

Abstract
Cytotoxic approaches to killing tumor cells, such as chemotherapeutic agents, γ-irradiation, suicide genes or immunotherapy, have been shown to induce cell death through apoptosis. The intrinsic apoptotic pathway is activated following treatment with cytotoxic drugs, and these reactions ultimately lead to the activation of caspases, which promote cell death in tumor cells. In addition, activation of the extrinsic apoptotic pathway with death-inducing ligands leads to an increased sensitivity of tumor cells toward cytotoxic stimuli, illustrating the interplay between the two cell death pathways. In contrast, tumor resistance to cytotoxic stimuli may be due to defects in apoptotic signaling. As a result of their importance in killing cancer cells, a number of apoptotic molecules are implicated in cancer therapy. The knowledge gleaned from basic research into apoptotic pathways from cell biological, structural, biochemical, and biophysical approaches can be used in strategies to develop novel compounds that eradicate tumor cells. In addition to current drug targets, research into molecules that activate procaspase–3 directly may show the direct activation of the executioner caspase to be a powerful therapeutic strategy in the treatment of many cancers.
Keywords: Caspase, apoptosis, programmed cell death, protease, zymogen, dimerization, active site formation, human disease, cancer treatment, dimer interface, proteolytic cleavage, active site loops, crystallography, enzyme activation
INTRODUCTION
Cell death can occur by many pathways, all of which have unique mechanisms and morphologies. The three major types of cell death include necrosis, oncosis and programmed cell death (Fig. 1). Necrosis was defined originally as the accidental death of cells and living tissue and generally is characterized by swelling of the cell, chromatin condensation and the eventual nuclear and cellular lysis that leads to inflammation [1]. Later, Majno and Joris disputed the earlier definition by stating that necrosis not a type of cell death. Rather, it represents the end stage of the cell death process and can occur in any pathway in the absence of phagocytosis [2] (see Fig. 1). Instead, “oncosis” was proposed as a term to refer to any type of cell death that is marked by cellular swelling. The morphology of oncosis also includes chromatin condensation (pyknosis) and nuclear fragmentation (karyorrhexis) with eventual nuclear fading (karyolysis) [3].
Fig. 1.

Cell death pathways. Morphological changes for three cell death pathways, apoptosis, oncosis, and autophagy. PCD refers to programmed cell death.
Programmed cell death (PCD) occurs through two distinct pathways that can be differentiated by the morphology of the cell in response to death signals: condensation prominent, called type I or apoptosis, and autophagy prominent, called type II. Type I PCD often found to occur during vertebrate development, especially in organ morphogenesis [4–7], and was characterized first by Kerr in 1972 as having morphological features that include cytoplasmic shrinkage, active membrane blebbing and condensed chromatin [8]. Apoptosis eventually leads to cell segregation and the formation of apoptotic bodies that contain intact organelles [8]. In addition, the nuclear DNA is degraded, the cytoskeleton is dismantled and cell progression is halted [9, 10]. Neighboring macrophages, which recognize phosphatidyl serine on the outside of the plasma membrane, then engulf the apoptotic bodies [8, 11]. In contrast, the most prominent features of autophagic PCD are formation of autophagic vacuoles that accompany the degradation of cytoplasmic materials, nuclear collapse and subsequent phagocytosis.
MECHANISMS OF APOPTOSIS
A family of cysteine-dependent aspartate-directed proteases, known as caspases, is intimately involved in apoptosis. The cleavage of key proteins in the cell by caspases leads to the aforementioned morphological and biochemical changes observed during apoptosis. Seven of the known eleven human caspases are involved in the execution of apoptosis (Fig. 2 and 3). The apoptotic caspases are divided into two groups, the initiators and the effectors, depending on their time of entry into the apoptotic cascade (Fig. 3). For example, the initiator caspases, such as caspases–2, –8, –9 and –10, have an early entry into the cascade, where they are responsible for activating the effector caspases–3, –6 and –7 (Fig. 4). The initiators are themselves activated either by so-called extrinsic or intrinsic mechanisms, as described below.
Fig. 2.

Sequence alignment of eleven human caspases. Amino acid residues in L1 (52–66) (yellow), L2 (163–175) (red), L3 (198–213) (blue), L4 (247–263) (brown) and L2′ (176′–192′) (cyan) of caspase–3 are shown by the solid lines, and the colors correspond to those in Figs. 5–8. The dark lines indicate regions of secondary structure. The catalytic cysteine (C163) is shown in bold. Boundaries observed in the crystal structures for the large subunit (S29-D175) and the small subunit (G177-H277) of caspase–3 are indicated by (⌊ ⌋ ). Two processing sites in procaspase–3 (D28 and D175) are indicated by (▾), where cleavage at D28 removes the prodomain and cleavage at D175 separates the two subunits, as described in the text. Sequence data were obtained using NCBI (http://www.ncbi.nlm.nih.gov/). Boundaries for the other caspases are approximately the same as those indicated for caspase-3. The sequence for caspase-10 is that of isoform 10/a.
Fig. 3.

Human caspase organization. Caspases are grouped on the left according to function, either initiators or effectors of apoptosis, and on the right according to the recognition sequence of the substrate. One should note that the Group I, inflammatory, caspases (–1, –4, and –5) are not included in this figure. Each caspase has an N-terminal prodomain, where some contain either a CARD (caspase recruitment domain) or DED (death effector domain), followed by the large subunit (Large), an intersubunit linker, and the small subunit (Small). The numbers on each caspase molecule refer to the length of each specific domain, which was determined using the NCBI domain organization database (http://www.ncbi.nlm.nih.gov/).
Fig. 4.

The caspase cascade. In the extrinsic apoptotic pathway, a death ligand (not shown) binds to a death receptor (green and purple), which signals an adaptor molecule to bind to the receptor via death domain (DD) interactions. The adaptor molecule interacts with the death effector domain (DED) of procaspases–2, –8, and –10, forming an activation complex. Dimerization (mechanism unknown) results in maturation and full activity. Caspases–8 and –10 then process effector caspases. Caspase–2 cleaves Bid (orange), a protein responsible for the increased permeability of the mitochondria. In the intrinsic apoptotic pathway, an increase in the permeability of the mitochondria, resulting from cleavage of Bid, reactive oxygen species (ROS) or chemotherapeutic agents, leads to an increase in the cytosolic concentration of cytochrome c (green) and the formation of the apoptosome (blue). The apoptosome is composed of Apaf-1 monomers that form a heptameric structure when cytochrome c binds, in an ATP-dependent manner, leading to interactions with the caspase recruitment domain (CARD) of procaspase–9. The interactions increase the local concentration of procaspase–9 monomers and thereby promote dimerization and activation. Caspase–9 then processes effector caspases, which leads to apoptosis. Inhibitors of apoptosis (IAPs), specifically XIAP, inhibit mature initiator or effector caspases (caspases –9, –3, and –7), and are themselves inhibited by Omi or Smac. Upon their release from the mitochondria, the nucleases Endo G and AIF migrate to the nucleus and cleave DNA.
The extrinsic pathway for initiator caspase activation ultimately is responsible for the elimination of unwanted cells that are produced during development or that have tumorogenic qualities. This pathway is initiated by ligation of a transmembrane death receptor in response to an extracellular signal, where the death receptors belong to the tumor necrosis factor receptor (TNFR) superfamily. Members of the TNFR family contain an intracellular domain, called a death domain (DD), that binds to a number of adaptor proteins with similar domains. These interactions effectively transmit the death signal from the exterior surface of the cell to the cytosol. The most characterized TNF receptors include CD95 (APO-1/Fas), TNF receptor 1 (TNFRI), TRAIL-R1 and TRAIL-R2. Their respective death ligands are FasL, TNFα and TRAIL (TNF-related apoptosis-inducing ligand) [12–14]. Upon ligation, the DD on the cytoplasmic tail of the death receptor recruits a multiprotein complex called the DISC (death inducing signaling complex). The DISC is thought to promote the dimerization of caspases–8 and –10 by increasing the local concentration of procaspase monomers, thereby increasing the probability of dimerization. This is referred to as the induced proximity model [15]. Following their activation, the initiator caspases cleave the zymogens of caspases–3, –6, or –7 (Fig. 4).
A fundamental difference exists between the caspase subfamilies, even though the structures of the mature caspases are quite similar. The initiator caspases exist in the cell as monomers that require a scaffold for dimerization. Once associated with the scaffold, the procaspase dimer has sufficient enzymatic activity for autolysis [16–19]. In contrast, the effector procaspases exist in the cell as stable dimers that have little enzymatic activity [20, 21]. As described below, the effector procaspases are inactive due to misaligned active sites that prevent efficient catalysis. Activation of the effector procaspases occurs after proteolytic cleavage, which allows the active site loops to rearrange, resulting in a large increase in activity (~200-fold) [21].
