

Abstract
Maitotoxin holds a special place in the annals of natural products chemistry as the largest and most toxic secondary metabolite known to date. Its fascinating, ladder-like, polyether molecular structure and diverse spectrum of biological activities elicited keen interest from chemists and biologists who recognized its uniqueness and potential as a probe and inspiration for research in chemistry and biology. Synthetic studies in the area benefited from methodologies and strategies that were developed as part of chemical synthesis programs directed toward the total synthesis of some of the less complex members of the polyether marine biotoxin class, of which maitotoxin is the flagship. This account focuses on progress made in the authors’ laboratories in the synthesis of large maitotoxin domains with emphasis on methodology development, strategy design, and structural comparisons of the synthesized molecules with the corresponding regions of the natural product. The article concludes with an overview of maitotoxin’s biological profile and future perspectives.
Keywords: biotoxins, maitotoxin, natural product, polyether, synthesis
1. Introduction
Despite its accidental occurrence, the synthesis of urea in 1828 by Friedrich Wöhler represents a milestone in the history of chemistry. [1] Its significance lies in the fact that it broke the barrier between inorganic and organic chemistry, buried vitalism (the dogma that organic molecules could only be synthesized by living systems), and inspired chemists to pursue, sharpen and apply the art of what became known as organic synthesis. Since then, countless natural and designed molecules with increasingly more complex structures have been synthesized in the laboratory by synthetic organic chemists. The lure of natural products as synthetic targets and their inspirational role in driving the art of total synthesis forward have been amply demonstrated in the intervening years since Wöhler’s synthesis of urea. No more dramatic has this inspiration been in terms of complexity than in the case of the marine natural product maitotoxin (1, Figure 1).[2] Indeed, the elegant molecular structure of maitotoxin represents the largest and most complex secondary metabolite ever isolated (32 rings, 98 stereocenters and one geometrical site). As if this was not enough, the appeal of maitotoxin as a substance for further investigation is amplified by its lethal potency (LD50 = 50 ng/kg)[3] and intriguing, albeit not fully understood, mechanism of action as a neurotoxin.
Figure 1.

Structures of maitotoxin (1), hemibrevetoxin (2), brevetoxin A (3), and brevetoxin B (4).
The story of maitototoxin began in 1971 with the observation by Yasumoto and co-workers of cytotoxicity in aqueous extracts of the gut of the surgeonfish Ctenochaetus striatus that was similar to those of other, previously detected polyether neurotoxins.[4] The chase for the active principle led to the isolation (from the dinoflagellate Gambierdiscus toxicus)[5] and structural elucidation (by the Yasumoto,[6] Tachibana,[7] and Kishi[8] groups) of maitotoxin. Fully assigned by 1996, the structure of this impressive molecule laid in the literature undisturbed until 2006 when Gallimore and Spencer noticed an “abnormality” within it that did not conform to their proposed biosynthetic hypothesis (see Figure 2).[9] Based on the previously proposed biosynthesis of brevetoxin B (4, see Figure 3),[10] and invoking polyepoxide cascade openings, this hypothesis required inversion of configuration at C51–C52 (JK ring junction; see Figure 2). It was this cloud of uncertainty cast over the structure of maitotoxin that prompted us to study this molecule. Our initial inquiry was directed toward the true structure of maitotoxin, but as we were pursuing this goal we became more intrigued by the molecule itself (its synthesis, its biology, and its mechanism of action). In this article, we summarize our investigations in this field, including computational studies, synthetic method development, large domain synthesis, 13 C NMR spectroscopic comparisons, and preliminary biological results.
Figure 2.

Postulated biosynthetic precursor (5) of maitotoxin (1) that brought into question the stereochemistry of the JK ring junction (C51 and C52) (Gallimore and Spencer, 2006).[9]
Figure 3.

Brevetoxin B biosynthetic hypothesis (Nakanishi et al., 1985, 1989).[10]
2. Methodology Development
The discovery of brevetoxin B (4, Figure 1) in 1981 heralded a new chapter in natural products chemistry and chemical synthesis.[11] Inspired by its fascinating molecular architecture, synthetic organic chemists began to ponder its synthesis, a goal that seemed at the time far into the future since no existing methods were up to the task. Despite the elegance of a possible biomimetic approach involving a polyepoxide opening cascade that was imagined (see Figure 3),[10c] we opted to initiate a program directed toward the discovery and invention of new methods for the construction of cyclic ethers of the type found in brevetoxin B, primarily due to the near impossibility of implementing a biomimetic strategy. Our ultimate goal was to assimilate these methods into a convergent strategy to accomplish the total synthesis of this and, as it turned out, other ladder-like polyether biotoxins that were subsequently discovered. Below we summarize some of the synthetic methods that were developed over the last three decades and subsequently refined, expanded, and applied by us and others in the total synthesis of several members of this growing class of marine biotoxins.
To address the construction of 6-membered cyclic ethers (tetrahydropyran systems) through regio- and stereoselective hydroxy epoxide openings, we devised a method involving substrates carrying a strategically placed vinyl moiety (activating group) adjacent to the far end of the epoxide group as shown in Scheme 1.[12] This design completely shut down the normally observed 5-exo cyclization in the absence of the olefinic bond (i.e., path b: 6 → 8 → 10, Scheme 1), resulting in the exclusive formation, under acidic conditions, of the pyran system through the now preferred 6-endo mode of ring closure (i.e., path a: 6 → 7 → 9, see Scheme 1). This reliable method has been, and continues to be, employed routinely in the total syntheses of polyether marine toxins as we shall see below.
Scheme 1.

The regio- and stereoselective 6-endo epoxide opening method for cyclic ether formation (Nicolaou et al., 1985).[12]
A second method for the synthesis of 6-membered cyclic ethers was also developed, this time involving intramolecular 1,4-addition of a hydroxyl group to a pendant α,β-unsaturated ester moiety.[13] Although previously demonstrated,[14] this process was streamlined and refined to deliver the desired cyclic ethers in a highly stereoselective manner. Thus, as shown in Scheme 2, under basic conditions hydroxy α,β-unsaturated substrate 11 underwent stereoselective cyclization, apparently through chair-like transition state 12, to afford bicyclic system 13 exclusively.
Scheme 2.

Intramolecular 1,4-addition of a hydroxyl group to an α,β-unsaturated ester as a means to form tetrahydropyran systems (Nicolaou et al., 1989).[13]
In order to develop a method for forming the oxocene ring of brevetoxin B and other subsequently discovered polyether structures, we called upon hydroxy dithioketals, such as 14, to serve as substrates in a cyclization expected to proceed through thionium species 16 (Scheme 3).[15] Thus, exposure of 14 to NCS-AgNO3-SiO2 under anhydrous basic conditions furnished O,S-mixed ketal 17, whose subsequent manipulation resulted in the formation of oxocene 18 (Ph3SnH, AIBN, Δ) or its methylated counterpart 19 (mCPBA; AlMe3). This powerful method, which was demonstrated to also work for other ring sizes and with hydroxy ketones (i.e. 15)[16] as substrates, found numerous applications in total synthesis, including brevetoxins A (3, Figure 1) and B (4, Figure 1), as well as in synthesis, including certain maitotoxin large domains (see below).
Scheme 3.

Cyclic ether formation reactions proceeding through cyclic O,S-acetals (Nicolaou et al., 1986).[15]
A direct method for the construction of oxepanes from hydroxy ketones was also developed using Lewis acid-catalyzed reductive cyclization conditions. Thus, as shown in Scheme 4, reaction of substrate 20 with Ph2MeSiH in the presence of TMSOTf led to tetracyclic bis(oxepane) system 21 in high yield and good diastereoselectivity.[17] This method was later extended to include silyl-protected substrates [18] and the synthesis of 6-membered ring ethers[19] (see Scheme 4b and 4c).
Scheme 4.

Cyclic ether formation through direct hydroxy ketone reductive cyclization.
In contrast to cyclic ethers, especially of medium ring size, lactones are generally more accessible and considerably more versatile in their chemistry. It was, therefore, reasoned that if lactones could be converted to cyclic ethers through short sequences, such methods may constitute convenient and practical entries into the latter class of building blocks. One way to achieve this goal was to add a nucleophile to the carbonyl group of the lactone and trap the resulting alkoxide as a stable intermediate for further elaboration to the desired cyclic ether. This scenario proved difficult to achieve due to the tendency of the alkoxide species to rupture into an open-chain system, thus leading to a missed opportunity for cyclic ether formation. In order to circumvent this problem, the lactone was converted to a thionolactone (i.e. 26); the latter suffered nucleophilic attack as expected to form the more stabilized thiolate anion (27), whose higher nucleophilicity allowed its trapping with MeI to form the stable cyclic O,S-ketal (28, see Scheme 5).[20] This intermediate could be processed reductively to the corresponding cyclic ether (29, Ph3SnH, AIBN, Δ) as shown in Scheme 5. This method was featured twice in our total synthesis of hemibrevetoxin (2, Figure 1).[21]
Scheme 5.

