

Abstract
Leishmania major is a protozoan parasite that causes skin ulcerations in cutaneous leishmaniasis. In the mammalian host, the parasite resides in professional phagocytes and has evolved to avoid killing by macrophages. We identified L. major genes encoding inhibitors of serine peptidases, ISPs, which are orthologues of bacterial ecotins and found that ISP2 inhibits trypsin-fold S1A family peptidases. Here we show that L. major mutants deficient in ISP2 and ISP3 (Δisp2/3) trigger higher phagocytosis by macrophages through a combined action of the complement type-3 receptor (CR3), toll-like receptor 4 (TLR4) and unregulated activity of neutrophil elastase (NE), leading to parasite killing. While all three components are required to mediate enhanced parasite uptake, only TLR4 and NE are necessary to promote parasite killing after infection. We found that the production of superoxide by macrophages in the absence of ISP2 is the main mechanism controlling the intracellular infection. Furthermore, we show that NE modulates macrophage infection in vivo, and that the lack of ISP leads to reduced parasite burdens at later stages of the infection. Our findings support the hypothesis that ISPs function to prevent the activation of TLR4 by NE during the Leishmania-macrophage interaction in order to promote parasite survival and growth.
INTRODUCTION
Leishmania is a flagelleted protozoan parasite that causes a broad spectrum of diseases ranging from skin ulcerations to visceral damage, depending on the species. In the sandfly, the parasite is found in two major forms; the logarithmitically growing procyclic promastigote and the non-dividing metacyclic promastigote, the later being the virulent disease-inducing form of Leishmania. In the mammalian host, Leishmania transform into the obligate intracellular amastigote stage that has evolved to survive within the phagolysosome of host phagocytic cells, primarily macrophages (Mø) (1). Different Leishmania species use several mechanisms to supress macrophage activation, enabling the formation of a suitable environment for intracellular survival. For example, the binding to and activation of the complement type −3 receptor (CR3) by L. major promastigotes leads to the inactivation of infected cells, contributing to their subsequent intracellular survival (2) and surface lipophosphoglycans (LPG) decrease phagosome fusion with late endocytic organelles and lysosomes, influencing the exposure of internalised promastigotes to hydrolases (3). L. major lines lacking LPG are highly susceptible to human complement, have lost the ability to inhibit phagolysosomal fusion transiently, and are oxidant sensitive, having drastically reduced capability to establish infections in macrophages (4, 5).
The exposure of infected macrophages to apoptotic neutrophils can also selectively alter the fate of intracellular L. major as neutrophil-macrophage interactions in BALB/c suceptible mice lead to the production of prostaglandin E2 and promote parasite growth, while such interactions in C57/B6 mice lead to TNF-α production and parasite killing (6). Recently, it was reported that the ability of neutrophils to induce parasite killing in macrophages of C57B6 mice is strikingly dependent on the activity of the serine peptidase neutrophil elastase, released by dying neutrophils (7). While macrophages are the final host cells for Leishmania during the chronic infection, neutrophils have been recognised as the first hosts to metacyclic promastigotes following inoculation by the sandfly bite (8). Leishmania is able to avoid killing by neutrophils, and the phagocytosed parasites reside temporarily as viable metacyclics inside vacuoles, before being released to infect macrophages. Alternatively, parasitised apoptotic neutrophils are taken up by standby macrophages, serving as a “Trojan horse” and promoting a silent transfer of parasites to their final host cell (9). In both settings, the infection of macrophages by metacyclic promastigotes remains the primary route to the establishment of the infection and the parasite factors acting on the Leishmania-macrophage interaction influence the outcome of the infection.
Recently, we implicated Leishmania ecotin-like inhibitors of serine peptidase (ISPs), as potential virulence factors playing a role in parasite uptake and intracellular survival in macropahges (10). L. major has three ISP genes, but apparently lacks genes encoding SPs from the S1A family, favoring the hypothesis that ISPs play a role in controlling host SPs (11). Indeed, ISP2 encodes a functional inhibitor of S1A family members neutrophil elastase (or human leukocyte elastase) (NE or HLE) and cathepsin G (12). These peptidases are microbicidal components of azurophil granules of neutrophils, a subset of lysosome-like organelles. They are also found in granules or at the cell surface of other cell types such as monocytic or basophilic/mast cell lines, and can be released to the extracellular environment under pathological conditions (13, 14). Considering that Leishmania is rapidly engulfed by professional phagocytes, neutrophils and macrophages, at the site of infection, NE and CG are candidate targets for ISPs.
L. major lacking ISP2 and ISP3 (Δisp2/3) were internalised more efficiently by BALB/c macrophages due to up-regulation of phagocytosis, driven by selective engagement of CR3 and unidentified SPs (10). In contrast, ISP deficiency resulted in the partial elimination of intracellular parasites 24 h post-infection, suggesting that ISPs are important for the initiation and persistence of infection of the mammalian host.
In the present study, we addressed the molecular mechanisms by which ISP2 modulates the intracellular development of L. major. We present evidence that ISP2 plays a role in oxidant resistance by controlling the activity of NE and preventing the activation of TLR4 mediated responses by L. major.
MATERIAL AND METHODS
Parasites
Leishmania major Friedlin (MHOM/JL/80/Friedlin) were grown as promastigotes in modified Eagle’s medium (designated HOMEM medium) supplemented with 10% heat inactivated foetal calf serum (FCS) at 25°C described previously (15). Parasite lines deficient in ISP2 and ISP3 (Δisp2/3) and re-expressing ISPs (Δisp2/3:ISP2/3) were generated by gene replacement and re-introduction, as described (10).
Infection assays
Peritoneal macrophages from C57/B6, C3HeN, C3HeJ (acquired from Instituto Oswaldo Cruz, FIOCRUZ, Rio de Janeiro) or TLR4 −/− (generated by S. Akira, Osaka University, Japan and donnated by Dr. Marcelo Bozza, Microbiology Institute, UFRJ, Brazil) mice were elicited with thioglycolate and cultured overnight in RPMI 1640 medium, 10% fetal calf serum before assayed as described (10). Briefly, interactions with stationary phase promastigotes were performed in RPMI, 0.1% bovine serum albumin (BSA) for 3 h and the cultures were washed before fixation and staining with Giemsa. The number of intracellular parasites was determined by counting at least 100 cells per replicate under the light microscope. Where indicated in the figure legends, aprotinin (Sigma-Aldrich, St., MO, USA), rISP2 (4 μg ml−1 each) or the irreversible inhibitor to neutrophil elastase OMeSuc-AAPV-CMK (10 μM, Calbiochem, Switzerland) were added to macrophages 5 min prior to addition of the parasites and remained during the 3 h of interaction. Anti-mouse CD11b monoclonal antibodies (M1/70), anti-mouse Toll-like 4 (TLR4) neutralizing monoclonal antibodies (MTS510) or rat IgG2b (BD Bioscience Pharmingen, San Jose, CA, USA) were incubated at 10 μg ml−1 or at 5 μg ml−1, according to figure legends, with macrophages in RMPI-FCS for 30 min, and removed by extensive washing before the addition of promastigotes in RPMI-BSA. Anti-mouse TLR4 activating monoclonal antibodies (TLR4/MD2, clone UT12) (Ebioscience, San Diego, CA, USA), or LPS were used at 5 μg ml−1 and 100 ng ml−1, respectively, and added to the cultures 5 min. prior to addition of parasites, remaining in the assay during the 3 h interaction. For the survival assays, the macrophages were infected in RPMI-BSA for 3h and after the removal of extracellular parasites by extensive washing, the cells were cultured at 37°C, in RPMI supplemented with 10% FCS for 24h, 48h or 72h. Where appropriate 50 μM EUK134 (Calbiochem, USA) or 2,000U. ml−1 catalase (Sigma, USA), were added after parasite internalisatuion and remained in the cultures at 37°C for 24 h 48 h or 70 h, before fixation and staining. In the survival assays testing the effects of the NE and TLR4 after parasite entry (Fig. 7C-D), the infections were performed in RPMI-BSA for 3 h. The extracellular parasites were removed and the cultures were subsequently supplied with NE (100 ng ml−1), anti-TLR4 activating antibodies (UT12) at 5 μg ml−1 or LPS 100 ng.ml−1 in RPMI-FCS and remained in the assay for 24 h. Alternatively, after removal of extracellular parasites, anti-TLR4 neutralizing antibodies (MTS510) were pre-incubated at 5 μg ml−1 with the cultures for 30 min. and removed by washing before the addition of NE.
Figure 7. NE promotes L. major uptake and intracellular killing through TLR4.