The caspase cascade also can be activated by the so-called intrinsic pathway, which is a mitochondria-dependent pathway that is activated by a variety of stimuli, including chemotherapeutic agents, cytokines, reactive oxygen species, and DNA damage [22] (see Fig. 4). In response to the apoptotic stimuli, the permeability of the mitochondrial membrane increases, and many apoptogenic factors are released, including cytochrome c, apoptosis inducing factor (AIF), Smac (second mitochondria derived activator of caspase)/DIABLO (direct IAP binding protein with low pI), Omi/HtrA2 (high temperature requirement A2) and endonuclease G [23] (Fig. 4). The increased permeability of the mitochondria is accomplished in part by the cleavage of Bid (BH3 interacting death agonist), a member of the Bcl-2 family of proteins, by caspase–2 [24] and possibly by caspases–8 and –10 [23, 25]. Caspase–2 is activated by a death receptor in a similar fashion as caspases–8 and –10 [26, 27]. The release of cytochrome c into the cytosol results in the activation of caspases–3, –6 and –7 through the formation of the apoptosome, which is a multiprotein complex comprised of cytochrome c, Apaf-1, and caspase–9 (Fig. 4). The apoptosome promotes dimerization and maturation of caspase–9, which in turn activates caspase–3. One should note that the two caspase activation pathways, intrinsic and extrinsic, are not mutually exclusive, and cross communication between the two pathways leads to a massive response to the initial signal, regardless of whether the initial signal was external or internal [28].
Smac/DIABLO and Omi/HtrA2 are proapoptotic molecules that are released from the intermembrane space of the mitochondria. Both proteins neutralize the inhibitory effects of inhibitors of apoptosis proteins (IAPs) [29, 30]. IAPs have evolved to protect cells against unwanted self-execution that may occur as a result of the premature activation of the caspase cascade. IAPs bind to and inhibit active caspases [31]. Thus, IAPs are a class of negative regulators of apoptosis, and they are neutralized by the binding of Smac/DIABLO Omi/HtrA2 (see Fig. 4). In contrast, AIF and endonuclease G mediate caspase-independent cell death upon their release from the mitochondria. Both proteins translocate into the nucleus and facilitate DNA fragmentation [32, 33], although the precise roleof AIF in apoptosis is still under debate [34, 35].
The killing of tumor cells by cytotoxic approaches such as chemotherapeutic agents, γ-irradiation, suicide genes or immunotherapy has been shown to induce cell death through apoptosis [22, 36–40]. Following treatment with cytotoxic drugs, the intrinsic apoptotic pathway is activated and promotes cell death in tumor cells. In addition, activation of the extrinsic pathway leads to increased sensitivity of tumor cells toward the cytotoxic stimuli [41]. In contrast, tumor resistance to cytotoxic stimuli may be due to defects in apoptotic signaling. In this review, we have focused on the apoptotic molecules that are implicated in cancer therapy. In addition to current therapeutic targets in the intrinsic and extrinsic apoptotic pathways, we suggest that research into molecules that bind to and activate procaspase–3 directly may prove to be a viable therapeutic strategy in cancer treatment.
CANCER DRUG TARGETS IN APOPTOSIS
Extrinsic Pathway
Activation of the extrinsic apoptotic pathway has been shown to increase the sensitivity of tumor cells to cytotoxic stimuli. For example, the CD95 receptor/ligand system is found in many cell types, including those in the immune system as well as in tumor cells [42]. FasL, the CD95 ligand, is produced by activated T cells and normally plays a major role in the regulation of the immune system by triggering autocrine suicide or paracrine death in lymphocytes or other target cells. Since CD95 is expressed on the surface of tumor cells, it has become a target of chemotherapy-induced tumor cell death in many studies [43–45]. The results of those studies showed that the treatment of tumor cells with anti-cancer drugs, such as doxorubicin or methotrexate (lymphoid leukemias) or bleomycin (hepatoma cells), increased the level of FasL expression, which in turn stimulated the CD95 pathway and activated the apoptotic cascade. In addition to upregulating FasL expression, the expression of the CD95 receptor also increased upon drug treatment [44]. Overall, these results indicate that the CD95 receptor/ligand system amplifies apoptosis induced by cytotoxic anticancer drugs.
Similarly to the CD95 system, the TRAIL receptor system has been implicated as a target in cancer therapy because TRAIL selectively kills tumor cells, but not normal cells, by inducing apoptosis. For example, TRAIL was administered intravenously to non-human primates, and no toxicity was observed, even at high doses [46]. In addition, TRAIL was administered to several human cell lines, including endothelial cells, smooth muscle cells, astrocytes, epithelial cells, endothelial cells and fibroblasts, and no cytotoxicity was observed [47]. TRAIL selectivity is a vital finding in cancer therapy as ligands for the CD95 and TNFR systems illicit severe toxicities in normal cells [14]. The mechanism by which TRAIL selects tumor cells over normal cells has not been defined but may be related to the ratios of pro- and anti-apoptotic molecules in normal cells versus tumor cells [48].
There some concern, however, about potential toxicity to brain tissue and hepatocytes in humans with increased doses of TRAIL [49, 50]. More recently, it has been suggested that TRAIL actually promotes cell survival and proliferation in some circumstances. For example, some cancer cells, such as acute leukemic cells, show resistance to TRAIL-induced apoptosis by activating the transcription factor NF-κB [51], which functions to activate the transcription of prosurvival genes, like cytokines or growth factors involved in proliferation [52]. This finding important because it suggests that TRAIL could induce tumorogenic proliferation in TRAIL resistant tumors. There are several possible reasons for the observed resistance to TRAIL-induced apoptosis. For example, resistant tumors may have a downregulated expression of death receptors. In addition, the decoy TRAIL receptor, TRAIL-R3, is overexpressed, concurrent with the lack of expression of the agonists TRAIL-R1 and TRAIL-R2, as seen in gastric carcinoma cells [53]. Interestingly, the loss of expression of the agonist TRAIL receptors could be due to their location on chromosome eight, as they are found in a region that has frequent loss of heterozygosity in tumors [48]. The lower expression of CD95 and TRAIL receptors may be caused by the hypermethylation of their gene promoters, which has been observed both in neuroblastoma and in colon carcinoma cells [54–56]. Finally, histone deacetylation may block transcription of the genes encoding these receptors by blocking transcription factor docking [57]. The addition of histone deacetylase inhibitor to resistant tumor cells restored chemosensitivity and suppressed tumor growth [58].
Caspases
The expression level of effector caspases in tumor cells is a major determinant in the effectiveness of anticancer agents. Expression levels of certain caspases in tumor cells may have an impact on their activity, since a lower protein concentration may lead to a decrease in apoptosis. For example, it has been observed that a frameshift mutation within exon three in the caspase–3 gene in MCF-7 breast carcinoma cells leads to a complete loss of functional protein for this vital executioner caspase [59]. Transfection of MCF-7 tumor cells with procaspase–3 was shown to increase sensitivity to cytotoxic drugs [60]. Indeed, there is a strong correlation between levels of procaspase–3 expression in several cancer cell lines (leukemia, lymphoma, melanoma, neuroblastoma, lines (leukemia, lymphoma, melanoma, neuroblastoma, breast, lung, adrenal, and renal cancers), where concentrations of the zymogen can vary up to five-fold compared to those of normal cells, and sensitivity to a cytotoxic drug [61]. In addition to changes in the concentrations of effector caspases, effects also are observed in initiator caspases. For example, hypermethylation of the caspase–8 promoter was observed in a number of tumor cells, including neuroblastoma, malignant brain tumors, Ewing tumors, and small lung cell carcinoma [62, 63]. Restoration of caspase–8 activity was accomplished either by gene transfer or by demethylation treatment, which sensitized resistant tumor cells to drug-induced apoptosis [63].
Intrinsic Pathway
The regulation of the intrinsic apoptotic pathway is governed by the Bcl-2 protein family [64, 65]. Members of the Bcl-2 family localize to the mitochondria and function either in pro- or anti-apoptotic pathways. The antiapoptotic members include Bcl-2, Bcl-X and Mcl-1, while the proapoptotic members include Bax, Bak and Bad. Other members are the BH3 domain only proteins, which are responsible for linking the extrinsic and the intrinsic pathways, and include Bid, Bim, Puma and Noxa [66]. Bcl-2 family members also have been implicated in autophagic cell death, which is a cellular response to nutrient deprivation and which is suppressed during tumorigenesis [67]. Because of their roles in apoptosis, autophagy and tumorigenesis, it has been suggested that Bcl-2 family proteins act as a master switch in life and death decisions in the cell [65]. As a result, Bcl-2 is the target of several cancer therapies [65].
Upon induction of apoptosis, the proapoptotic Bcl-2 proteins, such as Bax, translocate from the cytoplasm to the mitochondrial outer membrane where they oligomerize to form a pore that makes the mitochondria permeable [68, 69]. Formation of the pore leads to the release of cytochrome c (see Fig. 4). The BH3 domain only proteins, like Bid, are responsible for triggering pore formation by Bax, Bak and Bad. In contrast, the antiapoptotic Bcl-2 proteins are responsible for sequestering the BH3 domain only proteins to a stable mitochondrial complex so that Bax, Bak and Bad cannot form the mitochondrial pores. The antiapoptotic Bcl-2 proteins also disrupt cytochrome c release, thereby blocking apoptosis. This is accomplished by disrupting the mitochondrial voltage-dependent anion channel (VDAC) or the permeability transition pore complex (PTPC) made by proapoptotic members of the Bcl-2 family [70, 71]. In experimental systems, mutating or changing the expression levels of proapoptotic or antiapoptotic Bcl-2 family proteins drastically altered the drug response [72]. A chemoresistant phenotype has been observed in several cancer cells that have a high level of expression of antiapoptotic Bcl-2 proteins or have a reduced level of the proapoptotic Bax protein. In addition, a lack of Bax expression has been linked to TRAIL resistance in MMR (DNA mismatch repair)-deficient colon cancer cells because the reduced permeability of the mitochondria results in the deficient release of Smac/DIABLO from the mitochondria. After restoring Bax expression to an optimal level, Wu and colleagues observed that the tumor cells became sensitive to TRAIL induced apoptosis [73].
