

Abstract
Ubiquitination is a key event for protein degradation by the proteasome system, membrane protein internalization, and protein trafficking among cellular compartments. Few data are available on the role of the ubiquitin–proteasome system (UPS) in the trafficking of neuronal nicotinic acetylcholine receptors (nAChRs). Experiments conducted in neuron-like differentiated rat pheochromocytoma cells (PC12 cells) show that the α3, β2, and β4 nAChR subunits are ubiquitinated and that their ubiquitination is necessary for degradation. A 24-h treatment with the proteasome inhibitor PS-341 increased the total levels of α3 and the two β subunits in both whole cell lysates and fractions enriched for the ER/Golgi compartment. nAChR subunit upregulation was also detected in plasma membrane-enriched fractions. Inhibition of the lysosomal degradation machinery by E-64 had a significantly smaller effect on nAChR turnover. The present data, together with previous results showing that the α7 nAChR subunit is a target of the UPS, point to a prominent role of the proteasome in nAChR trafficking.
Keywords: Nicotinic acetylcholine receptor, Protein trafficking, Ubiquitin, Proteasome, Lysosome
Introduction
Neuronal nicotinic acetylcholine receptors (nAChRs) are pentameric ligand-gated ion channels formed, in most instances, by combinations of α and β subunits (De Biasi 2002; Le Novere et al. 2002). The central nervous system expresses nine α subunits (α2–α10) and three β subunits (β2–β4; Albuquerque et al. 2009) which can yield receptors with different subunit composition (Dani and De Biasi 2001; De Biasi and Salas 2008). The number of nAChRs presents at the plasma membrane impacts nicotinic cholinergic function in both physiological conditions and disease. For example, the expression of various nAChR subtypes is tightly regulated during development as nAChR activity can influence neurogenesis, cell migration and differentiation, and synaptogenesis (Dwyer et al. 2009). Prolonged exposure to nicotine leads to nAChR upregulation (Wang et al. 1998; Rezvani et al. 2007), a phenomenon thought to contribute to the addictive properties of tobacco (Dani and De Biasi 2001; De Biasi and Salas 2008). Finally, neurodegenerative diseases such as Alzheimer’s disease and dementia with Lewy body are accompanied by decreases in nAChR densities (Court et al. 1999, 2001). Understanding how nAChR levels are regulated is a critical step toward the design of drugs that address addiction and neurological disease.
Several factors control the abundance of plasma membrane receptors, including the rate and the efficiency of protein assembly, the endoplasmic reticulum-associated degradation (ERAD) system, and the rate of transport from endoplastic reticulum (ER) to plasma membrane (Yi and Ehlers 2005; Haas and Broadie 2008). Most of the information available on nAChR synthesis and trafficking comes from studies conducted in muscle nAChRs (Wang et al. 2002; Christianson and Green 2004; Lu et al. 2007; Wanamaker and Green 2007). After translation and post-translational modifications, chaperones such as the ER luminal binding protein, BiP (Blount and Merlie 1991; Paulson et al. 1991; Forsayeth et al. 1992), calnexin (Gelman et al. 1995; Keller et al. 1996), and RIC-3 (Castelan et al. 2008) promote proper folding and maturation of nAChR subunits. The process of folding and pentamer assembly is a prerequisite for the exit from the ER (Smith et al. 1987; Gu et al. 1991; Keller et al. 2001), and unassembled or misfolded receptors are targeted for degradation by the ERAD machinery (Merlie and Lindstrom 1983; Keller et al. 2001; Wanamaker et al. 2003). Nicotinic AChRs that pass the ERAD checkpoint are exported in coat protein complex II vesicles from the ER to the Golgi, presumably after masking of specific ER retention signals (Keller et al. 2001; Wang et al. 2002), and recognition of ER export motifs in properly folded and oligomerized nAChR subunits. Besides degrading misfolded proteins, proteasomes associated with ERAD function provide tight regulation of surface receptor densities by controlling the fraction of proteins that mature to multimers (Christianson and Green 2004; Meusser et al. 2005; Yan et al. 2005; Vembar and Brodsky 2008). The lysosomal degradation machinery is also present at the ER level and might participate in ERAD function (ERAD II; Fujita et al. 2007). Whether nAChR trafficking at the ER/Golgi level is controlled by proteasomes, lysosomes, or both is not completely understood.
In the present study, we examined the influence of proteasomes and lysosomes on the trafficking of nAChRs containing the α3, β2, or β4 subunits.
Results
We used differentiated PC12 (dPC12) cells to study the influence of both proteasomes and lysosomes on the turnover of the α3, β2, and β4 nAChR subunits. dPC12 cells were incubated for 24 h with either vehicle, the proteasome inhibitor PS-341, or the lysosome inhibitor E-64 (Ahlberg et al. 1985) in the presence of the protein synthesis blocker, emetine. To validate the system, the cell lysates were first subjected to western blotting with anti-ubiquitin antibodies. A typical ladder of high molecular weight, ubiquitinated proteins was detected in PS-341-treated cells (Fig. 1a). Quantification of the ubiquitin ladders from three different cell lysates confirmed a significant increase in ubiquitin signal in the cells treated with the proteasome inhibitor (F(2,6)=15, P≤0.005). Interestingly, a slight, although non-statistically significant, increase in the levels of ubiquitinated proteins could be also observed upon lysosomal inhibition (Fig. 1b). When we measured the chymotrypsin-like activity of the cell lysates (Kisselev and Goldberg 2005), we detected robust inhibition of chymotrypsin activity by PS-341 (~73%). Significant inhibition of chymotrypsin-like activity was also observed in the presence of E-64 (~11%; F(2,6)=470, P≤0.0001; Fig. 1c), likely reflecting a non-specific inhibitory effect of E-64 on proteasomal activity (Grinde and Seglen 1980).