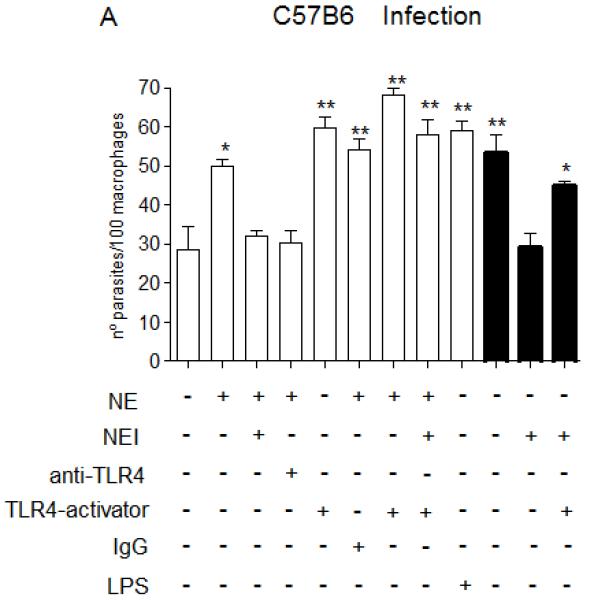
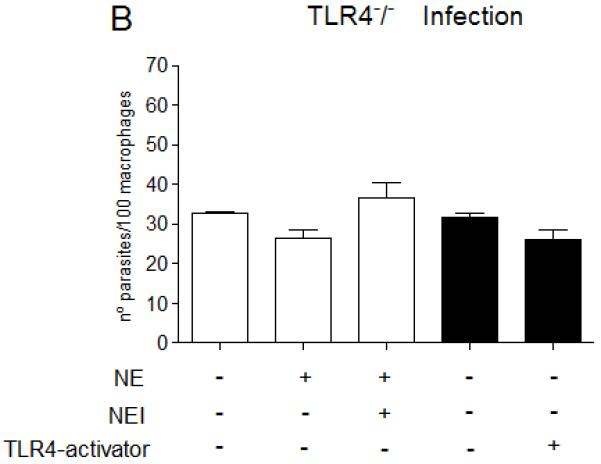
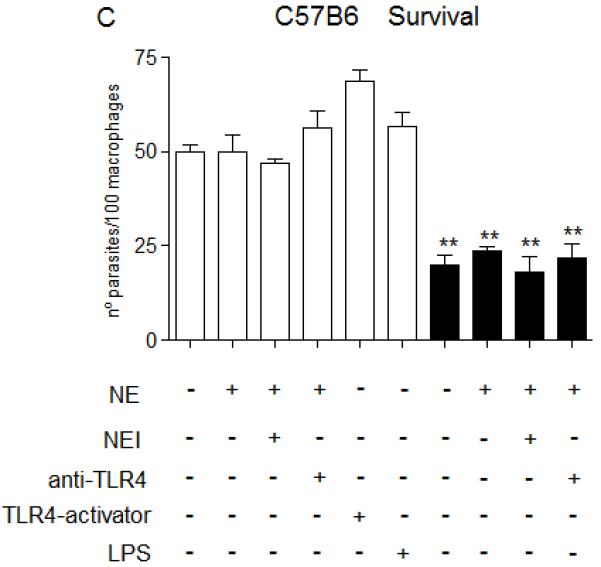
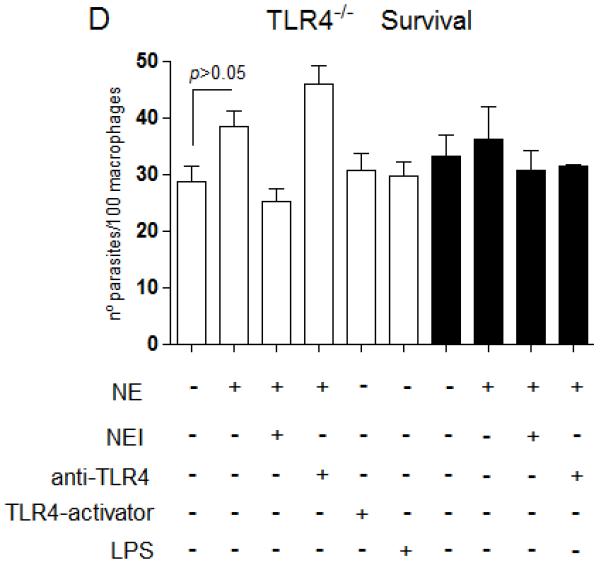
Stationary phase promastigotes were incubated with elicited peritoneal macrophages from C57/B6 (A and C), or from TLR4−/− mice (B and D) in RPMI-BSA, at a 3:1 parasite:macrophage ratio for 3 h. Extracellular parasites were removed and the cultures were fixed and stained. In A and B, the macrophage treatments were performed before addition of parasites. In C and D, infections were performed in RPMI-BSA for 3 h, extracellular parasites were removed and the treatments were added to the cultures thereafter, remaining for the following 24 h, before fixation and staining. In A and B, macrophages were pre-incubated with 5 μg.ml−1 of neutralizing anti-TLR4 (MTS510) or with control rat anti-mouse IgG2b for 30 min at 37°C, followed by three washes, before addition of promastigotes. Activating antibodies to TLR4 (UT12) or purified neutrophil elastase (NE) were added to the cultures 5 min prior to addition of parasites, at 5 μg ml−1 and 100 ng ml−1. NEI (inhibitor OMeSuc-AAPV-CMK), was pre-incubated with NE at 10 μM for 30 min prior to infection or added directly to the cultures 5 min prior to parasites, according to the indicated in the panels. LPS was used at 100 ng.ml−1 and added concomitantly to parasites. The experiments were performed in triplicates, two independent times. The graphs show one representative experiment, the data are means of triplicates ± SD. Key: white bars, wild type parasites; black bars, Δisp2/3; The asterisks indicate statistical significance from WT control at p< 0.001 (*) and at p< 0.0001 (**).
Immunofluorescence
Peritoneal macrophages from C57/B6, mice were elicited by injection of 1 ml of 1% thioglycolate in the peritoneal cavity and collected 3 days later as described above. The cells were washed, plated in a 16-well tissue culture slide (LabTek) at 5 × 104/well and cultivated overnight in RPMI supplemented with 10% of FCS (Invitrogen, UK). The culture was washed and fixed with 1% paraformaldeyde for 10 min at room temperature, followed by addition of 0.1 M Glycine and incubation for additional 10 min. The cells were washed with phosphate buffered saline (PBS) and blocked overnight in PBS containing 1% BSA. The blocking solution was removed and the cells were incubated with mouse anti-neutrophil elastase antibodies at 1:500 dilution (Santa Cruz Biotechnology), for 2 h, followed by washing with PBS and incubated with Cy3-conjugated goat anti-mouse IgG 1:1,000 dilution (Jackson, West Grove, PA, USA), for 1h. The slides were washed and incubated with rat anti-mouse CD11b monoclonal antibodies (M1/70) at 1:400 dilution, for 1 h (BD Bioscience Pharmingen, San Jose, CA, USA). After washing, the slides were incubated with anti-rat FITC at 1:2000 for 1 h (BDBiosciences). The step-wise incubations with antibodies to elastase were necessary to avoid cross-reaction of the anti-mouse IgG-Alexa 594 conjugate with the rat anti-CD11b antibodies that was observed in assays where concomitant labeling was performed (not shown). The slides were mounted in DAPCO/DAPI on coverslips and observed under the microscope. The fluorescence images were captured using a digital camera attached to an inverted microscope (Carl Zeiss Apotome), under an 100x objective. The fluorescences in the blue, green and red channels were acquired in 8 sequential optical z sections of 0.9 μm each.
Peptidase activity assays
The activities of human trypsin, chymotrypsin, neutrophil elastase and cathepsin G were tested in 50 mM Hepes, 150 mM NaCl, pH 7.5, 0.05% Igepal CA630, at room temperature, using 25 μM of the appropriate substrate, Bz-FR-AMC (trypsin), MeOSuc-AAPV-AMC (neutrophil elastase) and OMeSuc-AAPF-AMC (chymotrypsin and cathepsin G) (Calbiochem). Product formation was followed in continuous assays by measuring the fluorescence at 380 nm excitation, 440 emission, in a Hitachi F-4500 fluorimeter. Where indicated, the inhibitor MeOSuc-AAPV-CMK was added to the enzyme immediately before addition of the substrate. The initial velocities were calculated by linear regression of the substrate hydrolysis plots using at least 300 points.
In vivo infections
Stationary phase promastigotes were injected in the peritoneal cavity of C57B6 mice (2 × 107 parasites/animal, 3 mice per group) in PBS. After 3 h, the mice were sacrificed and the peritoneal cavity was washed with 5 mL of ice-cold RPMI. The collected exudates were centrifuged at 1,500 x rpm and 8 × 105 cells of each animal were plated separately on glass coverslips in 24-well tissue culture plates in RPMI-FCS 10%. The cells were left to adhere for 2 hs at 37°C, washed, fixed and Giemsa stained. Where indicated in the figures, the parasites were co-injected with 100 ng.ml−1 of NE or 5 μM of NE inhibitor (OMeSuc-AAPV-CMK). For the survival assays, the plated cells were cultivated for 24 hs before fixation and staining. No extracellular parasites were observed in the collected peritoneal exudates or in the plated cultures. For lesion progression, 2 × 106 parasites were injected in the footpads of mice (6 mice per group) and the size of the footpads were measured weekly with a dial caliper. The lesion size was estimated by calculating the differences of sizes of infected versus uninfected footpads of each mice and the graph represents the mean values of each mice group. Parasite loads were estimated by limiting dilution: popliteal lymph nodes were collected and submitted to rupture homogeneity with the back of a syringe through a nylon membrane in 6.4 ml of HOMEM medium supplemented with 10% FCS. One hundred microliters of the cell homogenates were submitted to 2 fold serial dilutions in 96-well plates. Plates were cultivated at 27°C for 5-7 days and then the wells inspected for parasite growth. The highest dilution well was used to estimate the number of parasites multiplied by the dilutions factors. Numbers are total per lymph node.
Statistical analyses
Significance of the data was assessed by analysis of variance, using the GraphPad Prism 4.0 Program. The data were analysed by One-way ANOVA using the Bonferroni post-test, at a significance level of 5%. In Fig. 2D the data were analysed by Two-way ANOVA using the Bonferroni post-test, at a significance level of 5%. The scores showing statistical significance are indicated in the figures with asterisks and the p values are indicated in the legends.
Figure 2. Neutrophil elastase is required for enhanced uptake and intracellular death of Δisp2/3 in macrophages.
