. doi: 10.1021/ja902797j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fersht AR, Shi JP, Knill-Jones J, Lowe DM, Wilkinson AJ, Blow DM, Brick P, Carter P, Waye MM, Winter G. Nature. 1985;314:235. doi: 10.1038/314235a0. [DOI] [PubMed] [Google Scholar]
- 43.Turner DH, Sugimoto N, Kierzek R, Dreiker SD. J Am Chem Soc. 1987;109:3783. [Google Scholar]
- 44.Seela F, Chen YM. Nucl Acids Res. 1995;23:2499. doi: 10.1093/nar/23.13.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seela F, Tranthi QH, Franzen D. Biochemistry. 1982;21:4338. doi: 10.1021/bi00261a024. [DOI] [PubMed] [Google Scholar]
- 46.Sistare MF, Codden SJ, Heimlich G, Thorp HH. J Am Chem Soc. 2000;122:4742. [Google Scholar]
- 47.Siegfried NA, Kierzek R, Bevilacqua PC. J Am Chem Soc. 2010;132:5342. doi: 10.1021/ja9107726. [DOI] [PubMed] [Google Scholar]
- 48.McConnell TS, Cech TR. Biochemistry. 1995;34:4056. doi: 10.1021/bi00012a024. [DOI] [PubMed] [Google Scholar]
- 49.Shan S, Herschlag D. Biochemistry. 1999;38:10958. doi: 10.1021/bi990388e. [DOI] [PubMed] [Google Scholar]
- 50.Sjogren AJ, Petterson E, Sjoberg BM, Stromberg R. Nucl Acids Res. 1997;25:648. doi: 10.1093/nar/25.3.648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shan S, Herschlag D. RNA. 2000;6:795. doi: 10.1017/s1355838200000649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Buchner EF. Ber Dt Chem Ges. 1897;30:117. [Google Scholar]
- 53.Begley T. The Organic Chemistry of Biological Pathways. Roberts & Company; 2005. [Google Scholar]
- 54.Fersht A. Structure and mechanism in protein science. W.H. Freeman and Company; New York: 1999. [Google Scholar]
- 55.Hedstrom L, Szilagyi L, Rutter WJ. Science. 1992;255:1249. doi: 10.1126/science.1546324. [DOI] [PubMed] [Google Scholar]
- 56.Lockless SW, Ranganathan R. Science. 1999;286:295. doi: 10.1126/science.286.5438.295. [DOI] [PubMed] [Google Scholar]
- 57.Oue S, Okamoto A, Yano T, Kagamiyama H. J Biol Chem. 1999;274:2344. doi: 10.1074/jbc.274.4.2344. [DOI] [PubMed] [Google Scholar]
- 58.Patten PA, Gray NS, Yang PL, Marks CB, Wedemayer GJ, Boniface JJ, Stevens RC, Schultz PG. Science. 1996;271:1086. doi: 10.1126/science.271.5252.1086. [DOI] [PubMed] [Google Scholar]
- 59.Das SR, Fong R, Piccirilli JA. Curr Opin Chem Biol. 2005;9:585. doi: 10.1016/j.cbpa.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 60.The second hydrogen bond with the 2′-OH of residue A261 that is formed in the closed complex is not formed in the open complex, but the 2.4 kcal/mol loss of binding energy associated with the removal of the exocyclic amino group of G suggests the loss of two hydrogen bonds, as described in ref. 26.
- 61.Wolfenden R. Mol Cell Biochem. 1974;3:207. doi: 10.1007/BF01686645. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.