Figure 1.

The proteasome controls the degradation of the α3, β2, and β4 nAChR subunits in PC12 cells. Differentiated PC12 cells were treated with the proteasome inhibitor, PS-341, or the lysosome inhibitor, E-64, in the presence of emetine, a protein synthesis inhibitor. a, b Probing of cell lysates with anti-ubiquitin yielded a classical ladder of ubiquitinated proteins (a) which indicated that PS-341 can significantly (b) increase the total levels of ubiquitinated proteins in PC12 cell lysates. c. PS-341 inhibited proteasomal chymotrypsin-like activity by ~73%. E-64 also induced a ~11% reduction in chymotrypsin-like activity. d–g Exposure of the cells to PS-341 (d) resulted in a 2-fold increase in total levels of α3 (e), β2 (f), and β4 (g) over the three sets of experiments we conducted to examine each nAChR subunit. There was a trend toward increased levels of subunits also in the presence of E-64, but the results did not achieve statistical significance. *p≤0.05; **p≤0.005; ***p≤0.001
To determine whether inhibition of the proteasome complex can prevent nAChR degradation, we probed Western blots from the same cell lysates with antibodies against α3, β2, or β4. As shown in Fig. 1d–g, PS-341 reduced nAChR subunit degradation compared to control, leading to an increase in total levels of α3 (F(2,6)=5, P≤0.05), β2 (F(2,6)=7.9, P≤0.05), and β4 (F(2,6)=28.5, P≤0.0005) nAChR subunits. Similar results were obtained in HEK and dPC12 cells using another proteasome inhibitor, MG132 (data not shown). Visual inspection of the blots suggested a potential inhibitory effect of E-64 on nAChR subunit degradation, in particular that of β2. However, quantification of the antibody signals in the three separate experiments performed did not reach statistical significance (Fig. 1e–g).
The same cell lysates used for the experiments described in Fig. 1 were used to precipitate the α3, β2, and β4 subunits via the corresponding primary antibody immobilized onto Protein A/G agarose. This method allows pull-down of both non-ubiquitinated and ubiquitinated subunits (Christianson and Green 2004). As seen in Fig. 2, a ladder of high-molecular-weight forms of ubiquitinated subunits appeared after Western blots were probed with anti-ubiquitin. PS-341 increased the ubiquitinated levels of all three subunits examined, but its effects were more robust on α3 and β2 than β4. Similar data (not shown) were obtained in HEK cells stably expressing α3β2 or α3β4 nAChRs. In those experiments, the p62-derived UBA domain was used to selectively pull down ubiquitinated nAChR subunits.
Figure 2.
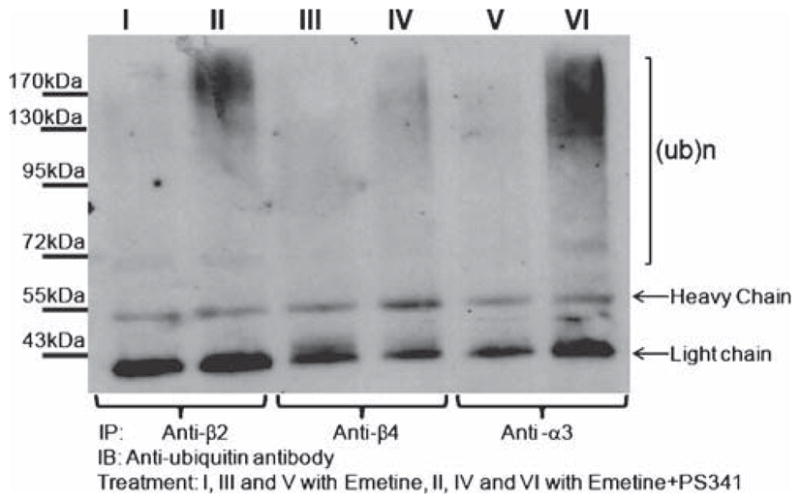
The α3, β2, and β4 nAChR subunits are ubiquitinated. Differentiated PC12 cells, which endogenously express α3β2 and α3β4 nAChRs, were treated with emetine for 24 h in the presence or absence of PS-341. Cells lysates were incubated with anti-α3, anti-β2, or anti-β4 nAChR antibodies immobilized on protein A/G agarose beads. After SDS-PAGE separation and blotting with anti-ubiquitin antibodies, we detected a ladder of ubiquitinated subunits. Ubiquitinated species appear as clusters of high molecular weight bands (marked with bracket)
As discussed previously, ERAD, located at the ER membrane and the cis-Golgi compartments (Haynes et al. 2002), is important for the degradation of muscle ubiquitinated nAChRs (Christianson and Green 2004). To verify the involvement of ERAD in the regulation of neuronal nAChR subunits, we examined whether proteasomal inhibition leads to accumulation of nAChR subunits within the ER/Golgi compartments. We also asked whether proteasomal inhibition affects nAChR levels at the plasma membrane. Lysates of dPC12 cells treated with either vehicle or PS-341 were subjected to a discontinuous iodixanol gradients fractionation (Koulen et al. 2002). The location of ER and Golgi was determined using antibodies against calreticulin (ER marker; data not shown, Khanna et al. 2004) and the cis-Golgi matrix protein, GM-130 (Nakamura et al. 1995). Alkaline phosphodiesterase