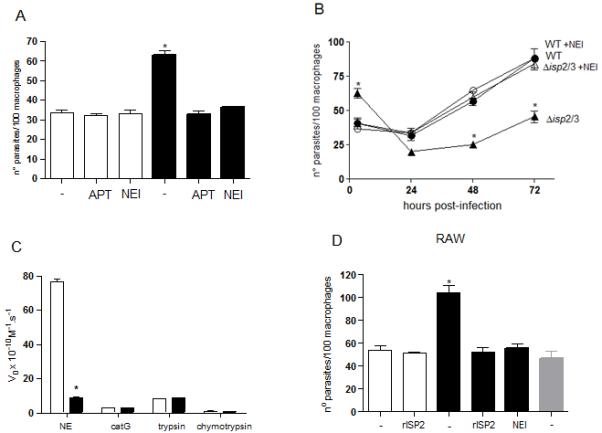
Stationary phase promastigotes were incubated with elicited peritoneal macrophages from C57/B6 mice (A and B) or with RAW 264.7 cells (D), in RPMI-BSA, for 3 hours. Where indicated, 10 μM of OMeSuc-AAPV-CMK (NEI) or 4 μg ml-1 of purified rISP2 or of aprotinin (APT) were added to the cultures 5 min prior to addition of parasites. Extracellular parasites were removed with three washes and the adhered cells were fixed and stained with Giemsa. Key: white bars, WT; black bars, Δisp2/3; grey bars, Δisp2/3:ISP2-ISP3. In B, macrophages were incubated with L. major for 3 h, the extracellular parasites were removed by extensive washing, the cultures were fixed at 3 h, 24 h, 48 h, 72 h and Giemsa stained. Where indicated, 10 μM OMeSuc-AAPV-CMK (NEI) was present during the 3 h infection and absent in the following 24-72 h. Key: filled circle, WT; open circle, WT + NEI, filled triangle, Disp2/3; open triangle, Disp2/3 + NEI. In A and C, the asterisk indicates statistical significance from all other experimental points at p< 0.001. In B, the asterisks indicate statistical significance from other experimental points at each time at p< 0.001 (*). The graphs show one representative experiment. (C) Selectivity of the inhibitor to neutrophil elastase. The peptidase activity of neutrophil elastase (NE), cathepsin G (catG), trypsin and chymotrypsin were tested in the absence or presence of 10 μM OMeSuc-AAPV-CMK in 50 mM Hepes, 150 mM NaCl, pH 7.5, using the appropriate substrates at 25 μM. The graph shows the initial velocities of substrate hydrolysis. Key: white bars, no inhibitor; black bars, with NEI.
RESULTS
Increased infectivity of L .major Δisp2/3 to C57/B6 -derived macrophage is mediated by serine peptidases and CR3
We have previously shown that L. major deficient in ISP2 and ISP3 (Δisp2/3) are phagocytosed by macrophages of susceptible BALB/c mice more efficiently than wild type L. major, and that this is mediated by unidentified serine peptidase(s) (SPs) acting at the initial stages of the parasite-host cell interaction (10). We next verified if similar characteristics were found with macrophages of resistant C57/B6 mice. Stationary phase Δisp2/3 promastigotes were internalized more efficiently by elicited peritoneal macrophages as compared to wild type (WT) or to the line re-expressing ISPs (Δisp2/3:ISP2/ISP3) (Fig 1A). The increase in macrophage infection was due to higher proportions of infected macrophages as well as increased numbers of parasites per infected cell. The internalisation returned to the levels of WT when the interaction was performed in the presence of recombinant ISP2 (r-ISP2) or of the inhibitor of trypsin-like petidases, aprotinin (APT) (Fig. 1A). The higher infection of Δisp2/3 also returned to the levels of WT when macrophages were pre-treated with antibodies that block CD11b, confirming that increased uptake of Δisp2/3 is mediated by CR3 in macrophages of both BALB/c (10) and C57/B6 mice (Fig. 1B). We next found that Δisp2/3 had reduced capacity to establish infection in macrophages of C57/B6 mice (Fig. 1C). After the initial higher infection, the numbers of intracellular Δisp2/3 was significantly reduced in the subsequent 72 h, as compared to WT or to the lines re-expressing ISP2/ISP3 (Fig. 1C). The addition of APT or r-ISP2 during the initial 3 h of interaction rescued the mutant parasites from cell death, resulting in numbers of intracellular Δisp2/3 similar to those of WT, 72 h after infection (Fig. 1C). However, blocking of CD11b at the entry stage had no effect on the subsequent survival and development of intracellular Δisp2/3 (Fig. 1D). These observations indicate that ISP2 and or ISP3 are required for the establishment of a productive infection in C57/B6 macrophages and that SP activity, but not CR3, compromises the capability of Δisp2/3 to survive in the host cell.
Figure 1. Enhanced uptake of Δisp2/3 by macrophages of C57/B6 mice is dependent on CD11b and serine peptidases.
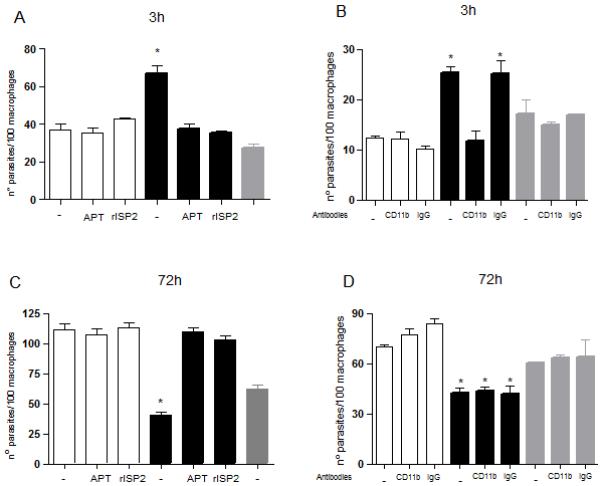
Stationary phase promastigotes were incubated with elicited peritoneal macrophages from C57/B6 mice in RPMI-BSA, for 3 h. Extracellular parasites were removed with three washes and the adhered cells were fixed and subsequently stained with Giemsa. In B and D, macrophages were pre-incubated with 10 μg ml−1 of anti-CD11b M1/70 (CD11b) or with control rat anti-mouse IgG2b (IgG) for 30 min at 37°C, followed by three washes, before addition of promastigotes. In C and D, the extracellular parasites were removed with three washes and the cells were cultured for an additional 70 h in RPMI-FCS, at 37°C before fixation and staining. The number of intracellular parasites was estimated by counting using light microscopy. Where indicated, the interaction was performed in the presence of 4 μg ml−1 of purified recombinant ISP2 (rISP2) or aprotinin (APT). The experiments were performed in triplicate, three independent times. The graphs show one representative experiment, the data are means of triplicates ± SD. Key: white bars, wild type parasites; black bars, Δisp2/3; grey bars, Δisp2/3:ISP2-ISP3. The asterisks indicate statistical significance from all other experimental points at p< 0.001.
Macrophage-associated neutrophil elastase modulates the entry and survival of L. major in macrophages
Two S1A family SPs are found in association with CR3 in polymorphonuclear leukocytes: neutrophil elastase (NE) (16) and factor Xa (17), so we tested if NE was involved in up-regulation of Leishmania uptake by macrophages using an irreversible NE inhibitor (Fig. 2). The infections with Δisp2/3 returned to the levels of those of WT in the presence of aprotinin (APT) or of the NE inhibitor (NEI) (Fig. 2A), suggesting that this peptidase is responsible for the increased internalisation of Δisp2/3. The analyses of the intracellular growth of parasites over a 3-day period revealed that the high numbers of intracellular Δisp2/3 observed at 3 h infection decreased drastically by 24 h, indicating parasite death (Fig. 2B, triangle). The remaining parasites recovered growth at 48 h, although at much lower rates as compared to WT (Fig. 2B, circle). The addition of the NE inhibitor during the initial 3 h interaction led to lower internalisation of Δisp2/isp3 but completely prevented their elimination in 24 h, and the intracellular parasites grew steadily over the 3-day period (Fig. 2B, open triangle). The NE inhibitor had no effect on the growth or survival of WT parasites (Fig. 2B, open circles). These results indicate that NE activity present during the early Δisp2/3-macrophage contact leads to subsequent parasite death and to delayed intracellular growth. The selectivity of the synthetic inhibitor used was tested against other S1A family SPs (Fig 2C). The activity of purified NE was readily abolished in the presence of the synthetic inhibitor the concentration used in the infection assays (Fig 2C). This inhibitor did not affect the activities of cathepsin G, trypsin or chymotrypsin, demonstrating selectivity to NE among the tested peptidases.
Δisp2/3 were more infective to RAW macrophage cell line than WT, and the increased infectivity was fully reversed in the presence of the NE inhibitor, suggesting that NE endogenously produced by monocytes/macrophages is sufficient to up-regulate the internalisation of the mutant parasites and that infection did not depend on the exposure of macrophages to neutrophils (Fig. 2D).
Although NE present in macrophages of C57/B6 mice is sufficient to promote uptake of Δisp2/3, WT parasites are refractory to this action (Fig.2A). We hypothesised that ISPs produced by WT L. major were sufficient to control the limited amount of NE activity found in macrophages of C57/B6 mice, preventing up-regulation of phagocytosis and guaranteeing parasite survival. To verify this, we added exogenous NE to macrophages during the interaction with WT L. major. NE promoted increased uptake of WT in a dose-dependent manner (Fig. 3A), and this effect was abolished when NE was irreversibly inhibited prior to addition to macrophages. In contrast, the presence of exogenous NE at concentrations (200 ng ml−1) that increased the internalisation of WT had no effect in the uptake of Δisp2/3, suggesting that, in the absence of ISP2 and ISP3, the NE present in macrophages is sufficient to promote maximal phagocytosis of Δisp2/3. Furthermore, there were reduced numbers of intracellular WT 72 h after infection when exogenous NE was present at the initial stages of parasite uptake (Fig. 3B). These results suggest that the control of NE activity by ISPs is necessary to keep the peptidase levels below the threshold necessary to activate parasite-killing mechanisms in macrophages.