IAPs
Inhibitor of apoptosis proteins (IAPs) are a family of apoptosis inhibitors, one of which (XIAP) specifically binds to and inhibits initiator and effector caspases to prevent premature apoptosis, either by binding to the active site, in the case of caspases–3 and –7, or the dimer interface, in the case of caspase–9 [74, 75] (Fig. 5 and 6). There are at least eight members of this family of proteins found in humans including X-linked IAP (XIAP), c-IAP1, c-IAP2, ILP-2, NAIP, ML-IAP, survivin and apollon. Of the aforementioned family members, XIAP may be the only member that binds caspases [76], and survivin and XIAP have the most potential as therapeutic targets in cancer cells [77]. IAPs, in general, are expressed at high levels in the majority of human cancers [75]. Several strategies have been employed to target IAP expression in cancer cells since survivin is found to be dramatically overexpressed in a majority of cancer cells, such as lung, breast, colon, stomach and pancreatic cancers, to name a few, implicating it in tumor malignancy [78]. Changes in survivin concentrations may be caused by dysregulation of transcription [78]. To this end, antisense oligonucleotides, ribozymes, siRNA and dominant negative survivin variants were used to downregulate the expression of survivin in cancer cells [79–82]. These studies, alone or coupled with chemotherapy, resulted in the suppression of tumor growth in models both in vivo and in vitro. Survivin is phosphorylated in vivo by the p34cdc2-cyclin B1 kinase complex during mitosis [31]. Without this phosphorylation event, survivin has no antiapoptotic activity [31]. The addition of cyclin-dependent kinase inhibitors, such as flavopiridol or purvanolol A, increased tumor cell response to taxol [83, 84]. In addition, small molecule antagonists of XIAP are in the forefront of current research [65]. One example involved Smac peptides harboring the N-terminal region of Smac (Ala-Val-Pro-Ile) that is essential for binding to IAPs. This molecule was shown to promote caspase activation and to sensitize various tumor cell lines to cytotoxic drugs [85].
Fig. 5.

Caspase–3 with XIAP BIR-2 bound. (A) Structure of caspase–3 (PDB entry 2J30). β-strands 1–8, α-helices 1–5, and active site loops L1 (yellow), L2 (red), L2′ (cyan), L3 (blue) and L4 (brown) are labeled. The prime (‘) indicates residues from the second monomer (large + small subunits). For clarity, only one active site is labeled. (B) BIR-2 from XIAP bound to caspase–3 active site (PDB entry 1I30). The active site loops of caspase–3 are labeled as in panel A. (C) Comparison of caspase–3 active site with tetrapeptide inhibitor bound (from panel A) or BIR-2 domain bound (from panel B). The lighter colors indicate the structure with BIR-2 bound, except for L2′, which is colored green. (D) Interactions of L2′ Asp179 or Asp181 with the P4 site of substrate/inhibitor (PDB entry 2J30, from panel A, and PDB entry 2DKO shown in green). Structures were generated using Pymol (Delano Scientific LLC, Palo Alto, CA).
Fig. 6.

Inhibited caspases. (A) Structure of caspase–9 with the BIR-3 domain of XIAP bound to the dimer interface (PDB entry 1NW9). Active site loops L1-L4 are labeled. Binding of the inhibitor prevents the procaspase–9 dimer from forming, resulting in a disorganized active site. (B) Structure of caspase–7 with FICA bound in the dimer interface (PDB entry 1SHL). Binding of inhibitor in the dimer interface displaces the side chain of Tyr223 and results in disorganized active site loops L2, L3 and L4. L2′ occupies the central cavity. For panels A and B, the secondary structure and active site loops are colored the same as those in Fig. 5, and side chains are colored using the cpk color mode. Structures were generated using Pymol (Delano Scientific LLC, Palo Alto, CA).
MOLECULES THAT SPECIFICALLY BIND TO CASPASES
A particularly attractive area of cancer therapy focuses on anti-apoptotic proteins that bind to caspases and suppress caspase activity [65]. For example, c-FLIP (cellular FLICE-like inhibitory protein) binds to caspases–8 or –10, typically suppressing the extrinsic apoptotic pathway (see Fig. 4). One drug that targets FLIP currently is in clinical trials [65]. In addition, TUCAN/CARD8 (tumor-upregulated CARD-containing agonist of caspase-9/caspase recruitment domain-containing protein 8) is overexpressed in some tumor cells and has been shown to bind to procaspase–9, preventing its activation by the apoptosome [86]. Using siRNA-mediated suppression of TUCAN expression in tumor cells, Yamamoto and coworkers showed that the overexpression of TUCAN is likely to be associated with chemoresistance of the tumor cells [87].
IAPs
Structural studies of several IAPs reveal a conserved motif that is found to repeat one to three times, depending on the IAP that is responsible for caspase inhibition [88–91]. The repeat domains, termed BIR (baculovirus IAP repeat), are zinc-binding proteins that are approximately eighty amino acids in length [77]. Most IAPs contain more than one BIR domain, although each domain in the protein has a different inhibitory function [77]. For example, XIAP is a 57 kDa protein that binds to and inhibits caspases–3, –7 and –9 and contains three BIR domains (BIR 1–3). Structural and biochemical studies have determined that the BIR-2 domain and the linker immediately N-terminal to BIR-2 are responsible for inhibiting caspases–3 and –7 [77, 92]. The linker region binds to the catalytic domain of caspase–3 and occludes the active site, thereby denying entry to incoming substrates (Fig. 5). The inhibition of effector caspases by the amino terminal BIR-2 linker is weak, however, and requires the bulkiness of the BIR-2 domain to promote strong inhibition. These findings, coupled with the crystal structure of the XIAP-BIR-2 domain bound to caspase–3 [92], suggest that two binding events occur to inhibit effector caspases by XIAP, the binding of the linker along with the binding of the BIR-2 domain [77]. Structural studies show that the active site loops of caspase–3 (Fig. 5A) are similar with a tetrapeptide inhibitor bound as with BIR-2 bound, with the exception of active site loop 2′ (L2′) (Fig. 5B and 5C). L2′ is sequestered by the C-terminal region of BIR-2, which may be important for caspase inhibition because Asp179 (or Asp181) of L2′ hydrogen bonds with the P4 site of the substrate. An analysis of caspase-3 structures shows that interactions between L2′ and P4 may occur directly (Asp181) or through a water molecule (Asp179) (Fig. 5D), indicating that the L2′ region of the protein is flexible. The structural insights suggest that one could create a novel inhibitor that interacts with the L2′-binding region of BIR-2, thereby competing for the binding of caspases–3 or –7. Indeed, McClendon and coworkers recently showed that peptide mimics are effective at binding to a similar region of BIR-3 [91].
The BIR-3 domain of XIAP binds to and inhibits caspase–9 in a manner different from the interactions of BIR-2 and caspase–3. The crystal structure of caspase–9 bound to the BIR-3 domain shows that the interactions between the two molecules occur in the dimer interface of caspase–9 [88] (Fig. 6A). The binding of BIR-3 to the interface effectively prevents the procaspase from forming a homodimer, which, in turn, prevents formation of the active site. Indeed, two of the five active site loops (L2 and L3) are either unstructured or misaligned in the caspase–9 monomer with BIR-3 bound (compare Fig. 5A and 6A). The structure also reveals a two-site mechanism with which BIR-3 inhibits caspase–9. In addition to the interface region, BIR-3 also interacts with the N-terminus of the small subunit. This region is important for active site formation because it interacts with the second active site in the dimeric structure, becoming active site loop 2′ (L2′, see Fig. 5A, for example). Without the interactions of the so-called loop bundle (L2, L4, and L2′), the active site cannot form properly [93]. As a result, BIR-3 locks caspase–9 into the inactive monomeric form with a misaligned active site [88].
CASPASE DIMER INTERFACE
A primary goal in therapeutic strategies for the treatment of cancer is the restoration of apoptosis in tumor cells. Recent efforts have focused on reestablishing the extrinsic or intrinsic apoptotic pathways, or both, by inhibiting antiapoptotic molecules, such as IAPs or c-FLIP, or by elevating levels of proapoptotic factors, such as Bcl-2 antagonists. In addition, a number of signal transduction proteins involved in apoptosis, such as p53 [94] and c-AKT [95], are attractive targets for inducing chemosensitivity by activating the apoptotic pathways indirectly. Current research strategies target multiple pathways to maximize the cytotoxic effects on tumor cells. For example, cocktails of doxorubicin, bleomycin, vinblastine, and dacarbazine (called ABVD) and of bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine and prednisone (BEACOPP), as well as others, have been shown to be effective against Hodgkin’s lymphoma [96, 97].
Interestingly, there have been no clinical drugs formulated that specifically target procaspase–3 in cancer therapy. The intent of such studies would be to activate the executioner caspase directly rather than relying on the aforementioned, more indirect, strategies. This is an important consideration because the levels of procaspase–3 are elevated in a number of cancer cell lines [61], but there are no deleterious effects on the cancer cells since the procaspase has little or no enzymatic activity. A number of inhibitors have been produced that bind to the active site of caspase–3 [98–100], and two molecules, described below, bind to the dimer interface and inhibit enzymatic activity [101]. However, as a general strategy for cancer therapy, the goal of drug development would be to identify molecules that bind to the procaspase–3 zymogen and that result in caspase–3 activation. This goal may be accomplished by identifying molecules that bind to the dimer interface. Recent biochemical and structural studies have shown that active site formation is coupled to dimerization via interactions between amino acids in the active site and those in the dimer interface, in a manner that is not fully understood [101, 102]. Two examples are described below that illustrate the communication between the dimer interface and the active site, in which molecules bound to the dimer interface prevent active site formation. In addition, two examples are described which demonstrate that targeting the dimer interface of procaspase–3 may be a viable strategy when considering new targets for cancer drug development. Included in this description are recent studies of a small molecule that binds to and activates procaspase–3, although at present the binding site on the protein has not been established.
