

Abstract
Sphingosine 1-phosphate (S1P) is a bioactive lipid that has been identified as an accelerant of cancer progression. The sphingosine kinases (SphKs) are the sole producers of S1P and thus SphK inhibitors may prove effective in cancer mitigation and chemosensitization. Of the two SphKs, SphK1 overexpression has been observed in a myriad of cancer cell lines and tissues, and has been recognized as the presumptive target over that of the poorly characterized SphK2. Herein, we present the design and synthesis of amidine-based nanomolar SphK1 subtype-selective inhibitors. A homology model of SphK1, trained with this library of amidine inhibitors, was then used to predict the activity of additional, more potent, inhibitors. Lastly, select amidine inhibitors were validated in human leukemia U937 cells, where they significantly reduced endogenous S1P levels at nanomolar concentrations.
Introduction
The scientific community has identified the sphingosine kinases (SphKs) as potential therapeutic targets for broad cancer mitigation and chemotherapeutic sensitization.1, 2 The SphKs are the sole producers of sphingosine 1-phosphate (S1P), which regulates cell survival, proliferation, neovascularization, and migration through five G protein coupled receptors (S1PR1–5) as well as through other intracellular mechanisms.3–7 Upregulation of the SphK1, the first of two SphK isoforms, is found in many cancers (brain,8, 9 bladder,10 breast,11, 12 colon,13, 14 gastric,15 head and neck,16, 17 leukemia,18 non-Hodgkin lymphoma,19 prostate,20, 21 skin,22 and squamous cell carcinoma;23 among others) and the overproduction of S1P has been shown to aid angiogenesis, tumorigenesis, and metastasis.
Because of its deregulation in cancer, SphK1 has been implicated as a potential oncogene;2, 24 however, no genetic mutations have yet been identified, indicating that malignancies may become dependant on SphK1 through a non-oncogene addiction.25 This theory is appealing due to the central role that S1P plays in the signal amplification of other known oncogenes. SphK1 expression and activation increases with mitogenic signaling from growth factors for a range of receptor tyrosine kinases26 (epidermal (EGF), vascular endothelial (VEGF), platelet derived (PDGF); among others), estrogen signaling,27 prolactin expression,28 and lysophosphatidic acid (LPA) signaling,29 which indicates SphK1 inhibitors may be capable of counteracting a range of oncogene-accelerated cancers. SphK1 expression has also been shown to protect rapidly dividing cells from hypoxia,30 autophagy,31 and chemotherapy.32 SphK1 siRNA has been shown to slow the rate of growth of cancer cells that have SphK1 overexpression.20, 21, 32, 33 Breast cancer,12 gastric cancer,15 and glioblastoma8, 9 patients with high levels of SphK1 have shorter life expectancies. The relationship between SphK1 and cell survival can be described as linear; with increased S1P facilitating more aggressive and chemotherapeutic resistant cells, and decreased S1P leading to a build up of ceramide, its biosynthetic precursor, and ceramide dependant apoptosis.34 Indeed, the sphingosine rheostat (Scheme 1) that governs cell fate by controlling the ratio of S1P to ceramide could be manipulated by applying the correct resistance at SphK1 with small molecule inhibitors that “dial-down” S1P concentrations.
Scheme 1.

The Sphingosine Rheostat.
To state that the less-inducible SphK2 is simply the housekeeping isoenzyme of SphK1 would be misleading. Unlike SphK1, which is cytosolic and when phosphorylated translocates to the inner leaflet of the cell membrane,35 SphK2 is predominately located on or in the organelles, such as the ER or the nucleus.36 Due to this location, S1P produced by SphK2 in the interior of the cell is not effectively positioned to enter into the inside-out S1P receptor signaling pathway occurring at the cell membrane, and therefore does not have the same proliferative effects.37 Instead, S1P synthesized in the nucleus by SphK2 causes histone deacetylase 1 and 2 (HDAC 1/2) inhibition, p21 gene expression, and cytostasis.7 SphK2 overexpression causes apoptosis, which is most likely due to its degradation by the proteasome and release of a short pro-apoptotic BH3-domain present in SphK2 that is absent in SphK1.38 The relationship between SphK2 and cell survival appears to be parabolic; where upregulation leads to its degradation and caspase-mediated apoptosis, moderate activity leads to p21 expression and cell cycle arrest, and downregulation leads to reduced p21 expression and apoptosis or proliferation depending on cell environment.1
If SphK inhibitors are to be used to mitigate the presentation of cancer or, to retard chemotherapeutic resistance, the question must be raised: Is it necessary to selectively inhibit one of the SphKs or inhibit both enzymes together? The inducibility of SphK1 by mitogenic factors is an indication of disease causing deregulation, however, siRNA experiments demonstrate that “knocking-down” SphK2 is more efficacious at retarding cell growth in two glioblastoma cell lines.9 It is possible that the inhibitor subtype selectivity necessary for effective treatment may be cancer dependent, and our research aim is to synthesize a spectrum of dual and selective SphK inhibitors.
Over the last few years several SphK inhibitors have appeared in the literature.1 A large portion of these are amino alcohol sphingosine analogs that compete for the substrate binding pocket,39–44 however, the ATP competitive SKI-II is one notable exception.45 Indeed, sphingosine kinase inhibitors with μM KI values have been effective in vivo in suppressing tumor growth in xenograft models39, 41, 46 and inhibited inflammation response in Crohn’s,47 inflammatory bowl,48 and sepsis49 disease models. However, there is still a need for a library of potent SphK inhibitors with a range of subtype selectivities that could elucidate the currently enigmatic differences between the SphKs in cancer disease states.
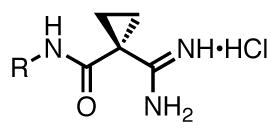
Previous work has led to the generation of sub-μM dual and selective SphK inhibitors 1 and 2, which were derivatives of the initial hit compound (S)-N-(1-amino-1-iminopropan-2-yl)-4-octylbenzamide hydrochloride (VPC94075) (Table 1).50 These amidine-based lipids were selective for the SphKs; they did not inhibit other lipid kinases, such as the diacylglycerol kinases (DGKs), or protein kinases, such as protein kinase C (PKC). They were, in our opinion, excellent starting points for drug optimization. The most interesting feature of the preliminary SAR was the selectivity for SphK1 induced simply by the direction of the amide functional group present in compounds 1 and 2. The amide-controlled selectivity was dependent on tail length, with a maximum effect only observed in the longer tailed derivatives. Potency and selectivity are affected by tail length and amide configuration as described in Figure 1. Shorter tails (C8 and C10) inhibit both SphK1 and SphK2 equally, but the maximum potency tail length of C12 differentiates dual inhibition and SphK1 selectivity based on amide direction before potencies drop off at longer tail lengths.
Table 1.
Previously described amidine-based sphingosine kinase inhibitors.
KI = [I]/(K’M/KM - 1); KM of Sphingosine at SphK1 = 10 μM; KM of Sphingosine at SphK2 = 5 μM
Selectivity = (KI/KM)SphK2/(KI/KM)SphK1
Figure 1.

Comparison between tail length and potency for the amide orientations in compounds 1 and 2. Compound 1 derivatives shown in red. Compound 2 derivatives shown in blue. SphK1 inhibition shown as solid lines. SphK2 inhibition shown as dotted lines.
These differences can be explained by the tail-binding region of the substrate pocket of SphK1 being larger than that of SphK2, which forces an altered binding position for the inhibitors and causes a repulsive electrostatic interaction for the amide configuration in compound 2. Seeking to exploit this tail length and amide derived selectivity; inhibitors with increased terminal steric bulk and amide rigid analogs derived from proline were synthesized and tested. Scheme 2 shows the individual head and tail optimizations and subsequent partnership to generate compound 38, which has a KI = 75 nM at SphK1 and is 80-fold selective over SphK2. The library of inhibitors synthesized was then used as a test set in the generation of a SphK1 homology model derived from the solved structure of diacylglycerol kinase β (DGKB).51 Lastly, a virtual library of possible linkers was docked into the SphK1 model and a class of heteroaromatic compounds with six fewer rotatable bonds was generated and synthesized. Biochemical evaluation led to the identification of the most potent inhibitors of SphK1 reported in the literature to date.52 Oxazole 56, which has a KI = 47 nM at SphK1 and 180-fold selectivity, and other amidine-based inhibitors described are shown to significantly reduce S1P concentrations in human leukemia U937 cells at nanomolar concentrations.
Scheme 2.

Outline of inhibitor optimization.
Results and Discussion
Tail Modifications
The tail region was defined to be everything distal to the amidine beyond the amide bond (Scheme 2). Three major modifications were made to the scaffold of compound 2: aryl deletion, the substitution of terminal ethers, and the substitution of terminal aromatics. The aryl deletion series was synthesized in two steps from the commercially available starting aliphatic amines and 1-cyano-1-cyclopropane. In the example shown in Scheme 3, tetradecylamine was coupled using PyBOP to form the nitrile 3a, and then transformed under base catalyzed Pinner conditions53 to yield the corresponding amidine 4a.
Scheme 3.

Example synthesis of an aryl deleted tail modification.
The ether tail derivatives were then examined and terminal steric bulk was built into the ether from the corresponding alcohol. In the example synthesis shown in Scheme 4, benzyl alcohol was coupled to 7-bromo-1-heptene using sodium hydride in DMF to form ether 5a. The terminal olefin was reduced to an alkylborane in situ using 9-BBN and then introduced to Suzuki conditions to be coupled with 1-bromo-4-nitrobenzene to form the aryl nitro 6a. On reduction to the aniline 7a with zinc dust and amide coupling facilitated by PyBOP to form nitrile 8a, our standard amidine formation lead to the final product 9a.
Scheme 4.

Example synthesis of an ether tail modification.
The non-ether aromatic tails were synthesized to compare the solubility effects of introducing an ether linkage in the middle of the tail region. In the example synthesis shown in Scheme 5, benzylmagnesium bromide was catalytically converted to its organocuprate with cuprous chloride, and coupled to 8-bromo-1-octene to form alkene 8a. This olefin was identical to that of compound 5a, with the exception of the ether linkage being substituted with a methylene, and was converted to its corresponding final product under similar chemical transformations.
Scheme 5.

Example synthesis of a non-ether tail modification.
The KI values of these tail derivatives were determined by a [γ-32P] ATP in vitro assay52 of SphK enzymatic activity and are shown in Table 2. The most striking observation about the aryl deletion series 4a-c was the lack of a potency response to changes in tail length. Unlike the aryl-containing analogs described in Figure 1, these saturated tails had a flat SAR in the low μM range, but did maintain SphK1 selectivity in the longer tailed 4b and 4c. It was hypothesized that these more hydrophobic compounds had strong affinities for the active site, but were so water insoluble that their active concentrations were small due to aggregation. The more soluble ether tails performed with a more consistent SAR, with the smaller terminal phenyl-containing 9a being less active than the cyclohexyl 9c by more than a log order (Table 2). The terminal cyclohexyl derivative 9c was synthesized to evaluate saturation as compared to the aromaticity of 9a, and the positive performance of 9c suggests a preference for the larger and more hydrophobic terminal cyclohexane. Adding further steric bulk in the adamantyl derivative 9e caused a loss of activity and selectivity, suggesting an alternative binding conformation for such a large substituent. Short and longer cyclohexyl-containing tails, 9b and 9d respectively, both performed more poorly than 9c indicating that is was the optimum length.
Table 2.
KI values for the tail modified compounds.
 | ||||
|---|---|---|---|---|
| Compound | Tail Group (R) | KI (μM)a |
SphK1 Selectivityb | |
| SphK1 | SphK2 | |||
| 2 |
|
0.3 | 6 | 40 |
| 4a |
|
6 | 7.5 | 2.5 |
| 4b |
|
5.4 | >100 | >37 |
| 4c |
|
9 | >100 | >22 |
| 9a |
|
5 | 38 | 15 |
| 14a |
|
0.50 | >10 | >40 |
| 14b |
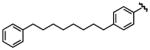
|
1.3 | >10 | 15 |
| 9b |
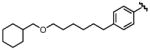
|
0.39 | 12 | 61 |
| 9c |
|
0.24 | 7 | 58 |
| 9d |
|
0.80 | >20 | >50 |
| 9e |
|
1.5 | 4.6 | 6.1 |
| 19a |
|
0.11 | 26 | 470 |
| 19b |
|
0.45 | 25 | 110 |
KI = [I]/(K’M/KM - 1); KM of Sphingosine at SphK1 = 10 μM; KM of Sphingosine at SphK2 = 5 μM
Selectivity = (KI/KM)SphK2/(KI/KM)SphK1
Unfortunately, compound 9c did not yield the substantial gains in potency or selectivity that were expected, but did increase water solubility to a CLogP = 3.61 versus a CLogP = 4.00 for compound 2.54 This added polar character allowed us to reconsider the aryl deletion series, and compounds 19a and 19b were then synthesized. Shown in Scheme 6 is the example synthesis of 19a; cyclohexylmethanol was coupled to 10-bromo-1-decene using sodium hydride in DMF to form ether 15a. The terminal olefin was converted to the primary alcohol 16a under hydroboration/oxidation conditions, and then displaced to the primary azide 17a through its mesylate. The azide 17a was reduced and ligated using Staudinger conditions55 to form nitrile 18a, before being converted to amidine 19a. Compound 19a proved to be both more potent, with a KI = 110 nM, and 470-fold selective for SphK1 over SphK2. The reduction in terminal ring size to the cyclopentyl 19b demonstrated that the steric bulk of the 6 membered saturated ring of 19a was optimal for both potency and selectivity (Table 2).
Scheme 6.

Example synthesis of ether containing/aryl deletion tail modified compounds.
Having achieved the design of a compound two and one half log orders selective for SphK1, our attention shifted to whether the bulkier tail design had aided selectivity in an amide-dependant manner. To test this relationship, the inverted amide derivatives of compounds 9c and 19a were synthesized. The synthesis of the aryl containing inverted amide is shown in Scheme 7; starting from the same terminal alkene used in the synthesis of 9c, the reduction of 5c to its alkylborane and coupling under Suzuki conditions to 4-bromobenzaldehyde gave the aryl aldehyde 20a. The aldehyde was then oxidized to benzoic acid 21a using Pinnick oxidation conditions.56 The carboxylic acid was coupled to 1-amino-1-cyclopropanecarbonitrile through its acid chloride. Nitrile 22a was then converted to its amidine to form the desired 23a. The synthesis of the non-aryl inverted amide analog 26 was relatively simple, starting with the Williamson ether coupling of cyclohexylmethanol and 11-bromoundecenoic acid (Scheme 8). The acid 24 was then coupled to 1-amino-1-cyclopropanecarbonitrile with PyBOP to form nitrile 25, and converted to the corresponding amidine 26.
Scheme 7.

Synthesis of 23a.
Scheme 8.

Synthesis of 26.
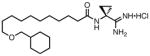
The results from the amide inversion experiments demonstrated that a cyclohexane at the tail terminus does itself increase selectivity for SphK1, as shown in the differences in activity between compounds 1 and 23a (Table 3). Again, substitution to the smaller cyclopentane reduced activity and selectivity. It was expected that a direct ether substitution in the tail of compound 1 would lead to reduced activity against both kinases equally due to its increased solubility in water; however, compound 23c lost potency disproportionately leading to a slight degree of SphK1 selectivity. The selectivity was due to the position of the ether linkage along the tail, and compound 30 was synthesized (Scheme SI-1) and evaluated to show no such change in selectivity compared to the saturated parent compound 1.
Table 3.
KI values for alternate amide configurations.
| Compound | Structure | KI (μM)a | SphK1 Selectiveb | |
|---|---|---|---|---|
| SphK1 | SphK2 | |||
| 1 |
|
0.2 | 0.5 | 5 |
| 23a |
|
0.13 | 15 | 240 |
| 23b |
|
0.17 | 18 | 210 |
| 23c |
|
1.3 | 40 | 62 |
| 30 |
|
4.0 | 10 | 5 |
| 26 |
|
0.095 | 12 | 250 |
KI = [I]/(K’M/KM - 1); KM of Sphingosine at SphK1 = 10 μM; KM of Sphingosine at SphK2 = 5 μM
Selectivity = (KI/KM)SphK2/(KI/KM)SphK1
An important subtlety of the tail modification data is that the deletion of the aromatic ring present in 9c, and replacement with a three carbon saturated spacer as in 19a increased both potency and selectivity (Table 2). However, the same conversion from 23a to 26, increased potency without such an obvious effect on selectivity. One explanation is that a saturated amide increases potency and accentuates the effect that amide already has on selectivity. On the other hand, a bulky substitution at the tail terminus, such as a cyclohexane, increases potency and selectivity regardless of amide orientation.
Head Group Modifications
An early examination of substitution alpha to the amidine showed that small substituents, such as methyl and cyclopropyl, were tolerated well by the enzyme.50 It was therefore desirable to test a bulkier cyclobutyl derivative, however, a ring expansion to the cyclobutyl would affect the angle of presentation of the amidine possibly hindering its function. More promising was a rigid analog design that restricted the dihedral angle between the position of the amide and that of the amidine. Restricting a bond between such functionally important groups should have an effect on selectivity and potency. Derivatives of both enantiomers of proline provided a synthetically useful avenue to rigidity, and would allow freedom of rotation about the amidine while restricting rotation of the amide.
The synthesis of the alpha, alpha-cyclobutyl analog 33 began with the conversion of cyclobutanone under Strecker conditions to 1-amino-1-cyclobutanecarbonitrile 31 (Scheme 9). Immediate acylation with 4-dodecylbenzoyl chloride to form nitrile 32, and conversion to its amidine gave compound 33. Next, the proline-based rigid analog syntheses began from the corresponding asymmetric amino acid (Scheme 10). L-proline was first N-Boc protected, before converting its carboxylic acid to the primary amide, and lastly dehydration of that amide to the nitrile in compound 34a. The Boc group was then deprotected and the free amine coupled using PyBOP to 4-dodecylbenzoic acid to form compound 35a. The nitrile was then converted to its amidine, and the synthesis was repeated for D-proline to produce both enantiomers.
Scheme 9.

Synthesis of the alpha, alpha-cyclobutyl head group analog 33.
Scheme 10.

Synthesis of the proline-based rigid analog 36a.
Table 4 shows the biological evaluation of the head group analogs. As suspected, the ring expansion from cyclopropane to the cyclobutane present in 33 worsened activity equally against both SphKs. The proline analogs 36a,b yielded selectivity as expected, with the (S) configuration derived from L-proline being 24-fold more selective for SphK1 while the (R) enantiomer was slightly SphK2 selective with less potency.
Table 4.
KI values for head group modifications.
| Compound | Structure | KI (μM)a | SphK1 Selectivityb | |
|---|---|---|---|---|
| SphK1 | SphK2 | |||
| 1 |
|
0.2 | 0.5 | 5 |
| 33 |
|
1.6 | 5 | 6.3 |
| 36a |
|
0.13 | 1.5 | 24 |
| 36b |
|
16 | 5 | 0.6 |
| 38 |
|
0.075 | 3 | 80 |
| 47 |
|
0.099 | 5.3 | 110 |
| 40 |
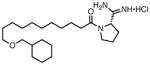
|
0.13 | 8 | 130 |
KI = [I]/(K’M/KM - 1); KM of Sphingosine at SphK1 = 10 μM; KM of Sphingosine at SphK2 = 5 μM
Selectivity = (KI/KM)SphK2/(KI/KM)SphK1
Compound 36a being more potent and selective for SphK1 than compound 1, a synthesis combining our best tail derivatives with a (S) proline head group was undertaken (Scheme 11). The aryl 38 and non-aryl 40 were synthesized and evaluated to have KI values of 75 nM and 130 nM respectively (Table 4). In previous series it was noted an increase in activity for the non-aryl over the aryl amide substitution (Tables 2 and 3). However, that relationship was for mono-nitrogen substitution on the amide bonds, while the proline derivatives are di-nitrogen substituted. For the proline aryl amides, A1,3 strain prohibits bond rotation about the carbonyl carbon aryl bond, effectively rigidifying two bonds as compared with compound 23a. The saturated 40, which is mono-substituted alpha to the carbonyl, has the ability to freely rotate, and has only one rigidified bond as compared with compound 26. The potency of the proline analogs is therefore dependent on a substitution alpha to the amide carbonyl that inhibits bond rotation, which prepays the cost of freezing that bond prior to reaching the enzyme active site.
Scheme 11.