Figure 3. Neutrophil elastase promotes enhanced uptake and subsequent death of L. major in macrophages.
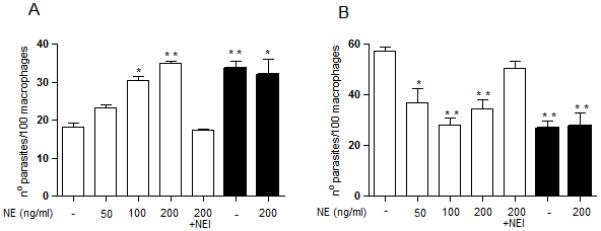
(A) Stationary phase promastigotes were incubated with elicited peritoneal macrophages from C57/B6 mice in RPMI-BSA, at a 3:1 parasite:macrophage ratio for 3 h. Where indicated, purified neutrophil elastase (NE) was incubated with the macrophages at 50, 100 or 200 ng ml−1 for 5 min prior to addition of parasites and remained during the 3 h interaction. For pre-inhibiton of NE (NEI), NE was diluted to 200 ng ml−1 in RPMI/BSA and incubated with 10 μM OMeSuc-AAPV-CMK, for 30 min at room temperature prior to addition to macrophages. Extracellular parasites were removed with three washes and the adhered cells were fixed and stained with Giemsa. The number of parasites was estimated by counting using light microscopy. In B, the extracellular parasites were removed with three washes after 3 h, and the cells were cultured for an additional 70 h in RPMI-FCS, at 37°C before fixation and staining. The experiments were performed in triplicate, two independent times. The graphs show one representative experiment, the data are means of triplicates ± SD. Key: white bars, WT; black bars, Δisp2/3. The asterisks indicate statistical significance from the infection with wild type parasites in medium at p< 0.01 (*) and p< 0.001 (**).
Localisation of CR3 and NE in macrophages
The physical association between NE and CD11b has been described in neutrophils, so we asked if this was also observed in monocytes/macrophages. Immunofluorescence of uninfected peritoneal macrophages showed that CD11b is found throughout the cell periphery (Fig. 4A). The evaluation of 0.9 μm optical sections of the cells revealed that staining with anti-CD11b antibodies is observed preferably at the lower sections (Fig. 4A), and very rarely at the upper sections (Fig. 4B). This suggests that CD11b is likely located at the regions of contact between the cell and the slide surface (Fig. 4A) and is present at very low levels at the apical surface (Fig. 4B). In contrast, NE was clearly observed in the upper sections (Fig. 4B). We did not observe co-localisation of CD11b and NE (Fig. 4 and B, merge) in uninfected macrophages, indicating that these molecules are not constitutively associated in macrophages. In contrast, we observed intense co-localisation of NE and CD11b in macrophages infected with Δisp2/3 (arrow), (Fig. 4C-D, arrowheads). The co-localisation was found exclusively at the apical sections, suggesting that the distribution of CD11b is altered upon infection, relocating to the apical regions of the cell, in close proximity to NE. CD11b was preferably observed at the basal sections of uninfected macrophages (Fig. 4C-D, asterisk) that were adjacent to an infected cell (Fig. 4C, arrow), suggesting that the re-distribution of CD11b to the upper region of macrophages requires contact with the parasite.
Figure 4. Sub-cellular localisation of CD11b and neutrophil elastase in macrophages.

Elicited macrophages from C57/B6 mice were cultured for 16 h in RPMI-FCS, washed and fixed with formaldehyde 1%. The monolayers were incubated with mouse anti-neutrophil elastase Abs washed and incubated with anti-mouse Cy3 (red). Subsequently, the cells were washed and incubated sequentially with rat anti-CD11b Abs and with anti-rat FITC (green). The monolayers were washed, stained with DAPI and mounted with DAPCO on converslips. The cells were sectioned 8 times in 0.9 μm sections, from bottom to top and sections number 1 (basal) are shown in panels A and C and sections number 5 (apical) and shown in panels B and D. Scale bar is 5 μm. A-B, uninfected macrophages; C-D, macrophages infected with Δisp2/3 for 45 min. The arrow points to the parasite, arrow heads indicate co-localization between CD11b and NE at the apical sections, the asterisk indicates an uninfected cell in the same field, showing CD11b at the basal section. The patterns represented in the figure were observed in two independent experiments.
Toll-like receptor 4 is required for enhanced uptake and elimination of Δisp2/3 Leishmania
Having determined that CR3 is not required for the elimination of intracellular parasites, we asked if an alternative cell surface receptor was recruited during the Δisp2/3-macrophage interaction, possibly contributing to parasite death. Recently, it has been shown that exogenous NE added to macrophages previously infected with L. major prevents parasite growth and that this effect is dependent on TLR4 (7).
We therefore investigated if TLR4 is involved in the phagocytosis and death of Δisp2/3 by blocking TLR4 with the monoclonal antibody MTS510 (Fig. 5). The neutralisation of TLR4 prior to the interaction with Leishmania reduced significantly the uptake of Δisp2/3 (Fig. 5A, black bars), while it had no effect in the internalisation of WT (Fig. 5A, white bars). The neutralisation of both CD11b and TLR4 had similar effects (Fig. 5A) to the neutralisation of either CD11b (Fig. 1C) or TLR4 alone, suggesting that those receptors act in a common pathway. As seen with the inhibition of NE, neutralisation of TLR4 at the entry stage prevented death of intracellular Δisp2/3 24 h after infection (Fig. 5B, black bars), as compared to macrophages that were pre-treated with control IgG. These results indicate that early mobilisation of TLR4 by Δisp2/3 is required for subsequent parasite death.
Figure 5. Toll-like receptor 4 is involved in enhanced uptake and elimination of Δisp2/3 in macrophages.
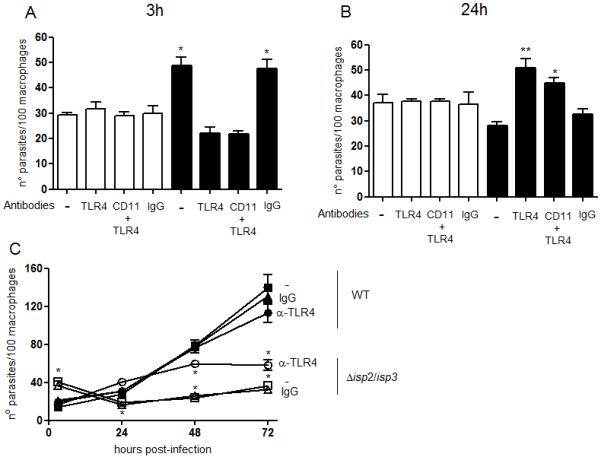
Stationary phase promastigotes were incubated with elicited peritoneal macrophages from C57/B6 mice, in RPMI-BSA, at a 3:1 parasite:macrophage ratio for 3 h. Where indicated, macrophages were pre-incubated with 10 μg ml−1 of anti-CD11b (M1/70), with 5 μg ml−1 of neutralising anti-TLR4 (MTS510) or with control rat anti-mouse IgG2b for 30 min in RPMI-FCS, followed by three washes, before addition of promastigotes. In A, the cultures were fixed and stained after the 3 h interaction. In B and C, the extracellular parasites were removed with three washes after 3h and the cells were cultured for an additional 20 h (B) or for 20, 48, 72h (C), in RPMI-FCS before fixation and staining. The number of parasites was estimated by counting using light microscopy. The experiments were performed in triplicate, two independent times. The graphs show one representative experiment, the data are means of triplicates ± SD. Key: white bars, WT; black bars, Δisp2/3, grey bars, Δisp2/3:ISP2-ISP3. In C, filled symbols WT; open symbols Δisp2/3; circle, pre-treatment of macrophages with anti-TLR4; triangle, pre-treatment with rat IgG2b. The asterisks indicate statistical significance from other experimental points at p< 0.01 in (A and B), the double asterisks indicate significance at p<0.001 from Δisp2/3; and from WT at p< 0.001 in (C).
When TLR4 was blocked before parasite entry, we observed a 2-fold increase in intracellular Δisp2/3 in 24 h, while the numbers of intracellular WT were similar during this time period (Fig 5B). This suggests that blockade of TLR4 promotes rapid intracellular growth of ISP-deficient parasites. To verify this further, we followed the effect of TLR4 neutralisation in the kinetics of Leishmania intracellular growth (Fig. 5C). Pre-treatment of the macrophages with anti-TLR4 had a minimal effect in the initial intracellular growth of WT parasites. In contrast, neutralisation of TLR4 not only prevented the death of Δisp2/3 in the 24 h following infection but also promoted growth in that time period, while pre-treatment with control IgG had no effect. Intracellular Δisp2/3 were unable to sustain growth in macrophages pre-treated with anti-TLR4 antibodies in the following 48 and 72 h.