activity was measured to identify plasma membrane enriched fractions (data not shown; Touster et al. 1970; Wibo et al. 1981). Figure 3a–c displays the results of separate fractionation experiments that were conducted to examine the effect of PS-341 on α3, β2, and β4, respectively. Fractions enriched for the ER/Golgi and the plasma membrane compartments were analyzed by Western blot analysis with antibodies against the α3, β2, and β4 nAChR subunits, respectively. Panels a–c in Fig. 3 show a significant upregulation of total subunit-protein levels in fractions enriched for the ER/Golgi compartment. In addition, PS-341 significantly upregulated the level of α3 and β2 nAChR subunits within relatively pure fraction for plasma membrane (lanes marked with asterisk in panels a and b). Exposure to PS-341 also caused an increase in GM-130 levels, which could be due to the ER stress response (Li et al. 2008). Finally, to determine whether ubiquitinated neuronal nAChR subunits sediment with fractions enriched for the ER/Golgi compartment, we combined the three fractions most enriched for the ER/Golgi and subjected them to p62 pull down followed by probing with anti-α3. As shown in Fig. 3d, we observed an accumulation of ubiquitinated α3 in the presence of PS-341. Thus, ubiquitination is the event that likely initiates the degradation of nAChR subunits by ERAD at the ER/Golgi level (Fig. 4).
Figure 3.

The proteasome inhibitor PS-341 increases the levels of the α3, β2, and β4 nAChR subunits in GM-130 positive cell fractions. Cell lysates were prepared from three batches of differentiated PC12 cells treated with vehicle or PS-341 for 24 h in the presence of emetine. Cell lysates were fractionated by centrifugation, using an iodixanol step gradient (8–34% linear Iodixanol gradient) to separate the ER/Golgi and plasma membrane compartments. Twenty fractions were collected from each cell lysate, and equal volumes of proteins from each fraction were analyzed by SDS-PAGE and immunoblotting. The blots were probed with antibodies against either α3 (a), β2 (b), or the β4 (c) nAChR subunit and the GM-130 Golgi marker (bottom blots in a–c). Cell treated with the proteasome inhibitor displayed nAChR upregulation in the fractions enriched with GM-130. The level of the α3 and β2, but not the β4, nAChR subunits was also increased in plasma membrane-enriched fractions (marked with asterisk). d Fractions enriched for the Golgi compartment (fractions 15–17) were subjected to p62-pull down followed by SDS-PAGE and probing with anti-α3 antibody. The data show an accumulation of ubiquitinated α3 in the presence of PS-341
Figure 4.
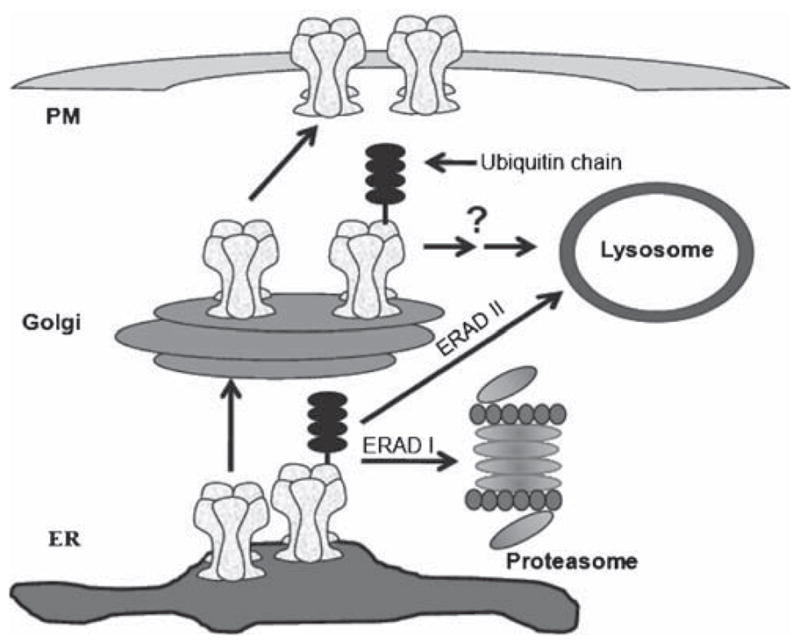
Protein degradation pathways and nAChR trafficking. Inhibition of the 26S proteasome can facilitate the trafficking of fully assembled nAChRs toward the plasma membrane. ERAD I is the main degradation pathway at the ER level while the lysosome might participate in nAChR regulation at the Golgi level and, to a lesser extent, at the ER level (ERAD II)
Discussion
Our results indicate that the UPS regulates the turnover of the α3, β2, and β4 nAChR subunits at the ERAD level. The selective proteasome inhibitor, PS-341 increased both total and ubiquitinated levels of nAChR subunits. Ubiquitinated nAChRs that cannot be degraded by the proteasome upon PS-341 exposure can be “recycled” via de-ubiquitination and spared from degradation (Rock et al. 1994; Mimnaugh et al. 1999), resulting in increased receptor levels. In addition, cell fractionation experiments showed that exposure to PS-341 leads to the accumulation of α3, β2, and β4 in fractions enriched for the cis-Golgi marker, GM-130. The increase in nAChR pool at the Golgi is likely due to a slower degradation rate of unassembled nAChR subunits at the ER level, which would promote receptor assembly and maturation (Claudio et al. 1989; Blount and Merlie 1990). The same mechanism has been previously suggested for muscle nicotinic receptors (Keller et al. 2001; Christianson and Green 2004).