Combination of tail and head group modifications.
The ether present in the tail increases its calculated water solubility, and in the case of 23c reduces activity versus its non-ether counterpart 1. A synthesis was then undertaken to eliminate the ether from compound 38 to investigate the limit of such solubility dependence. The synthesis of the non-ether 47 was completed (Scheme SI-2), and it was determined that its lower water solubility caused a decrease in activity (Table 4). The loss of activity for 47 and other compounds with high Clog P values suggests an ideal Clog P around 4.2.
In Silico Linker Screening
Crystal structures of kinases that bear close sequence homology to the ATP binding domain of the SphKs have been solved for YegS,57, 58 a bacterial lipid kinase, phosphofructokinase (PFK),59, 60 and DGKB.51 Of these structures, DGKB has the greatest overall sequence identity of 20% to SphK1. Cases of such low sequence identity are often referred to as “twilight zone” cases,61 and a 28 amino acid sequence that defines the substrate binding pocket of SphK1 has no meaningful sequence homology. It should be stated that modelers tread lightly in such situations, and any conclusions drawn should be supported by experimental data. However, the sequence homology between the two kinases suggests that SphK1 shares the basic quaternary structure of a beta-sandwich in DGKB, connected to the ATP binding domain through a hinge.
A homology model of SphK1 was generated (see supporting information for details pertaining to model generation and inhibitor docking) from the solved crystal structure of DGKB51 (Figure 2a). The current library of amidine inhibitors was docked into the SphK1 model (Figure 2b), and illuminated an interesting hypothesis of how the amidine may interact with the enzyme. The model suggests that the amidine interacts directly with ATP through a bidentate chelation of its gamma phosphate (Figure 2c and 2d). This supports a mechanism of inhibition where SphK first binds ATP and the inhibitor, and the amidine acts to stabilize the [SphK • ATP • I] complex. Using the test set of known amidine-based inhibitors enabled the virtual screening of theoretical amidine inhibitors and a prediction of their enzymatic activity.
Figure 2.

SphK homology model. (A) Crystal structure DGKB. (δ-helices = teal, β-sheets = lavender, ATP = yellow stick model, and Mg2+ = white sphere) (B) SphK1 homolog. (C) Theoretical binding orientation of compound 38 with ATP and protein backbone. (D) 2D representation of the amidine in compound 38 chelating ATP.
Long unrestricted alkyl chains have a large number of rotatable bonds, which add a large entropic cost when forced to lock into a single binding conformation. Our most potent compounds have between 11 and 15 rotatable bonds, thus it was desirable to reduce these large degrees a freedom by incorporating linker regions that are comprised of as many ring structures as possible. The SphK1 model suggests a tail binding region that is mostly comprised of hydrophobic surface area, indicating that this region of the pocket simply acts as a hydrocarbon ruler designed for sphingosine recognition. Therefore, without much possibility of polar interaction the ideal tail would be one that maximizes the energy associated with ligand and pocket desolvation. Assuming the binding positions of the amidine head group and the cyclohexyl tail fragments were accurate, several hundred possible linkers were created in silico, docked into the SphK1 homology model, and scored (see supporting information). These potential linker regions consisted of substituted benzenes, heteroaromatics, saturated rings, fused rings, and alkyl spacers in varying order, and scaffolds were chosen for both their predicted potencies as well as ease of synthesis. Figure 3 shows the general scaffold picked as a proof of principle for the linker region generation. It is a proline-based rigid analog series that includes a five-membered heterocycle with an aryl-aryl bond to another benzene that is meta substituted by a two carbon spacer to the terminal cyclohexane. The presence of a centralized heterocycle was ideal for solubility manipulation, and the synthesis of the X/Z imidazole, oxazole, and thiazole was undertaken to demonstrate a solubility/activity relationship. Figure 4 illustrates the linker generation process where the docking conformation of compound 38 (Figure 4a) was fragmented into an aryl amide head group and a cyclohexyl tail terminus (Figure 4b), and the in silico linker screening procedure led to a theoretical aromatic tail derivative (Figure 4c).
Figure 3.

Generic Scaffold for the Linker Design Proof of Principle.
Figure 4.

Progression of linker design. (A) Theoretical binding orientation of compound 38 chelating ATP (both drawn as stick models). (B) Deletion of aliphatic linker between the aryl amide and cyclohexyl group in 38. (C) Generation of a new linker (56) with a reduced number of rotatable bonds.
The synthesis of imidazole 53 began with the hydroboration of vinylcyclohexane and subsequent Suzuki coupling with 3-bromoacetophenone to form ketone 48 (Scheme 12). The ketone was then alpha brominated with molecular bromine and displaced by the cesium salt of mono tert-butyl protected terephthalic acid to yield ester 50. Compound 50 was then cyclized in refluxing xylenes with ammonium acetate to produce imidazole 51, which was deprotected and coupled to form nitrile 52. Standard Pinner conditions then yielded the desired imidazole containing amidine 53. The synthesis of oxazole 56 diverges form that of the imidazole at compound 50, which is cyclized in AcOH with ammonium acetate to yield the acid deprotected oxazole 54 in one step (Scheme 13). Amide followed by amidine formation then produced the oxazole containing amidine 56. Synthesis of the thiazole required the conversion of the mono tert-butyl protected terephthalic acid to its terminal amide using isobutylchloroformate and ammonia in methanol (Scheme 14). This terminal amide could then be transformed into the thioamide 57 using Lawesson’s reagent. Thioamide 57 was smoothly coupled then cyclized with the alpha bromoketone 49 to yield the thioazole 58. Tert-butyl deprotection, amide formation, and then amidine synthesis produced the desired thioazole containing amidine 60.
Scheme 12.

Synthesis of imidazole 53.
Scheme 13.

Synthesis of oxazole 56.
Scheme 14.

Synthesis of thiazole 60.
The SphK1 model predicted and in vitro determined KI values for the heterocycle series are listed in Table 5. All three heterocycles were predicted to geometrically fit in the substrate pocket, but the SphK1 model predicted a “Goldilocks” effect based on solubility, where the oxazole 56 with a Clog P of 4.24 should have the lowest KI value of 30 nM. The imidazole 53 and the thiazole 60 were predicted to have lesser potencies due to being too polar and hydrophobic respectively. On biological evaluation the model performed quite well, yielding the correct order of potency and predicting the actual KI value (47 nM) of the oxazole 56 within the 95% confidence limits. Indeed, the imidazole was the only compound of the three that had an experimentally determined KI value outside the 95% confidence limit, and this is probably due to the ratio of protonated versus neutral states. The pKa of the protonated imidazole ring is predicted to be around 7 in water, and if one assumes that the charged species has a KI > 10 μM, then that ratio would proportionally reduce the activity of compound 53. Comparing Clog P to reverse-phase HLPC retention time, which is a standard measure for comparing relative water solubilities, validates this reasoning (Figure SI-3). The retention times of the presented library of amidine containing inhibitors correlates well with Clog P (R2 = 0.71), and compound 53 is an outlier of this trend (see supporting information).
Table 5.
KI values for the heterocycle linker series.
| Compound | Structure | KI (μM)a | SphK1 Selectivityb | ||||
|---|---|---|---|---|---|---|---|
| Predicted SphK1 | 95% Confidence | SphK1 | SphK2 | ||||
| Lower Limit | Upper Limit | ||||||
| 38 |
|
- | - | - | 0.075 | 3.0 | 80 |
| 53 |
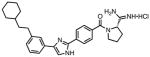
|
0.065 | 0.01 | 0.45 | 2.0 | 20 | 20 |
| 56 |
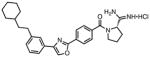
|
0.031 | 0.005 | 0.21 | 0.047 | 4.2 | 180 |
| 60 |
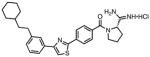
|
0.035 | 0.006 | 0.24 | 0.11 | 6.0 | 110 |
KI = [I]/(K’M/KM - 1); KM of Sphingosine at SphK1 = 10 μM; KM of Sphingosine at SphK2 = 5 μM
Selectivity = (KI/KM)SphK2/(KI/KM)SphK1
In Vitro Evaluation of Inhibitors in U937 Cells
To evaluate how well these amidine-based inhibitors penetrate and reduce endogenous S1P levels in living cells, U937 cells were pretreated with compounds 1, 19a, 38, and 56 for 2 hours (Table 6 and Figure 5). U937 cells are a human monoblastic leukemia cell line, whose S1P levels have been reduced by micromolar concentrations of the known sphingosine kinase inhibitor dimethyl sphingosine (DMS).40, 42 The amidine-based inhibitors indeed showed inhibition at concentrations near the KI values, all showed significant S1P reduction at 100 nM. At 10 nM concentrations, lower than the KI values of all the inhibitors, S1P reduction was still observed for compounds 19a and 38. In other experiments (not shown), it was determined that the decreased accumulation of S1P in U937 cells was the result of blockade of synthesis, rather than increased decay or export of S1P.
Table 6.
KI values for inhibitors used for S1P inhibition in living U937 cells.
| Compound | Structure | KI (μM)a | SphK1 Selectivityb | |
|---|---|---|---|---|
| SphK1 | SphK2 | |||
| 1 |
|
0.2 | 0.5 | 5 |
| 19a |
|
0.111 | 26 | 470 |
| 38 |
|
0.075 | 3.0 | 80 |
| 56 |
|
0.047 | 4.2 | 180 |
| 9ab |
|
1.4 | 31 | 44 |
| SKI-II |
|
12 | 33 | 6 |
KI = [I]/(K’M/KM - 1); KM of Sphingosine at SphK1 = 10 μM; KM of Sphingosine at SphK2 = 5 μM
Selectivity = (KI/KM)SphK2/(KI/KM)SphK1
Figure 5.