To investigate this further, we infected macrophages derived from C3HeJ mice (Fig. 6C and D), which are defective in TLR4 signaling, as compared to macrophages from the background mice strain C3HeN (Fig. 6A and B). As seen with macrophages from BALB/c (10) and C57/B6, Δisp2/3 were more infective to macrophages of CH3eN mice, as compared to WT (Fig. 6A), and this was reversed by rISP2 or by the NE inhibitor. We observed an effect of NEI in the phagocytosis of WT parasites by those macrophages (Fig. 6A), which might be a result of intrinsic differences in the contents of NE between different strains of mice. Δisp2/3 were poorly infective to TLR4-defective macrophages (Fig. 6C). rISP2 or NE inhibitor had no effect on the uptake of the mutant parasites in TLR4-defective macrophages, indicating that SP activity and TLR4 are acting in conjunction for the enhanced uptake of Δisp2/3. After 3 days, the numbers of intracellular Δisp2/3 in CH3eN-derived macrophages were reduced by 50% (Fig. 6B), as compared to the numbers 3 h after infection (Fig. 6A), indicating parasite death. Noteworthy, the numbers of intracellular Δisp2/3 at 3 days remained approximately half of that observed at 3h, indicating that parasite growth was severely compromised in C3HeN macrophages within 24-72 h. This is different from BALB/c macrophages, where intracellular Δisp2/3 were able to recover growth at 48 h, albeit slower than WT, resulting in similar numbers of intracellular parasites at 3 h or 3 days post-infection (10). Exposure to rISP2 or to the NE inhibitor partially rescued intracellular growth of Δisp2/3 in C3HeN macrophages, demonstrating that NE also modulates the intracellular growth of Δisp2/3 in macrophages of those mice. In constrast, even though Δisp2/3 were poorly internalised by TLR4-defective macrophages at 3 h (Fig. 6C), they grew efficiently in these macrophages, reaching numbers equivalent to those of WT at 3 days (Fig. 6D). The accelerated growth of Δisp2/3 in TLR4-defective macrophages implicates TLR4 in the control of Leishmania growth throughout the intracellular cycle. Importantly, intracellular Δisp2/3 are rescued from death in TLR4-defective macrophages to the same extent as in macrophages of C57/B6 mice that were exposed to NE inhibitor (Fig. 2B). Taken together these results define TLR4 as a receptor responsible for promoting the elimination of Δisp2/3 and restraining their intracellular growth in murine macrophages.
Figure 6. Functional TLR-4 is required for increased upatke and elimination of Δisp2/3 in macrophages.
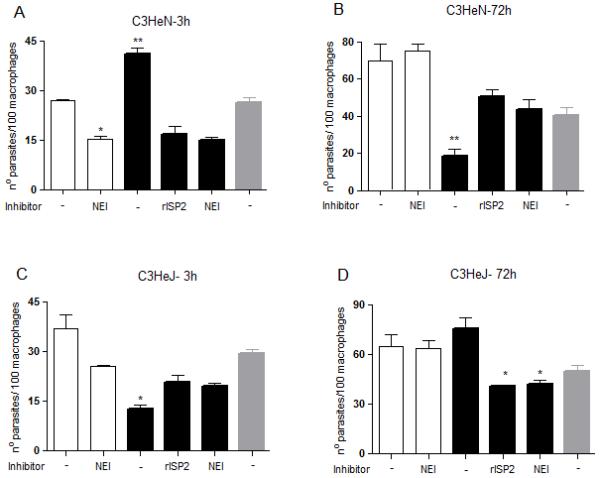
Stationary phase promastigotes were incubated with elicited peritoneal macrophages from from C3HeN (A and B) or C3HeJ mice (C and D), in RPMI-BSA, at a 3:1 parasite:macrophage ratio, for 3 hours. Where indicated, 10 μM OMeSuc-AAPV-CMK (NEI) or 4 μg ml−1 of purified rISP2 was added to the cultures 5 min prior to addition of parasites. In B and D, the extracellular parasites were removed with three washes and the cells were cultured for an additional 70 h in RPMI-FCS at 37°C before fixation and staining. The experiments were performed in triplicate, two independent times.The graphs show one representative experiment, the data are means of triplicates ± SD. Key: white bars, wild type parasites; black bars, Δisp2/3; grey bars, Δisp2/3:ISP2-ISP3. The asterisks indicate statistical significance from other experimental points at p< 0.01 in (C); from WT at p< 0.01 (*) and p< 0.001 (**) in (A); from WT at p< 0.001 and from Δisp2/3 + r-ISP2 at p< 0.05 in (B); from Δisp2/3 with no treatment at p< 0.01 in (D).
NE promotes Leishmania uptake and killing through TLR4
We next evaluated the effects of NE in the infection of macrophages of TLR4-deficient mice, as compared to the infection of macrophages of the genetic background mice, C57/B6 (Fig. 7). As shown previously in Fig. 3, the addition of exogenous NE to macrophages of C57/B6 mice increased the uptake of WT L. major at 3 h (Fig. 7A, white bars), which was abolished upon enzyme pre-inhibition with the NE synthetic inhibitor (NEI). Importantly, pre-treatment of macrophages with neutralising antibodies to TLR4 (α-TLR4), but not with control IgG, abolished the increase in phagocytosis provoked by exogenous NE, indicating that TLR4 is required for the effect of NE in parasite uptake. This supports the hypothesis that the effects of NE and TLR4 are not independent of each other. The engagement of TLR4 either by activating antibodies to TLR4 (UT12) or by LPS also promoted increased uptake of WT L. major, further confirming that the activation of this receptor leads to up-regulation of parasite phagocytosis. The concomitant addition of NE and activating antibodies to TLR4 led to a subtle, but significant increase in the up-regulation of phagocytosis, as compared to the up-regulation caused by either exogenous NE or TLR4-activating antibodies alone. Finally, pre-inhibition of exogenous NE was overcome by the activation of TLR4 with TLR4-activating antibodies, indicating that NE is acting upstream of TLR4 in the signaling pathway that leads to increased parasite internalisation (Fig. 7A). As shown previously, the NE inhibitor (NEI) reversed the enhanced phagocytosis of Δisp2/3 (Fig. 7A, black bars), linking increased uptake of the mutant line with NE activity. The negative effect of NEI on the uptake of Δisp2/3 was abolished by the activation of TLR4 with antibodies, corroborating that NE activity promotes parasite internalization through TLR4. Exogenous NE had no effect in the uptake of WT Leishmania by macrophages of TLR4 knock-out mice (TLR4−/−) (Fig. 7B, white bars), providing further evidence that TLR4 is required for the upregulation of Leishmania phagocytosis caused by NE. Δisp2/3 was as infective as WT in those macrophages (Fig. 7B, black bars), and this was unaffected by neutralising antibodies to TLR4, providing further support that ISPs control the TLR4-NE pathway.
Next, we asked if, in order to promote parasite killing, triggering of the TLR4-NE pathway was required to take place solely at the initial stages of the parasite-macrophage interaction. To address this, we infected macrophages of C57/B6 or TLR4−/− mice with WT Leishmania and triggered this pathway by adding exogenous NE or TLR4-activating antibodies after parasite entry (Fig. 7C-D, white bars). At 24 h post-infection, the numbers of intracellular WT parasites were equivalent in all tested conditions (Fig. 7C, white bars), indicating that the TLR4-NE pathway has little effect on parasite survival if triggered after parasite phagocytosis or that this pathway is unavailable for activation immediately after parasite uptake. We found similar results when analysing the survival of Δisp2/3, which were detected at lower numbers as compared to WT parasites at 24 h post-infection, a phenotype which was unaffected by inhibitors to NE or neutralization of TLR4 if incubated immediately after parasite entry (Fig. 7C, black bars). In contrast to what we observed in macrophages of C57B6 mice, those parasites were found at equal numbers to WT at 24 h post-infection in TLR4−/− macrophages (Fig. 7D, black bars). When added after parasite uptake, exogenous NE led to an unexpected subtle increase in the numbers of intracellular WT parasites in TLR4−/− macrophages after 24hrs, suggesting that NE might influence parasite survival through multiple mechanisms.
Superoxide mediates the killing of intracellular Δisp2/3 in macrophages
To determine if production of superoxide was the mechanism downstream of the TLR4-NE pathway, we verified the effect of the superoxide scavenger EUK134 in infected macrophages (Fig. 8). This compound did not affect the uptake of either WT or Δisp2/3 in macrophages at 3 h (Fig. 8A, 3h). As expected, in untreated macrophages, the numbers of intracellular Δisp2/3 dropped by 50% 24 h after infection (Fig.8 A, 24 h), as compared to the number of Δisp2/3 present at 3 h. In contrast, when the infected cultures were treated with EUK134 immediately after parasite entry, the numbers of Δisp2/3 at 24 h post-infection were similar to those at 3 h, indicating that superoxide strongly contributes to the killing of intracellular Δisp2/3. Treatment of macrophages infected with WT parasites with EUK134 had no effect on parasite survival. We followed the intracellular growth of L. major in macrophages treated with EUK134 showing that Δisp2/3 were rescued from death at 24 h, and that the growth was partially recovered in the following days (Fig. 8B, closed triangles). We further verified the involvement of reactive oxygen species by treatment of infected macrophages with catalase. As expected, Δisp2/3 were engulfed at higher numbers (Fig. 8C, 3h) and were partially eliminated in the following 24h (Fig. 8C, 24h). As observed with EUK 134, the addition of catalase immediately after the 3 h infection, rescued Δisp2/3 from death in the following 24 h, while it did not influence the survival of either WT or the re-expressing lines (Fig 8C, 24h catalase). Taken together, these results implicate superoxide as the main mechanism driving the intracellular elimination of ISP-deficient parasites by macrophages.
Figure 8. Superoxide mediates the elimination of intracellular Δisp2/3.