Besides facilitating nAChR trafficking in the secretory pathway, proteasomal inhibition augmented nAChR levels in plasma membrane-enriched fractions. This effect was particularly evident for the α3 and β2 subunits. Interestingly, nicotine contained in tobacco, which we showed to act as a partial proteasome inhibitor (Rezvani et al. 2007), can also upregulate β2-containing nAChRs at the plasma membrane. Its effects are robust on β2-containing but not for β4-containing nAChRs (Wang et al. 1998; Rezvani et al. 2009). Understanding why assembly and trafficking of β2-containing nAChRs is more susceptible to ERAD function will require further study. Differences in conformational structure between the β2 and β4 subunit could perhaps explain the phenomenon, as they could both influence the rate of nAChR maturation and determine which protein partner is recruited during trafficking (Phillips et al. 1997; Ficklin et al. 2005; Castillo et al. 2006; Brockhausen et al. 2008; Rezvani et al. 2009)
The lysosome can target ubiquitinated proteins for degradation (Komatsu et al. 2006) both at the ERAD II level and in the post Golgi compartment (Beck et al. 1999; Blondel et al. 2004; Pizzirusso and Chang 2004; Eimer et al. 2007). The autophagy/lysosome pathway, ERAD II, might function as support/alternative mechanism for the degradation of ubiquitinated proteins at the ER level (Komatsu et al. 2006; Fujita et al. 2007). We detected an apparent increase in the levels of total protein ubiquitination upon E-64 exposure. Although the phenomenon could be interpreted to reflect the block of ERAD II, we showed that E-64 induced ~11% reduction in chymotrypsin-like activity, pointing to a possibly nonspecific effect of the drug. The lysosome could still regulate nAChR subunit levels as AChRs exit the Golgi compartment or after they have been endocytosed. In fact, it has been shown that the Caenorhabditis elegans orthologs of the muscle nAChR subunits, levamisole-sensitive AChRs, are targeted by lysosomal degradation in the post-Golgi compartment (Eimer et al. 2007). Endocytosed plasma membrane nAChRs also seem to directly go to late endosomes for lysosomal degradation (Clementi et al. 1983; Hyman and Froehner 1983; Darsow et al. 2005; Kumari et al. 2008). AMPA receptors provide another example of ligand-gated ion channels that are regulated by both the proteasome and lysosome (Ehlers 2000; Patrick et al. 2003; Lee et al. 2004).
In summary, although we cannot completely rule out the involvement of the lysosomal degradation machinery, our data clearly show that the α3, β2, and β4 nAChRs are regulated by the proteasome at the ERAD level. These data, in addition to those previously published in this lab (Rezvani et al. 2007), suggest a general role of the UPS in the trafficking of neuronal nAChRs.
Materials and Methods
Antibodies
Polyclonal antibodies against the α3 (rabbit clone #sc-5590 for Western Blot and 1:500 dilution and goat clone #sc-1771 for immunoprecipitation) and β2 nAChR subunits (clone H-92, 1:500) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit polyclonal antibodies against the β4 nAChR subunit were purchased from Abnova (Walnut, CA). Anti-GM130 (BD Biosciences, San Jose, CA, 1:250) was used to locate the Golgi compartments. Monoclonal anti-ubiquitin was purchased from Cell signaling (Danvers, MA). A goat anti-mouse IgG HRP (Santa Cruz, 1:2,500), a mouse anti-rabbit IgG HRP (Santa Cruz, 1:2,500), and a donkey anti-goat IgG HRP (Santa Cruz, 1:2,500) were used as secondary antisera.
Cell Culture
PC12, a rat pheochromocytoma cell line (American Tissue Culture Collection, ATCC, Manassas, VA), was cultured according to ATCC guidelines. Differentiated PC12 cells (dPC12 cells) were obtained upon treatment with 100 ng/ml nerve growth factor (NGF, Sigma) for 2–7 days.