Thionolactones as precursors to cyclic ethers (Nicolaou et al., 1987).[20]
A novel bridging reaction of bis(thionolactones) was also developed as a means to construct polycyclic ethers of the type depicted in Scheme 5.[22] According to this method, treatment of the easily prepared bis(thionolactone) 30 with sodium naphthalenide gave the bis(radical anion) 31, which underwent intramolecular radical coupling to form a C–C bond across the macrocycle, furnishing a dianion, whose trapping with MeI led to bis(cyclic O,S-ketal) 33. Exposure of the latter to Ph3SnH and AIBN under thermal conditions afforded olefinic tetracyclic polyether 34 in high yield. A similar result was obtained from the same precursor (i.e., 30) under photoirradiation conditions through a pathway that involved stable dithiane system 32 as shown in Scheme 6.
Scheme 6.

The bis(thionolactone) bridging method for cyclic ether construction (Nicolaou et al., 1986).[22]
The success of the palladium-catalyzed cross-coupling reactions in synthesis[23] prompted us to seek suitable methods for their application to cyclic ether formation. Once again, lactones served as the starting materials for the developed technologies in this regard. Thus, in addition to ketene acetal triflates which were already in use,[24] we synthesized and employed cyclic ketene acetal phosphates (i.e., 35) in a Stille-type coupling reaction with vinyl stannanes to afford cyclic ethers through Pd(PPh3)4-catalyzed C–C bond formation (see Scheme 7a).[25] This concept was later expanded to include boranes (such as 38) as partners of the vinyl phosphates, leading to cyclic ethers through Suzuki-type cross-couplings (see Scheme 7b).[26]
Scheme 7.

Ketene acetal phosphates as substrates for palladium-catalyzed cross-couplings for the synthesis of cyclic ethers.
The advent of metathesis as a powerful tool in organic synthesis in the 1990s[7 provided inspiration for the reaction to be applied in cyclic ether formation.[28] Thus, following Grubbs’ pioneering studies that included simple cyclic ether formation,[29] we utilized olefinic esters (conveniently formed from cyclic ether building blocks through esterification) as substrates to synthesize growing polyether fragments through reaction with the Tebbe reagent[30] (see Scheme 8a).[31] This method was later extended to include Takai–Utimoto type reagents[32] as shown in Scheme 8b[33] and 8c.[34] Of particular interest in this regard is the observation made by Rainier and coworkers that the use of dibromoethane (rather than dibromomethane) can lead directly, under certain circumstances, to the targeted cyclic enol ether product (see 48 → 49, Scheme 8c). The variation of the outcomes of the three reactions in Scheme 8a–c reflects different mechanisms that most likely depend primarily on sterics. Thus, in the first two processes (Scheme 8a and 8b), the ester moiety interacts with the incipient carbene species (to form the enol ether which further reacts with the Tebbe reagent, see mechanism, Scheme 9) faster than with the olefinic bond. Whereas with the more hindered substrate 48 (Scheme 8c), the order of reactivity is reversed, leading to the observed cyclic enol ether directly through the mechanism depicted in Scheme 10 (for another example of this reaction, see Schemes 16 and 17 below).
Scheme 8.

Ester–olefin ring closures to form cyclic ethers.
Scheme 9.

Postulated mechanism for the Tebbe-promoted ester–olefin ring closure to form cyclic ethers (see Scheme 8a).
Scheme 10.

Postulated mechanism of the Takai–Utimoto based ester–olefin ring closure to form cyclic ethers (Scheme 8c).
Scheme 16.

Synthesis of the QRSTU maitotoxin domain 109 (a) and 13C chemical shift differences between 109 and maitotoxin (1) (b) (Nicolaou et al., 2010).[46]
Scheme 17.

Synthesis of the WXYZA′ maitotoxin domain 116 (a) and 13C chemical shift differences between 116 and maitotoxin (1) (b) (Nicolaou et al., 2011).[47]
Collectively, these new synthetic technologies enabled substantial progress to be made in the synthesis of the polyether marine biotoxins. Specifically, they were instrumental in our total syntheses of hemibrevetoxin (2)[21] and brevetoxins B (4)[10d, 35] and A (3)[36] (see Figure 1). They were also applied in other laboratories with equal success.[37] While these methods provided the means to construct the various rings of these neurotoxins, in most instances carbohydrates served as convenient starting materials as sources of chirality in the early synthetic campaigns in this area.
The challenge of maitotoxin (1) and the recent advances in asymmetric catalysis gave us the opportunity to contemplate yet another method for the enantioselective construction of cyclic ethers starting with prochiral building blocks. Specifically, we targeted enantiopure tetrahydropyran systems of the type found within the maitotoxin structure employing achiral furan derivatives as starting materials and two reliable reactions in sequence, namely the Noyori asymmetric transfer hydrogenation[38] (Noyori reduction) and the Achmatowicz rearrangement.[39] Thus, as shown in Scheme 11, acyl furans (61) undergo asymmetric phase transfer hydrogenation in the presence of ruthenium catalyst 62 to afford furanyl alcohols 63 in excellent yield and high enantiomeric excess. Oxidation of these hydroxy furan products, either with NBS or mCPBA, induces Achmatowicz rearrangement to yield the expanded ring systems 64, from which the hydroxyl group can be excised conveniently through reduction with Et3SiH and BF3•OEt2, generating 6-membered cyclic ether enones 65, and thence a variety of other tetrahydropyran derivatives. This methodology has been applied already to the construction of several maitotoxin ring systems, including the building blocks shown in Scheme 11b (66 – 71),[40–43] in multigram quantities.
Scheme 11.

General furan-based strategy for the construction of cyclic ethers (a) and maitotoxin building blocks prepared using this strategy (b) (Nicolaou et al., 2007, 2008, 2010, and 2011).[40–43]
More recently, we expanded the scope of the intramolecular 1,4-addition of hydroxy α,β-unsaturated systems as a method for forming cyclic ethers to include hydroxy ynones as substrates. Initially reported as a side reaction,[44] we adopted the AgOTf-promoted cyclization of these substrates (i.e., 72, Scheme 12) as a means to fuse a second cyclic ether onto a preexisting one. Containing an enone moiety, the resulting product (74) was then rapidly manipulated to afford the complex DE ring system 75 of maitotoxin as shown in Scheme 12.[42]
Scheme 12.
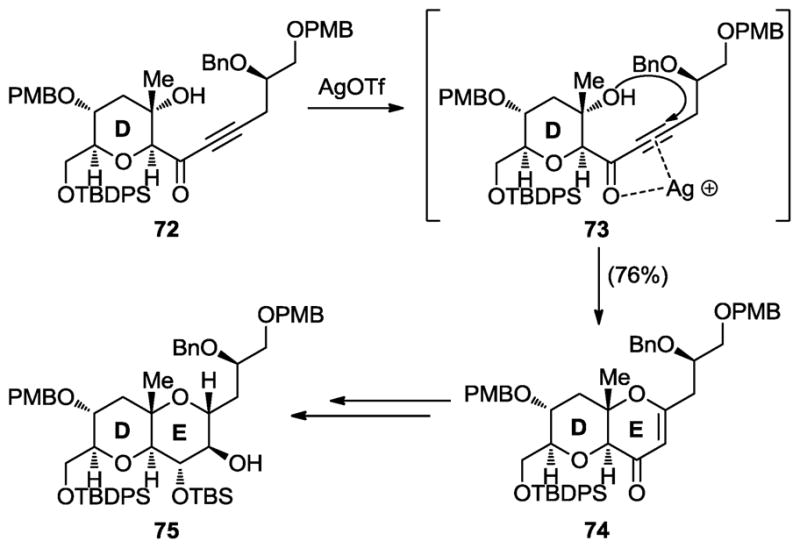
Silver-promoted, intramolecular, hydroxy ynone cyclization method for cyclic ether formation (Nicolaou et al., 2007, 2008, and 2010).[40–42]
3. Synthesis of Maitotoxin Domains
It was the Gallimore and Spencer questioning of the maitotoxin C51–C52 stereochemical configuration[9] that prompted us to initiate our investigations in the maitotoxin project. Our first foray into resolving the C51–C52 maitotoxin stereochemical controversy was computational in nature. Specifically, employing the Spartan’06 program, we calculated the 13C chemical shifts (δ) of the carbon atoms of the three diastereoisomers of the GHIJKLM domain of maitotoxin shown in Figure 4 (76: Kishi–Tachibana–Yasumoto structure; 77: revised C51–C52 configuration to fit the Gallimore and Spencer biosynthetic hypothesis; 78: further revised by us to fit both the biosynthetic hypothesis and the experimental NMR data) and compared them with the experimental values for the corresponding carbons of the natural product.[45] As shown graphically in Figure 2, the originally assigned structure (i.e., 76) exhibited the closest agreement between the calculated and experimental values, with structures 78 and 77 trailing in that order (the large deviations of the chemical shifts corresponding to the carbons at the two edges of the polyether ladder shown in this and subsequent figures are due to the structural variation at those sites between the fragments and the entire maitotoxin molecule).
Figure 4.