A Brief Description of the Caspase Dimer Interface and Active Site Formation
Structural studies of caspase–1, –7, and –9 provide clues to conformational changes that occur during maturation [103–107]. In general, the active site is misaligned in the zymogen, and cleavage of the intersubunit linker (see Fig. 3) allows for realignment of the active site loops. While the mechanistic details differ for caspases–7 and –1, both proteins follow the general scheme that the chain cleavage results in proper formation of the substrate binding pocket as well as formation of the loop bundle among L2, L4, and L2′ (Fig. 5A, for example). Formation of the loop bundle stabilizes the active site configuration and moves the catalytic cysteine into the S1 subsite where it is positioned to attack the substrate at the P1 site.
One should note that a numbering system was established by Fuentes-Prior and Salvesen using caspase-1 as a standard to which all other caspases are compared [108]. However, it is useful on occasion to compare the amino acid number for each caspase directly since the conversion from the caspase-1 numbering system is not always straightforward. Without intending to establish a new numbering system, we provide a direct comparison of the amino acids in each caspase (Fig. 2), and these values are used in (Figs. 5–8) and in the description below.
Fig. 8.
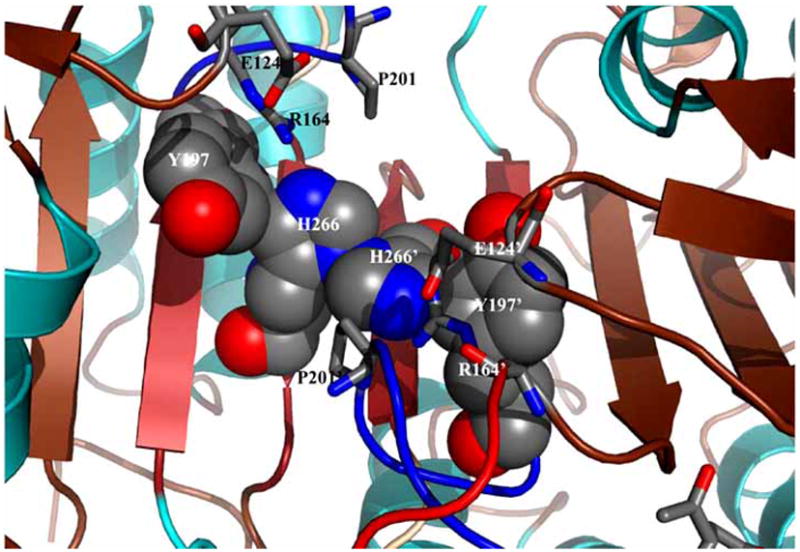
Model for inactivation of caspase–3 by the Val266His mutation. The space filling model, generated by replacing Val266 with His in silico, shows steric clashes between His266 and Tyr197 of one monomer and the same two residues of the second monomer. The overall effect of the Val266His mutant may result in a disorganized active site since it is predicted that the side-chain of Tyr197 cannot move toward the dimer interface and allow the substrate binding loop (L3) to insert properly. The secondary structure and active site loops are colored the same as those in Figs. 5–7, and side chains are colored using the cpk color mode. The structure was generated using Pymol (Delano Scientific LLC, Palo Alto, CA).
Maturation of caspase–3 is thought to be analogous to that of caspase–7. Prior to cleavage of the polypeptide chain in the intersubunit linker, one of the two linkers of the homodimer occupies the central cavity (between β strands 8 and 8′), whereas the other is positioned outside of the cavity. This effectively sequesters active site loop 2′ (L2′) away from the active site since it is part of the intersubunit linker in the zymogen. Following cleavage at Asp175, residues in active site loop 3 (L3) move into the central cavity and toward the dimer interface, resulting in formation of the substrate binding groove (Fig. 5A). In the procaspase, L3 occupies the substrate binding pocket because the loop is extended away from the protein. Upon cleavage of the intersubunit linker, the newly formed N-terminus of the small subunit is called L2′, and the newly formed C-terminus of the large subunit is called L2. L2′ leaves the central cavity and rotates approximately 180° to form new contacts in the loop bundle with L2, L3, and L4 from the opposing monomer (Fig. 5A).
Once L2′ rotates out of the central cavity, the N-terminal region of L3 can insert into the cavity, and a functional active site forms. More specifically, the arginine positioned next to the catalytic cysteine (Arg164 in caspase–3, see Fig. 2) rotates from a solvent exposed orientation in the substrate binding pocket toward the dimer interface and intercalates between Tyr197, on β-strand 7, and Pro201, on loop L3, to form an ordered lining to the central cavity (Fig. 7A). The reorientation of Arg164 on L2 toward the dimer interface and the accompanying loop movements lock the S1–S4 subsites into place and position the backbone amide of the catalytic cysteine into the correct orientation to form part of the oxyanion hole [103, 104]. Similar structural rearrangements are observed for caspase–1 as well [106] except that Arg286 (equivalent to Arg164 in caspase–3, see Fig. 2) forms a salt-bridge with Glu390 in the dimer interface (Fig. 7B).
Fig. 7.

Active site rearrangements of caspases–1, –3, and –9. (A) Caspase–3 (PDB entry 2J30): movements of Arg164 upon substrate binding result in intercalation of the side chain between Tyr197 and Pro201, from active site loop 3. The positive charge of Arg164 is neutralized by interactions with Glu124, which is situated on a loop above the dimer interface. (B) Caspase–1 (PDB entry 1SC3): movements of Arg286 upon substrate binding result in intercalation of the side chain between Cys331 and Pro335, from active site loop 3, forming a new salt bridge with Glu390 from the dimer interface. The red sphere indicates a water molecule between Glu390 and Glu390′. (C) Caspase–9 (PDB entry 1JXQ): upon inhibitor binding to one active site, loop rearrangements result in movement of the Tyr345 side chain, on β-strand 7, away from the active site and toward the protein interior, causing the side chain of Phe404 to move toward the dimer interface. Due to steric constraints in the interface, these movements can occur only in one heterodimer, so the second active site remains disorganized. (D) Interactions among amino acids in the elbow loop (Phe348-Phe351) and the second monomer (Phe246′, Pro338′, and Phe406′). For all panels, the secondary structure and active site loops are colored the same as those in Figs. 5 and 6, and side chains are colored using the cpk color mode. Structures were generated using Pymol (Delano Scientific LLC, Palo Alto, CA).
The effector caspases, such as caspases–3 and –7, do not have an equivalent salt bridge at the dimer interface, but the internalization of the arginine does occur. Like caspase–1, the comparable arginine in caspases–3 (Arg164) and –7 (Arg187), intercalates between Tyr197 (β-strand 7) and Pro201 (L3) (caspase–3 numbering, see Fig. 2) to stabilize active site loops 2 and 3. However, the positive charge on Arg164 is neutralized by Glu124, which is positioned on a loop just above the interface (Fig. 7A). This loop also contains the second residue involved in the catalytic dyad, His121, as well as Gly122, which forms part of the oxyanion hole. Overall, the conformational changes in active site loop 2 (L2) stabilize L2 and L3 and position L2, L2′, and L4 to form the loop bundle (Fig. 5A).
The formation of the active site of caspase–9 is unique from all other caspases because only one active site has the correct conformation to bind substrate. The other active site is not able to form due to steric constraints of residues in the dimer interface. The two active sites are in contact with one another through two tyrosine residues and two phenylalanine residues in the dimer interface. Phe404, on β-strand 8, interacts with Tyr345, on β-strand 7, as well as Phe404′ of the second monomer (Fig. 7C). Upon formation of one active site, Tyr345 rotates away from the active site and toward the dimer interface, causing Phe404 also to rotate toward the dimer interface. This movement creates a steric clash between the two phenylalanine residues across the dimer interface (Phe404 and Phe404′), leading to the inactivation of one of the active sites [107]. In order for the functional active site to form, the side chains of Ser344–Ser353, the so-called “elbow loop,” from one monomer, contact residues in the dimer interface and insert into the opposing monomer. Specifically, Phe348 and Phe351 from the elbow loop make contacts with a hydrophobic pocket lined with Pro338′ and Phe406′ in the opposing monomer (Fig. 7D). By stabilizing the elbow loop, the contacts enable L3 to move into the substrate binding conformation. Overall, the structural data for caspase–9 demonstrate that packing forces in the dimer interface affect active site formation, primarily through interactions between Tyr345 and Phe404 in each monomer and the equivalent interactions across the dimer interface.