S1P concentrations in human leukemia U937 cells dosed with SphK inhibitors. Error bars indicate 95% confidence limits. Concentrations are indicated by shading and patterns.
To compare these amidine based inhibitors to other known sphingosine kinase inhibitors, compounds 9ab44 and SKI-II45 were also examined in living U937 cells (Table 6 and Figure 5). Compound 9ab did not cause S1P reduction at 100 nM, which was expected given its KI values being 1.4 μM for SphK1 and 31 μM for SphK2.52 However, at a concentration of 1 μM, nearer to the KI value of compound 9ab at SphK1, a 40% reduction of S1P is observed. Comparing the KI values for 9ab versus those of the SphK1 selective compound 19a, 110 nM for SphK1 and 26 μM for SphK2 (Table 6), suggests that the observed reduction in S1P levels for 19a is accomplished through the inhibition of SphK1. SKI-II also fits this trend with a higher SphK1 KI value of 12 μM52 and no significant S1P reduction observed until 10 μM concentrations were applied.
A notable outlier in the series is the performance of oxazole 56 on whole cells. With the lowest KI value in the series (47 nM for SphK1), 56 should inhibit S1P production most successfully. Compound 56 does reduce S1P levels significantly, along with the other amidine inhibitors, at a concentration of 100 nM, but fails to outperform compounds 19a and 38 at 10 nM concentrations despite having the lowest KI value. This recombinant enzyme versus living cell deviation in activity is subtle and suggests differences in uptake or efflux. Interestingly, S1P reduction in U937 cells by these amidine-based inhibitors did not cause caspase-mediated apoptosis as previous reports have demonstrated with other SphK inhibitors (data not shown).40, 42 However, a more thorough investigation beyond the characterization of these inhibitors is needed to better understand these differences in cytotoxicity.
Conclusion
The role of the SphKs as the sole producers of S1P, a lipid promoter for tumorigenesis and angiogenesis, in the sphingosine rheostat illuminates the practicality of an anti-cancer strategy that targets their activity.1 Described herein is the optimization and SAR of amidine-based SphK1 subtype-selective inhibitors. The library of inhibitors evaluated were used as a test set in the generation of a SphK1 homology model from the crystal structure of DGKB, and used for the in silico design and synthesis of nanomolar SphK1 inhibitors. These inhibitors were found to significantly lower endogenous S1P levels in human leukemia U937 cells at 10 and 100 nM concentrations.
Experimental Section
Sphingosine Kinase Assay
Human SphK1 and mouse SphK2 cDNAs were used to generate mutant baculoviruses that encoded these proteins. Infection of Sf9 insect cells with the viruses for 72 h resulted in >1000-fold increases in SphK activity in 10000 g supernatant fluid from homogenized cell pellets. The enzyme assay conditions were exactly as described,52 except infected Sf9 cell extract containing 2–3 μg protein was used as a source of enzyme.
U937 Cell Culture Assay
U937 cells were grown according to previously described literature procedure.40 In general, cells were grown in RPMI 1640 media enriched with L-glutamine, 10% penicillin and streptomycin and 10% fetal bovine serum (FBS). Twenty-four hours before dosing with SphK inhibitors, the media was replaced with media containing 2% FBS. All cell cultures were grown at a stable temperature of 37ºC and the SphK inhibitors were dosed for 2 h.
S1P Extraction and LCMS Quantification
Extraction protocols and LCMS procedures were adapted from a previously reported study.62 Samples of pelleted cells (approximately 4 million) were taken up in 2 mL of 3:1 methanol:chloroform mixture and transferred to a capped glass vial. To this suspension was added 10 μL of internal standard solution containing 1 μM C17 S1P (purchased from Avanti Polar Lipids). The mixture was homogenized via sonication for 10 min and immediately incubated at 48°C for 16 h. After this time, the mixture was cooled to ambient temperature and 200 μL of 1 M KOH in methanol was added to the suspension. The samples were again sonicated and incubated at 37°C for an additional 2 h. After this time, the samples were neutralized through the addition of 30 μL of glacial acetic acid and transferred to 2 mL microcentrifuge tubes. Samples were then centrifuge at 10,000 x g for 10 min at 4°C. The supernatant fluid was collected in a separate glass vial and the pellets discarded. The resulting solution was evaporated (to a solid) with a stream of nitrogen. Immediately prior to LCMS analysis, the solid material was taken up in 300 μL of methanol and centrifuged at 12,000 x g for 12 min at 4°C. An auto-sampler vial was loaded with 150 μL of the resulting supernatant for LCMS analysis.
S1P analysis from cellular extracts was performed on an Applied Biosystems 4000 QTrap LC/MS/MS instrument. Chromatographic resolution of analytes was achieved with a Shimadzu LC-20AD system. A binary solvent gradient with a flow rate of 1 mL/min was used to separate sphingolipid analytes by reverse phase chromatography (Supelco Discovery C18 column; 50 mm, 2.1 mm (L., I.D.); 5 μm bead size). Mobile phase A was comprised of water: methanol: formic acid (79:20:1) and mobile phase B was comprised of methanol: formic acid (99:1). The run started with 100% A for 0.5 min. Solvent B was then increased linearly for 5.1 min to 100% of the total solvent composition and held at 100% for an additional 4.3 min. The column was finally re-equilibrated to 100% for 0.1 min and held for an additional 1 min. The following analytes (and fragmentation patterns) were monitored simultaneously for identification. C17S1P (366.4, 250.4); S1P (380.4, 264.4).
General Synthetic Materials and Methods
All nonaqueous reactions were carried out in oven or flame-dried glassware under an argon or nitrogen atmosphere with dry solvents and magnetic stirring, unless otherwise stated. The argon and nitrogen were dried by passing through a tube of Drierite. Anhydrous diethyl ether (Et2O), chloroform (CHCl3), Dimethyl sulfoxide (DMSO), toluene (PhMe), dichloromethane (CH2Cl2), methanol (MeOH), ethanol (EtOH), and tetrahydrofuran (THF) and N,N-dimethylformamide (DMF) were purchased from Aldrich or VMR Chemicals and used as received. THF was dried over activated molecular sieves (4 Å) prior to use. All other reagents were purchased from Acros chemicals and Aldrich chemicals. Except as indicated otherwise, reactions were monitored by thin layer chromatography (TLC) using 0.25 mm Whatman precoated silica gel plates. Flash chromatography was performed with the indicated solvents and Dynamic Adsorbents silica gel (particle size 0.023 – 0.040 mm). Proton (1H) and carbon (13C) NMR spectra were recorded on a Varian UnityInova 500/51 or Varian UnityInova 300/54 at 300K unless otherwise noted. Chemical shifts are reported in ppm (δ) values relative to the solvent as follows: CDCl3 (δ 7.24 for proton and δ 77.0 for carbon NMR), DMSO-d6 (δ 2.50 for proton and δ 39.5 for carbon NMR) CD3OD (δ 3.31 for proton and δ 47.6 for carbon NMR). All high-resolution mass spectrometry was carried out by the Mass Spectrometry Laboratory in the School of Chemical Sciences at the University of Illinois Urbana-Champagne (Urbana, IL).
TLC Stains
KMnO4; 3 g KMnO4 and 20 g K2CO3 in 300 mL water and 5 mL 5% NaOH. Seebach’s Dip; To a solution of 25 g phosphomolybdic acid and 7.5 g cerium (IV) sulfate in 479 mL water was added 25 mL conc. sulfuric acid dropwise. Ninhydrin; 1.5 g ninhydrin in 5 mL AcOH and 500 mL 95% EtOH. All stains required TLC development on a hot plate set to 80ºC.
Other abbreviations
1,1′-bis(diphenylphosphino)ferrocene (dppf), 4-dimethylaminopyridine (DMAP), 9-borabicyclo[3.3.1]nonane (9-BBN), acetic acid (AcOH), benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (PyBOP), di-tert-butyl dicarbonate (Boc2O), ethyl acetate (EtOAc), N,N-diisopropylethylamine (DIEA), tert-butanol (tBuOH), tetra-n-butylammonium bromide (TBAB), triethylamine (TEA), trifluoroacetic acid (TFA), trifluoroacetic anhydride (TFAA).
Liquid Chromatography and Mass Spectrometry for Evaluation of Chemical Purity
All compounds submitted for biological evaluation were determined to be > 95% pure by LCMS evaluation performed by the Mass Spectrometry Laboratory in the School of Chemical Sciences at the University of Illinois Urbana-Champagne (Urbana, IL). High performance liquid chromatography – mass spectrometry (LCMS) was carried out using an Agilent 2.1×50 mm C-18 column and a Micromass Q-tof Ultima mass spectrometer. Mobile phase A consisted of HPLC grade H2O and 0.01% TFA; mobile phase B consisted of MeCN and 0.01% TFA. LCMS identification and purity utilized a binary gradient starting with 90% A and 10% B and linearly increasing to 100% B over the course of 6 min, followed by an isocratic flow of 100% B for an additional 3 min. A flow rate of 0.5 mL/min was maintained throughout the HPLC method. The purity of all products was determined by integration of the total ion count (TIC) spectra and integration of the ultraviolet (UV) spectra at 214 nm. Retention times are abbreviated as tR; mass to charge ratios are abbreviated as m/z.
General Procedure A: Conversion of Nitriles to Amidines
To a solution of a nitrile (1.0 eq.) in MeOH (0.10 M) was added a 0.5 M solution of sodium methoxide in MeOH (0.50 eq.) at rt and then heated to 50ºC for 24 h. The intermediate imidate was detectable by TLC; however, being in equilibrium with the nitrile, full conversion does not occur. Ammonium chloride (2.0 eq.) was then added in one portion at that temperature and allowed to react until the imidate was completely consumed by TLC analysis. The reaction was then cooled to rt and evacuated to dryness to yield a crude solid. The solid was reconstituted with CHCl3 and filtered through a fine glass fritted funnel in order to remove excess ammonium chloride, and the filtrate was again evacuated to dryness. The material was then recrystallized in Et2O to yield the pure amidine hydrochloride salt. The yields varied greatly depending upon substrate, because amidine formation is dependant upon the equilibrium ratio between nitrile and imidate established under the sodium methoxide conditions.
General Procedure B: PyBOP Mediated Couplings of Amines and Anilines to Carboxylic Acids
To a suspension of an amine or aniline (1.0 eq.), carboxylic acid (1.0 eq.), and PyBOP (1.1 eq.), in CH2Cl2 at rt was added DIEA (4.0 eq.) and let stir for 4 h unless otherwise stated. The reaction was then evaporated to dryness and immediately purified by flash chromatography.
General Procedure C: Williamson Ether Synthesis
To a solution of an alcohol (2 eq.) in DMF (0.3 M) at 0ºC was added 60 % sodium hydride dispersed in mineral oil (2.0 eq.) at 0ºC, then let warm to rt, and then let react for 45 min. The alkyl bromide was then added in one portion and the reaction was stirred for 12 h. The reaction was quenched with sat. NaHCO3 (100x the volume of DMF) and extracted into EtOAc (100x the volume of DMF). The organic layer was washed 3x with neat water (100x the volume of DMF), dried with Na2SO4, evaporated to a yellow oil, and immediately purified by flash chromatography.
General Procedure D: Suzuki Coupling
To a solution of alkene (1.0 eq.) in THF (0.2 M) at rt was added a 0.5 M solution of 9-BBN in THF (2.0 eq.) and let stir until consumption of the alkene was evident by TLC analysis (4 h unless otherwise stated). The reaction was then treated with 3 M K3PO4(aq), and diluted with DMF (0.2 M relative to the starting alkene). The aryl bromide and PdCl2(dppf) were then sequentially added and the reaction was let stir for 16 h. The reaction was diluted with EtOAc (200x the volume of DMF) and washed 3x with neat water (100x the volume of DMF). The organic layer was then dried with Na2SO4, evaporated to a dark red oil, and immediately purified by flash chromatography.
General Procedure E: Aryl Nitro Reduction with Zinc Dust
To a solution of an aryl nitro (1.0 eq.) in AcOH (0.1 M) at rt was added zinc dust (10 eq.) in one portion and let stir for 16 h. The reaction was then diluted with EtOAc and filtered through Celite™. The filtrate was evaporated to dryness, coevaporated with PhMe to remove residual AcOH, and immediately purified by flash chromatography.
General Procedure F: Copper Mediated Alkane Synthesis
To a suspension of cuprous chloride (0.05 eq.) in Et2O (1.0 M relative to the alkyl halide) at −78ºC was added a 2.0 M solution of a Gignard reagent (2 eq.) in THF followed by the addition of an alkyl halide (Br or I) (1.0 eq.). The reaction was let warm to rt and held at that temperature until the starting alkene was judged consumed by TLC analysis (4 h unless otherwise stated). The reaction was then cooled to 0ºC and quenched with 1 N HCl (50x the volume of Et2O) and extracted into EtOAc (50x the volume of Et2O). The organic layer was then dried with Na2SO4, evaporated to a dark green oil, and immediately purified by flash chromatography.
General Procedure G: Hydroboration/Oxidation
To a solution of alkene (1.0 eq.) in THF (1.0 M) at rt was added a 0.5 M solution of 9-BBN in THF (2.0 eq.) and let stir until consumption of the alkene was evident by TLC analysis (4 h unless otherwise stated). The reaction was then cooled to 0ºC and then diluted with EtOH (10 eq.), 3 M NaOH(aq) (1.0 eq.), and slowly treated with 30% H2O2 in water (1.4 eq.) sequentially. The reaction was let warm to rt and then stir for 30 min before being quenched with 1 N HCl (equal to the total volume of THF) and extracted into EtOAc (10x the total volume of THF). The organic layer was then dried with Na2SO4, evaporated to a colorless oil, and immediately purified by flash chromatography.
General Procedure H: Alkyl Mesylate Formation
To a solution of an alcohol (1.0 eq.) in CH2Cl2 (0.3 M) at 0ºC was added TEA (2.0 eq.) then methanesulfonyl chloride (1.1 eq.). The reaction was let warm to rt and then heated to reflux for 30 min. The reaction was then cooled to rt, quenched with sat. NaHCO3 (10x the volume of CH2Cl2), and extracted into CHCl3 (200x the volume of CH2Cl2). The organic layer was then dried with Na2SO4, evaporated to a yellow oil, and immediately purified by flash chromatography.
General Procedure I: Alkyl Azide Formation
To a solution of an alkyl mesylate (1.0 eq.) in DMF (0.3 M) at rt was added sodium azide (1.1 eq.) and the reaction was heated to reflux for 12 h. The reaction was then cooled to rt, diluted with EtOAc (100x the volume of DMF), and washed 3x with sat. NaHCO3 (100x the volume of DMF). The organic layer was then dried with Na2SO4, evaporated to a yellow oil, and immediately purified by flash chromatography.
General Procedure J: Staudinger Reduction and Ligation
To a solution of an alkyl azide (1.0 eq.) and triphenylphosphine (1.1 eq.) in PhMe (0.3 M) was added neat water (2.0 eq.) and the reaction was heated to reflux for 30 min. The reaction was then cooled to rt and treated with a solution of 1-cyano-1-cyclopanecarboxylic acid (1.5 eq.), PyBOP (1.5 eq.), and TEA (2.0 eq.) in CH2Cl2 (1.5 M relative to the carboxylic acid). The reaction was let stir for 4 h, then evacuated to a dark red oil, and purified by flash chromatography.
General Procedure K: Pinnick Oxidation
To a solution of an aldehyde (1.0 eq.), NaH2PO4 (8.0 eq.), and 2-methyl-2-butene (10 eq.) in THF, water, and tBuOH (4:4:1) (0.04 M) at rt was added sodium chlorite (4 eq.) and let stir for 1 h. The reaction was diluted with EtOAc (10x the volume of the reaction’s mixture of solvents), and washed 3x with 1 N HCl (5x the volume of the reaction’s mixture of solvents). The organic layer was then dried with Na2SO4, and evaportated to a white solid. No further purification was necessary.
General Procedure L: Acid Chloride Formation
To a solution of a carboxylic acid (1.0 eq.) and DMF (0.05 eq.) in CH2Cl2 (0.1 M) at 0ºC was added oxalyl chloride (2.0 eq.) dropwise and let warm to rt. The reaction progresses to a yellow green color and after 3 h the reaction was evaporated to dryness, and then immediately purified by flash chromatography.
General Procedure M: Acid Chloride and Amine Coupling