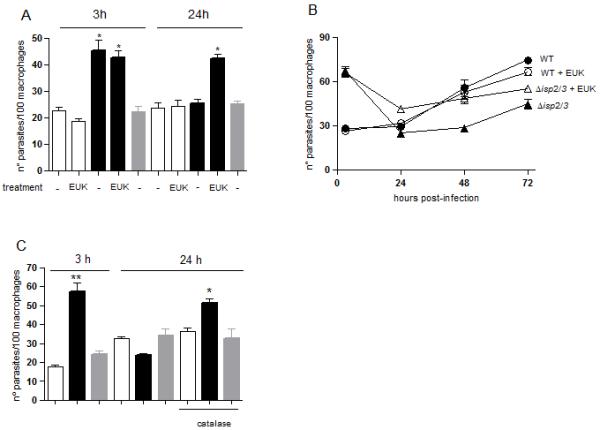
Stationary phase promastigotes were incubated with elicited peritoneal macrophages from C57/B6 mice, in RPMI-BSA, at a 3:1 parasite:macrophage ratio for 3 h. (A) Left panel (3 h): where indicated, the assays were performed in the presence of 50 mM of the superoxide scavenger EUK134, the extracellular parasites were removed after 3 h and the cells were fixed and stained with Giemsa. Right panel (24 h): Infections were performed in untreated macrophages for 3h, extracellular parasites were removed and the cells were cultured for an additional 21 h in RPMI-FCS at 37°C before fixation and staining. Where indicated, 50 mM EUK 134 was added to the infected cultures after removal of extracellular parasites and remained in the culture for 24 h. In B, the infections were performed for 3 h in untreated macrophages, the extracellular parasites were removed and the cells were cultured in RPMI-FCS, supplemented or not with 50 mM EUK 134 where indicated, at 37°C for an additional 45 or 70 h. In C, infections were performed in untreated macrophages for 3h, extracellular parasites were removed cells were fixed and stained (left panel, 3 h) or further cultured for 21 h in RPMI-FCS at 37°C (right panel, 24 h). Where indicated, 2000 units.ml−1 catalase were added to the infected cultures after removal of extracellular parasites and remained in the culture for the 21 h. Key: white bars, WT; black bars, Δisp2/3; grey bars, Δisp2/3:ISP2-ISP3. The experiments were performed in triplicate, three independent times. The graphs show one representative experiment, the data are means of triplicates ± SD. The asterisks indicate statistical significance from the remaining experimental points at p< 0.001 (A); from Δisp2/3 at p< 0.01 (*) and at p< 0.001 (**) in B, at p<0.01 (*) and p<0,0001 in C (**).
ISPs and NE modulate the infection in vivo
To evaluate parasite uptake by phagocytes at the intial stages of infection, we inoculated parasites in the peritoneal cavity of mice and collected the exudate after 3 hs. The majority of the collected cells were neutrophils, revealing early recrutiment of those cells to the site of infection, and no free parasites were observed in the exudates (not shown). Δisp2/3 were found at higher numbers than WT parasites and this was reversed when Δisp2/3 were incubated with the NE inhibitor (Fig 9A). Exogenous NE promoted higher uptake of WT parasites by macrophages in vivo, showing that this peptidase promotes the phagocytosis of L. major by macrophages at early stages of infection in mice. We found that a significant number of macrophages were infected by L. major a few hours after parasite inoculation, and no free parasites were observed, suggesting that they were rapidly engulfed by local cells (i.e., neutrophils, macrophages) or disseminated to other locations. The fate of the parasites internalised by macrophages in vivo was further evaluated by collecting the peritoneal exudates 3 h after parasite injection, followed by cultivation of the cells for 24 hs in vitro (Fig. 7B). We found approximately half intracellular Δisp2/3 as compared to WT, which was fully reversed when Δisp2/3 were injected together with the NE inhibitor. We observed no significant differences in the sizes of lesions in the footpad of C57B6 mice infected with either WT parasites and two independent clones of Δisp2/3 (Fig. 9C), suggesting that tissue infiltrate and/or inflammation was similar. However, there was a 10-fold reduction in the numbers of Δisp2/3 parasites in the lymph nodes of the infected mice as compared to WT.
Figure 9. In vivo parasite uptake and survival in phagocytes is modulated by NE.
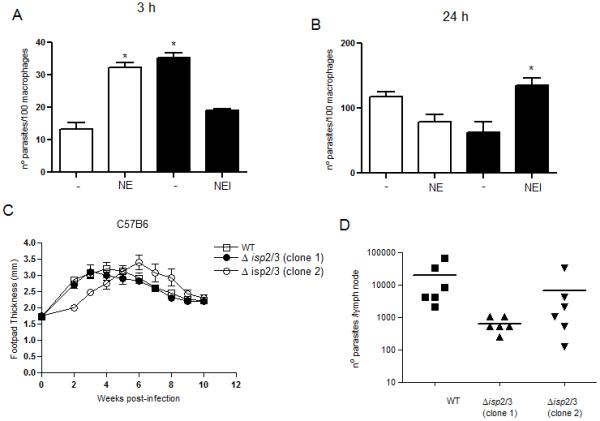
Stationary phase promastigotes were injected in the peritoneal cavity (A and B) or in the footpads (C and D) of C57B6 mice. In A and B, the peritoneal cavity exudates were collected 3 h after injection of parasites and the cells were plated in RPMI-FCS for 2h. The cultures were washed fixed and stained after 2 h (A) or 24 h (B), and the numbers of intracellular parasites in the adherent cells were determined. NE or NEI were injected concomitantly with parasites at 100 ng.ml−1 or 5 μM, respectively. In C, the sizes of footpad lesions were measured weekly and in D, the parasite burdens in the draining lymph nodes were determined by limiting dilution at week 10 post-infection. Two clones of Δisp2/3 generated independently were tested. The experiments were performed in 3 and 6 mice per group (And B) and (C and D), respectively. In A and B, the data are means ± SD. Key: white bars, WT; black bars, Δisp2/3. Asterisks indicate statistical significance at p<0.001 from WT in (A) and at p<0.05 from Δisp2/3 in B.
DISCUSSION
In the present study, we used parasites deficient in ISP2 and ISP3 as a tool to investigate the molecular pathways through which these natural inhibitors exert their role in the Leishmania-macrophage interaction. As the expression of ISP2 is abundant in WT, but ISP3 was never detected in the parasites, the phenotypes characterized in the mutant parasites are most likely related to the loss of ISP2 function. We observed that Δisp2/3 were internalised more efficiently than WT parasites by macrophages of C57/B6 mice, in a route dependent on CD11b, a sub-unit of CR3, and on NE activity.
The utilisation of a peptidyl synthetic irreversible inhibitor to NE enabled the identification of NE as the primary target of ISP2 in macrophages. The kinetics of intracellular parasite growth showed that, in addition to preventing death at 24 h post-infection, the inhibitor of NE rescued the capability of Δisp2/3 to grow inside macrophages. Although not reported in macrophages, studies in vivo showed that the phagocytosis of zymosan particles by neutrophils was diminished in NE-deficient animals, while the phagocytosis of fluorescein conjugated-E. coli particles was unchanged, suggesting that NE might affect phagocytosis in leukocytes in a selective way (18). Importantly, our data indicate that activation pathways triggered by NE at early stages, i.e. during parasite phagocytosis, are detrimental for the survival and intracellular development of amastigotes 3 days later. A deleterious effect of NE derived from apoptotic neutrophils on the survival of intracellular L. major in murine macrophages has been recently proposed (7). However, the role of NE in the uptake of L. major by macrophages was not addressed in that study, since exogenous NE was added after the infection of macrophages (7). In our model, the addition of exogenous NE to macrophages after parasite uptake had no effect in parasite survival within 24hs, while in the abovementioned study the authors observed that exogenous NE added after parasite phagocytosis, led to reduced growth of parasites 5-7 days later. The discrepancies between the two studies might result from the different time points used to evaluate parasite load. Importantly, we have shown that NE present at the surface of macrophages is readily available to influence parasite phagocytosis in the absence of neutrophils. Furthermore, our studies support the idea that, even in the absence of neutrophils, NE activity must be controlled during parasite-macrophage contact to avoid future elimination of intracellular Leishmania.
We showed that exogenous NE promotes increased phagocytosis of WT promastigotes, and partial elimination of intracellular amastigotes within 24 h. NE added exogenously did not affect the uptake or survival of Δisp2/isp3, suggesting that, in the absence of ISP2 and ISP3, the amounts of active NE present in macrophages is sufficient to promote parasite elimination. This is the first time that the involvement of serine peptidases from macrophages in the uptake and survival of L. major has been described and is important in light of the current view that NE secreted from neutrophils surrounding infected macrophages is the main mechanism of parasite elimination involving neutrophils. We showed that Δisp2/3 were likewise more infective to the murine macrophage cell line RAW, and increased infection was abolished by the inhibitor to NE, demonstrating that macrophages that had not been previously exposed to neutrophils are also capable of killing intracellular Leishmania through NE-dependent mechanisms. In this scenario, we postulate that, in the absence of neutrophils, ISP2 present in Leishmania might be sufficient to control local NE activity at the early stages of interaction with macrophages, preventing activation signals that would result in subsequent parasite death. We also observed a mild effect of NEI in the uptake of WT parasites by macrophages of C3HeN mice, suggesting that the amount of ISP2 might not be sufficient to inactivate all the NE present in macrophages from that strain of mice. However, we did not observe significant differences in the contents of NE of macrophages form the mouse strains used in this study (data not shown) by western blotting.