Dimer Interface Experiments that Suppress Caspase–3 Activity
FICA and DICA
Disulfide trapping (called “tethering”) experiments were performed on caspases–3, –7 and –1 to identify allosteric sites that potentially could be used to inhibit dimer formation [101, 109]. Tethering uses a library of small thiol-containing compounds that make disulfide bonds with naturally occurring cysteine residues in a protein. The modified cysteine generally is close to a small molecule binding site. Two classes of compounds were identified after screening thousands of molecules for binding to caspases–3 and –7. Representatives of these classes are FICA (5-fluoro-1H-indole-2-carboxylic acid (2-mercapto-ethyl) amide) and DICA (2-(2,4-dichlorophenoxy-N-(2-mercapto-ethyl)-acetamide). Both compounds bound to a single cysteine in the small subunit on β-strand 8, Cys264 in caspase–3, which sits close to the dimer interface and is 14 Å from the catalytic cysteine. Binding of the inhibitors did not cause dissociation of the dimer but did prevent binding of the substrate at the active site. Structural data showed that, in caspase–7, FICA interacts with Tyr223′ (β-strand 7) on the opposing monomer to which it binds and occupies the central cavity (Fig. 6B), whereas DICA interacts with Tyr223 in the same monomer to which it binds. Regardless, the same conformational changes occur at the active site in the presence of either FICA or DICA. By displacing Tyr223, Arg187, on L2, is pushed out of the central cavity and placed into a position that occludes substrate binding to the active site. In fact, the large conformational changes that occur as a result of inhibitor binding to the interface resemble the conformation of the zymogen rather than that of the active enzyme.
In the case of caspase–1, it was found that a thienopyrazole bound to Cys331, which is near the dimer interface on β-strand 7 and is in the same position as Tyr197 of caspase–3 or Tyr223 of caspase–7 (see Fig. 2) [109]. Like FICA in caspase–7, the caspase–1 inhibitor interacted with residues on the neighboring monomer. Overall, the binding of the tethered inhibitor resulted in a disorganized active site by causing the catalytic cysteine to be rotated away from the S1 subsite, the substrate-binding loop to be collapsed so that it could not interact productively with substrate, and the side chain of Arg286 to be rotated away from the dimer interface and toward the substrate-binding cleft. As with caspase–7, caspase–1 adopted a conformation closely resembling its ligand-free form when bound to the allosteric inhibitor. Current models suggest that binding of the inhibitor to the dimer interface shifts the equilibrium of species found in solution from the active form to the inactive form, underlining the coupling of the dimer interface to the active site.
Caspase-3 Interface Mutant Val266His
In an effort to examine amino acids in the dimer interface and their communication with active site residues, the central residue of the dimer interface of caspase–3, Val266 (Fig. 7A), was replaced with histidine or glutamate. While the Val266Glu mutant is described below, replacing Val266 with histidine abolished activity in both the zymogen and the mature caspase [102]. Limited proteolysis studies demonstrated that active site loop 3 (L3) was hyperexposed in the mutant at a site over 20 Å from the site of the mutation, demonstrating that L3 was not properly inserted into the substrate binding pocket in the mutant. Although the crystal structure of the mutant has not been determined, the Val266His mutation is thought to present similar packing problems as those found in caspase–9 (Fig. 8). In this model, steric constraints in the dimer interface prevent the side chain of Tyr197 from moving away from the active site, effectively preventing L3 from forming the substrate binding pocket.
Experiments that Activate the Caspase-3 Zymogen
In addition to replacing the central valine of caspase–3 with histidine, the residue also was replaced with glutamate. Remarkably, the Val266Glu mutation resulted in a pseudo-activation of the caspase–3 zymogen [102]. The enzymatic activity of the zymogen increased to a level comparable to that of the wild-type, mature, caspase–3, while cleavage of the polypeptide chain was not required for the activity increase. Overall, data from limited proteolysis studies indicate that the Val266Glu mutation in the dimer interface affects the intersubunit linker in a manner that allows the loop bundle to form between L2, L4 and L2′, independent of cleavage of the protein. Together, the dimer interface mutations of caspase–3 are important because they demonstrate that one can influence the activity of the zymogen, as well as that of the mature enzyme, by disrupting the interactions between amino acids in the interface and those in the active site. FICA and DICA, described above, likely mimic the packing effects of the Val266His mutation to abrogate caspase–3 activity. In contrast, molecules that mimic the effects of the Val266Glu mutation have yet to be identified, but the results show that the caspase–3 dimer interface should be considered a valid target when formulating drugs to activate the caspase–3 zymogen.
Recently, Hergenrother and colleagues screened over twenty thousand compounds for their ability to catalyze the activation of procaspase–3 to mature caspase–3 [61]. Four compounds were identified that resulted in procaspase–3 activation, and one compound demonstrated a dose-dependent effect. The compound is called PAC-1, for first procaspase-activating compound. Importantly, PAC-1 was able to induce apoptosis in a variety of cancer cell lines, and the data showed a strong correlation between the cellular concentration of procaspase–3 and PAC-1 sensitivity. Finally, tumor growth was retarded significantly in mice treated with PAC-1. Though these results are potentially very exciting and demonstrate that compounds that function to activate procaspase–3 directly can be effective in cancers, more studies need to be done to determine the mechanism of action.
CONCLUSIONS
Caspases are proteases that are intimately involved in the execution of apoptosis in normal cells. Cancer cells typically have gained the ability to circumvent apoptosis, so the proteins involved in the apoptotic cascades have become ideal targets for cancer therapy. Targeting the upstream proteins in these pathways can lead to resistance in cancer cells, thereby rendering cytotoxic drugs ineffective. On the other hand, directly targeting effector caspases, such as caspase-3, could lead to reduced resistance, and thus more effective therapy, since effector caspases ultimately are responsible for cellular suicide upon their activation. Because dimerization is necessary for proper active site formation of all caspases, the dimer interface should be considered a potential target for cancer therapy, where the identification of small molecules that bind to the dimer interfaces of procaspases, resulting in their activation, should be at the forefront of cancer research.
Acknowledgments
This work was supported by a grant from the National Institutes of Health (GM065970).
References
- 1.Wyllie AH, Kerr JF, Currie AR. Cell Death: The Significance of Apoptosis. Int Rev Cytol. 1980;68:251–306. doi: 10.1016/s0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]
- 2.Majno G, Joris I. Apoptosis, Oncosis and Necrosis. An Overview of Cell Death. Am J Pathol. 1995;146:3–15. [PMC free article] [PubMed] [Google Scholar]
- 3.Sanders EJ, Wride MA. Programmed Cell Death in Development. Int Rev Cytol. 1995;163:105–173. doi: 10.1016/s0074-7696(08)62210-x. [DOI] [PubMed] [Google Scholar]
- 4.Beaulaton J, Lockshin R. The Relation of Programmed Cell Death to Development and Reproduction: Comparative Studies and an Attempt at Classification. Int Rev Cytol. 1982;79:215–235. doi: 10.1016/s0074-7696(08)61675-7. [DOI] [PubMed] [Google Scholar]