To a solution of an acid chloride (1.0 eq.) in CH2Cl2 (0.3 M) at rt was added DIEA (4.0 eq.) followed by an amine HCl salt (1.5 eq.) and the reaction was left stirring for 12 h. The reaction was then evaporated to dryness and immediately purified by flash chromatography.
General procedure N: Deprotection of N-Boc and O-tBu Ester Protecting Groups
To a solution of either a N-Boc or O-tBu protecting group (1.0 eq.) in CH2Cl2 (0.2 M) at rt was added TFA (0.2 M) and the reaction was reacted until judged complete by TLC analysis (30 min unless otherwise stated). The reaction was then evaporated to dryness and taken on crude.
1-cyano-N-tetradecylcyclopropanecarboxamide (3a)
General procedure B was used to couple tetradecan-1-amine (213 mg, 1.00 mmol) and 1-cyano-1-cyclopanecarboxylic acid to yield the title compound. 99%. White solid. Rf = 0.50 (20% EtOAc in hexanes; KMnO4). 1H NMR (300 MHz, CDCl3) δ 6.41 (s, 1H), 3.27 (dt, J = 6.5, 5.3 Hz, 2H), 1.65 (dd, J = 8.1, 4.4 Hz, 2H), 1.52 (t, J = 6.9 Hz, 2H), 1.45 (dd, J = 8.0, 4.4 Hz, 2H), 1.36 – 1.11 (m, 22H), 0.86 (t, J = 6.7 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 165.22, 120.54, 40.89, 32.14, 29.87, 29.79, 29.71, 29.58, 29.45, 27.01, 22.91, 17.65, 14.34, 13.67.
1-cyano-N-hexadecylcyclopropanecarboxamide (3b)
General procedure B was used to couple hexadecan-1-amine (241 mg, 1.00 mmol) and 1-cyano-1-cyclopanecarboxylic acid to yield the title compound. 99%. White solid. Rf = 0.52 (20% EtOAc in hexanes; KMnO4). 1H NMR (500 MHz, CDCl3) δ 6.37 (s, 1H), 3.28 (dt, J = 6.6, 5.3 Hz, 2H), 1.66 (dd, J = 8.1, 4.3 Hz, 2H), 1.58 – 1.49 (m, 2H), 1.47 (dd, J = 8.1, 4.3 Hz, 2H), 1.39 – 0.99 (m, 24H), 0.87 (t, J = 6.9 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 164.99, 120.33, 40.68, 31.91, 29.67, 29.56, 29.48, 29.38, 29.22, 26.78, 22.68, 17.43, 14.12, 13.55.
1-cyano-N-octadecylcyclopropanecarboxamide (3c)
General procedure B was used to couple octadecan-1-amine (270 mg, 1.00 mmol) and 1-cyano-1-cyclopanecarboxylic acid to yield the title compound. 99%. White solid. Rf = 0.55 (20% EtOAc in hexanes; KMnO4). 1H NMR (500 MHz, CDCl3) δ 6.44 (s, 1H), 3.26 (dt, J = 6.6, 5.3 Hz, 2H), 1.65 (dd, J = 8.1, 4.3 Hz, 2H), 1.52 (dt, J = 13.7, 6.9 Hz, 2H), 1.45 (dd, J = 8.1, 4.3 Hz, 2H), 1.26 (m, 26H), 0.86 (t, J = 6.9 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 164.96, 120.28, 40.64, 31.91, 29.68, 29.56, 29.48, 29.36, 29.22, 26.78, 22.67, 17.40, 14.11, 13.42.
1-carbamimidoyl-N-tetradecylcyclopropanecarboxamide hydrochloride (4a)
General procedure A was used to convert 3a (306 mg, 1.00 mmol) to the title compound. 55%. White solid. 1H NMR (300 MHz, DMSO) δ 9.11 (s, 2H), 8.97 (s, 2H), 7.80 (t, J = 5.3 Hz, 1H), 3.01 (dd, J = 13.1, 6.5 Hz, 2H), 1.65 – 0.96 (m, 28H), 0.83 (t, J = 6.5 Hz, 3H). 13C NMR (75 MHz, DMSO) δ 168.54, 167.92, 40.13, 31.98, 29.74, 29.56, 29.48, 29.40, 29.09, 27.05, 22.78, 14.86, 14.65. LCMS: tR = 4.72; m/z = 324.3. HRMS m/z calc. for C19H38N3O (M+H), 324.3015; found, 324.3010.
1-carbamimidoyl-N-hexadecylcyclopropanecarboxamide hydrochloride (4b)
General procedure A was used to convert 3b (320 mg, 1.00 mmol) to the title compound. 46%. White solid. 1H NMR (300 MHz, DMSO) δ 9.16 (s, 2H), 8.96 (s, 2H), 7.82 (s, 1H), 3.01 (d, J = 5.1 Hz, 2H), 1.51 – 0.97 (m, 30H), 0.83 (s, 3H). 13C NMR (75 MHz, DMSO) δ 168.54, 167.92, 40.04, 31.98, 29.74, 29.56, 29.49, 29.40, 27.05, 22.79, 14.86, 14.65. LCMS: tR = 5.22; m/z = 352.3. HRMS m/z calc. for C21H42N3O (M+H), 352.3328; found, 352.3329.
1-carbamimidoyl-N-octadecylcyclopropanecarboxamide hydrochloride (4c)
General procedure A was used to convert 3c (362 mg, 1.00 mmol) to the title compound. 32%. White solid. 1H NMR (300 MHz, DMSO) δ 9.07 (s, 2H), 8.95 (s, 2H), 7.89 (s, 1H), 3.05 (d, J = 5.1 Hz, 2H), 1.58 – 0.95 (m, 32H), 0.83 (s, 3H). 13C NMR (75 MHz, DMSO) δ 168.64, 167.95, 40.09, 32.01, 29.78, 29.59, 29.54, 29.43, 27.12, 22.80, 14.92, 14.70. LCMS: tR = 6.22; m/z = 380.4. HRMS m/z calc. for C23H46N3O (M+H), 380.3641; found, 380.3633.
((hept-6-en-1-yloxy)methyl)benzene (5a)
General procedure C was used to couple benzyl alcohol and 7-bromohept-1-ene (1.00 mL, 6.52 mmol) to yield the title compound. 87%. Yellow oil. Rf = 0.88 (10% EtOAc in hexanes; KMnO4). 1H NMR (300 MHz, CDCl3) δ 7.57 – 7.19 (m, 5H), 5.89 (ddt, J = 16.9, 10.2, 6.7 Hz, 1H), 5.22 – 4.89 (m, 2H), 4.57 (s, 2H), 3.54 (t, J = 6.6 Hz, 2H), 2.14 (m, 2H), 1.82 – 1.61 (m, 2H), 1.59 – 1.40 (m, 4H). 13C NMR (75 MHz, CDCl3) δ 139.16, 139.01, 128.62, 127.86, 127.73, 114.65, 73.16, 70.66, 34.08, 29.97, 29.08, 26.04.
((hex-5-en-1-yloxy)methyl)cyclohexane (5b)
General procedure C was used to couple cyclohexylmethanol and 6-bromohex-1-ene (1.00 mL, 7.48 mmol) to yield the title compound. 70%. Clear and colorless oil. Rf = 0.39 (3% EtOAc in hexanes; KMnO4). 1H NMR (300 MHz, CDCl3) δ 5.76 (ddt, J = 16.9, 10.2, 6.7 Hz, 1H), 5.10 – 4.58 (m, 2H), 3.35 (t, J = 6.4, 2H), 3.15 (d, J = 6.5 Hz, 2H), 2.23 – 0.80 (m, 2H), 1.87 – 1.05 (m, 13H), 0.87 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 138.87, 114.59, 77.03, 71.03, 38.28, 33.80, 30.36, 29.42, 26.87, 26.10, 25.73.
((hept-6-en-1-yloxy)methyl)cyclohexane (5c)
General procedure C was used to couple cyclohexylmethanol and 7-bromohept-1-ene (1.00 mL, 6.52 mmol) to yield the title compound. 70%. Clear and colorless oil. Rf = 0.73 (5% EtOAc in hexanes; KMnO4). 1H NMR (300 MHz, CDCl3) δ 5.75 (ddt, J = 16.9, 10.2, 6.7 Hz, 1H), 5.07 – 4.70 (m, 2H), 3.32 (t, J = 6.6 Hz, 2H), 3.14 (d, J = 6.6 Hz, 2H), 2.15 – 1.90 (m, 2H), 1.84 – 0.98 (m, 15H), 0.98 – 0.65 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 139.04, 114.42, 77.03, 71.18, 38.27, 33.96, 30.37, 29.80, 28.98, 26.88, 26.10, 25.91.
((oct-7-en-1-yloxy)methyl)cyclohexane (5d)
General procedure C was used to couple cyclohexylmethanol and 8-bromooct-1-ene (1.00 mL, 5.98 mmol) to yield the title compound. 77%. Clear and colorless oil. Rf = 0.43 (3% EtOAc in hexanes; KMnO4). 1H NMR (300 MHz, CDCl3) δ 5.77 (ddt, J = 16.9, 10.2, 6.7 Hz, 1H), 5.13 – 4.71 (m, 2H), 3.35 (t, J = 6.6 Hz, 5H), 3.17 (d, J = 6.5 Hz, 2H), 2.12 – 1.91 (m, 2H), 1.86 – 1.01 (m, 17H), 0.98 – 0.74 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 139.18, 114.36, 77.03, 71.26, 38.29, 33.96, 30.39, 29.92, 29.19, 29.09, 26.89, 26.27, 26.11.
1-((hept-6-en-1-yloxy)methyl)adamantane (5e)
General procedure C was used to couple 1-adamantane-methanol and 7-bromohept-1-ene (1.00 mL, 6.52 mmol) to yield the title compound. 85%. Clear and colorless oil. Rf = 0.54 (5% EtOAc in hexanes; KMnO4). 1H NMR (300 MHz, CDCl3) δ 5.79 (ddt, J = 17.0, 10.2, 6.7 Hz, 1H), 5.09 – 4.77 (m, 2H), 3.36 (t, J = 6.5 Hz, 2H), 2.94 (s, 2H), 2.12 – 1.86 (m, 5H), 1.79 – 0.99 (m, 18H). 13C NMR (75 MHz, CDCl3) δ 139.14, 114.44, 82.10, 71.78, 39.97, 37.49, 34.00, 29.68, 29.00, 28.54, 25.90.
((hept-6-en-1-yloxy)methyl)cyclopentane (5f)
General procedure C was used to couple cyclopentylmethanol and 7-bromohept-1-ene (1.00 mL, 6.56 mmol) to yield the title compound. 79%. Clear and colorless oil. Rf = 0.69 (5% EtOAc in hexanes; KMnO4). 1H NMR (300 MHz, CDCl3) δ 5.98 – 5.57 (m, 1H), 5.10 – 4.79 (m, 2H), 3.38 (t, J = 6.6 Hz, 2H), 3.25 (d, J = 7.1, 2H), 2.24 – 1.95 (m, 3H), 1.88 – 1.01 (m, 14H). 13C NMR (75 MHz, CDCl3) δ 139.13, 114.45, 75.76, 71.17, 39.68, 33.97, 29.94, 29.81, 28.99, 25.92, 25.61.
7-butoxyhept-1-ene (5g)
General procedure C was used to couple butanol and 7-bromohept-1-ene (1.50 mL, 9.84 mmol) to yield the title compound. 75%. Clear and colorless oil. Rf = 0.74 (10% EtOAc in hexanes; KMnO4). 1H NMR (300 MHz, CDCl3) δ 5.73 (ddt, J = 16.9, 10.2, 6.7 Hz, 1H), 5.07 – 4.65 (m, 2H), 3.33 (m, 4H), 2.17 – 1.79 (m, 2H), 1.62 – 1.42 (m, 4H), 1.33 (m, 6H), 0.86 (t, J = 7.4 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 138.95, 114.38, 70.96, 70.75, 33.92, 32.05, 29.81, 28.95, 25.89, 19.53, 14.03.
1-(7-(benzyloxy)heptyl)-4-nitrobenzene (6a)
General procedure D was used to couple 5a (564 mg, 2.76 mmol) and 1-bromo-4-nitrobenzene to yield the title compound. 61%. Yellow solid. Rf = 0.16 (5% EtOAc in hexanes; Seebach’s Dip). 1H NMR (500 MHz, CDCl3) δ 8.13 (d, J = 7.9 Hz, 2H), 7.45 – 7.19 (m, 7H), 4.51 (s, 2H), 3.48 (t, J = 6.4 Hz, 2H), 2.70 (t, J = 7.7 Hz, 2H), 1.61 (m, 4H), 1.49 – 1.17 (m, 6H). 13C NMR (126 MHz, CDCl3) δ 150.79, 146.20, 138.67, 129.16, 128.36, 127.62, 127.50, 123.55, 72.88, 70.39, 35.83, 30.92, 29.74, 29.23, 29.11, 26.11.
1-(6-(cyclohexylmethoxy)hexyl)-4-nitrobenzene (6b)
General procedure D was used to couple 5b (1.02 g, 4.56 mmol) and 1-bromo-4-nitrobenzene to yield the title compound. 52%. Tan oil. Rf = 0.56 (5% EtOAc in hexanes; Seebach’s Dip). 1H NMR (300 MHz, CDCl3) δ 8.15 (d, J = 8.7 Hz, 2H), 7.27 (d, J = 7.8 Hz, 2H), 3.34 (t, J = 6.5 Hz, 2H), 3.15 (d, J = 6.6 Hz, 2H), 2.80 – 2.58 (m, 2H), 1.86 – 1.00 (m, 17H), 1.00 – 0.77 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 150.97, 146.39, 129.36, 123.74, 77.05, 71.10, 38.25, 35.99, 31.15, 30.38, 29.81, 29.20, 26.88, 26.21, 26.11.
1-(7-(cyclohexylmethoxy)heptyl)-4-nitrobenzene (6c)
General procedure D was used to couple 5c (951 mg, 4.52 mmol) and 1-bromo-4-nitrobenzene to yield the title compound. 63%. Tan oil. Rf = 0.61 (5% EtOAc in hexanes; Seebach’s Dip). 1H NMR (300 MHz, CDCl3) δ 8.07 (d, J = 8.7 Hz, 2H), 7.27 (d, J = 8.5 Hz, 2H), 3.33 (t, J = 6.5 Hz, 2H), 3.14 (d, J = 6.6 Hz, 2H), 2.75 – 2.53 (m, 2H), 1.84 – 0.96 (m, 19H), 0.95 – 0.70 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 150.99, 146.36, 129.33, 123.70, 77.03, 71.18, 38.25, 36.02, 31.13, 30.37, 29.89, 29.43, 29.32, 26.88, 26.28, 26.10.
1-(8-(cyclohexylmethoxy)octyl)-4-nitrobenzene (6d)
General procedure D was used to couple 5d (1.02 g, 4.56 mmol) and 1-bromo-4-nitrobenzene to yield the title compound. 60%. Tan oil. Rf = 0.64 (5% EtOAc in hexanes; Seebach’s Dip). 1H NMR (300 MHz, CDCl3) δ 8.12 (d, J = 8.6 Hz, 2H), 7.30 (d, J = 8.4 Hz, 2H), 3.36 (t, J = 6.6 Hz, 2H), 3.17 (d, J = 6.6 Hz, 2H), 2.79 – 2.57 (m, 2H), 1.85– 1.00 (m, 21H), 0.86 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 151.03, 146.41, 129.35, 123.76, 77.06, 71.26, 38.26, 36.07, 31.19, 30.39, 29.93, 29.56, 29.33, 26.89, 26.37, 26.11.
1-(((7-(4-nitrophenyl)heptyl)oxy)methyl)adamantane (6e)
General procedure D was used to couple 5e (671 mg, 2.77 mmol) and 1-bromo-4-nitrobenzene to yield the title compound. 59%. Tan oil. Rf = 0.74 (10% EtOAc in hexanes; Seebach’s Dip). 1H NMR (300 MHz, CDCl3) δ 8.15 (d, J = 6.6 Hz, 2H), 7.32 (m, J = 6.6 Hz, 2H), 3.36 (t, J = 6.5 Hz, 2H), 2.93 (s, 2H), 2.81 – 2.58 (m, 2H), 2.06 – 1.87 (m, 3H), 1.87 – 1.09 (m, 22H). 13C NMR (75 MHz, CDCl3) δ 151.01, 142.80, 132.25, 124.11, 82.13, 71.82, 39.98, 37.48, 36.09, 29.78, 29.46, 29.37, 28.54, 26.27.
4-(7-(benzyloxy)heptyl)aniline (7a)
General procedure E was used to convert 6a (215 mg, 0.657 mmol) to the title compound. 99%. Tan oil. Rf = 0.27 (20% EtOAc in hexanes; Seebach’s Dip). 1H NMR (300 MHz, CDCl3) δ 7.51 – 7.26 (m, 5H), 7.02 (d, J = 5.3 Hz, 2H), 6.66 (m, J = 5.3 Hz, 2H), 4.57 (s, 2H), 3.52 (m, 4H), 2.70 – 2.42 (m, 2H), 1.80 – 1.53 (m, 4H), 1.42 (t, J = 8.6 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 144.41, 139.03, 133.17, 129.42, 128.65, 127.94, 127.78, 115.48, 73.15, 70.80, 35.37, 32.09, 30.09, 29.69, 29.52, 26.48.
4-(6-(cyclohexylmethoxy)hexyl)aniline (7b)
General procedure E was used to convert 6b (252 mg, 0.789 mmol) to the title compound. 99%. Clear and colorless oil. Rf = 0.25 (20% EtOAc in hexanes; Seebach’s Dip). 1H NMR (300 MHz, CDCl3) δ 6.98 (d, J = 8.1 Hz, 2H), 6.62 (d, J = 8.1, 2H), 3.56 (s, 2H), 3.40 (t, J = 6.2, 2H), 3.21 (d, J = 6.5, 2H), 2.51 (t, J = 7.6 Hz, 2H), 1.93 – 1.04 (m, 17H), 1.04 – 0.73 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 144.33, 133.10, 129.36, 115.42, 77.07, 71.33, 38.30, 35.27, 32.04, 30.44, 29.95, 29.36, 26.96, 26.34, 26.17.
4-(7-(cyclohexylmethoxy)heptyl)aniline (7c)
General procedure E was used to convert 6c (663 g, 2.00 mmol) to the title compound. 99%. Clear and colorless oil. Rf = 0.34 (20% EtOAc in hexanes; Seebach’s Dip). 1H NMR (300 MHz, CDCl3) δ 6.99 (d, J = 8.1 Hz, 2H), 6.62 (d, J = 8.2 Hz, 2H), 3.58 (s, 2H), 3.40 (t, J = 6.6 Hz, 2H), 3.23 (d, J = 6.6 Hz, 2H), 2.62 – 2.41 (m, 2H), 1.97 – 1.04 (m, 19H), 1.06 – 0.83 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 144.36, 133.13, 129.37, 115.43, 77.10, 71.38, 38.32, 35.34, 32.07, 30.47, 30.02, 29.67, 29.50, 26.98, 26.44, 26.20.
4-(8-(cyclohexylmethoxy)octyl)aniline (7d)
General procedure E was used to convert 6d (572 mg, 1.65 mmol) to the title compound. 99%. Clear and colorless oil. Rf = 0.38 (20% EtOAc in hexanes; Seebach’s Dip). 1H NMR (300 MHz, CDCl3) δ 6.98 (d, J = 8.2 Hz, 2H), 6.62 (d, J = 8.3 Hz, 2H), 3.66 – 3.43 (m, 2H), 3.39 (t, J = 6.7 Hz, 2H), 3.21 (d, J = 6.6 Hz, 2H), 2.59 – 2.39 (m, 2H), 1.93 – 1.02 (m, 21H), 1.06 – 0.80 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 144.26, 133.25, 129.35, 115.43, 77.08, 71.37, 38.29, 35.32, 32.07, 30.43, 29.99, 29.70, 29.48, 26.94, 26.43, 26.16.
4-adamantan-1-ylmethoxy)heptyl)aniline (7e)
General procedure E was used to convert 6e (263 mg, 0.683 mmol) to the title compound. 99%. Clear and colorless oil. Rf = 0.31 (20% EtOAc in hexanes; Seebach’s Dip). 1H NMR (300 MHz, CDCl3) δ 6.96 (d, J = 7.8 Hz, 2H), 6.62 (d, J = 7.1 Hz, 2H), 3.54 (s, 2H), 3.37 (t, J = 6.6 Hz, 2H), 2.95 (d, J = 5.1 Hz, 2H), 2.49 (t, J = 6.3 Hz, 2H), 1.96 (s, 3H), 1.85 –1.19 (m, 22H). 13C NMR (75 MHz, CDCl3) δ 144.22, 133.30, 129.36, 115.44, 82.11, 71.96, 39.98, 37.49, 35.30, 32.02, 29.82, 29.62, 29.47, 28.54, 26.33.
N-(4-(7-(benzyloxy)heptyl)phenyl)-1-cyanocyclopropanecarboxamide (8a)
General procedure B was used to couple 7a (183 mg, 0.616 mmol) and 1-cyano-1-cyclopanecarboxylic acid to yield the title compound. 96%. Clear and colorless oil. Rf = 0.47 (20% EtOAc in hexanes; Seebach’s Dip). 1H NMR (300 MHz, CDCl3) δ 8.13 (s, 1H), 7.41 (d, J = 8.3 Hz, 2H), 7.38 – 7.24 (m, 5H), 7.16 (d, J = 8.3 Hz, 2H), 4.51 (s, 2H), 3.48 (t, J = 6.6 Hz, 2H), 2.68 – 2.42 (m, 2H), 1.78 (dd, J = 8.2, 4.3 Hz, 2H), 1.70 – 1.52 (m, 6H), 1.46 – 1.26 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 163.54, 140.25, 138.93, 134.78, 129.19, 128.59, 127.88, 127.72, 120.86, 120.31, 73.09, 70.69, 35.59, 31.61, 29.99, 29.56, 29.39, 26.39, 18.49, 14.34.
1-cyano-N-(4-(6-(cyclohexylmethoxy)hexyl)phenyl)cyclopropanecarboxamide (8b)
General procedure B was used to couple 7b (205 mg, 0.708 mmol) and 1-cyano-1-cyclopanecarboxylic acid to yield the title compound. 92%. Clear and colorless oil. Rf = 0.48 (20% EtOAc in hexanes; Seebach’s Dip). 1H NMR (300 MHz, CDCl3) δ 8.12 (s, 1H), 7.32 (d, J = 8.2 Hz, 2H), 7.13 (d, J = 8.2 Hz, 2H), 3.36 (t, J = 6.5 Hz, 2H), 3.17 (d, J = 6.5 Hz, 2H), 2.67 – 2.25 (m, 2H), 1.90 – 1.02 (m, 21H), 1.02 – 0.52 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 163.50, 140.17, 134.75, 129.15, 120.83, 120.27, 77.04, 71.22, 38.27, 35.52, 31.60, 30.40, 29.88, 29.26, 26.91, 26.27, 26.13, 18.47, 14.31.
1-cyano-N-(4-(7-(cyclohexylmethoxy)heptyl)phenyl)cyclopropanecarboxamide (8c)
General procedure B was used to couple 7c (288 mg, 0.950 mmol) and 1-cyano-1-cyclopanecarboxylic acid to yield the title compound. 93%. Clear and colorless oil. Rf = 0.49 (20% EtOAc in hexanes; Seebach’s Dip). 1H NMR (300 MHz, CDCl3) δ 8.15 (s, 1H), 7.38 (d, J = 8.2 Hz, 2H), 7.11 (d, J = 7.8 Hz, 2H), 3.35 (t, J = 6.6 Hz, 2H), 3.19 (t, J = 6.5 Hz, 2H), 2.55 (t, J = 7.6 Hz, 2H), 1.89 – 0.98 (m, 23H), 0.89 (dd, J = 21.8, 10.6 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 163.52, 140.17, 134.79, 129.11, 120.86, 120.25, 77.04, 71.27, 38.26, 35.57, 31.60, 30.40, 29.94, 29.55, 29.38, 26.91, 26.35, 26.13, 18.45, 14.29.
1-cyano-N-(4-(8-(cyclohexylmethoxy)octyl)phenyl)cyclopropanecarboxamide (8d)
General procedure B was used to couple 7d (160 mg, 0.504 mmol) and 1-cyano-1-cyclopanecarboxylic acid to yield the title compound. 86%. Clear and colorless oil. Rf = 0.62 (20% EtOAc in hexanes; Seebach’s Dip). 1H NMR (300 MHz, CDCl3) δ 8.04 (s, 1H), 7.39 (d, J = 8.4 Hz, 2H), 7.15 (d, J = 8.4 Hz, 2H), 3.36 (t, J = 6.6 Hz, 2H), 3.18 (d, J = 6.6 Hz, 2H), 2.73 – 2.31 (m, 2H), 1.89 – 1.02 (m, 25H), 1.02 – 0.68 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 163.47, 140.31, 134.69, 129.17, 120.78, 120.28, 77.05, 71.31, 38.26, 35.58, 31.64, 30.40, 29.95, 29.64, 29.38, 26.91, 26.39, 26.13, 18.44, 14.31.
N-(4-(7-adamantan-1-ylmethoxy)heptyl)phenyl)-1-cyanocyclopropanecarboxamide (8e)