Differences between the calculated 13C chemical shifts for compounds 76, 77, and 78 and those corresponding to the same region of maitotoxin (1).[45]
As supportive as these computational results were for the originally assigned configuration at the C51–C52 junction, they did not provide the compelling evidence we were seeking for the correct structure of maitotoxin. We were, therefore, tempted to embark on the synthesis of appropriate fragments of the molecule in the hope that the experimental 13C chemical shift values would provide a more definitive resolution to the stereochemical issue at hand.
We defined as our first synthetic target the GHIJK maitotoxin domain 88 (Scheme 13), possessing the originally assigned C51–C52 stereochemical configuration as the most likely correct structure based on the computational studies outlined above and shown graphically in Figure 4.[40] Employing some of the synthetic methods summarized in the previous section, domain 88 was synthesized as highlighted in Scheme 13a. The key ring-forming reactions in this synthesis were the Noyori reduction/Achmatowicz rearrangement[38,39] [rings J (79 → 67) and G (84 → 71)], the silver-catalyzed hydroxy ynone cyclization (ring K, 80 → 81), the Suzuki coupling with a vinyl triflate[23–25] (ring system GIJK, 82 → 85), and the hydroxy ketone cyclization/reductive deoxygenation[17–19] (ring H, 86 → 87). Comparison of the 13C chemical shifts of the synthetic GHIJK domain 88 with the values corresponding to the same carbons in natural maitotoxin revealed remarkable agreement as shown in Scheme 13b (with the exception of the edge carbons for the obvious reasons stated above), pointing to the validity of the Yasumoto–Kishi–Tachibana structure 1 for maitotoxin.
Scheme 13.

Synthesis of the GHIJK maitotoxin domain 88 (a) and 13C chemical shift differences between 88 and maitotoxin (1) (b) (Nicolaou et al., 2007).[40]
While the close agreement between the 13C NMR chemical shifts of synthetic GHIJK fragment 88 and the corresponding values for natural maitotoxin provided further support for the originally assigned structure of the latter structure at C51–C52, the skeptics may demand stronger evidence from a larger synthetic domain of maitotoxin. Encouraged by the performance of our synthetic technologies in delivering expediently the GHIJK fragment 88, and in order to provide such support, we targeted the maitotoxin GHIJKLMNO domain 95 (Scheme 14a).[41] The enantioselective synthesis of 95 was achieved starting from furan derivatives as summarized in Scheme 14a. Key reactions included a Noyori reduction/Achmatowicz rearrangement (79 → 66),[38,39] a silver-promoted hydroxy ynone cyclization (89 → 90), a Suzuki coupling (90 → 91),[23–25] a hydroxy ketone cyclization/reductive deoxygenation (91 → 92),[17–19] and a Horner–Wadworth–Emmons (HWE) olefination (92 → 94). Again, the results of 13C NMR chemical shift comparisons between synthetic GHIJKLMNO fragment 95 and maitotoxin were striking (see Scheme 14b), with the only deviations being those for the edge carbon atoms as expected. Given the centrality of the JK ring junction (C51–C52) within this rather large domain (GHIJKLMNO), the evidence for the accuracy of the originally assigned C51–C52 configuration is overwhelming.
Scheme 14.

Synthesis of the GHIJKLMNO maitotoxin domain 95 (a) and 13C chemical shift differences between 95 and maitotoxin (1) (b) (Nicolaou et al., 2008).[41]
Having settled the stereochemical configuration issue of the JK junction to our satisfaction, our focus then turned to other large domains of the maitotoxin molecule. Our reasons for pursuing the synthesis of such fragments included developing further synthetic technologies and strategies, rendering these otherwise unavailable maitotoxin domains readily available for biological investigations, confirming their stereochemical assignments, and scouting a possible synthetic pathway to the entire polycyclic framework of maitotoxin. First to be targeted was the ABCDEFG ring system 102 (Scheme 15a).[42] Commencing with furan derivatives (i.e., 84, 96), the developed strategy toward heptacyclic domain 102 (see Scheme 15a) featured two Noyori reductions/Achmatowicz rearrangements,[38,39] a Suzuki coupling employing a 9-BBN derivative and a vinyl phosphate,[23–25] a cyclic O,S-acetal formation/methylation,[15,16] a HWE olefination, and a hydroxy ketone cyclization/reductive deoxygenation,[17–19] all of which proceeded smoothly and through intermediates 68, 70, and 97 – 101. Although the originally assigned stereochemical configurations of this domain of maitotoxin had not been brought into question, we proceeded, nonetheless, to compare the 13C NMR chemical shifts of synthetic ABCDEFG fragment 102 (and all our subsequently synthesized large fragments) with those of the corresponding domain of maitotoxin in order to confirm the originally assigned structure of that region of the molecule. As seen in Scheme 15b, the agreement between the two structural motifs is compelling, providing strong support for the original assignment of this region of maitotoxin.
Scheme 15.

Synthesis of the ABCDEFG maitotoxin domain 102 (a) and 13C chemical shift differences between 102 and maitotoxin (1) (b) (Nicolaou et al., 2010).[42]
Our next target became the QRSTU domain 109 (Scheme 16a) of maitotoxin.[46] The main challenge presented by this pentacyclic system derived from the five rather congested methyl groups residing on its periphery. The designed convergent synthetic strategy (103 + 104 → 105 → 106 → 107 → 108 → 109, Scheme 16a) called upon an intramolecular ester olefin ring closure[29–33] (105 → 106) and a cyclic O,S-acetal formation/methylation[15,16] (107 → 108) to cast the final two rings of the molecule. Comparison of the 13C NMR data of synthetic QRSTU domain 109 with those of the corresponding region of maitotoxin revealed, as shown in Scheme 16b, excellent agreement between them, therefore providing confirming evidence for the originally assigned structure of this region of the natural product as well.
The next maitotoxin domain to be targeted was the WZYZA′ ring system 116 (Scheme 17a).[47] The convergent synthesis of this fragment mirrored the one employed to construct the QRSTU system 109 described above. Thus, coupling of 110 and 111 furnished ester 112, whose ester–olefin ring closure[29–33] led to cyclic enol ether 113. The latter was then swiftly converted to the desired compound (116) through intermediates 114 and 115 by a sequence involving a cyclic O,S-acetal formation/methylation[15,16] as shown in Scheme 17a. Once again, excellent correlations were observed for the 13C NMR chemical shifts of synthetic WZYZA′ domain 116 and those corresponding to the same region of maitotoxin (see Scheme 17b), lending strong support for its originally assigned structure.
The last remaining segment of maitotoxin, C′D′E′F′ ring system 122, was synthesized beginning with a furan derivative through a highly efficient linear synthesis as outlined in Scheme 18a (84 → 69 → 117 → 118 → 119 → 120 → 121 → 122).[43] This sequence employed a Noyori reduction/Achmatowicz rearrangement[38,39] (84 → 69), two intramolecular hydroxy epoxide openings[12] (69 → 117 and 120 → 121) and a ketyl radical conjugate addition to an α,β-unsaturated ester facilitated by SmI2[48] (118 → 119). As seen in Scheme 18b, the 13C NMR chemical shifts of synthetic C′D′E′F′ domain 122 were in excellent agreement with those of maitotoxin corresponding to the same carbons, with the exception of the edge carbons. This striking agreement leaves little doubt, if any, that the original assignment for this region of maitotoxin is also correct. Crystalline 122 yielded to X-ray crystallographic analysis, which revealed its interesting concavity, apparently caused by the three congested methyl groups as shown in Figure 5.[43]
Scheme 18.