Neutrophils are recruited to the site of tissue damage caused by sandfly probing and phagocytose 80-90% of parasites (8). While the phagocytosis of parasites acquired following release from infected neutrophils are likely influenced by ISP levels, parasites acquired via parasitised neutrophils, the “Trojan Horse” route (9), would not be influenced by ISP2 as proposed. We showed that the lack of ISP2 influences parasite uptake by macrophages in vivo, in the presence of additional cells either resident in the tissue or recruited early to the site of infection. ISP2 might also influence the phagocytosis by and/or the ability of Leishsmania to survive inside neutrophils, exerting a further role in the establishment of the initial infection. In vivo imaging studies showed that at least a few parasites are incorporated into the vacuoles of dermal dendritic cells (DCs) minutes following parasite injection (19). It remains to be determined if ISPs also influences the internalisation of Leishmania by DCs and additionally modulate DC function through the local inhibition of NE.
Our immunofluorescence studies revealed NE at the surface of elicited macrophages. It was shown that sub-populations of the human pro-monocytic cell line U937 express NE at distinct sub-cellular sites, i.e., granule-like compartments or at the cell surface (20). Cell surface NE is available for binding to the inhibitors alpha-1-proteinase and alpha-1-antitrypsin, and behaves as a receptor (14). We seldomly detected co-localisation of NE and CD11b in the elicited macrophages, having observed a clear pattern of CD11b distribution at the edge of the cell, at basal regions, while NE was also observed at the apical region of the cell. Importantly, there was co-localisation between CD11b and NE in macrophages exposed to Δisp2/3, indicating that both molecules are in close proximity at the site of parasite entry. Furthermore, there was an exclusive clear pattern of CD11b located at the upper regions of infected cells, suggesting that the parasite induces the surface re-distribution of this receptor in macrophages. Our results suggest that NE present at the surface of elicited macrophages is active since it is subject to modulation by rISP2, aprotinin or the synthetic NE inhibitor. Alternatively, the enzyme could be produced in an inactive form and fully activated upon interaction with the parasite, via an unidentified mechanism. In either case, NE is clearly available for binding to and regulation by ISP2. In monocytes, the association of active cell surface NE to inhibitors induces enzyme clustering with CD4 and CXCR4 in a plasma membrane configuration, demonstrating that ligand binding to cell surface NE is capable of changing its distribution, promoting association with different cell surface receptors (14). At the moment, we do not know how NE and CD11b or TLR4 interact to promote the up-regulation of phagoctyosis, but our results of co-localization support the hypothesis that Leishmania induces the rearrangement of cell surface components in macrophages, bringing CR3 and NE to a common signaling platform.
We found that the neutralisation of TLR4 by antibodies or the infection of macrophages deficient in functional TLR4 or lacking TLR4 reverted increased phagocytosis of Δisp2/3 and prevented parasite elimination in the following 24 h, similarly to pre-treatment with NE inhibitors. We also showed that the engagement of TLR4 by exogenous agents such as LPS or activating antibodies promoted enhanced uptake of WT parasites, showing that the activation of this receptor modulates the phagocytosis of L. major. Importantly, the effects of exogenous NE on the increased uptake and fate of internalised WT parasites was completely prevented when TLR4 is previously blocked, establishing that NE and TLR4 act in a common pathway. This hypothesis is further supported by the finding that the activation of TLR4 by antibodies can bypass the detrimental effect of NE inhibition in the upregulation of parasite phagocytosis, observed either with Δisp2/3 or with WT in the presence of exogenous NE. It is tempting to speculate that, in order to act in conjunction, CR3, TLR4 and NE join at a cell surface signaling platform to mediate such events. A link between TLR4-mediated responses and NE activity was previously suggested in RAW cells where pretreatment with a NE inhibitor before exposure to LPS resulted in a significant dose-dependent decrease in MIP-2 production, comparable to that seen following pretreatment with TLR-4 antibody (21). We observed that the neutralisation of TLR-4 before the Leishmania-macrophage contact exerted a protective effect to Δisp2/3 only until 24 h post-infection, but the subsequent intracellular growth was practically arrested after 48-72 h. In contrast, Δisp2/3 grew efficiently macrophages defective in TLR-4 signaling, increasing approximately 6-fold in 3 days, as compared to an approximate 2-fold increase in the numbers of WT, in the same period. These results suggest that TLR4 is not only implicated in parasite elimination at short times post-infection (24 h), but has an additional role in controlling amastigote growth at later stages of the parasite intracellular life cycle.
The activation of TLR4 after parasite internalisation had no effect on the outcome of infection 24 h later (Fig 7), suggesting that the TLR4-NE pathway must be activated concomitantly to parasite phagocytosis in order to promote intracellular killing. It has been reported that phagosome maturation in macrophages is influenced by TLR-signaling coupled to phagocytosis (22). Furthermore, it has been proposed that intraphagosomal sensing and modulation of the nascent phagosome by TLR acts through inducible phagosome maturation following proinflamatory stimuli (23). Although the requirement of functional TLR2 or TLR4 for phagosome maturation is controversial (24), it is possible that TLR signaling might affect the maturation of the parasitophorous vacuole, significantly influencing intracellular parasite growth. We showed that whilst ISP2-3-deficient mutants had normal sized lesions in C57/B6 mice, there was a significant decrease in parasite numbers in those mice compared to wild type, suggesting that the initial modulation of NE and TLR4 by ISP influences the outcome of anti-parasite immunity.
The superoxide scavenger EUK134 or treatment of macrophages with catalase rescued Δisp2/3 from intracellular death after 24 h, establishing that reactive oxygen species (ROS) are responsible for the killing of intracellular ISP2-deficient parasites. This is important since it is known that Leishmania have evolved multiple ways to avoid ROS production by macrophages, causing superoxide to play a limited role in the control of Leishmania infection in mice (25) or in human macrophages (26). Our findings suggest that ISP2 has an important role in the control of ROS production during infection. L. major lacking all phosphoglycans (LPG and PPG), reside in fusogenic phagosomes of macrophages where they are subsequently destroyed, although they can survive in macrophages defective in oxidative burst (27). Although we cannot discount that other pathways that contribute to the downregulation of ROS production are affected in Δisp2/3, this seems unlikely because parasite death is completely reversed by NE inhibitors or in macrophages defective in TLR4 signaling. Furthermore, we found no differences in the production of nitric oxide by macrophages infected with WT or with Δisp2/3 (not shown).
In epithelial cells, stimulation with NE is known to induce ROS (28) as part of an intricate cascade leading to gene expression (29). Although the mechanisms underlying triggering of TLR4 by NE in epithelial cells are not fully understood, a recent study provides strong evidence that this is done indirectly, by promoting the dimerisation between the epidermal growth factor receptor (EGFR) and TLR4, subsequently promoting TLR4 activation (30). At present, we cannot discriminate if ISP2 acts by preventing NE-induced association between CD11b, TLR4 and other additional receptor(s) in a common signaling platform or if other Leishmania factors promote receptor association, but their activation is subsequently impaired due to NE inhibition by ISP2. In those lines, we cannot exclude that the deletion of isp2 and isp3 caused additional changes in the parasite resulting in new ligands that promote the mobilisation of additional receptors.
In conclusion, we propose that ISP2 prevents the activation of a TLR4-NE signaling cascade during early parasite-macrophage contact, leading to down-regulation of parasite phagocytosis but exerting a beneficial effect for intracellular amastigote survival and growth in macrophages, due to the inhibiton of ROS production. It remains to be determined if ISP2 plays an additional role in controlling the continuous activation of the TLR4-NE route after parasite engulfment, helping to ensure adequate amastigote growth at later stages of infection.
ACKNOWLEDGMENTS
We thank Edna Lopes for technical assistance in parasite culture. We are indebted to Prof. George A dosReis for the use of the animal facilities. We thank Alexandre Morrot for helpful discussions.
This work was supported by the Wellcome Trust (081877), FAPERJ and CNPq. APCAL is a CNPq fellow; RAP is a member of the Instituto Nacional de Ciência e Tecnologia de Biologia Estrutural e Bioimagem.