General procedure B was used to couple 7e (83 mg, 0.233 mmol) and 1-cyano-1-cyclopanecarboxylic acid to yield the title compound. 85%. Clear and colorless oil. Rf = 0.52 (20% EtOAc in hexanes; Seebach’s Dip). 1H NMR (300 MHz, CDCl3) δ 8.01 (s, 1H), 7.39 (d, J = 8.4 Hz, 2H), 7.15 (d, J = 8.3 Hz, 2H), 3.36 (t, J = 6.6 Hz, 2H), 2.94 (d, J = 6.7 Hz, 2H), 2.70 – 2.43 (m, 2H), 1.95 (s, 3H), 1.85 – 1.28 (m, 26H). 13C NMR (75 MHz, CDCl3) δ 163.45, 140.38, 134.60, 129.23, 120.71, 120.32, 82.10, 71.90, 39.96, 37.47, 35.58, 34.30, 31.62, 29.79, 29.54, 29.39, 28.52, 26.30, 18.48, 14.34.
N-(4-(7-(benzyloxy)heptyl)phenyl)-1-carbamimidoylcyclopropanecarboxamide hydrochloride (9a)
General procedure A was used to convert 8a (230 mg, 0.590 mmol) to the title compound. 52%. White solid. 1H NMR (500 MHz, DMSO) δ 9.54 (s, 1H), 9.15 (s, 2H), 8.88 (s, 2H), 7.44 (d, J = 8.3 Hz, 2H), 7.37 – 7.20 (m, 5H), 7.10 (d, J = 8.3 Hz, 2H), 4.40 (s, 2H), 3.38 (t, J = 6.4 Hz, 2H), 2.49 (t, J = 7.0 Hz, 2H), 1.66 – 1.35 (m, 8H), 1.26 (m, 6H). 13C NMR (126 MHz, CDCl3) δ 172.78, 171.30, 143.91, 143.23, 141.18, 133.40, 132.57, 132.49, 126.14, 76.96, 74.74, 39.69, 36.13, 34.34, 33.81, 33.71, 30.84, 19.96. LCMS: tR = 4.23; m/z = 408.3. HRMS m/z calc. for C25H34N3O2 (M+H), 408.2651; found, 408.2646.
1-carbamimidoyl-N-(4-(6-(cyclohexylmethoxy)hexyl)phenyl)cyclopropanecarboxamide hydrochloride (9b)
General procedure A was used to convert 8b (248 mg, 0.648 mmol) to the title compound. 41%. White solid. 1H NMR (300 MHz, DMSO) δ 9.67 (s, 1H), 9.03 (s, 4H), 7.47 (d, J = 8.4 Hz, 2H), 7.10 (d, J = 8.4 Hz, 2H), 3.27 (t, J = 6.3 Hz, 2H), 3.10 (d, J = 6.4 Hz, 2H), 2.49 (t, J = 5.0 Hz, 2H), 1.40 (m, 21H), 0.85 (dd, J = 21.4, 11.0 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 173.21, 171.45, 143.36, 141.52, 133.61, 126.28, 81.17, 75.51, 42.96, 39.92, 36.41, 35.01, 34.58, 33.81, 31.61, 30.96, 30.79, 20.13. LCMS: tR = 4.44; m/z = 400.3. HRMS m/z calc. for C24H38N3O2 (M+H), 400.2964; found, 400.2958.
1-carbamimidoyl-N-(4-(7-(cyclohexylmethoxy)heptyl)phenyl)cyclopropanecarboxamide hydrochloride (9c)
General procedure A was used to convert 8c (352 mg, 0.888 mmol) to the title compound. 43%. White solid. 1H NMR (300 MHz, DMSO) δ 9.71 (s, 1H), 9.12 (s, 4H), 7.47 (d, J = 8.4 Hz, 2H), 7.09 (d, J = 8.4 Hz, 2H), 3.27 (t, J = 6.4 Hz, 2H), 3.12 (d, J = 5.9 Hz, 2H), 2.49 (t, J = 7.4 Hz, 2H), 1.78 – 0.97 (m, 23H), 0.85 (dd, J = 21.6, 10.9 Hz, 2H). 13C NMR (75 MHz, DMSO) δ 168.53, 166.67, 138.61, 136.79, 128.84, 121.51, 76.43, 70.77, 38.21, 35.19, 31.64, 30.27, 29.84, 29.32, 29.23, 26.86, 26.34, 26.05, 15.36. LCMS: tR = 4.65; m/z = 414.3. HRMS m/z calc. for C25H40N3O2 (M+H), 414.3121; found, 414.3111.
1-carbamimidoyl-N-(4-(8-(cyclohexylmethoxy)octyl)phenyl)cyclopropanecarboxamide hydrochloride (9d)
General procedure A was used to convert 8d (178 mg, 0.434 mmol) to the title compound. 50%. White solid. 1H NMR (500 MHz, DMSO) δ 9.65 (s, 1H), 9.22 (s, 2H), 8.99 (s, 2H), 7.46 (d, J = 8.4 Hz, 2H), 7.11 (d, J = 8.3 Hz, 2H), 3.29 (t, J = 6.3 Hz, 2H), 3.11 (d, J = 6.4 Hz, 2H), 2.50 (t, J = 7.4 Hz, 2H), 1.78 – 0.99 (m, 25H), 0.99 – 0.74 (m, 2H). 13C NMR (126 MHz, DMSO) δ 168.19, 166.47, 138.44, 136.51, 128.62, 121.33, 76.20, 70.55, 37.98, 35.00, 31.39, 30.03, 29.62, 29.24, 28.98, 26.63, 26.14, 25.82, 15.16. LCMS: tR = 4.86; m/z = 428.3. HRMS m/z calc. for C26H42N3O2 (M+H), 428.3277; found, 428.3277.
N-(4-(7-((adamantan-1-ylmethoxy)heptyl)phenyl)-1-carbamimidoylcyclopropanecarboxamide hydrochloride (9e)
General procedure A was used to convert 8e (79 mg, 0.176 mmol) to the title compound. 22%. White solid. 1H NMR (500 MHz, DMSO) δ 9.56 (s, 1H), 9.17 (s, 2H), 8.88 (s, 2H), 7.44 (d, J = 8.5 Hz, 2H), 7.10 (d, J = 8.5 Hz, 2H), 3.28 (t, J = 6.4 Hz, 2H), 2.88 (s, 2H), 2.50 (m, 2H), 1.89 (m, 2H), 1.65 (d, J = 12.1 Hz, 3H), 1.60 – 1.25 (s, 26H). 13C NMR (126 MHz, DMSO) δ 176.39, 168.07, 167.14, 166.50, 163.30, 138.48, 136.45, 134.79, 128.61, 121.35, 81.37, 71.06, 37.16, 34.94, 34.08, 31.37, 29.93, 29.44, 28.97, 28.04, 26.04, 15.18. LCMS: tR = 5.65; m/z = 466.4. HRMS m/z calc. for C29H45N3O2 (M+H), 466.3434; found, 466.3424.
non-8-en-1-ylbenzene (10a)
General procedure F was used to couple benzylmagnesium bromide and 8-bromooct-1-ene (1.00 mL, 5.98 mmol) to yield the title compound. 84%. Clear and colorless oil. Rf = 0.75 (hexanes; KMnO4). 1H NMR (300 MHz, CDCl3) δ 7.50 – 6.98 (m, 5H), 5.84 (ddt, J = 16.9, 10.2, 6.7 Hz, 1H), 5.20 – 4.82 (m, 2H), 2.78 – 2.47 (m, 2H), 1.89 (dt, J = 7.0, 6.8 Hz, 2H), 1.64 (p, J = 7.0 Hz, 2H), 1.55 – 1.21 (m, 8H). 13C NMR (75 MHz, CDCl3) δ 143.09, 139.38, 128.63, 128.45, 125.81, 114.67, 114.42, 36.26, 34.09, 31.80, 29.56, 29.35, 29.20.
oct-7-en-1-ylbenzene (10b)
General procedure F was used to couple benzylmagnesium bromide and 7-bromohept-1-ene (1.00 mL, 6.52 mmol) to yield the title compound. 84%. Clear and colorless oil. Rf = 0.75 (hexanes; KMnO4). 1H NMR (300 MHz, CDCl3) δ 7.65 – 7.15 (m, 5H), 5.97 (ddt, J = 16.9, 10.0, 6.7 Hz, 1H), 5.13 (m, 2H), 2.98 – 2.56 (m, 2H), 2.21 (dt, J = 6.9, 6.5 Hz, 2H), 1.97 – 1.67 (m, 2H), 1.67 – 1.28 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 143.17, 139.42, 128.76, 128.59, 125.95, 114.60, 36.37, 34.20, 31.88, 29.56, 29.41, 29.26.
1-nitro-4-(9-phenylnonyl)benzene (11a)
General procedure D was used to couple 10a (971 mg, 4.80 mmol) and 1-bromo-4-nitrobenzene to yield the title compound. 59%. Tan oil. Rf = 0.67 (10% EtOAc in hexanes; Seebach’s Dip). 1H NMR (300 MHz, CDCl3) δ 8.18 (d, J = 8.6 Hz, 2H), 7.42 – 7.30 (m, 4H), 7.30 – 7.18 (m, 3H), 2.72 (m, 4H), 1.66 (m, 24.4 Hz, 4H), 1.39 (m, 10H). 13C NMR (75 MHz, CDCl3) δ 151.12, 146.49, 143.12, 129.45, 128.70, 128.55, 125.90, 123.81, 36.33, 36.15, 31.87, 31.31, 29.82, 29.75, 29.66, 29.53.
1-nitro-4-(8-phenyloctyl)benzene (11b)
General procedure D was used to couple 10b (1.00 g, 5.31 mmol) and 1-bromo-4-nitrobenzene to yield the title compound. 71%. Tan oil. Rf = 0.49 (5% EtOAc in hexanes; Seebach’s Dip). 1H NMR (300 MHz, CDCl3) δ 8.16 (d, J = 8.8 Hz, 2H), 7.39 – 7.27 (m, 4H), 7.27 – 7.15 (m, 3H), 2.72 – 2.60 (m, 4H), 1.78 – 1.48 (m, 4H), 1.40 (d, J = 19.9 Hz, 8H). 13C NMR (75 MHz, CDCl3) δ 151.09, 146.46, 143.08, 129.42, 128.67, 128.52, 125.87, 123.81, 36.25, 36.12, 31.78, 31.26, 29.67, 29.63, 29.55, 29.45.
4-(9-phenylnonyl)aniline (12a)
General procedure E was used to convert 11a (426 mg, 1.42 mmol) to the title compound. 99%. Tan oil. Rf = 0.33 (20% EtOAc in hexanes; Seebach’s Dip). 1H NMR (300 MHz, CDCl3) δ 7.48 – 7.20 (m, 5H), 7.08 (d, J = 8.3 Hz, 2H), 6.74 (d, J = 8.3 Hz, 2H), 3.58 (s, 2H), 2.87 – 2.66 (m, 2H), 2.66 – 2.53 (m, 2H), 1.86 – 1.57 (m, 4H), 1.42 (s, 10H). 13C NMR (75 MHz, CDCl3) δ 144.38, 143.27, 133.31, 129.46, 128.76, 128.58, 125.91, 115.54, 36.36, 35.45, 32.21, 31.90, 29.91, 29.71, 29.65.
4-(8-phenyloctyl)aniline (12b)
General procedure E was used to convert 11b (572 mg, 1.65 mmol) to the title compound. 99%. Tan oil. Rf = 0.31 (20% EtOAc in hexanes; Seebach’s Dip). 1H NMR (300 MHz, CDCl3) δ 7.52 – 7.20 (m, 5H), 7.11 (d, J = 8.3 Hz, 2H), 6.73 (d, J = 8.3 Hz, 2H), 3.81 – 3.37 (m, 2H), 2.77 (m, 2H), 2.63 (m, 2H), 1.72 (m, 4H), 1.43 (d, J = 19.9 Hz, 8H). 13C NMR (75 MHz, CDCl3) δ 144.45, 143.29, 133.31, 129.50, 128.81, 128.63, 125.96, 115.58, 36.40, 35.49, 32.25, 31.95, 29.90, 29.75, 29.69.
1-cyano-N-(4-(9-phenylnonyl)phenyl)cyclopropanecarboxamide (13a)
General procedure B was used to couple 12a (251 mg, 0.850 mmol) and 1-cyano-1- cyclopanecarboxylic acid to yield the title compound. 94%. Clear and colorless oil. Rf = 0.56 (20% EtOAc in hexanes; Seebach’s Dip). 1H NMR (300 MHz, CDCl3) δ 8.12 (s, H), 7.44 (d, J = 8.4 Hz, 2H), 7.37 – 7.09 (m, 7H), 2.62 (m, 4H), 1.80 (dd, J = 8.2, 4.4 Hz, 2H), 1.74 – 1.47 (m, 6H), 1.34 (s, 10H). 13C NMR (75 MHz, CDCl3) δ 163.53, 143.15, 140.34, 134.78, 129.22, 128.66, 128.49, 125.83, 120.84, 120.33, 36.26, 35.64, 31.79, 31.70, 29.76, 29.59, 29.48, 18.50, 14.37.
1-cyano-N-(4-(8-phenyloctyl)phenyl)cyclopropanecarboxamide (13b)
General procedure B was used to couple 12b (485 mg, 1.72 mmol) and 1-cyano-1- cyclopanecarboxylic acid to yield the title compound. 91%. Clear and colorless oil. Rf = 0.54 (20% EtOAc in hexanes; Seebach’s Dip). 1H NMR (300 MHz, CDCl3) δ 8.19 (s, 1H), 7.45 (d, J = 8.4 Hz, 2H), 7.39 – 7.28 (m, 2H), 7.28 – 7.12 (m, 5H), 2.64 (m, 4H), 1.81 (dd, J = 8.2, 4.4 Hz, 2H), 1.75 – 1.50 (m, 6H), 1.50 – 1.21 (m, 8H). 13C NMR (75 MHz, CDCl3) δ 163.59, 143.14, 140.29, 134.85, 129.22, 128.68, 128.52, 125.86, 120.91, 120.35, 36.27, 35.65, 31.81, 31.72, 29.72, 29.60, 29.49, 18.52, 14.38.
1-carbamimidoyl-N-(4-(9-phenylnonyl)phenyl)cyclopropanecarboxamide hydrochloride (14a)
General procedure A was used to convert 13a (312 mg, 0.803 mmol) to the title compound. 44%. White solid. 1H NMR (500 MHz, DMSO) δ 9.61 (s, 1H), 9.21 (s, 2H), 8.97 (s, 2H), 7.46 (d, J = 8.3 Hz, 2H), 7.29 – 7.19 (m, 2H), 7.18 – 7.05 (m, 5H), 2.58 – 2.48 (m, 4H), 1.69 – 1.31 (m, 8H), 1.23 (s, 10H). 13C NMR (126 MHz, DMSO) δ 168.18, 166.48, 142.74, 138.45, 136.48, 128.68, 128.64, 126.01, 121.35, 35.59, 34.97, 31.44, 29.39, 29.26, 29.06, 29.00, 15.16. LCMS: tR = 4.79; m/z = 406.3. HRMS m/z calc. for C26H36N3O (M+H), 406.2858; found, 406.2852.
1-carbamimidoyl-N-(4-(9-phenylnonyl)phenyl)cyclopropanecarboxamide hydrochloride (14b)
General procedure A was used to convert 13b (560 mg, 1.50 mmol) to the title compound. 39%. White solid. 1H NMR (300 MHz, DMSO) δ 10.50 (s, 2H), 9.62 (s, 1H), 8.89 (s, 2H), 7.45 (d, J = 7.9 Hz, 2H), 7.17 (m, 7H), 2.71 – 2.24 (m, 4H), 1.92 – 0.92 (m, 14H). 13C NMR (75 MHz, DMSO) δ 170.61, 168.67, 166.78, 142.98, 138.63, 136.80, 128.91, 128.87, 126.25, 123.40, 121.59, 35.82, 35.20, 31.67, 30.30, 29.49, 29.30, 29.24, 16.07, 15.23. LCMS: tR = 4.51; m/z = 392.3. HRMS m/z calc. for C25H34N3O (M+H), 392.2702; found, 392.2695.
((dec-9-en-1-yloxy)methyl)cyclohexane (15a)
General procedure C was used to couple cyclohexylmethanol and 10-bromodec-1-ene (4.35 mL, 21.7 mmol) to yield the title compound. 74%. Clear and colorless oil. Rf = 0.86 (10% EtOAc in hexanes; KMnO4). 1H NMR (300 MHz, CDCl3) δ5.74 (ddt, J = 16.9, 10.2, 6.7 Hz, 1H), 5.13 – 4.74 (m, 2H), 3.36 (t, J = 6.6 Hz, 2H), 3.17 (d, J = 6.6 Hz, 2H), 2.02 (dt, J = 9.9, 4.0 Hz, 2H), 1.90 – 0.99 (m, 21H), 0.98 – 0.70 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 139.30, 114.31, 77.05, 71.32, 38.29, 34.03, 30.40, 29.97, 29.66, 29.30, 29.14, 26.90, 26.40, 26.12.
((dec-9-en-1-yloxy)methyl)cyclopentane (15b)
General procedure C was used to couple cyclopentylmethanol and 10-bromodec-1-ene (1.00 mL, 4.98 mmol) to yield the title compound. 76%. Clear and colorless oil. Rf = 0.85 (10% EtOAc in hexanes; KMnO4). 1H NMR (300 MHz, CDCl3) δ 5.80 (ddt, J = 16.9, 10.2, 6.7 Hz, 1H), 5.17 – 4.75 (m, 2H), 3.39 (t, J = 6.7 Hz, 2H), 3.26 (d, J = 7.1 Hz, 2H), 2.15 (dp, J = 15.0, 7.4 Hz, 1H), 2.03 (m, 2H), 1.82 – 0.70 (m, 21H). 13C NMR (75 MHz, CDCl3) δ 139.39, 114.32, 75.77, 71.29, 39.67, 34.04, 29.94, 29.83, 29.67, 29.31, 29.14, 26.40, 25.62.
10-(cyclohexylmethoxy)decan-1-ol (16a)
General procedure G was used to convert 15a (4.00 g, 15.8 mmol) to the title compound. 81%. Clear and colorless oil. Rf = 0.68 (30% EtOAc in hexanes; Seebach’s Dip). 1H NMR (500 MHz, CDCl3) δ 3.64 (s, 1H), 3.42 (t, J = 6.7 Hz, 2H), 3.23 (t, J = 6.7 Hz, 2H), 3.04 (d, J = 6.6 Hz, 2H), 1.67 – 1.48 (m, 5H), 1.48 – 1.32 (m, 5H), 1.24 – 0.93 (m, 13H), 0.85 – 0.66 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 76.66, 70.97, 62.19, 37.84, 32.53, 30.00, 29.54, 29.49, 29.46, 29.38, 26.54, 26.03, 25.75.
10-(cyclopentylmethoxy)decan-1-ol (16b)
General procedure G was used to convert 15b (902 mg, 3.78 mmol) to the title compound. 85%. Clear and colorless oil. Rf = 0.65 (30% EtOAc in hexanes; Seebach’s Dip). 1H NMR (300 MHz, CDCl3) δ 3.69 (s, 1H), 3.38 (t, J = 6.7 Hz, 2H), 3.24 (t, J = 5.9 Hz, 2H), 3.05 (d, J = 6.3 Hz, 2H), 2.14 (sept., J = 7.4 Hz, 1H), 1.96 – 0.49 (m, 24H).
(((10-azidodecyl)oxy)methyl)cyclohexane (17a)
General procedure H was used to convert 16a (2.35 g, 8.69 mmol) to 10-(cyclohexylmethoxy)decyl methanesulfonate. 98%. Clear and colorless oil. Rf = 0.26 (10% EtOAc in hexanes; Seebach’s Dip). 1H NMR (500 MHz, CDCl3) δ 4.07 (t, J = 6.6 Hz, 2H), 3.23 (t, J = 6.5 Hz, 2H), 3.05 (d, J = 6.6, 2H), 2.86 (s, 3H), 1.69 – 1.48 (m, 7H), 1.48 – 1.35 (m, 3H), 1.35 – 0.94 (m, 15H), 0.86 – 0.68 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 76.61, 70.88, 70.15, 37.94, 36.99, 30.02, 29.62, 29.33, 29.30, 29.23, 28.98, 28.89, 26.56, 26.04, 25.78, 25.28. General procedure I was then used to convert the alkyl mesylate (2.05 g, 5.88 mmol) to the title compound. 94%. Clear and colorless oil. Rf = 0.93 (10% EtOAc in hexanes; Seebach’s Dip). 1H NMR (500 MHz, CDCl3) δ 3.30 (t, J = 6.6 Hz, 2H), 3.17 (t, J = 7.0 Hz, 2H), 3.12 (d, J = 6.6 Hz, 2H), 1.74 – 1.56 (m, 5H), 1.56 – 1.40 (m, 5H), 1.35 – 1.00 (m, 15H), 0.94 – 0.76 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 76.71, 70.98, 51.33, 38.03, 30.10, 29.69, 29.42, 29.37, 29.07, 28.78, 26.63, 26.12, 25.85.
(((10-azidodecyl)oxy)methyl)cyclopentane (17b)
General procedure H was used to convert 16b (904 mg, 3.21 mmol) to 10-(cyclohexylmethoxy)decyl methanesulfonate. Rf = 0.22 (10% EtOAc in hexanes; Seebach’s Dip). The alkyl mesylate was not purified by flash chromatography in the example, and was taken on to the next step crude. This method was later shown to be inferior to that of the synthesis of 17a. General procedure I was then used to convert the alkyl mesylate to the title compound. 63% over 2 steps. Clear and colorless oil. Rf = 0.93 (10% EtOAc in hexanes; Seebach’s Dip). 1H NMR (300 MHz, CDCl3) δ 3.38 (t, J = 6.6 Hz, 2H), 3.29 – 3.14 (m, 4H), 2.24 – 1.94 (m, 1H), 1.93 – 0.55 (m, 24H).
1-cyano-N-(10-(cyclohexylmethoxy)decyl)cyclopropanecarboxamide (18a)
General procedure J was used to couple 17a (1.55 g, 5.25 mmol) and 1-cyano-1- cyclopanecarboxylic acid to yield the title compound. 82%. Clear and colorless oil. Rf = 0.56 (20% EtOAc in hexanes; Seebach’s Dip). 1H NMR (500 MHz, CDCl3) δ 6.59 (t, J = 5.4 Hz, 1H), 3.25 (t, J = 6.6 Hz, 2H), 3.14 (dt, J = 6.6, 5.4 Hz, 2H), 3.06 (d, J = 6.6 Hz, 2H), 1.67 – 1.35 (m, 12H), 1.33 (m, 2H), 1.26 – 0.95 (m, 15H), 0.79 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 164.86, 120.06, 76.64, 70.92, 40.48, 37.95, 30.04, 29.63, 29.38, 29.33, 29.26, 29.11, 26.68, 26.57, 26.06, 25.79, 17.22, 13.28.
1-cyano-N-(10-(cyclopentylmethoxy)decyl)cyclopropanecarboxamide (18b)
General procedure J was used to couple 17b (569 mg, 2.02 mmol) and 1-cyano-1- cyclopanecarboxylic acid to yield the title compound. 84%. Clear and colorless oil. Rf = 0.54 (20% EtOAc in hexanes; Seebach’s Dip). 1H NMR (300 MHz, CDCl3) δ 6.52 (s, 1H), 3.32 (t, J = 6.6 Hz, 2H), 3.19 (d, J = 7.1 Hz, 2H), 2.07 (sept., J = 7.4 Hz, 1H), 1.77 – 0.99 (m, 28H).
1-carbamimidoyl-N-(10-(cyclohexylmethoxy)decyl)cyclopropanecarboxamide hydrochloride (19a)
General procedure A was used to convert 18a (1.56 g, 4.30 mmol) to the title compound. 44%. White solid. 1H NMR (500 MHz, DMSO) δ 9.04 (s, 4H), 7.83 (t, J = 5.6 Hz, 1H), 3.28 (t, J = 6.5 Hz, 2H), 3.11 (d, J = 6.4 Hz, 2H), 3.02 (dt, J = 6.7, 5.6 Hz, 2H), 1.71 – 1.54 (m, 5H), 1.53 – 1.00 (m, 24H), 0.94 – 0.76 (m, 2H). 13C NMR (126 MHz, DMSO) δ 168.34, 167.68, 76.20, 70.56, 37.97, 30.03, 29.64, 29.47, 29.42, 29.32, 29.23, 26.82, 26.62, 26.17, 25.81, 14.62. LCMS: tR = 4.29; m/z = 380.3. HRMS m/z calc. for C22H42N3O2 (M+H), 380.3277; found, 380.3268.
1-carbamimidoyl-N-(10-(cyclohexylmethoxy)decyl)cyclopropanecarboxamide hydrochloride (19b)
General procedure A was used to convert 18b (591 mg, 1.70 mmol) to the title compound. 29%. White solid. 1H NMR (500 MHz, DMSO) δ 9.08 (s, 4H), 7.78 (t, J = 5.4 Hz, 1H), 3.34 – 3.28 (m, 2H), 3.18 (d, J = 7.0 Hz, 2H), 3.03 (td, J = 6.8, 5.4 Hz, 2H), 2.12 – 1.95 (m, 1H), 1.68 – 1.57 (m, 2H), 1.57 – 1.31 (m, 10H), 1.31 – 1.10 (m, 16H). 13C NMR (126 MHz, DMSO) δ 168.15, 167.73, 74.92, 70.52, 39.37, 29.63, 29.54, 29.46, 29.41, 29.34, 29.22, 28.91, 26.81, 26.17, 25.42, 16.85, 14.62. LCMS: tR = 4.08; m/z = 366.3. HRMS m/z calc. for C21H40N3O2 (M+H), 366.3121; found, 366.3111.
4-(7-(cyclohexylmethoxy)heptyl)benzaldehyde (20a)