- 5.Schweichel J, Merker H. The Morphology of Various Types of Cell Death in Prenatal Tissues. Teratology. 1973;7:253–266. doi: 10.1002/tera.1420070306. [DOI] [PubMed] [Google Scholar]
- 6.Zakeri Z, Bursch W, Tenniswood M, Lockshin R. Cell Death: Programmed Apoptosis, Necrosis or Other? Cell Death Differ. 1995;2:83–92. [PubMed] [Google Scholar]
- 7.Hinchliffe J. In: Cell Death in Biology and Pathology. Bowen I, Lockshin R, editors. Chapman and Hall; New York: 1981. pp. 35–78. [Google Scholar]
- 8.Kerr JFR, Wyllie AH, Currie AR. Apoptosis: A Basic Biological Phenomenon with Wide-Ranging Implications in Tissue Kinetics. Brit J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaufmann SH. Induction of Endonucleolytic DNA Cleavage in Human Acute Myelogenous Leukemia Cells by Etoposide, Camptothecin, and Other Cytotoxic Anticancer Drugs: A Cautionary Note. Cancer Res. 1989;49:5870–5878. [PubMed] [Google Scholar]
- 10.Canman CE, Tange H-Y, Normolle DP, Lawrence TS, Maybaum J. Variations in Patterns of DNA Damage Induced in Human Colorectal Tumor Cells by 5-Fluorodeoxyuridine: Implications for Mechanisms of Resistance and Cytotoxicity. Proc Natl Acad Sci USA. 1992;89:10474–10478. doi: 10.1073/pnas.89.21.10474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. Exposure of Phosphatidylserine on the Surface of Apoptotic Lymphocytes Triggers Specific Recognition and Removal by Macrophages. J Immunol. 1992;148:2207–2216. [PubMed] [Google Scholar]
- 12.Ashkenazi A, Dixit VM. Death Receptors: Signaling and Modulation. Science. 1998;281:1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- 13.Ashkenazi A, Dixit VM. Apoptosis Control by Death and Decoy Receptors. Curr Opin Cell Biol. 1999;11:255–260. doi: 10.1016/s0955-0674(99)80034-9. [DOI] [PubMed] [Google Scholar]
- 14.Walczak H, Krammer PH. The Cd95 (Apo-1/Fas) and the Trail (Apo-2l) Apoptosis Systems. Exp Cell Res. 2000;256:58–66. doi: 10.1006/excr.2000.4840. [DOI] [PubMed] [Google Scholar]
- 15.Muzio M, Stockwell BR, Stennicke H, Salvesen GS, Dixit VM. An Induced Proximity Model for Capase-8 Activation. J Biol Chem. 1998;273:2926–2930. doi: 10.1074/jbc.273.5.2926. [DOI] [PubMed] [Google Scholar]
- 16.Stennicke HR, Deveraux QL, Humke EW, Reed JC, Dixit VM, Salvesen GS. Caspase-9 Can Be Activated without Proteolytic Processing. J Biol Chem. 1999;274:8359–8362. doi: 10.1074/jbc.274.13.8359. [DOI] [PubMed] [Google Scholar]
- 17.Zou H, Li Y, Liu X, Wang X. An Apaf-1 Cytochrome C Multimeric Complex Is a Functional Apoptosome That Activates Procaspase-9. J Biol Chem. 1999;274:11549–11556. doi: 10.1074/jbc.274.17.11549. [DOI] [PubMed] [Google Scholar]
- 18.Medema JP, Scaffidi C, Kischkel FC, Shevchenko A, Mann M, Krammer PH, Peter ME. Flice Is Activated by Association with the Cd95 Death-Inducing Signaling Complex (Disc) EMBO J. 1997;16:2794–2804. doi: 10.1093/emboj/16.10.2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Srinivasula SM, Hegde R, Saleh A, Datta P, Shiozaki E, Chai J, Lee R-A, Robbins PD, Fernandes-Alnemri T, Shi Y, Alnemri ES. A Conserved Xiap-Interaction Motif in Caspase-9 and Smac/Diablo Regulates Caspase Activity and Apoptosis. Nature. 2001;410:112–116. doi: 10.1038/35065125. [DOI] [PubMed] [Google Scholar]
- 20.Pop C, Chen Y-R, Smith B, Bose K, Bobay B, Tripathy A, Franzen S, Clark AC. Removal of the Pro-Domain Does Not Affect the Conformation of the Procaspase-3 Dimer. Biochemistry. 2001;40:14224–14235. doi: 10.1021/bi011037e. [DOI] [PubMed] [Google Scholar]
- 21.Bose K, Pop C, Feeney B, Clark AC. An Uncleavable Procaspase-3 Mutant Has a Lower Catalytic Efficiency but an Active Site Similar to That of Mature Caspase-3. Biochemistry. 2003;42:12298–12310. doi: 10.1021/bi034998x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lowe SW, Lin AW. Apoptosis in Cancer. Carcinogenesis. 2000;21:485–495. doi: 10.1093/carcin/21.3.485. [DOI] [PubMed] [Google Scholar]
- 23.van Loo G, Saelens X, van Gurp M, MacFarlane M, Martin S, Vandenabeele P. The Role of Mitochondrial Factors in Apoptosis: A Russian Roulette with More Than One Bullet. Cell Death Differ. 2002;9:1031–1042. doi: 10.1038/sj.cdd.4401088. [DOI] [PubMed] [Google Scholar]
- 24.Guo Y, Srinivasula SM, Druilhe A, Fernandes-Alnemri T, Alnemri ES. Caspase-2 Induces Apoptosis by Releasing Proapoptotic Proteins from Mitochondria. J Biol Chem. 2002;277:13430–13437. doi: 10.1074/jbc.M108029200. [DOI] [PubMed] [Google Scholar]
- 25.Roy S, Nicholson DW. Cross-Talk in Cell Death Signaling. J Exp Med. 2000;192:21–26. [PubMed] [Google Scholar]
- 26.Duan H, Dixit VM. Raidd Is a New ‘Death’ Adaptor Molecule. Nature. 1997;385:86–89. doi: 10.1038/385086a0. [DOI] [PubMed] [Google Scholar]
- 27.Baliga BC, Read SH, Kumar S. The Biochemical Mechanism of Caspase-2 Activation. Cell Death Differ. 2004;11:1234–1241. doi: 10.1038/sj.cdd.4401492. [DOI] [PubMed] [Google Scholar]
- 28.Fulda S, Debatin K-M. Extrinsic Versus Intrinsic Apoptosis Pathways in Anticancer Chemotherapy. Oncogene. 2006;25:4798–4811. doi: 10.1038/sj.onc.1209608. [DOI] [PubMed] [Google Scholar]
- 29.Du C, Fang M, Li Y, Li L, Wang X. Smac, a Mitochondrial Protein That Promotes Cytochrome C-Dependent Caspase Activation by Eliminating Iap Inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 30.Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, Moritz RL, Simpson RJ, Vaux DL. Identification of Diablo, a Mammalian Protein That Promotes Apoptosis by Binding to and Antagonizing Iap Proteins. Cell. 2000;102:43–53. doi: 10.1016/s0092-8674(00)00009-x. [DOI] [PubMed] [Google Scholar]
- 31.Zangemeister-Wittke U, Simon H-U. An Iap in Action: The Multiple Roles of Survivin in Differentiation, Immunity and Malignancy. Cell Cycle. 2004;3:1121–1123. [PubMed] [Google Scholar]
- 32.Nagata S. Apoptotic DNA Fragmentation. Exp Cell Res. 2000;256:12–18. doi: 10.1006/excr.2000.4834. [DOI] [PubMed] [Google Scholar]
- 33.Li LY, Luo X, Wang X. Endonuclease G Is an Apoptotic Dnase When Released from Mitochondria. Nature. 2001;412:95–99. doi: 10.1038/35083620. [DOI] [PubMed] [Google Scholar]
- 34.Klein JA, Longo-Guess CM, Rossman MP, Seburn KL, Hurd RE, Frankel WN, Bronson RT, Ackerman SL. The Harlequin Mouse Mutation Down-Regulates Apoptosis-Inducing Factor. Nature. 2002;419:367–374. doi: 10.1038/nature01034. [DOI] [PubMed] [Google Scholar]
- 35.Hansen TM, Nagley P. Aif: A Multifunctional Cog in the Life and Death Machine. Science STKE. 2003;193:pe31. doi: 10.1126/stke.2003.193.pe31. [DOI] [PubMed] [Google Scholar]
- 36.Debatin K-M. In: Drug Resistance Updates. Broxterman H, editor. Churchill Livingstone; Edinburgh: 1999. pp. 85–90. [Google Scholar]
- 37.Debatin K-M. Anticancer Drugs, Programmed Cell Death and the Immune System: Defining New Roles in an Old Play. J Natl Cancer Inst. 1997;89:750–753. doi: 10.1093/jnci/89.11.750. [DOI] [PubMed] [Google Scholar]
- 38.Herr I, Debatin K-M. Cellular Stress Response and Apoptosis in Cancer Therapy. Blood. 2001;98:2603–2614. doi: 10.1182/blood.v98.9.2603. [DOI] [PubMed] [Google Scholar]
- 39.Kaufmann SH, Earnshaw WC. Induction of Apoptosis by Cancer Chemotherapy. Exp Cell Res. 2000;256:42–49. doi: 10.1006/excr.2000.4838. [DOI] [PubMed] [Google Scholar]
- 40.Solary E, Droin N, Bettaieb A, Corcos L, Dimanche-Boitrel M, Garrido C. Positive and Negative Regulation of Apoptotic Pathways by Cytotoxic Agents in Hematological Malignancies. Leukemia. 2000;14:1833–1849. doi: 10.1038/sj.leu.2401902. [DOI] [PubMed] [Google Scholar]
- 41.Debatin K-M. Apoptosis Pathways in Cancer and Cancer Therapy. Cancer Immunol Immun. 2004;53:153–159. doi: 10.1007/s00262-003-0474-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krammer PH. Cd95’s Deadly Mission in the Immune System. Nature. 2000;407:789–795. doi: 10.1038/35037728. [DOI] [PubMed] [Google Scholar]
- 43.Friesen C, Herr I, Krammer PH, Debatin K-M. Involvement of the Cd95 (Apo-1/Fas) Receptor/Ligand System in Drug-Induced Apoptosis in Leukemia Cells. Nat Med. 1996;2:574–577. doi: 10.1038/nm0596-574. [DOI] [PubMed] [Google Scholar]