REFERENCES
- (1).Kima PE. The amastigote forms of Leishmania are experts at exploiting host cell processes to establish infection and persist. Int J Parasitol. 2007;37:1087–1096. doi: 10.1016/j.ijpara.2007.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Mosser DM, Edelson PJ. The third component of complement (C3) is responsible for the intracellular survival of Leishmania major. Nature. 1987;327:329–331. doi: 10.1038/327329b0. [DOI] [PubMed] [Google Scholar]
- (3).Dermine JF, Scianimanico S, Privé C, Descoteaux A, Desjardins M. Leishmania promastigotes require lipophosphoglycan to actively modulate the fusion properties of phagosomes at an early step of phagocytosis. Cell Microbiol. 2000;2:115–126. doi: 10.1046/j.1462-5822.2000.00037.x. [DOI] [PubMed] [Google Scholar]
- (4).Späth GF, Epstein L, Leader B, Singer SM, Avila HA, Turco SJ, Beverley SM. Lipophosphoglycan is a virulence factor distinct from related glycoconjugates in the protozoan parasite Leishmania major. Proc Natl Acad Sci USA. 2000;97:9258–9263. doi: 10.1073/pnas.160257897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Späth GF, Garraway LA, Turco SJ, Beverley SM. The role(s) of lipophosphoglycan (LPG) in the establishment of Leishmania major infections in mammalian hosts. Proc Natl Acad Sci USA. 2003;100:9536–9541. doi: 10.1073/pnas.1530604100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Ribeiro-Gomes FL, Otero AC, Gomes NA, Moniz-De-Souza MC, Cysne-Finkelstein L, Arnholdt AC, Calich VL, Coutinho SG, Lopes MF, DosReis GA. Macrophage interactions with neutrophils regulate Leishmania major infection. J Immunol. 2004;172:4454–4462. doi: 10.4049/jimmunol.172.7.4454. [DOI] [PubMed] [Google Scholar]
- (7).Ribeiro-Gomes FL, Moniz-de-Souza MC, Alexandre-Moreira MS, Dias WB, Lopes MF, Nunes MP, Lungarella G, DosReis GA. Neutrophils activate macrophages for intracellular killing of Leishmania major through recruitment of TLR4 by neutrophil elastase. J Immunol. 2007;179:3988–3994. doi: 10.4049/jimmunol.179.6.3988. [DOI] [PubMed] [Google Scholar]
- (8).Peters NC, Egen JG, Secundino N, Debrabant A, Kimblin N, Kamhawi S, Lawyer P, Fay MP, Germain RN, Sacks D. In vivo imaging reveals an essential role for neutrophils in leishmaniasis transmitted by sand flies. Science. 2008;321:970–974. doi: 10.1126/science.1159194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).van Zandbergen G, Klinger M, Mueller A, Dannenberg S, Gebert A, Solbach W, Laskay T. Cutting edge: neutrophil granulocyte serves as a vector for Leishmania entry into macrophages. J Immunol. 2004;173:6521–6525. doi: 10.4049/jimmunol.173.11.6521. [DOI] [PubMed] [Google Scholar]
- (10).Eschenlauer SC, Faria MS, Morrison LS, Bland N, Ribeiro-Gomes FL, DosReis GA, Coombs GH, Lima APCA, Mottram JC. Influence of parasite encoded inhibitors of serine peptidases in early infection of macrophages with Leishmania major. Cell Microbiol. 2009;11:106–120. doi: 10.1111/j.1462-5822.2008.01243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Ivens AC, Peacock CS, Worthey EA, Murphy L, Aggarwal G, Berriman M, Sisk E, Rajandream MA, Adlem E, Aert R, Anupama A, Apostolou Z, Attipoe P, Bason M, Bauser N,C, Beck A, Beverley SM, Bianchettin G, Borzym K, Bothe G, Bruschi CV, Collins M, Cadag E, Ciarloni L, Clayton C, Coulson RM, Cronin A, Cruz AK, Davies RM, De Gaudenzi J, Dobson DE, Duesterhoeft A, Fazelina G, Fosker N, Frasch AC, Fraser A, Fuchs M, Gabel C, Goble A, Goffeau A, Harris D, Hertz-Fowler C, Hilbert H, Horn D, Huang Y, Klages S, Knights A, Kube M, Larke N, Litvin L, Lord A, Louie T, Marra M, Masuy D, Matthews K, Michaeli S, Mottram JC, Müller-Auer S, Munden H, Nelson S, Norbertczak H, Oliver K, O’neil S, Pentony, Pohl TM, Price C, Purnelle B, Quail MA, Rabbinowitsch E, Reinhardt R, Rieger M, Rinta J, Robben J, Robertson L, Ruiz JC, Rutter S, Saunders D, Schäfer M, Schein J, Schwartz DC, Seeger K, Seyler A, Sharp S, Shin H, Sivam D, Squares R, Squares S, Tosato V, Vogt C, Volckaert G, Wambutt R, Warren T, Wedler H, Woodward J, Zhou S, Zimmermann W, Smith DF, Blackwell JM, Stuart KD, Barrell B, Myler PJ. The genome of the kinetoplastid parasite, Leishmania major. Science. 2005;309:436–442. doi: 10.1126/science.1112680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Pham CTN. Neutrophil serine proteases: specific regulators of inflammation. Nat Rev Immunol. 2006;6:541–550. doi: 10.1038/nri1841. [DOI] [PubMed] [Google Scholar]
- (13).Gullberg U, Lindmark A, Lindgren G, Persson AM, Nilsson E, Olsson I. Carboxyl-terminal prodomain-deleted human leukocyte elastase and cathepsin G are efficiently targeted to granules and enzymatically activated in the rat basophilic/mast cell line RBL. J Biol Chem. 1995;270:12912–12918. doi: 10.1074/jbc.270.21.12912. [DOI] [PubMed] [Google Scholar]
- (14).Bristow CL, Mercatante DR, Kole R. HIV-1 preferentially binds receptors co-patched with cell surface elastase. Blood. 2003;102:4479–4486. doi: 10.1182/blood-2003-05-1635. [DOI] [PubMed] [Google Scholar]
- (15).Besteiro S, Williams RAM, Morrison LS, Coombs GH, Mottram JC. Endosome sorting and autophagy are essential for differentiation and virulence of Leishmania major. J Biol Chem. 2006;281:11384–11396. doi: 10.1074/jbc.M512307200. [DOI] [PubMed] [Google Scholar]
- (16).Cai TQ, Wright SD. Human leukocyte elastase is an endogenous ligand for the integrin CR3 (CD11b/CD18, Mac-1, alpha M beta 2) and modulates polymorphonuclear leukocyte adhesion. J Exp Med. 1996;184:1213–1223. doi: 10.1084/jem.184.4.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Altieri DC, Morrissey JH, Edgington TS. Adhesive receptor Mac-1 coordinates the activation of factor X on stimulated cells of monocytic and myeloid differentiation: an alternative initiation of the coagulation protease cascade. Proc Natl Acad Sci USA. 1988;85:7462–7466. doi: 10.1073/pnas.85.20.7462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Young RE, Thompson RD, Larbi KY, La M, Roberts CE, Shapiro SD, Perretti M, Nourshargh S. Neutrophil elastase (NE)-deficient mice demonstrate a nonredundant role for NE in neutrophil migration, generation of proinflammatory mediators, and phagocytosis in response to zymosan particles in vivo. J Immunol. 2004;172:4493–44502. doi: 10.4049/jimmunol.172.7.4493. [DOI] [PubMed] [Google Scholar]
- (19).Ng LG, Hsu A, Mandell MA, Roediger B, Hoeller C, Mrass P, Iparraguirre A, Cavanagh LL, Triccas JA, Beverley SM, Scott P, Weninger W. Migratory dermal dendritic cells act as rapid sensors of protozoan parasites. PLoS Pathog. 2008;4:e1000222. doi: 10.1371/journal.ppat.1000222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Bristow CL, Wolkowicz R, Trucy M, Franklin A, Di Meo F, Kozlowski MT, Winston R, Arnold RR. NF-kappaB signaling, elastase localization, and phagocytosis differ in HIV-1 permissive and nonpermissive U937 clones. J Immunol. 2008;180:492–499. doi: 10.4049/jimmunol.180.1.492. [DOI] [PubMed] [Google Scholar]
- (21).Tsujimoto H, Ono S, Majima T, Kawarabayashi N, Takayama E, Kinoshita M, Seki S, Hiraide H, Moldawer LL, Mochizuki H. Neutrophil elastase, MIP-2, and TLR-4 expression during human and experimental sepsis. Shock. 2005;23:39–44. doi: 10.1097/01.shk.0000145936.31967.d7. [DOI] [PubMed] [Google Scholar]
- (22).Blander JM. Signaling and phagocytosis in the orchestration of host defense. Cell Microbiol. 2007;9:290–299. doi: 10.1111/j.1462-5822.2006.00864.x. [DOI] [PubMed] [Google Scholar]
- (23).Blander JM. Coupling Toll-like receptor with phagocytosis: potentiation of antigen presentation. Trends Immunol. 2007;28:19–25. doi: 10.1016/j.it.2006.11.001. [DOI] [PubMed] [Google Scholar]
- (24).Russell DG, Yates RM. TLR signaling and phagosome maturation: an alternative viewpoint. Cell Microbiol. 2007;9:849–850. doi: 10.1111/j.1462-5822.2007.00920.x. [DOI] [PubMed] [Google Scholar]
- (25).Blos M, Schleicher U, Soares Rocha FJ, Meissner U, Rollinghoff M, Müller M, Werner-Felmayer G, Bogdan C. Organ-specific and stage-dependent control of Leishmania major infection by inducible nitric oxide synthase and phagocyte NADPH oxidase. Eur J Immunol. 2003;33:1224–1234. doi: 10.1002/eji.200323825. [DOI] [PubMed] [Google Scholar]
- (26).Gantt KR, Goldman TL, Miller MA, McCormick ML, Jeronimo SM, Nascimento ET, Britigan BE, Wilson ME. Oxidative responses of human and murine macrophages during phagocytosis of Leishmania chagasi. J Immunol. 2001;167:893–901. doi: 10.4049/jimmunol.167.2.893. [DOI] [PubMed] [Google Scholar]
- (27).Späth GF, Lye LF, Segawa H, Sacks DL, Turco SJ, Beverley SM. Persistence without pathology in phosphoglycan-deficient Leishmania major. Science. 2003;301:1241–1243. doi: 10.1126/science.1087499. [DOI] [PubMed] [Google Scholar]
- (28).Aoshiba K, Yasuda K, Yasui S, Tamaoki J, Nagai A. Serine proteases increase oxidative stress in lung cells. Am J Physiol Lung Cell Mol Physiol. 2001;281:556–564. doi: 10.1152/ajplung.2001.281.3.L556. [DOI] [PubMed] [Google Scholar]
- (29).Shao MX, Nadel JA. Neutrophil elastase induces MUC5AC mucin production in human airway epithelial cells via a cascade involving protein kinase C, reactive oxygen species, and TNF-alpha-converting enzyme. J Immunol. 2005;175:4009–4016. doi: 10.4049/jimmunol.175.6.4009. [DOI] [PubMed] [Google Scholar]
- (30).Bergin DA, Greene CM, Sterchi EE, Kenna C, Geraghty P, Belaaouaj A, Taggart CC, O’Neill SJ, McElvaney NG. Activation of the epidermal growth factor receptor (EGFR) by a novel metalloprotease pathway. J Biol Chem. 2008;283:31736–31744. doi: 10.1074/jbc.M803732200. [DOI] [PubMed] [Google Scholar]