To prepare cell lysates, cell pellets were rinsed three times with phosphate buffer serum (pH=7.4) before addition of lysis buffer (20 mM Tris-HCl, pH 7.2; 1 ml/100 mm plate) containing EDTA (1 mM), NaN3 (1 mM), β-mercaptoethanol (1 mM), NP40 (0.1% v/v), glycerol (10% v/v), and a tissue extract protease inhibitor cocktail (Sigma). Cell extracts were prepared by sonication on ice followed by centrifugation for 10 min at 13,000 rpm with a table-top centrifuge. All cell extracts were subjected to Western blot analysis or iodixanol gradient fractination after protein concentrations were determined with a BCA Protein Assay Kit (Pierce, Biotechnology, Rockford, IL).
Drug Treatments
For the degradation assays, the proteasomal inhibitor PS-341 (0.5 μM; a gift from Dr John F. de Groot, MD Anderson Cancer Center) or the lysosomal inhibitor E-64 (10 μM; Research products international, Mt. Prospect, IL) were used in the presence of the protein synthesis blocker emetine (150 μM, Calbiochem, San Diego, CA).
Immunoprecipitation and p62 Pull-Down Assays
Immuno-precipitations were conducted as described previously (Christianson and Green 2004; Karan et al. 2005). Pull-down assays were conducted with the agarose-immobilized p62-derived UBA domain (Biomol) as previously described (Rezvani et al. 2007).
Iodixanol Gradient Analysis
Iodixanol gradient fractionation was conducted according to the method described by Koulen et al. (Koulen et al. 2002). This method enabled us to have an optimized separation of ER, Golgi, and plasma membrane compartment.
Statistics
An automated digitizing system (UN-Scan-it gel, version 6.1, Orem, Utah) was used to quantify the intensity of Western blot bands. Statistical analysis was conducted using one-way analysis of variance and the multiple comparison Newman–Keuls test, when appropriate. A p value equal or less than 0.05 was considered statistically significant. All data are reported as mean ± SEM.
Acknowledgments
This work was supported by grants from the National Institute on Drug Abuse (DA017173 & DA024385) to MDB.
Footnotes
Proceedings of the XIII International Symposium on Cholinergic Mechanisms
Contributor Information
Khosrow Rezvani, Department of Neuroscience, Baylor College of Medicine, Houston, TX 77030, USA.
Yanfen Teng, Department of Neuroscience, Baylor College of Medicine, Houston, TX 77030, USA.
Mariella De Biasi, Email: debiasi@bcm.tmc.edu, Department of Neuroscience, Baylor College of Medicine, Houston, TX 77030, USA. Graduate Program in Translational Biology and Molecular Medicine, Baylor College of Medicine, Houston, TX 77030, USA.
References
- Ahlberg J, Berkenstam A, Henell F, Glaumann H. Degradation of short and long lived proteins in isolated rat liver lysosomes. Effects of pH, temperature, and proteolytic inhibitors. Journal of Biological Chemistry. 1985;260:5847–5854. [PubMed] [Google Scholar]
- Albuquerque EX, Pereira EF, Alkondon M, Rogers SW. Mammalian nicotinic acetylcholine receptors: From structure to function. Physiological Reviews. 2009;89:73–120. doi: 10.1152/physrev.00015.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck T, Schmidt A, Hall MN. Starvation induces vacuolar targeting and degradation of the tryptophan permease in yeast. Journal of Cell Biology. 1999;146:1227–1238. doi: 10.1083/jcb.146.6.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondel MO, Morvan J, Dupre S, Urban-Grimal D, Haguenauer-Tsapis R, Volland C. Direct sorting of the yeast uracil permease to the endosomal system is controlled by uracil binding and Rsp5p-dependent ubiquitylation. Molecular Biology of the Cell. 2004;15:883–895. doi: 10.1091/mbc.E03-04-0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blount P, Merlie JP. Mutational analysis of muscle nicotinic acetylcholine receptor subunit assembly. Journal of Cell Biology. 1990;111:2613–2622. doi: 10.1083/jcb.111.6.2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blount P, Merlie JP. BIP associates with newly synthesized subunits of the mouse muscle nicotinic receptor. Journal of Cell Biology. 1991;113:1125–1132. doi: 10.1083/jcb.113.5.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockhausen J, Cole RN, Gervasio OL, Ngo ST, Noakes PG, Phillips WD. Neural agrin increases postsynaptic ACh receptor packing by elevating rapsyn protein at the mouse neuromuscular synapse. Dev Neurobiol. 2008;68:1153–1169. doi: 10.1002/dneu.20654. [DOI] [PubMed] [Google Scholar]
- Castelan F, Castillo M, Mulet J, Sala S, Sala F, Dominguez Del Toro E, et al. Molecular characterization and localization of the RIC-3 protein, an effector of nicotinic acetylcholine receptor expression. Journal of Neurochemistry. 2008;105:617–627. doi: 10.1111/j.1471-4159.2007.05169.x. [DOI] [PubMed] [Google Scholar]