- 44.Muller M, Strand S, Hug H, Heinemann E-M, Walczak H, Hofmann WJ, Stremmel W, Krammer PH, Galle PR. Drug-Induced Apoptosis in Hepatoma Cells Is Mediated by the Cd95 (Apo-1/Fas) Receptor/Ligand System and Involves Activation of Wild-Type P53. J Clin Invest. 1997;99:403–413. doi: 10.1172/JCI119174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Muller M, Wilder S, Bannasch D, Israeli D, Lehlbach K, Li-Weber M, Friedman SL, Galle PR, Stremmel W, Oren M, Krammer PH. P53 Activates the Cd95 (Apo-1/Fas) Gene in Response to DNA Damage by Anticancer Drugs. J Exp Med. 1998;188:2033–2045. doi: 10.1084/jem.188.11.2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ashkenazi A, Pai RC, Fong S, Leung S, Lawrence DA, Marsters SA, Blackie C, Chang L, McMurtrey AE, Hebert A, DeForge L, Koumenis IL, Lewis D, Harris L, Bussiere J, Koeppen H, Shahrokh Z, Schwall RH. Safety and Antitumor Activity of Recombinant Soluable Apo2 Ligand. J Clin Invest. 1999;104:155–162. doi: 10.1172/JCI6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lawrence DA, Shahrokh Z, Marsters SA, Achilles K, Shih D, Mounho B, Hillan K, Totpal K, DeForge L, Schow P, Hooley J, Sherwood S, Pai RC, Leung S, Khan L, Gliniak B, Bussiere J, Smith CA, Strom SS, Kelley S, Fox JA, Thomas D, Ashkenazi A. Differential Hepatocyte Toxicity of Recombinant Apo2l/Trail Versions. Nat Med. 2001;7:383–385. doi: 10.1038/86397. [DOI] [PubMed] [Google Scholar]
- 48.LeBlanc HN, Ashkenazi A. Apo-2l/Trail and Its Death and Decoy Receptors. Cell Death Differ. 2003;10:66–75. doi: 10.1038/sj.cdd.4401187. [DOI] [PubMed] [Google Scholar]
- 49.Jo M, Kim T-H, Seol D-W, Esplen JE, Dorko K, Billiar TR, Strom SC. Apoptosis Induced in Normal Human Hepatocytes by Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand. Nat Med. 2000;6:564–567. doi: 10.1038/75045. [DOI] [PubMed] [Google Scholar]
- 50.Nitsch R, Bechmann I, Deisz RA, Haas D, Lehmann T-N, Wendling U, Zipp F. Human Brain-Cell Death Induced by Tumour-Necrosis-Factor-Related Apoptosis-Inducing Ligand (Trail) Lancet. 2000;356:827–828. doi: 10.1016/S0140-6736(00)02659-3. [DOI] [PubMed] [Google Scholar]
- 51.Ehrhardt H, Fulda S, Schmid I, Hiscott J, Debatin K-M, Jeremias I. Trail Induced Survival and Proliferation in Cancer Cells Resistant Towards Trail-Induced Apoptosis Mediated by Nf-Kb. Oncogene. 2003;22:3842–3852. doi: 10.1038/sj.onc.1206520. [DOI] [PubMed] [Google Scholar]
- 52.Karin M, Cao Y, Greten FR, Li Z-W. Nf-Kb in Cancer: From Innocent Bystander to Major Culprit. Nat Rev Cancer. 2002;2:301–310. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- 53.Sheikh MS, Huang Y, Fernandez-Salas EA, El-Deiry W, Friess H, Amundson S, Yin J, Meltzer SJ, Holbrook NJ, Fornace AJJ. The Antiapoptotic Decoy Receptor Trid/Trail-R3 Is a P53-Regulated DNA Damage Inducible Gene That Is Overexpressed in Primary Tumors of the Gastrointestinal Tract. Oncogene. 1999;18:4153–4159. doi: 10.1038/sj.onc.1202763. [DOI] [PubMed] [Google Scholar]
- 54.Baylin SB. Mechanisms Underlying Epigenetically Mediated Gene Silencing in Cancer. Semin Cancer Biol. 2002;12:331–337. doi: 10.1016/s1044-579x(02)00053-6. [DOI] [PubMed] [Google Scholar]
- 55.van Noesel MM, van Bezouw S, Voute P, Herman JG, Pieters R, Verstee R. Clustering of Hypermethylated Genes in Neuroblastoma. Genes, Chromosomes Canc. 2003;38:226–233. doi: 10.1002/gcc.10278. [DOI] [PubMed] [Google Scholar]
- 56.Petak I, Danam R, Tillman D, Vernes R, Howell S, Berczi L, Kopper L, Brent T, Houghton J. Hypermethylation of the Gene Promoter and Enhancer Region Can Regulate Fas Expression and Sensitivity in Colon Carcinoma. Cell Death Differ. 2003;10:211–217. doi: 10.1038/sj.cdd.4401132. [DOI] [PubMed] [Google Scholar]
- 57.Marks PA, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone Deacetylases and Cancer: Causes and Therapies. Nat Rev Cancer. 2001;1:194–202. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- 58.Maecker HL, Yun Z, Maecker HT, Giaccia AJ. Epigenetic Changes in Tumor Fas Levels Determine Immune Escape and Response to Therrapy. Cancer Cell. 2002;2:139–148. doi: 10.1016/s1535-6108(02)00095-8. [DOI] [PubMed] [Google Scholar]
- 59.Janicke RU, Sprengart ML, Wati MR, Porter AG. Caspase-3 Is Required for DNA Fragmentation and Morphological Changes Associated with Apoptosis. J Biol Chem. 1998;273:9357–9360. doi: 10.1074/jbc.273.16.9357. [DOI] [PubMed] [Google Scholar]
- 60.Yang X-H, Sladek TL, Liu X, Butler BR, Froelich CJ, Thor AD. Reconsititution of Caspase 3 Sensitizes Mcf-7 Breast Cancer Cells to Doxorubicin- and Etoposide-Induced Apoptosis. Cancer Res. 2001;61:348–354. [PubMed] [Google Scholar]
- 61.Putt KS, Chen GW, Pearson JM, Sandhorst JS, Hoagland MS, Kwon J-T, Hwang S-K, Jin H, Churchwell MI, Cho H-H, Doerge DR, Helferich WG, Hergenrother PJ. Small-Molecule Activation of Procaspase-3 to Caspase-3 as a Personalized Anticancer Strategy. Nat Chem Biol. 2006;2:543–550. doi: 10.1038/nchembio814. [DOI] [PubMed] [Google Scholar]
- 62.Teitz T, Wei T, Valentine MB, Vanin EF, Grenet J, Valentine VA, Behm FG, Look AT, Lahti JM, Kidd VJ. Caspase 8 Is Deleted or Silenced Preferentially in Childhood Neuroblastomas with Amplification of Mycn. Nat Med. 2000;6:529–535. doi: 10.1038/75007. [DOI] [PubMed] [Google Scholar]
- 63.Fulda S, Kufer MU, Meyer E, van Valen F, Dockhorn-Dworniczak B, Debatin K-M. Sensitization for Death Receptor- or Drug-Induced Apoptosis by Re-Expression of Caspase-8 through Demethylation or Gene Transfer. Oncogene. 2001;20:5865–5877. doi: 10.1038/sj.onc.1204750. [DOI] [PubMed] [Google Scholar]
- 64.Adams J, Cory S. The Bcl-2 Apoptotic Switch in Cancer Development and Therapy. Oncogene. 2007;26:1324–1337. doi: 10.1038/sj.onc.1210220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reed JC. Proapoptotic Multidomain Bcl-2/Bax-Family Proteins: Mechanisms, Physiological Roles, and Therapeutic Opportunities. Cell Death Differ. 2006;18:1378–1386. doi: 10.1038/sj.cdd.4401975. [DOI] [PubMed] [Google Scholar]
- 66.Antonsson B, Martinou J-C. The Bcl-2 Protein Family. Exp Cell Res. 2000;256:50–57. doi: 10.1006/excr.2000.4839. [DOI] [PubMed] [Google Scholar]
- 67.Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, Tsujimoto Y. Role of Bcl-2 Family Proteins in a Non-Apoptotic Programmed Cell Death Dependent on Autophagy Genes. Nat Cell Biol. 2004;6:1221–1228. doi: 10.1038/ncb1192. [DOI] [PubMed] [Google Scholar]
- 68.Reed JC. Proapoptotic Multidomain Bcl-2/Bax-Family Proteins: Mechanisms, Physiological Roles, and Therapeutic Opportunities. Cell Death Differ. 2006;13:1378–1386. doi: 10.1038/sj.cdd.4401975. [DOI] [PubMed] [Google Scholar]
- 69.Kroemer G, Galluzzi L, Brenner C. Mitochondrial Membrane Permeabilization in Cell Death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- 70.Kroemer G, Reed JC. Mitochondrial Control of Cell Death. Nat Med. 2000;6:513–519. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- 71.Martinou JC, Green DR. Breaking the Mitochondrial Barrier. Nat Rev Mol Cell Biol. 2001;2:63–67. doi: 10.1038/35048069. [DOI] [PubMed] [Google Scholar]
- 72.Minn AJ, Rudin CM, Boise LH, Thompson CB. Expression of Bcl-Xl Can Confer a Multidrug Resistance Phenotype. Blood. 1995;86:1903–1910. [PubMed] [Google Scholar]
- 73.Deng Y, Lin Y, Wu X. Trail-Induced Apoptosis Requires Bax-Dependent Mitochondrial Release of Smac/Diablo. Gene Dev. 2002;16:33–45. doi: 10.1101/gad.949602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Deveraux QL, Reed JC. Iap Family Proteins-Suppressors of Apoptosis. Gene Dev. 1999;13:239–252. doi: 10.1101/gad.13.3.239. [DOI] [PubMed] [Google Scholar]
- 75.Salvesen GS, Duckett CS. Iap Proteins: Blocking the Road to Death’s Door. Nat Rev Mol Cell Biol. 2002;3:401–410. doi: 10.1038/nrm830. [DOI] [PubMed] [Google Scholar]
- 76.Eckelman BP, Salvesen GS, Scott FL. Human Inhibitor of Apoptosis Proteins: Why Xiap Is the Black Sheep of the Family. EMBO Rep. 2006;7:988–994. doi: 10.1038/sj.embor.7400795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schimmer A, Dalili S, Batey R, Riedl SJ. Targeting Xiap for the Treatment of Malignancy. Cell Death Differ. 2006;13:179–188. doi: 10.1038/sj.cdd.4401826. [DOI] [PubMed] [Google Scholar]