Synthesis of the C′D′E′F′ maitotoxin domain 122 (a) and 13C chemical shift differences between 122 and maitotoxin (1) (b) (Nicolaou et al., 2010).[43]
Figure 5.
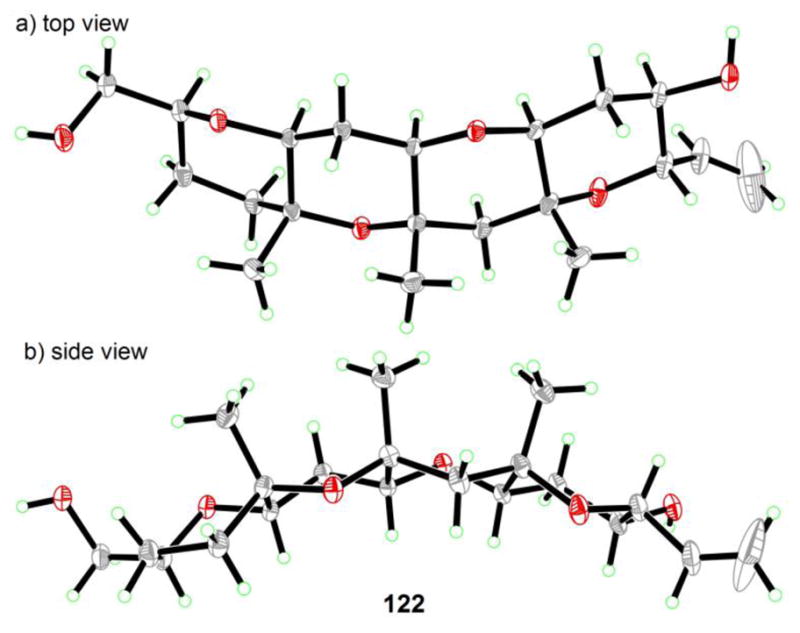
Concavity of the C′D′E′F′ domain 122 as revealed by X-ray crystallographic analysis (Nicolaou et al., 2011).[43]
4. Biological Activity and Mechanism of Action of Maitotoxin
As the largest and most toxic secondary metabolite isolated to date, maitotoxin (1) commands considerable attention from both chemists and biologists alike. In contrast to brevetoxin B (4), whose mechanism of action involves binding to a known voltage-sensitive sodium channel,[49] thereby inducing sodium ion influx into cells, maitotoxin’s target is unknown. The latter has been implicated in calcium ion influx into cells,[50] although it does not cause calcium ion influx into artificial phospholipid vesicles.[51] This observation suggests a more complex mechanism of action, leaving a gap in our understanding of the basis of its neurotoxicity and multitude of other physiological effects. Maitotoxin has been observed, for example, to cause muscle contraction,[52] norepinephrine,[53] dopamine[54] and insulin secretion,[55] phosphoinositide breakdown,[56] arachidonic acid release,[57] and acrosome reaction in sperm.[58] While direct analogy may suggest that maitotoxin binds to a voltage-sensitive calcium ion channel, electrophysiological experiments indicate that maitotoxin’s target is voltage independent.[59] Consistent with this notion is the observed induction of non-selective influx of ions into cells by maitotoxin.[60]
An intriguing model for the mode of action of maitotoxin has been advanced by Murata and Yasumoto on the basis of NMR spectroscopic studies (see Figure 6).[61] According to their hypothesis, maitotoxin anchors itself into the cell membrane with its lipophilic domain (i.e., RSTUVWXYZA′B′C′D′E′F′ ring system) spanning the lipophilic layer, while the hydrophilic domain of the molecule (i.e., ABCDEFGHIJKLMNOPQ) remains outside the cell. A molecular assembly consisting of several maitotoxin molecules and its biological target within the membrane was suggested to form a pore for non-selective ion influx, which in turn activates a voltage-sensitive calcium channel, leading to the observed physiological effects of maitotoxin. Unfortunately, due to the extreme natural scarcity of this biotoxin, this hypothesis remains without experimental support. During the course of our own efforts described above, we have had the opportunity to evaluate the biological properties of two of our synthesized maitotoxin domains, namely ring systems ABCDEFG (102) and QRSTU (109), in collaboration with Thomas Murray and coworkers. Neither of these fragments induces calcium influx into cerebral cortical neurons. [62] These findings are in line with the proposed binding mode described above. Further biological investigation with other synthetic maitotoxin fragments (i.e., 88, 116, 122) and larger domains to be synthesized in the future are warranted and will be pursued as part of the maitotoxin program.
Figure 6.
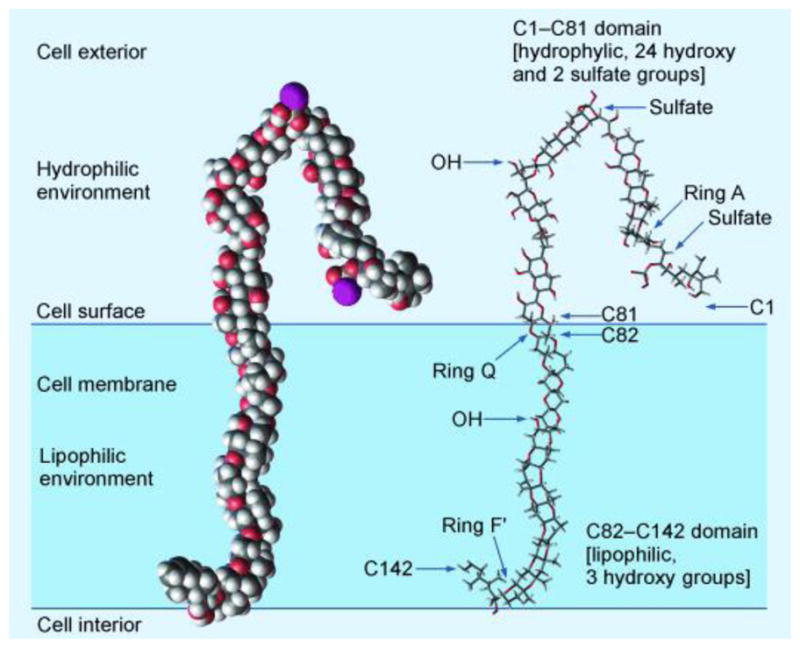
The Murata–Yasumoto hypothetical model of the mode of action of maitotoxin.[62]
5. Conclusion and Future Perspectives
With its magnificent molecular architecture and potent biological effects, maitotoxin has been providing intrigue and inspiration to scientists for over 30 years since its detection in the gut of the surgeonfish Ctenochaetus striatus.[4] Thus far, this fascination led to its structural elucidation, including absolute configuration, through a combination of NMR spectroscopic, mass spectroscopic and synthetic studies.[6–8] Significant strides have also been made with regard to understanding its biological properties and mechanism of action.[51–63] The scarcity of maitotoxin, however, prevents its full biological investigation and hampers the identification of its target and the complete elucidation of its mechanism of action. On the other hand, synthetic studies inspired by the molecule of maitotoxin culminated in the synthesis of several of its polycyclic frameworks, including the ABCDEFG (102),[42] GHIJKLMNO (95),[41] QRSTU (109),[46] WXYZA′ (116),[47] and C′D′E′F′ (122)[43] domains (see Figure 7). The availability of these synthetic materials allowed re-confirmation of the originally assigned structure of maitotoxin, which had been challenged, in the meantime, and provided the foundation for further developments in the field. Hinging primarily on chemical synthesis, these developments may include biological tools to facilitate investigations in neurobiology, responses to environmental challenges, and construction of larger fragments of this extraordinary molecule. With regard to the latter, the highlighted bonds (C12–C13, C36–C37, C64–C65 and C135–C136, Figure 7) and rings (P, V and B′, Figure 7) may serve as the connecting bridges that, upon construction, should lead to a variety of desired domains for biological investigation.
Figure 7.

Stuctures of maitotoxin (1) and synthesized domains ABCDEFG (102), GHIJKLMNO (95), QRSTU (109), WXYZA′ (116), and C′D′E′F′ (122).
Acknowledgments
We thank Drs. D.-H. Huang, G. Siuzdak, and R. Chadha for spectroscopic, mass spectrometric, and X-ray crystallographic assistance, respectively. Financial support for this work was provided by the National Institutes of Health (U.S.A.) and The Skaggs Institute for Chemical Biology.
Abbreviations
- AIBN
2,2′-azobis(2-methylpropionitrile)
- 9-BBN
9-borabicyclo[3.3.1]nonane
- Bn
benzyl
- cat
catalytic
- Cp
cyclopentadienyl
- mCPBA
meta-chlorperbenzoic acid
- CSA
camphor sulfonic acid
- HWE
Horner–Wadsworth–Emmons
- LD50
median lethal dose
- MOM
methoxymethyl
- MS
molecular sieves
- NAP
naphthyl
- NBS
N-bromosuccinimide
- NCS
N-chlorosuccinimide
- NMR
nuclear magnetic resonance
- Piv
trimethylacetyl
- PMB
para-methoxybenzyl
- TBAF
tetra-n-butylammonium fluoride
- TBDPS
tert-butyldiphenylsilyl
- TBS
tert-butyldimethylsilyl
- TES
triethylsilyl
- Tf
trifluoromethanesulfonyl
- TIPS
triisopropylsilyl
- TMEDA
tetramethylethylenediamine
- TMS
trimethylsilyl
- Ts
para-toluenesulfonyl
Biographies

K. C. Nicolaou, born in Cyprus and educated in UK and USA, is Chairman of the Department of Chemistry at The Scripps Research Institute where he holds the Darlene Shiley Chair in Chemistry and the Aline W. & L.S. Skaggs Professorship in Chemical Biology, and is Distinguished Professor of Chemistry at the University of California, San Diego. The impact of his work in chemistry, biology and medicine flows from his contributions to chemical synthesis as described in over 700 publications. His dedication to chemical education is reflected in his Classics in Total Synthesis series and Molecules That Changed the World books and his training of hundreds of graduate students and postdoctoral fellows. He is a Member of the National Academy of Sciences, USA, and the German Academy of Sciences, Leopoldina, and Corresponding Member of the Academy of Athens.