- Castillo M, Mulet J, Gutierrez LM, Ortiz JA, Castelan F, Gerber S, et al. Role of the RIC-3 protein in trafficking of serotonin and nicotinic acetylcholine receptors. Journal of Molecular Neuroscience. 2006;30:153–156. doi: 10.1385/JMN:30:1:153. [DOI] [PubMed] [Google Scholar]
- Christianson JC, Green WN. Regulation of nicotinic receptor expression by the ubiquitin-proteasome system. EMBO Journal. 2004;23:4156–4165. doi: 10.1038/sj.emboj.7600436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claudio T, Paulson HL, Green WN, Ross AF, Hartman DS, Hayden D. Fibroblasts transfected with Torpedo acetylcholine receptor beta-, gamma-, and delta-subunit cDNAs express functional receptors when infected with a retroviral alpha recombinant. Journal of Cell Biology. 1989;108:2277–2290. doi: 10.1083/jcb.108.6.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clementi F, Sher E, Erroi A. Acetylcholine receptor degradation: Study of mechanism of action of inhibitory drugs. European Journal of Cell Biology. 1983;29:274–280. [PubMed] [Google Scholar]
- Court J, Spurden D, Lloyd S, McKeith I, Ballard C, Cairns N, et al. Neuronal nicotinic receptors in dementia with Lewy bodies and schizophrenia: Alpha-bungarotoxin and nicotine binding in the thalamus. Journal of Neurochemistry. 1999;73:1590–1597. doi: 10.1046/j.1471-4159.1999.0731590.x. [DOI] [PubMed] [Google Scholar]
- Court J, Martin-Ruiz C, Piggott M, Spurden D, Griffiths M, Perry E. Nicotinic receptor abnormalities in Alzheimer’s disease. Biological Psychiatry. 2001;49:175–184. doi: 10.1016/s0006-3223(00)01116-1. [DOI] [PubMed] [Google Scholar]
- Dani JA, De Biasi M. Cellular mechanisms of nicotine addiction. Pharmacology, Biochemistry and Behavior. 2001;70:439–446. doi: 10.1016/s0091-3057(01)00652-9. [DOI] [PubMed] [Google Scholar]
- Darsow T, Booker TK, Pina-Crespo JC, Heinemann SF. Exocytic trafficking is required for nicotine-induced up-regulation of alpha 4 beta 2 nicotinic acetylcholine receptors. Journal of Biological Chemistry. 2005;280:18311–18320. doi: 10.1074/jbc.M501157200. [DOI] [PubMed] [Google Scholar]
- De Biasi M. Nicotinic mechanisms in the autonomic control of organ systems. Journal of Neurobiology. 2002;53:568–579. doi: 10.1002/neu.10145. [DOI] [PubMed] [Google Scholar]
- De Biasi M, Salas R. Influence of neuronal nicotinic receptors over nicotine addiction and withdrawal. Experimental Biology and Medicine (Maywood) 2008;233:917–929. doi: 10.3181/0712-MR-355. [DOI] [PubMed] [Google Scholar]
- Dwyer JB, McQuown SC, Leslie FM. The dynamic effects of nicotine on the developing brain. Pharmacology & Therapeutics. 2009;122(2):125–139. doi: 10.1016/j.pharmthera.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron. 2000;28:511–525. doi: 10.1016/s0896-6273(00)00129-x. [DOI] [PubMed] [Google Scholar]
- Eimer S, Gottschalk A, Hengartner M, Horvitz HR, Richmond J, Schafer WR, et al. Regulation of nicotinic receptor trafficking by the transmembrane Golgi protein UNC-50. EMBO Journal. 2007;26(20):4313–4323. doi: 10.1038/sj.emboj.7601858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficklin MB, Zhao S, Feng G. Ubiquilin-1 regulates nicotine-induced up-regulation of neuronal nicotinic acetylcholine receptors. Journal of Biological Chemistry. 2005;280:34088–34095. doi: 10.1074/jbc.M506781200. [DOI] [PubMed] [Google Scholar]
- Forsayeth JR, Gu Y, Hall ZW. BiP forms stable complexes with unassembled subunits of the acetylcholine receptor in transfected COS cells and in C2 muscle cells. Journal of Cell Biology. 1992;117:841–847. doi: 10.1083/jcb.117.4.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita E, Kouroku Y, Isoai A, Kumagai H, Misutani A, Matsuda C, et al. Two endoplasmic reticulum-associated degradation (ERAD) systems for the novel variant of the mutant dysferlin: Ubiquitin/proteasome ERAD(I) and autophagy/lysosome ERAD(II) Human Molecular Genetics. 2007;16:618–629. doi: 10.1093/hmg/ddm002. [DOI] [PubMed] [Google Scholar]
- Gelman MS, Chang W, Thomas DY, Bergeron JJ, Prives JM. Role of the endoplasmic reticulum chaperone calnexin in subunit folding and assembly of nicotinic acetylcholine receptors. Journal of Biological Chemistry. 1995;270:15085–15092. doi: 10.1074/jbc.270.25.15085. [DOI] [PubMed] [Google Scholar]
- Grinde B, Seglen PO. Differential effects of proteinase inhibitors and amines on the lysosomal and non-lysosomal pathways of protein degradation in isolated rat hepatocytes. Biochimica et Biophysica Acta. 1980;632:73–86. doi: 10.1016/0304-4165(80)90250-0. [DOI] [PubMed] [Google Scholar]