General procedure D was used to couple 5c (1.08 g, 5.12 mmol) and 4-bromobenzaldehyde to yield the title compound. 77%. Clear and colorless oil. Rf = 0.60 (10% EtOAc in hexanes; Seebach’s Dip). 1H NMR (300 MHz, CDCl3) δ 9.94 (t, J = 2.0 Hz, 1H), 7.76 (d, J = 8.2 Hz, 2H), 7.30 (d, J = 8.0 Hz, 2H), 3.34 (t, J = 6.6 Hz, 2H), 3.16 (d, J = 6.6 Hz, 2H), 2.74 – 2.52 (m, 2H), 1.84 – 1.03 (m, 19H), 0.87 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 191.83, 150.40, 134.57, 129.96, 129.17, 76.94, 71.13, 38.22, 36.29, 31.16, 30.32, 29.88, 29.43, 29.35, 26.84, 26.27, 26.07.
4-(7-(cyclopentylmethoxy)heptyl)benzaldehyde (20b)
General procedure D was used to couple 5f (500 mg, 2.55 mmol) and 4-bromobenzaldehyde to yield the title compound. 77%. Clear and colorless oil. Rf = 0.72 (10% EtOAc in hexanes; Seebach’s Dip). 1H NMR (300 MHz, CDCl3) δ 9.89 (s, 1H), 7.72 (d, J = 8.0 Hz, 2H), 7.26 (d, J = 8.1 Hz, 2H), 3.33 (t, J = 6.6 Hz, 2H), 3.19 (d, J = 7.1 Hz, 2H), 2.68 – 2.49 (m, 2H), 2.08 (sept., J = 7.4 Hz, 1H), 1.77 – 1.37 (m, 10H), 1.28 – 1.06 (m, 8H). 13C NMR (75 MHz, CDCl3) δ 191.97, 150.49, 134.59, 130.01, 129.22, 75.69, 71.13, 39.65, 36.34, 31.18, 29.88, 29.78, 29.45, 29.37, 26.29, 25.60.
4-(7-butoxyheptyl)benzaldehyde (20c)
General procedure D was used to couple 5g (1.26 g, 7.39 mmol) and 4-bromobenzaldehyde to yield the title compound. 89%. Clear and colorless oil. Rf = 0.48 (10% EtOAc in hexanes; Seebach’s Dip). 1H NMR (500 MHz, CDCl3) δ 9.83 (s, 1H), 7.66 (d, J = 8.2 Hz, 2H), 7.19 (d, J = 8.2 Hz, 2H), 3.26 (m, 4H), 2.70 – 2.35 (m, 2H), 1.60 – 1.06 (m, 14H), 0.77 (t, J = 7.5 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 191.53, 150.12, 134.33, 129.69, 128.90, 70.68, 70.46, 36.03, 31.78, 30.90, 29.65, 29.18, 29.08, 26.02, 19.29, 13.84.
N-(1-cyanocyclopropyl)-4-(7-(cyclohexylmethoxy)heptyl)benzamide (22a)
General procedure K was used to convert 20a (1.25 g, 3.94 mmol) to is corresponding carboxylic acid 21a, which was taken on to the next reaction crude. General procedure I was then used to convert the crude carboxylic acid to its acid chloride and was used immediately after purification. General procedure M was used to couple the acid chloride and 1-amino-1-cyclopropanecarbonitirle to yield the title compound. 57% over 3 steps. While solid. Rf = 0.52 (40% EtOAc in hexanes; Seebach’s Dip). 1H NMR (300 MHz, CDCl3) δ 7.68 (dd, J = 8.3, 2.7 Hz, 2H), 7.34 – 7.12 (m, 2H), 6.97 (d, J = 20.0 Hz, 1H), 3.36 (td, J = 6.6, 2.9 Hz, 2H), 3.18 (dd, J = 6.6, 2.8 Hz, 2H), 2.63 (dd, J = 10.1, 5.2 Hz, 2H), 1.87 – 0.99 (m, 25H), 0.99 – 0.73 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 168.63, 148.03, 130.24, 128.87, 127.66, 120.65, 77.06, 71.26, 38.22, 36.03, 31.30, 30.37, 29.90, 29.49, 29.37, 26.87, 26.31, 26.09, 21.11, 16.99.
N-(1-cyanocyclopropyl)-4-(7-(cyclopentylmethoxy)heptyl)benzamide (22b)
General procedure K was used to convert 20b (536 mg, 1.77 mmol) to is corresponding carboxylic acid 21b, which was taken on to the next reaction crude. General procedure I was then used to convert the crude carboxylic acid to its acid chloride and was used immediately after purification. General procedure M was used to couple the acid chloride and 1-amino-1-cyclopropanecarbonitirle to yield the title compound. 61% over 3 steps. While solid. Rf = 0.48 (40% EtOAc in hexanes; Seebach’s Dip). 1H NMR (300 MHz, CDCl3) δ 7.91 (s, 1H), 7.71 (d, J = 8.2 Hz, 2H), 7.15 (d, J = 8.2 Hz, 2H), 3.36 (t, J = 9.6 Hz, 2H), 3.22 (d, J = 7.2 Hz, 2H), 2.58 (t, J = 6.6 Hz, 2H), 2.18 – 2.00 (m, 1H), 1.80 – 0.98 (m, 22H). 13C NMR (75 MHz, CDCl3) δ 168.68, 147.90, 130.29, 128.80, 127.71, 120.70, 75.76, 71.20, 39.63, 36.02, 31.29, 29.87, 29.81, 29.49, 29.36, 26.31, 25.59, 21.11, 16.92.
4-(7-butoxyheptyl)-N-(1-cyanocyclopropyl)benzamide (22c)
General procedure K was used to convert 20c (1.40 g, 5.07 mmol) to is corresponding carboxylic acid 21c, which was taken on to the next reaction crude. General procedure I was then used to convert the crude carboxylic acid to its acid chloride and was used immediately after purification. General procedure M was used to couple the acid chloride and 1-amino-1- cyclopropanecarbonitirle to yield the title compound. 78% over 3 steps. While solid. Rf = 0.32 (40% EtOAc in hexanes; Seebach’s Dip). 1H NMR (300 MHz, CDCl3) δ 8.20 (s, 1H), 7.73 (d, J = 8.1 Hz, 2H), 7.13 (d, J = 8.1 Hz, 2H), 3.57 – 3.13 (m, 4H), 2.53 (m, 2H), 1.64 – 1.42 (m, 8H), 1.42 – 1.13 (m, 10H), 0.86 (t, J = 7.3 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 169.04, 147.95, 130.24, 128.83, 127.77, 120.80, 71.05, 70.83, 36.01, 32.02, 31.29, 29.92, 29.48, 29.36, 26.31, 21.15, 19.56, 16.87, 14.16.
N-(1-carbamimidoylcyclopropyl)-4-(7-(cyclohexylmethoxy)heptyl)benzamide hydrochloride (23a)
General procedure A was used to convert 22a (890 mg, 2.25 mmol) to the title compound. 71%. White solid. 1H NMR (500 MHz, DMSO) δ 9.12 (s, 1H), 8.86 (s, 2H), 8.57 (s, 2H), 7.82 (d, J = 8.2 Hz, 2H), 7.26 (d, J = 8.2 Hz, 2H), 3.28 (t, J = 6.4 Hz, 2H), 3.11 (d, J = 6.4 Hz, 2H), 2.60 (t, J = 7.5 Hz, 2H), 1.78 – 0.52 (m, 25H). 13C NMR (126 MHz, CDCl3) δ 177.08, 172.73, 151.72, 135.97, 133.16, 133.08, 80.94, 75.27, 42.72, 40.10, 37.82, 35.92, 34.78, 34.34, 33.80, 33.69, 31.37, 30.85, 30.55, 23.17. LCMS: tR = 4.36; m/z = 414.3. HRMS m/z calc. for C25H40N3O2 (M+H), 414.3121; found, 414.3111.
N-(1-carbamimidoylcyclopropyl)-4-(7-(cyclopentylmethoxy)heptyl)benzamide hydrochloride (23b)
General procedure A was used to convert 22b (413 mg, 1.08 mmol) to the title compound. 73%. White solid. 1H NMR (500 MHz, DMSO) δ 9.15 (s, 1H), 8.94 (s, 2H), 8.60 (s, 2H), 7.81 (d, J = 7.6 Hz, 2H), 7.26 (d, J = 7.6 Hz, 2H), 3.30 (t, J = 5.6 Hz, 2H), 3.17 (d, J = 7.0 Hz, 2H), 2.60 (t, J = 7.4 Hz, 2H), 2.13 – 1.94 (m, 1H), 1.75 – 1.01 (m, 22H). 13C NMR (126 MHz, DMSO) δ 172.42, 167.98, 146.94, 131.23, 128.41, 128.35, 74.92, 70.49, 35.35, 33.06, 31.16, 29.59, 29.54, 29.05, 28.95, 26.10, 25.42, 18.41. LCMS: tR = 4.29; m/z = 400.3. HRMS m/z calc. for C24H38N3O2 (M+H), 400.2964; found, 400.2957.
4-(7-butoxyheptyl)-N-(1-carbamimidoylcyclopropyl)benzamide hydrochloride (23c)
General procedure A was used to convert 22c (791 mg, 2.22 mmol) to the title compound. 60%. White solid. 1H NMR (500 MHz, DMSO) δ 9.14 (s, 1H), 8.75 (s, 4H), 7.80 (d, J = 7.7 Hz, 2H), 7.26 (d, J = 7.3 Hz, 2H), 3.29 (m, 4H), 2.60 (t, J = 7.4 Hz, 2H), 1.75 – 0.93 (m, 18H), 0.93 – 0.48 (m, 3H). 13C NMR (126 MHz, DMSO) δ 172.38, 167.98, 146.95, 131.22, 128.41, 128.34, 70.33, 70.04, 35.35, 33.07, 31.79, 31.17, 29.64, 29.07, 28.94, 26.11, 19.35, 18.40, 14.23. LCMS: tR = 4.08; m/z = 374.3. HRMS m/z calc. for C22H36N3O2 (M+H), 374.2808; found, 374.2802.
11-(cyclohexylmethoxy)undecanoic acid (24)
General procedure C was used to couple cyclohexylmethanol and 11-bromoundecenoic acid (1.00 g, 3.77 mmol), with TBAB (121 mg, 0.38 mmol, 0.1 eq.) being added along with the 11-bromoundecenoic acid in one portion, to yield the title compound. 74%. Clear and colorless oil. Rf = 0.80 (60% EtOAc in hexanes; KMnO4). 1H NMR (500 MHz, CDCl3) δ 6.46 (s, 1H), 3.30 (d, J = 6.6 Hz, 2H), 2.21 (t, J = 7.5 Hz, 2H), 1.72 – 0.95 (m, 27H), 0.93 – 0.65 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 177.87, 76.69, 71.00, 40.15, 34.00, 30.03, 29.52, 29.45, 29.36, 29.32, 29.18, 29.02, 26.52, 25.78, 24.73.
N-(1-cyanocyclopropyl)-11-(cyclohexylmethoxy)undecanamide (25)
General procedure B was used to couple 24 (358 mg, 3.02 mmol) and 1-amino-1- cyclopropanecarbonitirle to yield the title compound. 81%. White solid. Rf = 0.51 (20% EtOAc in hexanes; Seebach’s Dip). 1H NMR (500 MHz, CDCl3) δ 7.23 – 7.13 (m, 1H), 3.34 (t, J = 6.7 Hz, 2H), 3.16 (d, J = 6.6 Hz, 2H), 2.16 (t, J = 7.6 Hz, 2H), 1.78 – 0.99 (m, 31H), 0.86 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 174.62, 120.27, 76.79, 71.07, 37.95, 35.86, 30.12, 29.68, 29.49, 29.42, 29.37, 29.28, 29.14, 26.62, 26.11, 25.83, 25.28, 16.58.
N-(1-carbamimidoylcyclopropyl)-11-(cyclohexylmethoxy)undecanamide hydrochloride (26)
General procedure A was used to convert 25 (791 mg, 2.22 mmol) to the title compound. 68%. White solid. 1H NMR (500 MHz, DMSO) δ 9.26 – 7.91 (m, 5H), 3.28 (t, J = 6.4 Hz, 2H), 3.10 (d, J = 6.4 Hz, 2H), 2.09 (t, J = 7.5 Hz, 2H), 1.72 – 0.64 (m, 33H). 13C NMR (126 MHz, DMSO) δ 174.53, 172.36, 76.19, 70.55, 37.96, 35.41, 32.38, 30.02, 29.63, 29.49, 29.44, 29.26, 29.12, 26.61, 26.15, 25.81, 24.92, 18.36. LCMS: tR = 4.36; m/z = 380.3. HRMS m/z calc. for C22H42N3O2 (M+H), 380.3277; found, 380.3271.
1-aminocyclobutanecarbonitrile hydrochloride (31)
To a solution of cyclobutanone (930 mg, 1.0 eq.) in 2N NH3/MeOH (0.2 M), was added potassium cyanide (2.0 eq.) and ammonium chloride (2.0 eq.). The reaction was run under ammonia gas (1 atm) at rt for 17 h. The reaction was then evaporated and the inorganic salts precipitated in CHCl3 and removed via filtration through a fine frit. The solution was diluted in MeOH (100 mL), cooled to 0 °C, and treated with 12.1 N HCl (2.0 eq.). The reaction was let stir for 5 min and then evaporated to dryness to yield the title compound. 60%. White solid. 1H NMR (300 MHz, CD3OD) δ 2.73 – 2.47 (m, 2H), 2.29 (dt, J = 12.7, 8.8 Hz, 2H), 2.19 – 1.93 (m, 2H). 13C NMR (75 MHz, CD3OD) δ 123.66, 37.44, 35.80, 15.47.
N-(1-cyanocyclobutyl)-4-dodecylbenzamide (32)
General procedure L was used to couple 31 (87 mg, 0.90 mmol) and 4-dodecylbenzoyl chloride to yield the title compound. 30%. Clear and colorless oil. Rf = 0.48 (25% EtOAc in hexanes; Seebach’s Dip). 1H NMR (300 MHz, CDCl3) δ 8.44 (d, J = 8.2 Hz, 2H), 7.99 (d, J = 8.7 Hz, 2H), 7.26 (s, 1H), 3.71 – 3.52 (m, 2H), 3.44 – 3.33 (m, 2H), 3.21 (dd, J = 21.0, 9.6 Hz, 2H), 3.11 – 2.78 (m, 2H), 2.35 (m, 2H), 2.16 – 1.91 (m, 18H), 1.62 (t, J = 6.6 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 166.71, 147.92, 130.05, 128.73, 127.21, 120.88, 48.00, 35.86, 34.14, 31.90, 31.16, 29.63, 29.56, 29.45, 29.34, 29.22, 22.68, 16.17, 14.13.
N-(1-carbamimidoylcyclobutyl)-4-dodecylbenzamide (33)
General procedure A was used to convert 32 (100 mg, 0.27 mmol) to the title compound. 7%. White solid. 1H NMR (500 MHz, DMSO) δ 9.15 (s, 1H), 8.82 (s, 4H), 7.88 (d, J = 8.2 Hz, 2H), 7.30 (d, J = 8.2 Hz, 2H), 2.68 – 2.56 (m, 4H), 2.42 (dd, J = 17.6, 11.1 Hz, 2H), 2.10 – 1.96 (m, 1H), 1.96 – 1.82 (m, 1H), 1.55 (pent., J = 6.8 Hz, 2H), 1.25 (m, 18H), 0.85 (t, J = 6.8 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 172.70, 166.46, 146.63, 130.64, 128.08, 127.95, 57.31, 34.98, 31.32, 31.01, 30.80, 29.03, 28.85, 28.73, 28.58, 22.12, 15.05, 13.99. LCMS: tR = 5.29; m/z = 386.3. HRMS m/z calc. for C24H40N3O (M+H), 386.3171; found, 386.3178.
(S)-tert-butyl 2-cyanopyrrolidine-1-carboxylate (34a)
To a solution of L-proline (600 mg, 5.21 mmol) in dioxane (7 mL) and 10% Na2CO3(aq) (14 mL) was added Boc2O (2.0 eq.) at rt and let stir for 16 h. The reaction was washed 2x with hexanes (100 mL), then acidified with 1 N HCl (100 mL) and extracted 3x with EtOAc (200 mL). The EtOAc layer was dried with Na2SO4 and evaporated to a white solid. The crude white solid was then dissolved in CH2Cl2 (0.3 M) at 0ºC and treated with TEA (3.0 eq.) then isobutyl chloroformate (1.1 eq.). The reaction turned turbid after the addition and was let warm to rt before slowly clearing over time. After 1 h at rt, the reaction was treated with 2 M NH3 in MeOH (2.0 eq.) and let stir for 6 h. The reaction was evaporated to a white solid and taken on crude. The crude solid was dissolved in DMF (0.1 M) and 2,4,6-collidine (8 eq.) at 0ºC and then treated with cyanuric chloride (3.15 eq.) and let warm to rt. The reaction was let stir for 12 h. The reaction was then extracted with EtOAc (20x the volume of DMF) and washed 3x with sat. NaHCO3 (10x the volume of DMF), 3x with 1 N HCl (10x the volume of DMF), and once with brine (10x the volume of DMF). The organic layer was then dried with Na2SO4, evaporated to a yellow oil, and immediately purified by flash chromatography to yield the title compound. 98%. Clear and colorless oil. Rf = 0.37 (20% EtOAc in hexanes; Ninhydrin). 1H NMR (300 MHz, CDCl3) δ 4.46 – 4.10 (m, 1H), 3.30 (dd, J = 13.9, 7.8 Hz, 1H), 3.16 (dd, J = 17.5, 8.1 Hz, 1H), 2.15 – 1.95 (m, 2H), 1.95 – 1.76 (m, 2H), 1.39 – 1.21 (m, 9H). 13C NMR (75 MHz, CDCl3) δ 153.01, 119.28, 81.08, 47.23, 45.79, 31.68, 30.84, 28.29, 24.73, 23.87.
(S)-1-(4-dodecylbenzoyl)pyrrolidine-2-carbonitrile (35a)
General procedure N was use to deprotect 34 (400 mg, 2.05 mmol). General procedure B was used to couple the deprotected 34 and 4-dodecylbenzoic acid to yield the title compound. 57%. White solid. Rf = 0.75 (40% EtOAc in hexanes; Seebach’s Dip). 1H NMR (500 MHz, CDCl3) δ 7.50 (d, J = 8.0 Hz, 2H), 7.22 (d, J = 7.6 Hz, 2H), 4.96 – 4.84 (m, 1H), 3.73 – 3.63 (m, 1H), 3.62 – 3.52 (m, 1H), 2.68 – 2.58 (m, 2H), 2.39 – 2.26 (m, 1H), 2.24 – 2.10 (m, 1H), 2.06 – 1.96 (m, 1H), 1.67 – 1.55 (m, 1H), 1.39 – 1.18 (m, 20H), 0.87 (t, J = 6.9 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 170.18, 146.28, 128.40, 127.61, 127.17, 118.73, 49.51, 46.87, 35.84, 31.91, 31.20, 30.30, 29.65, 29.56, 29.46, 29.34, 29.23, 25.58, 22.68, 14.12.
(S)-1-(4-dodecylbenzoyl)pyrrolidine-2-carboximidamide hydrochloride (36a)
General procedure A was used to convert 35a (213 mg, 0.519 mmol) to the title compound. 64%. White solid. 1H NMR (500 MHz, CD3OD) δ 7.63 (d, J = 8.0 Hz, 2H), 7.30 (d, J = 8.0 Hz, 2H), 4.73 (t, J = 7.5 Hz, 1H), 3.87 (dd, J = 15.4, 9.0 Hz, 1H), 3.73 – 3.60 (m, 1H), 2.67 (t, J = 7.6 Hz, 2H), 2.54 (dd, J = 13.3, 6.6 Hz, 1H), 2.12 – 1.93 (m, 1H), 1.71 – 1.56 (m, 1H), 1.34 – 1.28 (m, 18H), 0.90 (t, J = 6.9 Hz, 3H). 13C NMR (126 MHz, CD3OD) δ 171.79, 146.52, 131.91, 128.06, 127.63, 58.94, 50.59, 35.36, 31.66, 31.14, 31.03, 29.33, 29.28, 29.15, 29.06, 28.88, 25.27, 22.32, 13.03. LCMS: tR = 5.04; m/z = 386.3. HRMS m/z calc. for C24H40N3O (M+H), 386.3171; found, 386.3175.
(R)-1-(4-dodecylbenzoyl)pyrrolidine-2-carbonitrile (35b)
General procedure N was use to deprotect ent-34a (275 mg, 1.40 mmol). General procedure B was used to couple the deprotected ent-34a and 4-dodecylbenzoic acid to yield the title compound. 53%. White solid. Rf = 0.75 (40% EtOAc in hexanes; Seebach’s Dip). 1H and 13C NMR data were identical to that of 35a.
(R)-1-(4-dodecylbenzoyl)pyrrolidine-2-carboximidamide hydrochloride (36b)
General procedure A was used to convert 35b (213 mg, 0.519 mmol) to the title compound. 73%. White solid. 1H and 13C NMR data were identical to that of 36a.
(S)-1-(4-(7-(cyclohexylmethoxy)heptyl)benzoyl)pyrrolidine-2-carbonitrile (37)
General procedure N was use to deprotect 34 (179 mg, 0.911 mmol). General procedure B was used to couple the deprotected 34 and 21a to yield the title compound. 61%. Clear and colorless oil. Rf = 0.38 (40% EtOAc in hexanes; Seebach’s Dip). 1H NMR (300 MHz, CDCl3) δ 7.48 (d, J = 8.1 Hz, 2H), 7.20 (d, J = 8.0 Hz, 2H), 4.99 – 4.68 (m, 1H), 3.72 – 3.44 (m, 2H), 3.35 (t, J = 6.6 Hz, 2H), 3.16 (d, J = 6.6 Hz, 2H), 2.59 (t, J = 9.7 Hz, 2H), 2.42 – 1.90 (m, 4H), 1.57 (m, 10H), 1.40 – 1.01 (m, 9H), 1.01 – 0.72 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 170.19, 146.40, 133.46, 128.66, 127.79, 118.72, 77.05, 71.25, 58.68, 49.97, 38.24, 36.02, 31.33, 31.12, 30.37, 29.92, 29.50, 29.36, 26.88, 26.31, 26.10.
(S)-1-(4-(7-(cyclohexylmethoxy)heptyl)benzoyl)pyrrolidine-2-carboximidamide hydrochloride (38)
General procedure A was used to convert 37 (213 mg, 0.519 mmol) to the title compound. 85%. White solid. 1H NMR (500 MHz, DMSO) δ 9.11 (s, 2H), 8.85 (s, 2H), 7.61 (d, J = 7.8 Hz, 2H), 7.25 (d, J = 7.7 Hz, 2H), 4.61 (t, J = 6.7 Hz, 1H), 3.81 (dd, J = 14.4, 7.4 Hz, 1H), 3.39 (m, 1H), 3.28 (t, J = 8.5, 2H), 3.09 (d, J = 6.5 Hz, 2H), 2.57 (m, 2H), 2.35 – 1.87 (m, 4H), 1.71 – 1.35 (m, 9H), 1.35 – 0.96 (m, 10H), 0.86 (m, 2H). 13C NMR (126 MHz, DMSO) δ 171.55, 169.93, 145.60, 133.21, 128.38, 128.30, 76.19, 70.53, 58.49, 50.66, 37.97, 35.39, 31.40, 31.13, 30.03, 29.59, 29.04, 26.62, 26.07, 25.81, 25.67. LCMS: tR = 4.65; m/z = 428.3. HRMS m/z calc. for C26H42N3O2 (M+H), 428.3277; found, 428.3274.
(S)-1-(11-(cyclohexylmethoxy)undecanoyl)pyrrolidine-2-carbonitrile (39)
General procedure N was use to deprotect 34 (371 mg, 1.89 mmol). General procedure B was used to couple the deprotected 34 and 24 to yield the title compound. 84%. Clear and colorless oil. Rf = 0.29 (40% EtOAc in hexanes; Seebach’s Dip). 1H NMR (300 MHz, CDCl3) δ 4.64 (d, J = 6.0 Hz, 1H), 3.53 (dd, J = 8.3, 7.1 Hz, 1H), 3.42 – 3.22 (m, 3H), 3.09 (d, J = 6.5 Hz, 2H), 2.44 – 1.84 (m, 6H), 1.78 – 1.35 (m, 6H), 1.35 – 0.96 (m, 19H), 0.81 (dd, J = 21.9, 10.6 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 172.23, 114.29, 77.45, 71.23, 46.51, 46.38, 38.19, 34.59, 30.31, 30.22, 29.89, 29.68, 29.55, 29.44, 26.83, 26.31, 26.05, 25.30, 24.56.