Robert J. Aversa was born in Philadelphia, Pennsylvania, in 1984. He received his BA in chemistry and biochemistry from Cornell University in 2006, where he carried out research under the tutelage of Professor Tadhg P. Begley. He is currently pursuing his PhD degree at The Scripps Research Institute in the laboratories of Professor K. C. Nicolaou, where he has been working toward the total synthesis of maitotoxin. In 2010, he received the Roche Excellence in Chemistry award in recognition of this work
References
- 1.Wöhler F. Annalen der Physik. 1828;88:253–256. [Google Scholar]
- 2.Murata M, Yasumoto Y. Nat Prod Rep. 2000;17:293–314. doi: 10.1039/a901979k. [DOI] [PubMed] [Google Scholar]
- 3.Takahashi M, Ohizumi Y, Yasumoto T. J Biol Chem. 1982;257:7287–7289. [PubMed] [Google Scholar]
- 4.a) Yasumoto T, Bagnis R, Randal JE, Banner AH. Nippon Suisan Gakkaishi. 1971;37:724–734. [Google Scholar]; b) Yasumoto T, Bagnis R, Vernoux JP. Nippon Suisan Gakkaishi. 1976;42:359–365. [Google Scholar]
- 5.a) Yasumoto T, Nakajima I, Bagnis R, Adachi R. Nippon Suisan Gakkaishi. 1977;43:1021–1026. [Google Scholar]; b) Yokoyama A, Murata M, Oshima Y, Iwashita T, Yasumoto T. J Biochem. 1988;104:184–187. doi: 10.1093/oxfordjournals.jbchem.a122438. [DOI] [PubMed] [Google Scholar]
- 6.a) Murata M, Iwashita T, Yokoyama A, Sasaki M, Yasumoto T. J Am Chem Soc. 1992;114:6594–6596. [Google Scholar]; b) Murata M, Naoki H, Iwashita T, Matusunaga S, Sasaki M, Yokoyama A, Yasumoto T. J Am Chem Soc. 1993;115:2060–2062. [Google Scholar]; c) Murata M, Naoki H, Matsunaga S, Satake M, Yasumoto T. J Am Chem Soc. 1994;116:7098–7107. [Google Scholar]; d) Satake M, Ishida S, Yasumoto T, Murata M, Utsumi H, Hinomoto T. J Am Chem Soc. 1995;117:7019–7020. [Google Scholar]; e) Matsumori N, Kaneno D, Murata M, Nakamura H, Tachibana K. J Org Chem. 1999;64:866–876. doi: 10.1021/jo981810k. [DOI] [PubMed] [Google Scholar]
- 7.a) Sasaki M, Matsumori N, Maruyama T, Nonomura T, Murata M, Tachibana K, Yasumoto T. Angew Chem. 1996;108:1782–1785. [Google Scholar]; Angew Chem Int Ed Engl. 1996;35:1672–1675. [Google Scholar]; b) Matsumori N, Nonomura T, Sasaki M, Murata M, Tachibana K, Satake M, Yasumoto T. Tetrahedron Lett. 1996;37:1269–1272. [Google Scholar]; c) Sasaki M, Matsumori N, Murata M, Tachibana K, Yasumoto T. Tetrahedron Lett. 1995;36:9011–9014. [Google Scholar]; d) Sasaki M, Nonomura T, Murata M, Tachibana K. Tetrahedron Lett. 1994;35:5023–5026. [Google Scholar]; e) Sasaki M, Nonomura T, Murata M, Tachibana K. Tetrahedron Lett. 1995;36:9007–9010. [Google Scholar]; f) Nonomura T, Sasaki M, Matsumori N, Murata M, Tachibana K, Yasumoto T. Angew Chem. 1996;108:1786–1789. [Google Scholar]; Angew Chem Int Ed Engl. 1996;35:1675–1678. [Google Scholar]
- 8.a) Zheng W, DeMattei JA, Wu JP, Duan JJW, Cook LR, Oinuma H, Kishi Y. J Am Chem Soc. 1996;118:7946–7968. [Google Scholar]; b) Cook LR, Oinuma H, Semones MA, Kishi Y. J Am Chem Soc. 1997;119:7928–7937. [Google Scholar]; c) Kishi Y. Pure Appl Chem. 1998;70:339–344. [Google Scholar]
- 9.Gallimore AR, Spencer JB. Angew Chem. 2006;118:4514–4421. doi: 10.1002/anie.200504284. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2006;45:4406–4413. doi: 10.1002/anie.200504284. [DOI] [PubMed] [Google Scholar]
- 10.a) Nakanishi K. Toxicon. 1985;23:473–479. doi: 10.1016/0041-0101(85)90031-5. [DOI] [PubMed] [Google Scholar]; b) Lee MS, Qin GW, Nakanishi K, Zagorski MG. J Am Chem Soc. 1989;111:6234–6241. [Google Scholar]; c) Nicolaou KC. NIH application GM31398-01 (submission date: February 24, 1982) [Google Scholar]; d) Nicolaou KC. Angew Chem. 1996;108:644–663. [Google Scholar]; Angew Chem Int Ed Engl. 1996;35:588–607. [Google Scholar]
- 11.Lin YY, Risk M, Ray SM, Van Engen D, Clardy J, Golik J, James JC, Nakanishi K. J Am Chem Soc. 1981;103:6773–6775. [Google Scholar]
- 12.a) Nicolaou KC, Duggan ME, Hwang CK, Somers PK. J Chem Soc, Chem Commun. 1985:1359–1362. [Google Scholar]; b) Nicolaou KC, Prasad CVC, Somers PK, Hwang CK. J Am Chem Soc. 1989;111:5330–5334. [Google Scholar]; c) Nicolaou KC, Prasad CVC, Somers PK, Hwang CK. J Am Chem Soc. 1989;111:5335–5340. [Google Scholar]
- 13.Nicolaou KC, Hwang CK, Duggan ME. J Am Chem Soc. 1989;111:6682–6690. [Google Scholar]
- 14.Giannis A, Sandhoff K. Carb Res. 1987;171:201–210. [Google Scholar]
- 15.a) Nicolaou KC, Duggan ME, Hwang CK. J Am Chem Soc. 1986;108:2468–2469. doi: 10.1021/ja00269a067. [DOI] [PubMed] [Google Scholar]; b) Nicolaou KC, Prasad CVC, Hwang C-K, Duggan ME, Veale CA. J Am Chem Soc. 1989;111:5321–5330. [Google Scholar]
- 16.For an application of this process on related substrates, see: Nicolaou KC, Veale CA, Hwang CK, Hutchinson J, Prasad CVC, Ogilvie WW. Angew Chem. 1991;103:304–308.Angew Chem Int Ed Engl. 1991;30:299–303.Fuwa H, Sasaki M, Tachibana K. Tetrahedron Lett. 2000;41:8371–8375.Fuwa H, Sasaki M, Tachibana K. Tetrahedron. 2001;57:3019–3033.
- 17.a) Nicolaou KC, Hwang CK, Nugiel DA. Angew Chem. 1988;100:1413–1415. [Google Scholar]; Angew Chem Int Ed. 1988;27:1362–1364. [Google Scholar]; b) Nicolaou KC, Hwang CK, Nugiel DA. J Am Chem Soc. 1989;111:4136–1437. [Google Scholar]
- 18.Evans PA, Cui J, Gharpure SJ, Hinkle RJ. J Am Chem Soc. 2003;125:11456–11457. doi: 10.1021/ja036439j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sato K, Sasaki M. Tetrahedron. 2007;63:5977–6003. [Google Scholar]
- 20.a) Nicolaou KC, McGarry DG, Somers PK, Veale CA, Furst GT. J Am Chem Soc. 1987;109:2504–2506. [Google Scholar]; b) Nicolaou KC, McGarry DG, Somers PK, Kim BH, Ogilvie WW, Yiannikouros G, Prasad CVC, Vaele CA, Hark RR. J Am Chem Soc. 1990;112:6263–6276. [Google Scholar]
- 21.a) Nicolaou KC, Reddy KR, Skokotas G, Sato F, Xiao XY. J Am Chem Soc. 1992;114:7935–7936. [Google Scholar]; b) Nicolaou KC, Reddy KR, Skokotas G, Sato F, Xiao XY, Hwang CK. J Am Chem Soc. 1993;115:3558–3575. [Google Scholar]