- Gu Y, Forsayeth JR, Verrall S, Yu XM, Hall ZW. Assembly of the mammalian muscle acetylcholine receptor in transfected COS cells. Journal of Cell Biology. 1991;114:799–807. doi: 10.1083/jcb.114.4.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas KF, Broadie K. Roles of ubiquitination at the synapse. Biochimica et Biophysica Acta. 2008;1779:495–506. doi: 10.1016/j.bbagrm.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes CM, Caldwell S, Cooper AA. An HRD/DER-independent ER quality control mechanism involves Rsp5p-dependent ubiquitination and ER-Golgi transport. Journal of Cell Biology. 2002;158:91–101. doi: 10.1083/jcb.200201053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman C, Froehner SC. Degradation of acetylcholine receptors in muscle cells: Effect of leupeptin on turnover rate, intracellular pool sizes, and receptor properties. Journal of Cell Biology. 1983;96:1316–1324. doi: 10.1083/jcb.96.5.1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karan G, Yang Z, Howes K, Zhao Y, Chen Y, Cameron DJ, et al. Loss of ER retention and sequestration of the wild-type ELOVL4 by Stargardt disease dominant negative mutants. Molecular Vision. 2005;11:657–664. [PubMed] [Google Scholar]
- Keller SH, Lindstrom J, Taylor P. Involvement of the chaperone protein calnexin and the acetylcholine receptor beta-subunit in the assembly and cell surface expression of the receptor. Journal of Biological Chemistry. 1996;271:22871–22877. doi: 10.1074/jbc.271.37.22871. [DOI] [PubMed] [Google Scholar]
- Keller SH, Lindstrom J, Ellisman M, Taylor P. Adjacent basic amino acid residues recognized by the COP I complex and ubiquitination govern endoplasmic reticulum to cell surface trafficking of the nicotinic acetylcholine receptor alpha-Subunit. Journal of Biological Chemistry. 2001;276:18384–18391. doi: 10.1074/jbc.M100691200. [DOI] [PubMed] [Google Scholar]
- Khanna R, Lee EJ, Papazian DM. Transient calnexin interaction confers long-term stability on folded K+ channel protein in the ER. Journal of Cell Science. 2004;117:2897–2908. doi: 10.1242/jcs.01141. [DOI] [PubMed] [Google Scholar]
- Kisselev AF, Goldberg AL. Monitoring activity and inhibition of 26S proteasomes with fluorogenic peptide substrates. Methods in Enzymology. 2005;398:364–378. doi: 10.1016/S0076-6879(05)98030-0. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- Koulen P, Cai Y, Geng L, Maeda Y, Nishimura S, Witzgall R, et al. Polycystin-2 is an intracellular calcium release channel. Nature Cell Biology. 2002;4:191–197. doi: 10.1038/ncb754. [DOI] [PubMed] [Google Scholar]
- Kumari S, Borroni V, Chaudhry A, Chanda B, Massol R, Mayor S, et al. Nicotinic acetylcholine receptor is internalized via a Rac-dependent, dynamin-independent endocytic pathway. Journal of Cell Biology. 2008;181:1179–1193. doi: 10.1083/jcb.200709086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Novere N, Corringer PJ, Changeux JP. The diversity of subunit composition in nAChRs: Evolutionary origins, physiologic and pharmacologic consequences. Journal of Neurobiology. 2002;53:447–456. doi: 10.1002/neu.10153. [DOI] [PubMed] [Google Scholar]
- Lee SH, Simonetta A, Sheng M. Subunit rules governing the sorting of internalized AMPA receptors in hippocampal neurons. Neuron. 2004;43:221–236. doi: 10.1016/j.neuron.2004.06.015. [DOI] [PubMed] [Google Scholar]
- Li X, Zhang Y, Hu Y, Chang M, Liu T, Wang D, et al. Chaperone proteins identified from synthetic proteasome inhibitor-induced inclusions in PC12 cells by proteomic analysis. Acta Biochim Biophys Sin (Shanghai) 2008;40:406–418. doi: 10.1111/j.1745-7270.2008.00416.x. [DOI] [PubMed] [Google Scholar]
- Lu Z, Je HS, Young P, Gross J, Lu B, Feng G. Regulation of synaptic growth and maturation by a synapse-associated E3 ubiquitin ligase at the neuromuscular junction. Journal of Cell Biology. 2007;177:1077–1089. doi: 10.1083/jcb.200610060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlie JP, Lindstrom J. Assembly in vivo of mouse muscle acetylcholine receptor: Identification of an alpha subunit species that may be an assembly intermediate. Cell. 1983;34:747–757. doi: 10.1016/0092-8674(83)90531-7. [DOI] [PubMed] [Google Scholar]
- Meusser B, Hirsch C, Jarosch E, Sommer T. ERAD: The long road to destruction. Nature Cell Biology. 2005;7:766–772. doi: 10.1038/ncb0805-766. [DOI] [PubMed] [Google Scholar]