(S)-1-(11-(cyclohexylmethoxy)undecanoyl)pyrrolidine-2-carboximidamide hydrochloride (40)
General procedure A was used to convert 39 (433 mg, 1.15 mmol) to the title compound. 53%. White solid. 1H NMR (500 MHz, DMSO) δ 8.89 (s, 4H), 4.45 (dd, J = 9.0, 4.0 Hz, 1H), 3.68 (ddd, J = 9.3, 7.2, 4.8 Hz, 1H), 3.41 (dd, J = 16.6, 7.3 Hz, 1H), 3.34 (s, 1H), 3.27 (t, J = 8.6 Hz, 2H), 3.09 (d, J = 8.5 Hz, 2H), 2.40 – 2.07 (m, 4H), 2.04 – 0.98 (m, 27H), 0.93 – 0.68 (m, 2H). 13C NMR (126 MHz, DMSO) δ 172.43, 171.78, 76.20, 70.56, 57.22, 47.31, 37.97, 33.86, 31.25, 30.03, 29.47, 29.39, 29.30, 29.23, 26.62, 25.81, 24.72, 24.22. LCMS: tR = 4.51; m/z = 394.3. HRMS m/z calc. for C23H44N3O2 (M+H), 394.3434; found, 394.3425.
1-(3-(2-cyclohexylethyl)phenyl)ethanone (48)
To a 0.5 M solution of 9-BBN (1.5 eq.) was added vinylcyclohexane (1.5 mL, 11.3 mmol, 1.5 eq.) and let stir for 12 h. The reaction was then treated with 3N NaOH(aq) (1.7 eq.), followed by the solid additions of 3-bromoacetophenone (1.00 mL, 7.56 mmol, 1.0 eq.) and Pd(PPh3)4 sequentially. The reaction was then heated to reflux for 1 h, then cooled to rt and extracted 2x into EtOAc (400 mL). The organic layer was then dried with Na2SO4, evaporated to a black oil, and immediately purified by flash chromatography to yield the title compound. 82%. Clear and colorless oil. Rf = 0.58 (10% EtOAc in hexanes; Seebach’s Dip). 1H NMR (300 MHz, CDCl3) δ 7.85 – 7.58 (m, 2H), 7.42 – 7.07 (m, 2H), 2.59 (t, J = 7.3 Hz, 2H), 2.51 (s, 3H), 1.88 – 1.53 (m, 5H), 1.53 – 1.35 (m, 2H), 1.32 – 1.00 (m, 4H), 0.88 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 198.29, 143.87, 137.36, 133.36, 128.63, 128.19, 126.02, 39.53, 37.54, 33.46, 33.33, 26.85, 26.79, 26.52.
2-bromo-1-(3-(2-cyclohexylethyl)phenyl)ethanone (49)
To a solution of 48 (2.14 g, 9.28 mmol) in CHCl3 (40 mL, 0.23 M) at 40ºC was added Br2 (1.0 eq.) dropwise. The reaction was complete immediately after the addition of the bromine, and was cooled to rt and quenched with sat. NaHCO3 (100 mL). The mixture was then extracted 3x with CHCl3 (100 mL). The organic layer was then dried with Na2SO4, evaporated to a black oil, and immediately purified by flash chromatography to yield the title compound. 92%. Yellow oil. Rf = 0.71 (10% EtOAc in hexanes; Seebach’s Dip). 1H NMR (300 MHz, CDCl3) δ 7.90 – 7.66 (m, 2H), 7.45 – 7.28 (m, 2H), 4.42 (s, 2H), 2.74 – 2.56 (m, 2H), 1.80 – 1.54 (m, 5H), 1.47 (dt, J = 10.4, 7.1 Hz, 2H), 1.32 – 1.01 (m, 4H), 0.89 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 191.54, 144.30, 134.30, 134.21, 129.04, 128.95, 126.56, 39.45, 37.51, 33.49, 33.32, 31.67, 26.89, 26.56.
4-(tert-butoxycarbonyl)benzoic acid
To a mixture of terephthalic acid (6.64 g, 40.0 mmol), Boc2O (1.0 eq.), and DMAP (0.25 eq.) was added tBuOH (60 mL) and THF (20 mL) and heated to reflux for 24 h. The reaction was then evaporated to dryness and immediately purified by flash chromatography to yield the title compound. 36%. White solid. Rf = 0.50 (5% MeOH in CHCl3; KMnO4). Rotomer A: 1H NMR (300 MHz, DMSO) δ 8.20 – 7.74 (m, 4H), 1.54 (s, 9H), 1.41 – 1.17 (m, 2H). Rotomer B: 1H NMR (300 MHz, DMSO) δ 8.20 – 7.74 (m, 4H), 1.41 – 1.17 (m, 9H). Rotomer ratio A/B = 1.95.
tert-butyl (2-(3-(2-cyclohexylethyl)phenyl)-2-oxoethyl) terephthalate (50)
A solution of 4-(tert-butoxycarbonyl)benzoic acid (1.10 g, 4.95 mmol) and Cs2CO3 (0.51 eq.) in EtOH (80 mL, 0.06 M) was sonicated for 6 m at rt. The solvent was then evaporated and dried under vacuum for 20 min. The cesium carboxylate salt was then dissolved in DMF (30 mL) and 49 (1.2 eq.) was transferred by cannula in DMF (10 mL) at rt. The reaction was let stir for 12 h before being diluted with EtOAc (400 mL) and washed 3x with neat water (100 mL). The organic layer was then dried with Na2SO4, evaporated to a yellow oil, and immediately purified by flash chromatography to yield the title compound. 99%. Tan oil. Rf = 0.43 (10% EtOAc in hexanes; Seebach’s Dip). 1H NMR (300 MHz, CDCl3) δ 8.11 (d, J = 6.8 Hz, 2H), 8.01 (d, J = 6.8 Hz, 2H), 7.73 (m, 2H), 7.47 – 7.04 (m, 2H), 5.55 (s, 2H), 2.62 (t, J = 6.9 Hz, 2H), 1.84 – 0.52 (m, 22H). 13C NMR (75 MHz, CDCl3) δ 192.11, 165.50, 164.95, 144.34, 136.30, 134.28, 133.00, 129.95, 129.60, 128.98, 127.82, 125.37, 81.86, 67.01, 39.41, 37.49, 33.44, 33.30, 28.31, 26.83, 26.50.
tert-butyl 4-(4-(3-(2-cyclohexylethyl)phenyl)-1H-imidazol-2-yl)benzoate (51)
To a round bottom flask, fitted with a deans-stark trap containing 4 Å molecular sieves, was added 50 (1.00 g, 2.22 mmol) and ammonium acetate (5.0 eq.) and the mixture was dissolved in xylenes (30 mL, 0.07 M). The reaction was then heated to reflux for 7 h. The reaction was then cooled to rt, evaporated to a black oil, and immediately purified by flash chromatography to yield the title compound. 36%. Yellow solid. Rf = 0.05 (10% EtOAc in hexanes; Seebach’s Dip). 1H NMR (300 MHz, CDCl3) δ 8.00 (d, J = 8.4 Hz, 2H), 87.92 (d, J = 8.4 Hz, 2H), 7.62 (s, 1H), 7.52 (d, J = 7.7 Hz, 1H), 7.42 (s, 1H), 7.32 –7.15 (m, 1H), 7.05 (d, J = 7.6 Hz, 1H), 2.70 – 2.39 (m, 2H), 1.85 – 1.01 (m, 20H), 1.01 –v0.69 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 165.90, 146.77, 144.07, 134.13, 131.60, 130.22, 128.88, 127.64, 125.44, 122.74, 81.65, 39.61, 37.65, 33.56, 33.48, 28.42, 26.91, 26.56.
(S)-1-(4-(4-(3-(2-cyclohexylethyl)phenyl)-1H-imidazol-2-yl)benzoyl)pyrrolidine-2-carbonitrile (52)
General procedure N was used to deprotect the tBu ester on compound 51 (193 mg, 0.448 mmol) to yield the corresponding carboxylic acid. In a separate flask, general procedure N was used to deprotect 34 (88 mg, 0.448 mmol). General procedure B was used to couple the deprotected 51 and 34 to yield the title compound. 84%. Yellow solid. Rf = 0.57 (EtOAc; Seebach’s Dip). 1H NMR (300 MHz, DMSO) δ 12.81 (s, 1H), 8.22 – 7.90 (m, 2H), 7.90 – 7.34 (m, 5H), 7.26 (t, J = 6.8 Hz, 1H), 7.03 (d, J = 6.5 Hz, 1H), 4.88 (dd, J = 7.5, 5.2 Hz, 1H), 4.09 (ddd, J = 12.3, 7.6, 3.3 Hz, 1H), 3.98 – 3.40 (m, 3H), 3.08 – 2.66 (m, 2H), 2.66 – 2.51 (m, 2H), 2.39 – 0.56 (m, 13H). 13C NMR (75 MHz, DMSO) δ 168.98, 145.55, 143.45, 142.40, 134.95, 133.13, 129.09, 128.77, 128.44, 125.29, 124.94, 122.55, 120.15, 115.64, 49.93, 47.44, 39.89, 37.49, 33.45, 31.92, 30.28, 26.89, 26.52, 25.84.
(S)-1-(4-(4-(3-(2-cyclohexylethyl)phenyl)-1H-imidazol-2-yl)benzoyl)pyrrolidine-2-carboximidamide hydrochloride (53)
General procedure A was used to convert 52 (140 mg, 0.310 mmol) to the title compound. 81%. Yellow solid. 1H NMR (500 MHz, DMSO) δ 12.85 (s, 1H), 9.07 (s, 2H), 8.74 (s, 2H), 8.07 (d, J = 8.2 Hz, 2H), 7.80 (d, J = 8.2 Hz, 2H), 7.65 (m, 2H), 7.25 (t, J = 7.6 Hz, 1H), 7.03 (d, J = 7.2 Hz, 1H), 4.62 (t, J = 7.5 Hz, 1H), 3.93 – 3.70 (m, 1H), 3.47 (dd, J = 19.6, 12.3 Hz, 1H), 2.67 – 2.53 (m, 2H), 2.37 (dd, J = 14.8, 8.7 Hz, 1H), 2.02 – 0.92 (m, 14H). 13C NMR (126 MHz, DMSO) δ 171.36, 169.60, 145.32, 143.20, 142.05, 134.89, 134.67, 132.81, 129.15, 128.91, 126.78, 124.71, 122.30, 120.07, 58.61, 50.66, 39.71, 37.25, 33.22, 31.42, 26.64, 26.28, 25.93. LCMS: tR = 3.79; m/z = 470.3. HRMS m/z calc. for C29H36N5O (M+H), 470.2920; found, 470.2916.
4-(4-(3-(2-cyclohexylethyl)phenyl)oxazol-2-yl)benzoic acid (54)
To a round bottom flask containing 50 (1.10 g, 2.44 mmol) and ammonium acetate (5.0 eq.) was added AcOH (20 mL, 0.12 M). The reaction was then heated to reflux for 16 h. The reaction was then cooled to rt, evaporated to dryness, and immediately purified by flash chromatography to yield the title compound. 25%. Yellow solid. Rf = 0.58 (EtOAc; Seebach’s Dip). Compound was crude by NMR and taken on to the next step without further purification.
(S)-1-(4-(4-(3-(2-cyclohexylethyl)phenyl)oxazol-2-yl)benzoyl)pyrrolidine-2-carbonitrile (55)
General procedure N was used to deprotect 34 (88 mg, 0.448 mmol). General procedure B was used to couple the deprotected 34 and crude 54 to yield the title compound. 81%. Yellow solid. Rf = 0.51 (50% EtOAc in hexanes; Seebach’s Dip). 1H NMR (300 MHz, CDCl3) δ 8.15 (d, J = 8.0 Hz, 2H), 7.96 (s, 1H), 7.81 – 7.52 (m, 4H), 7.30 (dt, J = 6.6, 4.9 Hz, 1H), 7.14 (d, J = 7.6 Hz, 1H), 5.03 – 4.69 (m, 1H), 3.91 – 3.26 (m, 2H), 2.77 – 2.43 (m, 2H), 2.43 – 0.62 (m, 17H). 13C NMR (75 MHz, CDCl3) δ 169.25, 160.94, 144.13, 142.74, 136.60, 134.30, 130.90, 129.79, 128.93, 128.65, 128.28, 126.69, 125.81, 123.13, 118.81, 49.69, 47.18, 39.68, 37.67, 33.54, 30.48, 26.92, 26.57, 25.82.
(S)-1-(4-(4-(3-(2-cyclohexylethyl)phenyl)oxazol-2-yl)benzoyl)pyrrolidine-2-carboximidamide hydrochloride (56)
General procedure A was used to convert 55 (169 mg, 0.373 mmol) to the title compound. 75%. Yellow solid. 1H NMR (500 MHz, DMSO) δ 9.23 (s, 2H), 9.03 (s, 2H), 8.78 (s, 1H), 8.12 (d, J = 8.4 Hz, 2H), 7.90 (d, J = 8.4 Hz, 2H), 7.73 – 7.60 (m, 2H), 7.34 (t, J = 7.6 Hz, 1H), 7.16 (d, J = 7.6 Hz, 1H), 4.67 (t, J = 7.5 Hz, 1H), 3.99 – 3.69 (m, 1H), 3.50 – 3.37 (m, 1H), 2.61 (t, J = 9.1 Hz, 2H), 2.44 – 2.30 (m, 1H), 2.01 – 1.79 (m, 3H), 1.74 (d, J = 13.0 Hz, 2H), 1.69 – 1.54 (m, 3H), 1.54 – 1.41 (m, 2H), 1.30 – 1.00 (m, 4H), 1.00 – 0.75 (m, 2H). 13C NMR (126 MHz, DMSO) δ 171.39, 169.19, 160.68, 143.73, 141.87, 137.70, 136.51, 130.93, 129.22, 129.15, 128.68, 126.16, 125.49, 123.13, 58.50, 50.57, 37.24, 33.18, 33.02, 31.46, 26.62, 26.25, 25.61. LCMS: tR = 4.79; m/z = 471.3. HRMS m/z calc. for C29H35N4O2 (M+H), 471.2760; found, 471.2759.
tert-butyl 4-carbamothioylbenzoate (57)
4-(tert-butoxycarbonyl)benzoic acid (3.24 g, 14.6 mmol) was then dissolved in CH2Cl2 (0.3 M) at 0ºC and treated with TEA (3.0 eq.) then isobutyl chloroformate (1.1 eq.). The reaction turned turbid after the addition and was let warm to rt before slowly clearing over time. After 1 h at rt, the reaction was treated with 2 N NH3 in MeOH (2.0 eq.) and let stir for 6 h. The reaction was evaporated to a white solid and purified by flash chromatography to yield the corresponding amide. 66%. White solid. Rf = 0.39 (60% EtOAc in hexanes; UV). 1H NMR (300 MHz, CDCl3) δ 8.02 (d, J = 8.1 Hz, 2H), 7.84 (d, J = 8.1 Hz, 2H), 6.48 (s, 2H), 1.59 (s, 9H). 13C NMR (75 MHz, CDCl3) δ 169.25, 165.10, 137.02, 135.22, 129.87, 127.47, 81.99, 28.35. The amide was then dissolved in THF (0.5 M) and treated with Lawesson’s reagent (0.6 eq.) in one portion. The reaction was let stir for 3 h before being evaporated to dryness and purified by flash chromatography to yield the title compound. 92%. Yellow/green solid. Rf = 0.93 (50% EtOAc in hexanes; Seebach’s Dip). 1H NMR (500 MHz, DMSO) δ 10.06 (s, 1H), 9.65 (s, 1H), 8.12 – 7.55 (m, 4H), 1.53 (s, 9H). 13C NMR (126 MHz, DMSO) δ 199.69, 164.80, 143.60, 133.51, 129.12, 127.88, 81.62, 28.18.
tert-butyl 4-(4-(3-(2-cyclohexylethyl)phenyl)thiazol-2-yl)benzoate (58)
To a solution of 57 (1.95 g, 8.22 mmol) and KHCO3 (1.1 eq.) in THF (45 mL) at −5ºC was added 49 (2.05 g, 6.63 mmol) in THF (5 mL) and let warm to rt. The reaction was let stir for 2 h and then cooled to −5ºC again and treated with TEA (2.2 eq.) and TFAA (1.1 eq.). The reaction was let warm to rt and stir for 16 h. The reaction was diluted with CHCl3 (400 mL) and quenched with water (100 mL). The organic layer was then dried with Na2SO4, evaporated to dryness, and immediately purified by flash chromatography to yield the title compound. 80%. Yellow solid. Rf = 0.88 (10% EtOAc in hexanes; Seebach’s Dip). 1H NMR (300 MHz, CDCl3) δ 8.18 – 7.95 (m, 4H), 7.84 (s, 1H), 7.81 – 7.64 (m, 1H), 7.45 (s, 1H), 7.33 (t, J = 7.6 Hz, 1H), 7.17 (d, J = 7.6 Hz, 1H), 2.67 (t, J = 8.6 Hz, 2H), 1.94 – 1.44 (m, 15H), 1.44 – 1.06 (m, 5H), 0.96 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 166.61, 165.35, 157.22, 143.99, 137.37, 135.15, 134.43, 133.24, 130.52, 128.94, 126.92, 126.47, 124.04, 113.70, 81.48, 77.68, 39.74, 37.70, 33.60, 33.48, 28.43, 26.99, 26.65.
(S)-1-(4-(4-(3-(2-cyclohexylethyl)phenyl)thiazol-2-yl)benzoyl)pyrrolidine-2-carbonitrile (59)
General procedure N was used to deprotect the tBu ester on compound 58 (2.10 mg, 4.69 mmol) to yield the corresponding carboxylic acid. In a separate flask, general procedure N was used to deprotect 34 (919 mg, 4.69 mmol). General procedure B was used to couple the deprotected 58 and 34 to yield the title compound. 65%. Yellow solid. Rf = 0.36 (50% EtOAc in hexanes; Seebach’s Dip). 1H NMR (300 MHz, CDCl3) δ 8.02 (d, J = 8.1 Hz, 2H), 7.79 (s, 1H), 7.73 (d, J = 7.8 Hz, 1H), 7.65 (d, J = 7.5 Hz, 2H), 7.48 (s, 1H), 7.30 (t, J = 7.6 Hz, 1H), 7.14 (d, J = 7.7 Hz, 1H), 4.82 (s, 1H), 3.85 – 3.35 (m, 2H), 2.64 (t, J = 5.3 Hz, 2H), 2.40 – 1.81 (m, 4H), 1.81 – 1.39 (m, 7H), 1.39 – 1.02 (m, 4H), 1.02 – 0.74 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 169.21, 168.71, 166.42, 157.05, 144.05, 136.02, 134.33, 128.95, 128.71, 128.51, 126.69, 123.97, 118.92, 113.77, 49.74, 47.23, 39.72, 37.64, 33.55, 33.43, 30.42, 26.93, 26.59,25.83.
(S)-1-(4-(4-(3-(2-cyclohexylethyl)phenyl)thiazol-2-yl)benzoyl)pyrrolidine-2-carboximidamide hydrochloride (60)
General procedure A was used to convert 59 (682 mg, 1.50 mmol) to the title compound. 74%. White solid. 1H NMR (500 MHz, DMSO) δ 9.23 (s, 2H), 9.02 (s, 2H), 8.23 (s, 1H), 8.15 – 8.00 (m, 2H), 7.94 – 7.60 (m, 4H), 7.37 (t, J = 6.5 Hz, 1H), 7.17 (d, J = 6.4 Hz, 1H), 4.66 (s, 1H), 3.84 (dd, J = 34.3, 15.3 Hz, 1H), 3.53 – 3.20 (m, 2H), 2.62 (t, J = 5.6 Hz, 2H), 2.53 – 2.09 (m, 2H), 2.08 – 1.31 (m, 9H), 1.31 – 0.96 (m, 4H), 0.89 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 176.16, 173.99, 171.05, 160.83, 148.44, 142.06, 139.74, 138.98, 133.98, 133.47, 131.24, 131.00, 128.82, 124.56, 120.56, 63.26, 55.32, 41.95, 37.93, 37.79, 36.20, 31.37, 31.00, 30.38. LCMS: tR = 5.08; m/z = 487.3. HRMS m/z calc. for C29H35N4OS (M+H), 487.2532; found, 487.2530.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health (R01 GM067958 to KRL and TLM). The authors thank Jose L. Tomsig, Department of Pharmacology, University of Virginia, for S1P/LCMS quantification. AJK would like to offer special thanks to the Jefferson Scholars Foundation for financial support.
Abbreviations List
- 9-BBN
9-borabicyclo[3.3.1]nonane
- AcOH
acetic acid
- DGK
diacylglycerol kinase
- DMF
dimethyl formamide
- DMS
dimethyl sphingosine
- EGF
epidermal growth factor
- HDAC
histone deacetylase
- LPA
lysophosphatidic acid
- MOE
molecular operating environment
- PDGF
platelet derived growth factor
- PFK
phosphofructokinase
- PKC
protein kinase C
- PyBOP
benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate
- S1P
sphingosine 1-phosphate
- S1PR
sphingosine 1-phosphate receptor
- SAR
structure activity relationship
- SphK
sphingosine kinase
- VEGF
vascular endothelial growth factor
Footnotes
Supporting Information Available: Includes the synthetic schemes for compounds 30 and 47, the tabulated results form the inhibitor docking studies, and the SphK1/DGKB sequence alignment. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Pyne NJ, Pyne S. Sphingosine 1-phosphate and cancer. Nat Rev Cancer. 2010;10:489–503. doi: 10.1038/nrc2875. [DOI] [PubMed] [Google Scholar]
- 2.Shida D, Takabe K, Kapitonov D, Milstien S, Spiegel S. Targeting sphk1 as a new strategy against cancer. Curr Drug Targets. 2008;9:662–673. doi: 10.2174/138945008785132402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spiegel S, Milstien S. Sphingosine-1-phosphate: An enigmatic signalling lipid. Nat Rev Mol Cell Biol. 2003;4:397–407. doi: 10.1038/nrm1103. [DOI] [PubMed] [Google Scholar]
- 4.Ogretmen B, Hannun YA. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat Rev Cancer. 2004;4:604–616. doi: 10.1038/nrc1411. [DOI] [PubMed] [Google Scholar]
- 5.Hannun YA, Obeid LM. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat Rev Mol Cell Biol. 2008;9:139–150. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- 6.Rosen H, Gonzalez-Cabrera PJ, Sanna MG, Brown S. Sphingosine 1-phosphate receptor signaling. Annu Rev Biochem. 2009;78:743–768. doi: 10.1146/annurev.biochem.78.072407.103733. [DOI] [PubMed] [Google Scholar]