- 22.a) Nicolaou KC, Hwang C-K, Duggan ME, BalReddy K, Marron BE, McGarry DG. J Am Chem Soc. 1986;108:6800–6802. [Google Scholar]; b) Nicolaou KC, Hwang CK, Duggan ME, Carroll PJ. J Am Chem Soc. 1987;109:3801–3802. [Google Scholar]; c) Nicolaou KC, Hwang CK, DeFrees SA, Stylianides NA. J Am Chem Soc. 1988;110:4868–4869. [Google Scholar]; d) Nicolaou KC, Hwang CK, Marron BE, DeFrees SA, Couladouros EA, Abe Y, Carroll PJ, Snyder JP. J Am Chem Soc. 1990;112:3040–3054. [Google Scholar]; e) Nicolaou KC, DeFrees SA, Hwang CK, Stylianides N, Carroll PJ, Snyder JP. J Am Chem Soc. 1990;112:3029–3039. [Google Scholar]
- 23.For selected reviews on cross coupling reactions, see: de Meijere A, Diederich F, editors. Metal-Catalyzed Cross-Coupling Reactions. 2. Wiley-VCH; Weinheim: 2004. p. 938.Hegedus LS. Transition Metals in the Synthesis of Complex Organic Molecules. 2. University Science Books; Sausalito: 1999. p. 352.Negishi E, de Meijere A, editors. Handbook of Organopalladium Chemistry for Organic Synthesis. Wiley Interscience; New York: 2002. p. 3350.Miyaura N, editor. Topics in Current Chemistry. 219. Springer-Verlag; Berlin: 2002. Cross-Coupling Reactions: A Practical Guide; p. 248.Nicolaou KC, Bulger PG, Sarlah D. Angew Chem. 2005;117:4516–4563. doi: 10.1002/anie.200500368.Angew Chem Int Ed. 2005;44:4442–4489. doi: 10.1002/anie.200500368.
- 24.a) Tsushima K, Araki K, Murai A. Chem Lett. 1989:1313–1316. [Google Scholar]; b) Tsushima K, Murai A. Chem Lett. 1990:761–764. [Google Scholar]
- 25.Nicolaou KC, Shi GQ, Gunzner JL, Gärtner P, Yang Z. J Am Chem Soc. 1997;119:5467–5468. [Google Scholar]
- 26.Sasaki M, Fuwa H, Ishikawa M, Tachibana K. Org Lett. 1999;1:1075–1077.For a review on the B-alkyl Suzuki reaction see: Chemler SR, Trauner D, Danishefsky SJ. Angew Chem. 2001;113:4676–4698. doi: 10.1002/1521-3773(20011217)40:24<4544::aid-anie4544>3.0.co;2-n.Angew Chem Int Ed. 2001;40:4544–4568. doi: 10.1002/1521-3773(20011217)40:24<4544::aid-anie4544>3.0.co;2-n.
- 27.For selected reviews on olefin metathesis, see: Grubbs RH, Chang S. Tetrahedron. 1998;54:4413–4450.Fürstner A. Angew Chem. 2000;112:3140–3171.Angew Chem Int Ed. 2000;39:3012–3043.Schmidt B, Hermanns J. Top Organomet Chem. 2004;13:223–267.Connon SJ, Blechert S. Top Organomet Chem. 2004;11:93–124.Nicolaou KC, Bulger PG, Sarlah D. Angew Chem. 2005;117:4564–4601. doi: 10.1002/anie.200500368.Angew Chem Int Ed. 2005;44:4490–4527. doi: 10.1002/anie.200500369.Schrock RR, Czekelius C. Adv Synth Catal. 2007;349:55–77.Mori M. Adv Synth Catal. 2007;349:121–135.
- 28.For a review of applications of olefin metathesis in the synthesis of cyclic ethers, see: Clark JS. Chem Commun. 2006:3571–3581. doi: 10.1039/b601839d.
- 29.a) Fu GC, Grubbs RH. J Am Chem Soc. 1992;114:5426–5427. [Google Scholar]; b) Fu GC, Nguyn ST, Grubbs RH. J Am Chem Soc. 1993;115:9856–9857. [Google Scholar]; c) Fujimura O, Fu GC, Grubbs RH. J Org Chem. 1994;59:4029–4031. [Google Scholar]
- 30.a) Tebbe FN, Parshall GW, Reddy GS. J Am Chem Soc. 1978;100:3611–3613. [Google Scholar]; b) Pine SH, Zahler R, Evans DA, Grubbs RH. J Am Chem Soc. 1980;102:3270–3272. [Google Scholar]
- 31.a) Nicolaou KC, Postema MHD, Claiborne CF. J Am Chem Soc. 1996;118:1565–1566. [Google Scholar]; b) Nicolaou KC, Postema MHD, Yue EW, Nadin A. J Am Chem Soc. 1996;118:10335–10336. [Google Scholar]
- 32.Takai K, Kakiuchi T, Kataoka Y, Utimoto K. J Org Chem. 1994;59:2668–2670. [Google Scholar]
- 33.a) Clark JS, Kettle JG. Tetrahedron Lett. 1997;38:123–126. [Google Scholar]; b) Clark JS, Kettle JG. Tetrahedron Lett. 1997;38:127–130. [Google Scholar]
- 34.a) Johnson HWB, Majumder U, Rainer JD. Chem Eur J. 2006;12:1747–1753. doi: 10.1002/chem.200500994. [DOI] [PubMed] [Google Scholar]; b) Majumder U, Rainer JD. Tetrahedron Lett. 2005;46:7209–7211. [Google Scholar]
- 35.a) Nicolaou KC, Duggan ME, Hwang CK. J Am Chem Soc. 1989;111:6666–6675. [Google Scholar]; b) Nicolaou KC, Duggan ME, Hwang CK. J Am Chem Soc. 1989;111:6676–6682. [Google Scholar]; c) Nicolaou KC, Hwang CK, Duggan ME. J Am Chem Soc. 1989;111:6682–6690. [Google Scholar]; d) Nicolaou KC, Nugiel DA, Couladouros E, Hwang CK. Tetrahedron. 1990;46:4517–4552. [Google Scholar]; e) Nicolaou KC, Tiebes J, Theodorakis EA, Rutjes FPJT, Koide K, Sato M, Untersteller E. J Am Chem Soc. 1994;116:9371–9372. [Google Scholar]; f) Nicolaou KC, Theodorakis EA, Rutjes FPJT, Tiebes J, Sato M, Untersteller E, Xiao XY. J Am Chem Soc. 1995;117:1171–1172. [Google Scholar]; g) Nicolaou KC, Rutjes FPJT, Theodorakis EA, Tiebes J, Sato M, Untersteller E. J Am Chem Soc. 1995;117:1173–1174. [Google Scholar]; h) Nicolaou KC, Hwang C-K, Duggan ME, Nugiel DA, Abe Y, Bal Reddy K, DeFrees SA, Reddy DR, Awartani RA, Conley SR, Rutjes FPJT, Theodorakis EA. J Am Chem Soc. 1995;117:10227–10238. [Google Scholar]; i) Nicolaou KC, Theodorakis EA, Rutjes FPJT, Sato M, Tiebes J, Xiao XY, Hwang CK, Duggan ME, Yang Z, Couladouros EA, Sato F, Shin J, He HM, Bleckman T. J Am Chem Soc. 1995;117:10239–10251. [Google Scholar]; j) Nicolaou KC, Rutjes FPJT, Theodorakis EA, Tiebes J, Sato M, Untersteller E. J Am Chem Soc. 1995;117:10252–10263. [Google Scholar]
- 36.a) Nicolaou KC, McGarry DG, Somers PK. J Am Chem Soc. 1990;112:3696–3697. [Google Scholar]; b) Nicolaou KC, Prasad CVC, Ogilvie WW. J Am Chem Soc. 1990;112:4988–4989. [Google Scholar]; c) Nicolaou KC, Yang Z, Shi G-Q, Gunzner JL, Agrios KA, Gärtner P. Nature. 1998;392:264–269. doi: 10.1038/32623. [DOI] [PubMed] [Google Scholar]; d) Nicolaou KC, Bunnage ME, McGarry DG, Shi S, Somers PK, Wallace PA, Chu XJ, Agrios KA, Gunzner JG, Yang Z. Chem Eur J. 1999;5:599–617. [Google Scholar]; e) Nicolaou KC, Wallace PA, Shi S, Ouellette MA, Bunnage ME, Gunzner JL, Agrios KA, Shi GQ, Gärtner P, Yang Z. Chem Eur J. 1999;5:618–627. [Google Scholar]; f) Nicolaou KC, Shi GQ, Gunzner JL, Gärtner P, Wallace PA, Ouelette MA, Shi S, Bunnage ME, Agrios KA, Veale CA, Hwang CK, Hutchinson J, Prasad CVC, Ogilvie WW, Yang Z. Chem Eur J. 1999;5:628–645. [Google Scholar]; g) Nicolaou KC, Gunzner JL, Shi GQ, Agrios KA, Gärtner P, Yang Z. Chem Eur J. 1999;5:646–658. [Google Scholar]