- Mimnaugh EG, Bonvini P, Neckers L. The measurement of ubiquitin and ubiquitinated proteins. Electrophoresis. 1999;20:418–428. doi: 10.1002/(SICI)1522-2683(19990201)20:2<418::AID-ELPS418>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Nakamura N, Rabouille C, Watson R, Nilsson T, Hui N, Slusarewicz P, et al. Characterization of a cis-Golgi matrix protein, GM130. Journal of Cell Biology. 1995;131:1715–1726. doi: 10.1083/jcb.131.6.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick GN, Bingol B, Weld HA, Schuman EM. Ubiquitin-mediated proteasome activity is required for agonist-induced endocytosis of GluRs. Current Biology. 2003;13:2073–2081. doi: 10.1016/j.cub.2003.10.028. [DOI] [PubMed] [Google Scholar]
- Paulson HL, Ross AF, Green WN, Claudio T. Analysis of early events in acetylcholine receptor assembly. Journal of Cell Biology. 1991;113:1371–1384. doi: 10.1083/jcb.113.6.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips WD, Vladeta D, Han H, Noakes PG. Rapsyn and agrin slow the metabolic degradation of the acetylcholine receptor. Molecular and Cellular Neurosciences. 1997;10:16–26. doi: 10.1006/mcne.1997.0634. [DOI] [PubMed] [Google Scholar]
- Pizzirusso M, Chang A. Ubiquitin-mediated targeting of a mutant plasma membrane ATPase, Pma1–7, to the endosomal/vacuolar system in yeast. Molecular Biology of the Cell. 2004;15:2401–2409. doi: 10.1091/mbc.E03-10-0727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezvani K, Teng Y, Shim D, De Biasi M. Nicotine regulates multiple synaptic proteins by inhibiting proteasomal activity. Journal of Neuroscience. 2007;27:10508–10519. doi: 10.1523/JNEUROSCI.3353-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezvani K, Teng Y, Pan Y, Dani J, Lindstrom J, García Gras EA, et al. UBXD4, a UBX containing protein, regulates the cell surface number and the subunit stability of alpha 3-containing nicotinic acetylcholine receptors. Journal of Neuroscience. 2009;29(21):6883–6896. doi: 10.1523/JNEUROSCI.4723-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock KL, Gramm C, Rothstein L, Clark K, Stein R, Dick L, et al. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell. 1994;78:761–771. doi: 10.1016/s0092-8674(94)90462-6. [DOI] [PubMed] [Google Scholar]
- Smith MM, Lindstrom J, Merlie JP. Formation of the alpha-bungarotoxin binding site and assembly of the nicotinic acetylcholine receptor subunits occur in the endoplasmic reticulum. Journal of Biological Chemistry. 1987;262:4367–4376. [PubMed] [Google Scholar]
- Touster O, Aronson NN, Jr, Dulaney JT, Hendrickson H. Isolation of rat liver plasma membranes. Use of nucleotide pyrophosphatase and phosphodiesterase I as marker enzymes. Journal of Cell Biology. 1970;47:604–618. doi: 10.1083/jcb.47.3.604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vembar SS, Brodsky JL. One step at a time: Endoplasmic reticulum-associated degradation. Nature Reviews. Molecular Cell Biology. 2008;9:944–957. doi: 10.1038/nrm2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanamaker CP, Green WN. ER chaperones stabilize nicotinic receptor subunits and regulate receptor assembly. Journal of Biological Chemistry. 2007;282(43):31113–31123. doi: 10.1074/jbc.M705369200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanamaker CP, Christianson JC, Green WN. Regulation of nicotinic acetylcholine receptor assembly. Annals of the New York Academy of Sciences. 2003;998:66–80. doi: 10.1196/annals.1254.009. [DOI] [PubMed] [Google Scholar]
- Wang F, Nelson ME, Kuryatov A, Olale F, Cooper J, Keyser K, et al. Chronic nicotine treatment up-regulates human alpha3 beta2 but not alpha3 beta4 acetylcholine receptors stably transfected in human embryonic kidney cells. Journal of Biological Chemistry. 1998;273:28721–28732. doi: 10.1074/jbc.273.44.28721. [DOI] [PubMed] [Google Scholar]
- Wang JM, Zhang L, Yao Y, Viroonchatapan N, Rothe E, Wang ZZ. A transmembrane motif governs the surface trafficking of nicotinic acetylcholine receptors. Nature Neuroscience. 2002;5:963–970. doi: 10.1038/nn918. [DOI] [PubMed] [Google Scholar]
- Wibo M, Thines-Sempoux D, Amar-Costesec A, Beaufay H, Godelaine D. Analytical study of microsomes and isolated subcellular membranes from rat liver VIII. Subfractionation of preparations enriched with plasma membranes, outer mitochondrial membranes, or Golgi complex membranes. Journal of Cell Biology. 1981;89:456–474. doi: 10.1083/jcb.89.3.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan FF, Lin CW, Cartier EA, Shyng SL. Role of ubiquitin-proteasome degradation pathway in biogenesis efficiency of {beta}-cell ATP-sensitive potassium channels. American Journal of Physiology Cell Physiology. 2005;289:C1351–C1359. doi: 10.1152/ajpcell.00240.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi JJ, Ehlers MD. Ubiquitin and protein turnover in synapse function. Neuron. 2005;47:629–632. doi: 10.1016/j.neuron.2005.07.008. [DOI] [PubMed] [Google Scholar]