- 7.Hait NC, Allegood J, Maceyka M, Strub GM, Harikumar KB, Singh SK, Luo C, Marmorstein R, Kordula T, Milstien S, Spiegel S. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science. 2009;325:1254–1257. doi: 10.1126/science.1176709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li J, Guan HY, Gong LY, Song LB, Zhang N, Wu J, Yuan J, Zheng YJ, Huang ZS, Li M. Clinical significance of sphingosine kinase-1 expression in human astrocytomas progression and overall patient survival. Clin Cancer Res. 2008;14:6996–7003. doi: 10.1158/1078-0432.CCR-08-0754. [DOI] [PubMed] [Google Scholar]
- 9.Van Brocklyn JR, Jackson CA, Pearl DK, Kotur MS, Snyder PJ, Prior TW. Sphingosine kinase-1 expression correlates with poor survival of patients with glioblastoma multiforme: Roles of sphingosine kinase isoforms in growth of glioblastoma cell lines. J Neuropathol Exp Neurol. 2005;64:695–705. doi: 10.1097/01.jnen.0000175329.59092.2c. [DOI] [PubMed] [Google Scholar]
- 10.Dyrskjøt L, Kruhøffer M, Thykjaer T, Marcussen N, Jensen JL, Møller K, Ørntoft TF. Gene expression in the urinary bladder: A common carcinoma in situ gene expression signature exists disregarding histopathological classification. Cancer Res. 2004;64:4040–4048. doi: 10.1158/0008-5472.CAN-03-3620. [DOI] [PubMed] [Google Scholar]
- 11.Ruckhäberle E, Rody A, Engels K, Gaetje R, von Minckwitz G, Schiffmann S, Grösch S, Geisslinger G, Holtrich U, Karn T, Kaufmann M. Microarray analysis of altered sphingolipid metabolism reveals prognostic significance of sphingosine kinase 1 in breast cancer. Breast cancer research and treatment. 2008;112:41–52. doi: 10.1007/s10549-007-9836-9. [DOI] [PubMed] [Google Scholar]
- 12.Erez-Roman R, Pienik R, Futerman AH. Increased ceramide synthase 2 and 6 mrna levels in breast cancer tissues and correlation with sphingosine kinase expression. Biochem Biophys Res Commun. 2010;391:219–223. doi: 10.1016/j.bbrc.2009.11.035. [DOI] [PubMed] [Google Scholar]
- 13.Kohno M, Momoi M, Oo ML, Paik JH, Lee YM, Venkataraman K, Ai Y, Ristimaki AP, Fyrst H, Sano H, Rosenberg D, Saba JD, Proia RL, Hla T. Intracellular role for sphingosine kinase 1 in intestinal adenoma cell proliferation. Mol Cell Biol. 2006;26:7211–7223. doi: 10.1128/MCB.02341-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kawamori T, Kaneshiro T, Okumura M, Maalouf S, Uflacker A, Bielawski J, Hannun YA, Obeid LM. Role for sphingosine kinase 1 in colon carcinogenesis. FASEB J. 2009;23:405–414. doi: 10.1096/fj.08-117572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li W, Yu CP, Xia J-t, Zhang L, Weng GX, Zheng H-q, Kong Q-l, Hu L-j, Zeng MS, Zeng Y-x, Li M, Li J, Song LB. Sphingosine kinase 1 is associated with gastric cancer progression and poor survival of patients. Clin Cancer Res. 2009;15:1393–1399. doi: 10.1158/1078-0432.CCR-08-1158. [DOI] [PubMed] [Google Scholar]
- 16.Pyeon D, Newton MA, Lambert PF, den Boon JA, Sengupta S, Marsit CJ, Woodworth CD, Connor JP, Haugen TH, Smith EM, Kelsey KT, Turek LP, Ahlquist P. Fundamental differences in cell cycle deregulation in human papillomavirus-positive and human papillomavirus-negative head/neck and cervical cancers. Cancer Res. 2007;67:4605–4619. doi: 10.1158/0008-5472.CAN-06-3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ginos MA, Page GP, Michalowicz BS, Patel KJ, Volker SE, Pambuccian SE, Ondrey FG, Adams GL, Gaffney PM. Identification of a gene expression signature associated with recurrent disease in squamous cell carcinoma of the head and neck. Cancer Res. 2004;64:55–63. doi: 10.1158/0008-5472.can-03-2144. [DOI] [PubMed] [Google Scholar]
- 18.Andersson A, Ritz C, Lindgren D, Edén P, Lassen C, Heldrup J, Olofsson T, Råde J, Fontes M, Porwit-Macdonald A, Behrendtz M, Höglund M, Johansson B, Fioretos T. Microarray-based classification of a consecutive series of 121 childhood acute leukemias: Prediction of leukemic and genetic subtype as well as of minimal residual disease status. Leukemia. 2007;21:1198–1203. doi: 10.1038/sj.leu.2404688. [DOI] [PubMed] [Google Scholar]
- 19.Bayerl MG, Bruggeman RD, Conroy EJ, Hengst JA, King TS, Jimenez M, Claxton DF, Yun JK. Sphingosine kinase 1 protein and mrna are overexpressed in non-hodgkin lymphomas and are attractive targets for novel pharmacological interventions. Leuk Lymphoma. 2008;49:948–954. doi: 10.1080/10428190801911654. [DOI] [PubMed] [Google Scholar]
- 20.Akao Y, Banno Y, Nakagawa Y, Hasegawa N, Kim TJ, Murate T, Igarashi Y, Nozawa Y. High expression of sphingosine kinase 1 and s1p receptors in chemotherapy-resistant prostate cancer pc3 cells and their camptothecin-induced up-regulation. Biochem Biophys Res Commun. 2006;342:1284–1290. doi: 10.1016/j.bbrc.2006.02.070. [DOI] [PubMed] [Google Scholar]
- 21.Pchejetski D, Golzio M, Bonhoure E, Calvet C, Doumerc N, Garcia V, Mazerolles C, Rischmann P, Teissié J, Malavaud B, Cuvillier O. Sphingosine kinase-1 as a chemotherapy sensor in prostate adenocarcinoma cell and mouse models. Cancer Res. 2005;65:11667–11675. doi: 10.1158/0008-5472.CAN-05-2702. [DOI] [PubMed] [Google Scholar]
- 22.Talantov D, Mazumder A, Yu JX, Briggs T, Jiang Y, Backus J, Atkins D, Wang Y. Novel genes associated with malignant melanoma but not benign melanocytic lesions. Clin Cancer Res. 2005;11:7234–7242. doi: 10.1158/1078-0432.CCR-05-0683. [DOI] [PubMed] [Google Scholar]
- 23.Nindl I, Dang C, Forschner T, Kuban RJ, Meyer T, Sterry W, Stockfleth E. Identification of differentially expressed genes in cutaneous squamous cell carcinoma by microarray expression profiling. Mol Cancer. 2006;5:30. doi: 10.1186/1476-4598-5-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xia P, Gamble JR, Wang L, Pitson SM, Moretti PA, Wattenberg BW, D’Andrea RJ, Vadas MA. An oncogenic role of sphingosine kinase. Curr Biol. 2000;10:1527–1530. doi: 10.1016/s0960-9822(00)00834-4. [DOI] [PubMed] [Google Scholar]
- 25.Vadas M, Xia P, McCaughan G, Gamble J. The role of sphingosine kinase 1 in cancer: Oncogene or non-oncogene addiction? Biochim Biophys Acta. 2008;1781:442–447. doi: 10.1016/j.bbalip.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 26.Alvarez SE, Milstien S, Spiegel S. Autocrine and paracrine roles of sphingosine-1-phosphate. Trends Endocrinol Metab. 2007;18:300–307. doi: 10.1016/j.tem.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 27.Sukocheva O, Wadham C, Holmes A, Albanese N, Verrier E, Feng F, Bernal A, Derian CK, Ullrich A, Vadas MA, Xia P. Estrogen transactivates egfr via the sphingosine 1-phosphate receptor edg-3: The role of sphingosine kinase-1. J Cell Biol. 2006;173:301–310. doi: 10.1083/jcb.200506033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Döll F, Pfeilschifter J, Huwiler A. Prolactin upregulates sphingosine kinase-1 expression and activity in the human breast cancer cell line mcf7 and triggers enhanced proliferation and migration. Endocr Relat Cancer. 2007;14:325–335. doi: 10.1677/ERC-06-0050. [DOI] [PubMed] [Google Scholar]
- 29.Shida D, Fang X, Kordula T, Takabe K, Lépine S, Alvarez SE, Milstien S, Spiegel S. Cross-talk between lpa1 and epidermal growth factor receptors mediates up-regulation of sphingosine kinase 1 to promote gastric cancer cell motility and invasion. Cancer Res. 2008;68:6569–6577. doi: 10.1158/0008-5472.CAN-08-0411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ahmad M, Long JS, Pyne NJ, Pyne S. The effect of hypoxia on lipid phosphate receptor and sphingosine kinase expression and mitogen-activated protein kinase signaling in human pulmonary smooth muscle cells. Prostaglandins Other Lipid Mediat. 2006;79:278–286. doi: 10.1016/j.prostaglandins.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 31.Lavieu G, Scarlatti F, Sala G, Carpentier S, Levade T, Ghidoni R, Botti J, Codogno P. Regulation of autophagy by sphingosine kinase 1 and its role in cell survival during nutrient starvation. J Biol Chem. 2006;281:8518–8527. doi: 10.1074/jbc.M506182200. [DOI] [PubMed] [Google Scholar]
- 32.Guillermet-Guibert J, Davenne L, Pchejetski D, Saint-Laurent N, Brizuela L, Guilbeau-Frugier C, Delisle MB, Cuvillier O, Susini C, Bousquet C. Targeting the sphingolipid metabolism to defeat pancreatic cancer cell resistance to the chemotherapeutic gemcitabine drug. Mol Cancer Ther. 2009;8:809–820. doi: 10.1158/1535-7163.MCT-08-1096. [DOI] [PubMed] [Google Scholar]
- 33.Baran Y, Salas A, Senkal CE, Gunduz U, Bielawski J, Obeid LM, Ogretmen B. Alterations of ceramide/sphingosine 1-phosphate rheostat involved in the regulation of resistance to imatinib-induced apoptosis in k562 human chronic myeloid leukemia cells. J Biol Chem. 2007;282:10922–10934. doi: 10.1074/jbc.M610157200. [DOI] [PubMed] [Google Scholar]
- 34.Cuvillier O, Pirianov G, Kleuser B, Vanek PG, Coso OA, Gutkind S, Spiegel S. Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature. 1996;381:800–803. doi: 10.1038/381800a0. [DOI] [PubMed] [Google Scholar]
- 35.Pitson SM, Moretti PAB, Zebol JR, Lynn HE, Xia P, Vadas MA, Wattenberg BW. Activation of sphingosine kinase 1 by erk1/2-mediated phosphorylation. EMBO J. 2003;22:5491–5500. doi: 10.1093/emboj/cdg540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maceyka M, Sankala H, Hait NC, Le Stunff H, Liu H, Toman R, Collier C, Zhang M, Satin LS, Merrill AH, Milstien S, Spiegel S. Sphk1 and sphk2, sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J Biol Chem. 2005;280:37118–37129. doi: 10.1074/jbc.M502207200. [DOI] [PubMed] [Google Scholar]
- 37.Sankala HM, Hait NC, Paugh SW, Shida D, Lépine S, Elmore LW, Dent P, Milstien S, Spiegel S. Involvement of sphingosine kinase 2 in p53-independent induction of p21 by the chemotherapeutic drug doxorubicin. Cancer Res. 2007;67:10466–10474. doi: 10.1158/0008-5472.CAN-07-2090. [DOI] [PubMed] [Google Scholar]
- 38.Liu H, Toman RE, Goparaju SK, Maceyka M, Nava VE, Sankala H, Payne SG, Bektas M, Ishii I, Chun J, Milstien S, Spiegel S. Sphingosine kinase type 2 is a putative bh3-only protein that induces apoptosis. J Biol Chem. 2003;278:40330–40336. doi: 10.1074/jbc.M304455200. [DOI] [PubMed] [Google Scholar]
- 39.Endo K, Igarashi Y, Nisar M, Zhou QH, Hakomori S. Cell membrane signaling as target in cancer therapy: Inhibitory effect of n,n-dimethyl and n,n,n-trimethyl sphingosine derivatives on in vitro and in vivo growth of human tumor cells in nude mice. Cancer Res. 1991;51:1613–1618. [PubMed] [Google Scholar]
- 40.Paugh SW, Paugh BS, Rahmani M, Kapitonov D, Almenara JA, Kordula T, Milstien S, Adams JK, Zipkin RE, Grant S, Spiegel S. A selective sphingosine kinase 1 inhibitor integrates multiple molecular therapeutic targets in human leukemia. Blood. 2008;112:1382–1391. doi: 10.1182/blood-2008-02-138958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.French KJ, Zhuang Y, Maines LW, Gao P, Wang W, Beljanski V, Upson JJ, Green CL, Keller SN, Smith CD. Pharmacology and antitumor activity of abc294640, a selective inhibitor of sphingosine kinase-2. J Pharmacol Exp Ther. 2010;333:129–139. doi: 10.1124/jpet.109.163444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wong L, Tan SSL, Lam Y, Melendez AJ. Synthesis and evaluation of sphingosine analogues as inhibitors of sphingosine kinases. J Med Chem. 2009;52:3618–3626. doi: 10.1021/jm900121d. [DOI] [PubMed] [Google Scholar]
- 43.Xiang Y, Hirth B, Kane JL, Liao J, Noson KD, Yee C, Asmussen G, Fitzgerald M, Klaus C, Booker M. Discovery of novel sphingosine kinase-1 inhibitors. Part 2 Bioorg Med Chem Lett. 2010;20:4550–4554. doi: 10.1016/j.bmcl.2010.06.019. [DOI] [PubMed] [Google Scholar]
- 44.Xiang Y, Asmussen G, Booker M, Hirth B, Kane JL, Liao J, Noson KD, Yee C. Discovery of novel sphingosine kinase 1 inhibitors. Bioorg Med Chem Lett. 2009;19:6119–6121. doi: 10.1016/j.bmcl.2009.09.022. [DOI] [PubMed] [Google Scholar]
- 45.French KJ, Schrecengost RS, Lee BD, Zhuang Y, Smith SN, Eberly JL, Yun JK, Smith CD. Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer Res. 2003;63:5962–5969. [PubMed] [Google Scholar]
- 46.Kapitonov D, Allegood JC, Mitchell C, Hait NC, Almenara JA, Adams JK, Zipkin RE, Dent P, Kordula T, Milstien S, Spiegel S. Targeting sphingosine kinase 1 inhibits akt signaling, induces apoptosis, and suppresses growth of human glioblastoma cells and xenografts. Cancer Res. 2009;69:6915–6923. doi: 10.1158/0008-5472.CAN-09-0664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maines LW, Fitzpatrick LR, Green CL, Zhuang Y, Smith CD. Efficacy of a novel sphingosine kinase inhibitor in experimental crohn’s disease. Inflammopharmacology. 2010;18:73–85. doi: 10.1007/s10787-010-0032-x. [DOI] [PubMed] [Google Scholar]
- 48.Maines LW, Fitzpatrick LR, French KJ, Zhuang Y, Xia Z, Keller SN, Upson JJ, Smith CD. Suppression of ulcerative colitis in mice by orally available inhibitors of sphingosine kinase. Dig Dis Sci. 2008;53:997–1012. doi: 10.1007/s10620-007-0133-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Puneet P, Yap CT, Wong L, Lam Y, Koh DR, Moochhala S, Pfeilschifter J, Huwiler A, Melendez AJ. Sphk1 regulates proinflammatory responses associated with endotoxin and polymicrobial sepsis. Science. 2010;328:1290–1294. doi: 10.1126/science.1188635. [DOI] [PubMed] [Google Scholar]
- 50.Mathews TP, Kennedy AJ, Kharel Y, Kennedy PC, Nicoara O, Sunkara M, Morris AJ, Wamhoff BR, Lynch KR, Macdonald TL. Discovery, biological evaluation, and structure-activity relationship of amidine based sphingosine kinase inhibitors. J Med Chem. 2010;53:2766–2778. doi: 10.1021/jm901860h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miller DJ, Jerga A, Rock CO, White SW. Analysis of the staphylococcus aureus dgkb structure reveals a common catalytic mechanism for the soluble diacylglycerol kinases. Structure. 2008;16:1036–1046. doi: 10.1016/j.str.2008.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kharel Y, Mathews TP, Kennedy AJ, Houck JD, Lynch KR. A rapid assay for assessment of sphingosine kinase inhibitors and substrates. Anal Biochem. 2011;411:230–235. doi: 10.1016/j.ab.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schaefer F, Petters G. Base-catalyzed reaction of nitriles with alcohols. A convenient route to imidates and amidine salts The Journal of Organic Chemistry. 1961;26:412–418. [Google Scholar]
- 54.Wildman S, Crippen G. Prediction of physicochemical parameters by atomic contributions. Journal of chemical information and computer sciences. 1999;39:868–873. [Google Scholar]
- 55.Kohn M, Breinbauer R. The staudinger ligation - a gift to chemical biology. Angew Chem Int Edit. 2004;43:3106–3116. doi: 10.1002/anie.200401744. [DOI] [PubMed] [Google Scholar]
- 56.Bal B, Childers W, Pinnick H. Oxidation of alpha, beta-unsaturated aldehydes. Tetrahedron. 1981;37:2091–2096. [Google Scholar]
- 57.Nichols CE, Lamb HK, Lockyer M, Charles IG, Pyne S, Hawkins AR, Stammers DK. Characterization of salmonella typhimurium yegs, a putative lipid kinase homologous to eukaryotic sphingosine and diacylglycerol kinases. Proteins. 2007;68:13–25. doi: 10.1002/prot.21386. [DOI] [PubMed] [Google Scholar]
- 58.Bakali HMA, Herman MD, Johnson KA, Kelly AA, Wieslander A, Hallberg BM, Nordlund P. Crystal structure of yegs, a homologue to the mammalian diacylglycerol kinases, reveals a novel regulatory metal binding site. J Biol Chem. 2007;282:19644–19652. doi: 10.1074/jbc.M604852200. [DOI] [PubMed] [Google Scholar]
- 59.Cabrera R, Baez M, Pereira HM, Caniuguir A, Garratt RC, Babul J. The crystal complex of phosphofructokinase-2 of escherichia coli with fructose-6-phosphate: Kinetic and structural analysis of the allosteric atp inhibition. Journal of Biological Chemistry. 2011;286:5774–5783. doi: 10.1074/jbc.M110.163162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Martinez-Oyanedel J, McNae IW, Nowicki MW, Keillor JW, Michels PAM, Fothergill-Gilmore LA, Walkinshaw MD. The first crystal structure of phosphofructokinase from a eukaryote: Trypanosoma brucei. J Mol Biol. 2007;366:1185–1198. doi: 10.1016/j.jmb.2006.10.019. [DOI] [PubMed] [Google Scholar]
- 61.Blake JD, Cohen FE. Pairwise sequence alignment below the twilight zone. J Mol Biol. 2001;307:721–735. doi: 10.1006/jmbi.2001.4495. [DOI] [PubMed] [Google Scholar]
- 62.Merrill AH, Stokes TH, Momin A, Park H, Portz BJ, Kelly S, Wang E, Sullards MC, Wang MD. Sphingolipidomics: A valuable tool for understanding the roles of sphingolipids in biology and disease. J Lipid Res. 2009;50 (Suppl):S97–102. doi: 10.1194/jlr.R800073-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.