- 37.Kadota I, Jung-Youl P, Koumura N, Pollaud G, Matsukawa Y, Yamamoto Y. Tetrahedron Lett. 1995;36:5777–5780.Kadota I, Yamamota Y. J Org Chem. 1998;63:6597–6606.Nakata T, Nomura S, Matsukura H. Tetrahedron Lett. 1996;37:213–216.Nakata T, Nomura S, Matsukura H, Morimoto M. Tetrahedron Lett. 1996;37:217–220.Morimoto M, Matsukura H, Nakata T. Tetrahedron Lett. 1996;37:6365–6368.Mori Y, Yaegashi K, Furukawa H. J Am Chem Soc. 1997;119:4557–4558.Mori Y, Yaegashi K, Furukawa H. J Org Chem. 1998;63:6200–6209. doi: 10.1021/jo980320p.Rainer JD, Allwein SP. J Org Chem. 1998;63:5310–5311.Rainer JD, Allwein SP. Tetrahedron Lett. 1998;39:9601–9604.Rainier JD, Allwein SP, Cox JM. Org Lett. 2000;2:231–234. doi: 10.1021/ol991371r.Rainier JD, Allwein SP, Cox JM. J Org Chem. 2001;66:1380–1386. doi: 10.1021/jo001514j.Holland JM, Lewis M, Nelson A. Angew Chem. 2001;113:4206–4208.Angew Chem Int Ed. 2001;40:4082–4084.Holland JM, Lewis M, Nelson A. J Org Chem. 2003;68:747–753. doi: 10.1021/jo026456b.Zakarian A, Batch A, Holton RA. J Am Chem Soc. 2003;125:7822–7824. doi: 10.1021/ja029225v.Fujiwara K, Sato D, Watanabe M, Morishita H, Murai A, Kawai H, Suzuki T. Tetrahedron Lett. 2004;45:5243–5246.Kadota I, Abe T, Ishitsuka Y, Touchy AS, Nagata R, Yamamota Y. Tetrahedron Lett. 2007;48:219–221.Matsuo G, Matsukura H, Hori N, Nakata T. Tetrahedron Lett. 2000;41:7673–7676.Matsuo G, Hori N, Matsukura H, Nakata T. Tetrahedron Lett. 2000;41:7677–7680.Matsukura H, Hori N, Matsuo G, Nakata T. Tetrahedron Lett. 2000;41:7681–7684.Matsuo G, Kawamura K, Hori N, Matsukura H, Nakata T. J Am Chem Soc. 2004;126:14374–14376. doi: 10.1021/ja0449269.Crimmins MT, Zuccarello JL, Ellis JM, McDougall PJ, Haile PA, Parrish JD, Emmite KA. Org Lett. 2009;11:489–492. doi: 10.1021/ol802710u.Crimmins MT, Ellis JM, Emmite KA, Haile PA, McDougall PJ, Parrish JD, Zuccarello JL. Chem Eur J. 2009:9223–9234. doi: 10.1002/chem.200900776.Crimmins MT, Zuccarello JL, McDougall PJ, Ellis JM. Eur J Chem. 2009;15:9235–9244. doi: 10.1002/chem.200900777.Oishi T, Uehara H, Nagumo Y, Shoji M, Le Brazidec JY, Kosaka M, Hirama M. Chem Commun. 2001:381–382.Imai H, Uehara H, Inoue M, Oguri H, Oishi T, Hirama M. Tetrahedron Lett. 2001;42:6219–6222.Hirama M, Oishi T, Uehara H, Inoue M, Maruyama M, Oguri H, Satake M. Science. 2001;294:1904–1907. doi: 10.1126/science.1065757.Maruyama M, Inoue M, Oishi T, Oguri H, Ogasawara Y, Shindo Y, Hirama M. Tetrahedron. 2002;58:1835–1851.Inoue M, Uehara H, Maruyama M, Hirama M. Org Lett. 2002;4:4551–4554. doi: 10.1021/ol027105m.Uehara H, Oishi T, Inoue M, Shoji M, Nagumo Y, Kosaka M, Le Brazidec JY, Hirama M. Tetrahedron. 2002;58:6493–6512.Tatami A, Inoue M, Uehara H, Hirama M. Tetrahedron Lett. 2003;44:5229–5233.Oguri H, Hirama M, Tsumuraya T, Fujii I, Maruyama M, Uehara H, Nagumo Y. J Am Chem Soc. 2003;125:7608–7612. doi: 10.1021/ja034990a.Inoue M, Yamashita S, Tatami A, Miyazaki K, Hirama M. J Org Chem. 2004;69:2797–2804. doi: 10.1021/jo049877x.Inoue M, Hirama M. Acc Chem Res. 2004;37:961–986. doi: 10.1021/ar0301577.Inoue M, Hirama M. Synlett. 2004;4:577–595.Inoue M, Miyazaki K, Uehara H, Maruyama M, Hirama M. Proc Natl Acad Sci U S A. 2004;101:12013–12018. doi: 10.1073/pnas.0401684101.Fuwa H, Sasaki M, Tachibana K. Tetrahedron Lett. 2000;41:8371–8375.Fuwa H, Sasaki M, Tachibana K. Tetrahedron. 2001;57:3019–3033.Fuwa H, Sasaki M, Tachibana K. Org Lett. 2001;3:3549–3552. doi: 10.1021/ol0166597.Fuwa H, Kainuma N, Tachibana K, Sasaki M. J Am Chem Soc. 2002;124:14983–14992. doi: 10.1021/ja028167a.Kadota I, Park CH, Ohtaka M, Oguro N, Yamamoto Y. Tetrahedron Lett. 1998;39:6365–6368.Kadota I, Kadowaki C, Yoshida N, Yamamoto Y. Tetrahedron Lett. 1998;39:6369–6372.Kadota I, Ohno A, Matsukura Y, Yamamoto Y. Tetrahedron Lett. 1998;39:6373–6376.Kadota I, Ohno A, Matsuda K, Yamamoto Y. J Am Chem Soc. 2001;123:6702–6703. doi: 10.1021/ja010911o.Kadota I, Kadowaki C, Park CH, Takamura H, Sato K, Chan PWH, Thorand S, Yamamoto Y. Tetrahedron. 2002;58:1799–1816.Kadota I, Takamura H, Sato K, Yamamoto Y. J Org Chem. 2002;67:3494–3498. doi: 10.1021/jo025566f.Kadota I, Ohno A, Matsuda K, Yamamoto Y. J Am Chem Soc. 2002;124:3562. doi: 10.1021/ja025523g.Kadota I, Takamura H, Sato K, Ohno A, Matsuda K, Yamamoto Y. J Am Chem Soc. 2003;125:46–47. doi: 10.1021/ja028726d.Kadota I, Takamura H, Sato K, Ohno A, Matsuda K, Satake M, Yamamoto Y. J Am Chem Soc. 2003;125:11893–11899. doi: 10.1021/ja036984k.Cox JM, Rainer JD. Org Lett. 2001;3:2919–2922.Majumder U, Cox JM, Rainier JD. Org Lett. 2003;5:913–916. doi: 10.1021/ol034100w.Johnson HWB, Majumder U, Rainer JD. J Am Chem Soc. 2005;127:848–849. doi: 10.1021/ja043396d.Majumder U, Cox JM, Johnson HWB, Rainier JD. Chem Eur J. 2006;12:1747–1753. doi: 10.1002/chem.200500994.Johnson HWB, Majumder U, Rainier JD. Chem Eur J. 2006;12:1736–1746. doi: 10.1002/chem.200500993.Sasaki M, Tsukano C, Tachibana K. Org Lett. 2002;4:1747–1750. doi: 10.1021/ol025814u.Sasaki M, Tsukano C, Tachibana K. Tetrahedron Lett. 2003;44:4351–4354.Tsukano C, Sasaki M. J Am Chem Soc. 2003;125:14294–14295. doi: 10.1021/ja038547b.Tsukano C, Ebine M, Sasaki M. J Am Chem Soc. 2005;127:4326–4335. doi: 10.1021/ja042686r.Tsukano C, Sasaki M. Tetrahedron Lett. 2006;47:6803–6807.Fuwa H, Ebine M, Bourdelais AJ, Baden DG, Sasaki M. J Am Chem Soc. 2006;128:16989–16999. doi: 10.1021/ja066772y.Sakamoto Y, Matsuo G, Matsukura H, Nakata T. Org Lett. 2001;3:2749–2752. doi: 10.1021/ol016355k.Morita M, Ihiyama S, Koshino H, Nakata T. Org Lett. 2008;10:1675. doi: 10.1021/ol800267x.Morita M, Haketa T, Koshino H, Nakata T. Org Lett. 2008;10:1679–1682. doi: 10.1021/ol800268c.Satoh M, Koshino H, Nakata T. Org Lett. 2008;10:1683–1685. doi: 10.1021/ol8002699.Oishi T, Hasegawa F, Torikai K, Konoki K, Matsumori N, Murata M. Org Lett. 2008;10:3599–3602. doi: 10.1021/ol801369g.For a review summarizing many of these efforts, see: Nicolaou KC, Frederick MO, Aversa RJ. Angew Chem. 2008;120:7292–7335. doi: 10.1002/anie.200801696.Angew Chem Int Ed. 2008;47:7182–7225. doi: 10.1002/anie.200801696.
- 38.Fujii A, Hashiguchi S, Uematsu N, Ikariya T, Noyori R. J Am Chem Soc. 1996;118:2521–2522. [Google Scholar]
- 39.a) Achmatowicz O, Bielski R. Carbohydr Res. 1977;55:165–176. doi: 10.1016/s0008-6215(00)84452-3. [DOI] [PubMed] [Google Scholar]; b) Guo H, O’Doherty GA. Angew Chem. 2007;119:5298–5300. [Google Scholar]; Angew Chem Int Ed. 2007;46:5206–5208. doi: 10.1002/anie.200701354. [DOI] [PubMed] [Google Scholar]; c) Zhou M, O’Doherty GA. J Org Chem. 2007;72:2485. doi: 10.1021/jo062534+. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Guo H, O’Doherty GA. Org Lett. 2006;8:1609–1612. doi: 10.1021/ol0602811. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Harris JM, Keranen MD, Nguyen H, Young VG, O’Doherty GA. Carbohydr Res. 2000;328:17–36. doi: 10.1016/s0008-6215(00)00031-8. [DOI] [PubMed] [Google Scholar]; f) Li M, Scott J, O’Doherty GA. Tetrahedron Lett. 2004;45:1005–1009. [Google Scholar]; g) Krishna UM, Srikanth GSC, Trivedi GK. Tetrahedron Lett. 2003;44:8227–8228. [Google Scholar]; h) Henderson JA, Jackson KL, Phillips AJ. Org Lett. 2007;9:5299–5302. doi: 10.1021/ol702559e. [DOI] [PubMed] [Google Scholar]
- 40.Nicolaou KC, Cole KP, Frederick MO, Aversa RJ, Denton RM. Angew Chem. 2007;119:9031–9035. doi: 10.1002/anie.200703742. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2007;46:8875–8879. doi: 10.1002/anie.200703742. [DOI] [PubMed] [Google Scholar]
- 41.Nicolaou KC, Frederick MO, Burtoloso A, Denton RM, Rivas F, Cole KP, Aversa RJ, Gibe R, Umezawa T, Suzuki T. J Am Chem Soc. 2008;130:7466–7476. doi: 10.1021/ja801139f. [DOI] [PubMed] [Google Scholar]
- 42.Nicolaou KC, Aversa RJ, Jin J, Rivas F. J Am Chem Soc. 2010;132:6855–6861. doi: 10.1021/ja102260q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nicolaou KC, Seo J-H, Nakamura T, Aversa RJ. J Am Chem Soc. doi: 10.1021/ja109531d. ASAP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang C, Forsyth CJ. Org Lett. 2006;8:2997–3000. doi: 10.1021/ol0609457. [DOI] [PubMed] [Google Scholar]
- 45.Nicolaou KC, Frederick MO. Angew Chem. 2007;119:5372–5376. [Google Scholar]; Angew Chem Int Ed. 2007;46:5278–5282. doi: 10.1002/anie.200604656. [DOI] [PubMed] [Google Scholar]
- 46.Nicolaou KC, Gelin CF, Seo JH, Huang Z, Umezawa T. J Am Chem Soc. 2010;132:9900–9907. doi: 10.1021/ja103708j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nicolaou KC, Baker TM, Nakamura T. J Am Chem Soc. doi: 10.1021/ja109533y. ASAP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.a) Hori N, Matsukura H, Matsuo G, Nakata T. Tetrahedron Lett. 1999;40:2811–2814. [Google Scholar]; b) Hori N, Matsukura H, Nakata T. Org Lett. 1999;1:1099–1101. [Google Scholar]; c) Matsuo G, Hori N, Nakata T. Tetrahedron Lett. 1999;40:8859–8862. [Google Scholar]; d) Hori N, Matsukura H, Matsuo G, Nakata T. Tetrahedron. 2002;58:1853–1864. [Google Scholar]
- 49.a) Trainer VL, Baden DG, Catterall WA. J Biol Chem. 1994;269:19904–19909. [PubMed] [Google Scholar]; b) Gawley RE, Rein KS, Jeglitsch G, Adams DJ, Theodorakis EA, Tiebes J, Nicolaou KC, Baden DG. Chem Biol. 1995;2:533–541. doi: 10.1016/1074-5521(95)90187-6. [DOI] [PubMed] [Google Scholar]; c) Catterall WA, Risk M. Mol Pharmacol. 1981;19:345–348. [PubMed] [Google Scholar]; d) Bidard JN, Vijverberg HP, Frelin C, Chunge E, Legrand AM, Bagnis R, Lazdunski M. J Biol Chem. 1984;259:8353–8357. [PubMed] [Google Scholar]
- 50.a) Takahashi M, Ohizumi Y, Yasumoto T. J Biol Chem. 1982;257:7287–7289. [PubMed] [Google Scholar]; b) Gusovsky F, Daly JW. Biochem Pharmacol. 1990;39:1633–1639. doi: 10.1016/0006-2952(90)90105-t. [DOI] [PubMed] [Google Scholar]; c) Ueda H, Tamura S, Fukushima N, Takagi H. Eur J Pharmacol. 1986;122:379–380. doi: 10.1016/0014-2999(86)90421-8. [DOI] [PubMed] [Google Scholar]; d) Konoki K, Hashimoto M, Nonomura T, Sasaki M, Murata M, Tachibana K. J Neurochem. 1998;70:409–416. doi: 10.1046/j.1471-4159.1998.70010409.x. [DOI] [PubMed] [Google Scholar]
- 51.Murata M, Gusovsky F, Yasumoto T, Daly JW. Eur J Pharmacol. 1992;227:43–49. doi: 10.1016/0922-4106(92)90140-q. [DOI] [PubMed] [Google Scholar]
- 52.Ohizumi Y, Kajiwara A, Yasumoto T. J Pharmacol Exp Ther. 1983;227:199–204. [PubMed] [Google Scholar]
- 53.Takahashi M, Tatsumi M, Ohizumi Y, Yasumoto T. J Biol Chem. 1983;258:10944–10949. [PubMed] [Google Scholar]
- 54.Taglialatela M, Amoroso S, Yasumoto T, DiRenzo G, Annunziato L. Brain Res. 1986;381:356–358. doi: 10.1016/0006-8993(86)90088-0. [DOI] [PubMed] [Google Scholar]
- 55.Soergel DG, Gusovsky F, Yasumoto T, Daly JW. J Pharmacol Exp Ther. 1990;255:1360–1365. [PubMed] [Google Scholar]
- 56.Gusovsky F, Yasumoto T, Daly JW. FEBS Lett. 1989;243:307–312. doi: 10.1016/0014-5793(89)80151-6. [DOI] [PubMed] [Google Scholar]
- 57.Choi OH, Padgett WL, Nishizawa Y, Gusovsky F, Yasumoto T, Daly JW. Mol Pharmacol. 1990;37:222–230. [PubMed] [Google Scholar]
- 58.Treviño CL, De La Vega-Beltrán JL, Nishigaki T, Felix R, Darszon A. J Cell Physiol. 2006;206:449–456. doi: 10.1002/jcp.20487. [DOI] [PubMed] [Google Scholar]
- 59.Escobar LI, Salvador C, Martinez M, Vaca L. Neurobiology. 1998;6:59–74. [PubMed] [Google Scholar]
- 60.a) Nishio M, Muramatsu I, Yasumoto T. Eur J Pharmacol. 1996;297:293–298. doi: 10.1016/0014-2999(95)00751-2. [DOI] [PubMed] [Google Scholar]; b) Wisnoskey BJ, Estacion M, Schilling WP. Am J Physiol: Cell Physiol. 2004;287:C345–C356. doi: 10.1152/ajpcell.00473.2003. [DOI] [PubMed] [Google Scholar]
- 61.a) Murata M, Matsumori N, Konoki K, Oishi T. Bull Chem Soc Jpn. 2008;81:307–319. [Google Scholar]; b) Konoki K, Hashimoto M, Murata M, Tachibana K. Chem Res Toxicol. 1999;12:993–1001. doi: 10.1021/tx990014m. [DOI] [PubMed] [Google Scholar]
- 62.Unpublished results.