- 78.Altieri DC. Validating Survivin as a Cancer Therapeutic Target. Nat Rev Cancer. 2003;3:46–54. doi: 10.1038/nrc968. [DOI] [PubMed] [Google Scholar]
- 79.Williams NS, Gaynor RB, Scoggin S, Verma U, Gokaslan T, Simmang C, Fleming J, Tavana D, Frenkel E, Becerra C. Identification and Validation of Genes Involved in the Pathogenesis of Colorectal Cancer Using Cdna Microarrays and Rna Interference. Clin Cancer Res. 2003;9:931–946. [PubMed] [Google Scholar]
- 80.Altieri DC. Survivin, Versatile Modulation of Cell Division and Apoptosis in Cancer. Oncogene. 2003;22:8581–8589. doi: 10.1038/sj.onc.1207113. [DOI] [PubMed] [Google Scholar]
- 81.Blanc-Brude OP, Mesri M, Wall NR, Plescia J, Dohi T, Altieri DC. Therapeutic Targeting of Survivin Pathway in Cancer: Initiation of Mitochondrial Apoptosis and Suppression of Tumor-Associated Angiogenesis. Clin Cancer Res. 2003;9:2683–2692. [PubMed] [Google Scholar]
- 82.Grossman D, Kim PJ, Schechner JS, Altieri DC. Inhibition of Melanoma Tumor Growth in Vivo by Survivin Targeting. Proc Natl Acad Sci USA. 2001;98:635–640. doi: 10.1073/pnas.230450097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.O’Connor DS, Wall NR, Porter ACG, Altieri DC. A P34(Cdc2) Survival Checkpoint in Cancer. Cancer Cell. 2002;2:43–54. doi: 10.1016/s1535-6108(02)00084-3. [DOI] [PubMed] [Google Scholar]
- 84.Wall NR, O’Connor DS, Plescia J, Pommier Y, Altieri DC. Suppression of Survivin Phosphorylation on Thr34 by Flavopiridol Enhances Tumor Cell Apoptosis. Cancer Res. 2003;63:230–235. [PubMed] [Google Scholar]
- 85.Fulda S, Wick W, Weller M, Debatin K-M. Smac Agonist Sensitize for Apo2l/Trailor Anticancer Drug-Induced Apoptosis and Induce Regression of Malignant Glioma. in Vivo Nat Med. 2002;8:808–815. doi: 10.1038/nm735. [DOI] [PubMed] [Google Scholar]
- 86.Pathan N, Marusawa H, Krajewska M, Matsuzawa S-I, Kim H, Okada K, Torii S, Kitada S, Krejewski S, Welsh K, Pio F, Godzik A, Reed JC. Tucan, and Antiapoptotic Caspase-Associated Recruitment Domain Family Protein Overexpressed in Cancer. J Biol Chem. 2001;276:32220–32229. doi: 10.1074/jbc.M100433200. [DOI] [PubMed] [Google Scholar]
- 87.Yamamoto M, Torigoe T, Kamiguchi K, Hirohashi Y, Nakanishi K, Nabeta C, Asanuma H, Tsuruma T, Sato T, Hata F, Ohmura T, Yamaguchi K, Kurotaki T, Hirata K, Sato N. A Novel Isoform of Tucan Is Overexpressed in Human Cancer Tissues and Suppresses Both Caspase-8- and Caspase-9-Mediated Apoptosis. Cancer Res. 2005;65:8706–8714. doi: 10.1158/0008-5472.CAN-04-4649. [DOI] [PubMed] [Google Scholar]
- 88.Shiozaki EN, Chai J, Rigotti DJ, Riedl SJ, Li P, Srinivasula SM, Alnemri ES, Fairman R, Shi Y. Mechanism of Xiap-Mediated Inhibition of Caspase-9. Mol Cell. 2003;11:519–527. doi: 10.1016/s1097-2765(03)00054-6. [DOI] [PubMed] [Google Scholar]
- 89.Chai J, Shiozaki E, Srinivasula SM, Wu Q, Dataa P, Alnemri ES, Shi Y. Structural Basis of Caspase-7 Inhibition by Xiap. Cell. 2001;104:769–780. doi: 10.1016/s0092-8674(01)00272-0. [DOI] [PubMed] [Google Scholar]
- 90.Sun C, Nettesheim D, Liu Z, Olejniczak ET. Solution Structure of Human Survivin and Its Binding Interface with Smac/Diablo. Biochemistry. 2005;44:11–17. doi: 10.1021/bi0485171. [DOI] [PubMed] [Google Scholar]
- 91.Wist AD, Gu L, Reidl SJ, Shi Y, McLendon GL. Structure-Based Study of the Smac-Binding Pocket within the Bir3 Domain of Xiap. Bioorgan Med Chem. 2007;15:2935–2943. doi: 10.1016/j.bmc.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 92.Riedl SJ, Renatus M, Schwarzenbacher R, Zhou Q, Sun C, Fesik SW, Liddington RC, Salvesen GS. Structural Basis for the Inhibition of Caspase-3 by Xiap. Cell. 2001;104:791–800. doi: 10.1016/s0092-8674(01)00274-4. [DOI] [PubMed] [Google Scholar]
- 93.Feeney B, Pop C, Swartz P, Mattos C, Clark AC. Role of Loop Bundle Hydrogen Bonds in the Maturation and Activity of (Pro)Caspase-3. Biochemistry. 2006;45:13249–13263. doi: 10.1021/bi0611964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Vassilev LT. Small-Molecule Antagonists of P53-Mdm2 Binding. Cell Cycle. 2004;3:419–421. [PubMed] [Google Scholar]
- 95.Mitsiades CS, Mitsiades N, Koutsilieris M. The Akt Pathway: Molecular Targets for Anti-Cancer Drug Development. Curr Cancer Drug Targets. 2004;4:235–256. doi: 10.2174/1568009043333032. [DOI] [PubMed] [Google Scholar]
- 96.Diehl V, Stein H, Hummel M, Zollinger R, Connors JM. Hodgkin’s Lymphoma: Biology and Treatment Strategies for Primary, Refractory, and Relapsed Disease. Hematology. 2003:225–247. doi: 10.1182/asheducation-2003.1.225. [DOI] [PubMed] [Google Scholar]
- 97.Diehl V, Behringer K. Could Beacopp Be the New Standard for the Treatment of Advanced Hodgkin’s Lymphoma? Cancer Invest. 2006;24:461–465. doi: 10.1080/07357900600705789. [DOI] [PubMed] [Google Scholar]
- 98.Ganesan R, Jelakovic S, Campbell AJ, Li ZZ, Asgian JL, Powers JC, Grutter MG. Exploring the S4 and S1 Prime Subsite Specificities in Caspase-3 with Aza-Peptide Epoxide Inhibitors. Biochemistry. 2006;45:9059–9067. doi: 10.1021/bi060364p. [DOI] [PubMed] [Google Scholar]
- 99.Ekici OD, Gotz MG, James KE, Li ZZ, Rukamp BJ, Asgian JL, Caffrey CR, Hansell E, Dvorak J, McKerrow JH, Potempa J, Travis J, Mikolajczyk J, Salvesen GS, Powers JC. Aza-Peptide Michael Acceptors: A New Class of Inhibitors Specific for Caspases and Other Clan Cd Cysteine Proteases. J Med Chem. 2004;47:1889–1892. doi: 10.1021/jm049938j. [DOI] [PubMed] [Google Scholar]
- 100.James KE, Asgian JL, Li ZZ, Ekici OD, Rubin JR, Mikolajczyk J, Salvesen GS, Powers JC. Design, Synthesis and Evaluation of Aza-Peptide Epoxides as Selective and Potent Inhibitors of Caspases-1, -3, -6, and -8. J Med Chem. 2004;47:1553–1574. doi: 10.1021/jm0305016. [DOI] [PubMed] [Google Scholar]
- 101.Hardy JA, Lam J, Nguyen JT, O’Brien T, Wells JA. Discovery of an Allosteric Site in the Caspases. Proc Natl Acad Sci USA. 2004;101:12461–12466. doi: 10.1073/pnas.0404781101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pop C, Feeney B, Tripathy A, Clark AC. Mutations in the Procaspase-3 Dimer Interface Affect the Activity of the Zymogen. Biochemistry. 2003;42:12311–12320. doi: 10.1021/bi034999p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chai J, Wu Q, Shiozaki E, Srinivasula SM, Alnemri ES, Shi Y. Crystal Structure of a Procaspase-7 Zymogen: Mechanisms of Activation and Substrate Binding. Cell. 2001;107:399–407. doi: 10.1016/s0092-8674(01)00544-x. [DOI] [PubMed] [Google Scholar]
- 104.Riedl SJ, Fuentes-Prior P, Renatus M, Kairies N, Krapp S, Huber R, Salvesen GS, Bode W. Structural Basis for the Activation of Human Procaspase-7. Proc Natl Acad Sci USA. 2001;98:14790–14795. doi: 10.1073/pnas.221580098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wilson KP, Black J-AF, Thomson JA, Kim EE, Griffith JP, Navia MA, Murcko MA, Chambers SP, Aldape RA, Raybuck SA, Livingston DJ. Structure and Mechanism of Interleukin-1β Converting Enzyme. Nature. 1994;370:270–275. doi: 10.1038/370270a0. [DOI] [PubMed] [Google Scholar]
- 106.Romanowski MJ, Scheer JM, O’Brien T, McDowell RS. Crystal Structures of Ligand-Free and Malonate-Bound Human Caspase-1: Implications for the Mechanism of Substrate Binding. Structure. 2004;12:1361–1371. doi: 10.1016/j.str.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 107.Renatus M, Stennicke HR, Scott FL, Liddington RC, Salvesen GS. Dimer Formation Drives the Activation of the Cell Death Protease Caspase 9. Proc Natl Acad Sci USA. 2001;98:14250–14255. doi: 10.1073/pnas.231465798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fuentes-Prior P, Salvesen GS. The Protein Structures That Shape Caspase Activity, Specificity Activation and Inhibition. Biochem J. 2004;384:201–232. doi: 10.1042/BJ20041142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Scheer JM, Romanowski MJ, Wells JA. A Common Allosteric Site and Mechanism in Caspases. Proc Natl Acad Sci USA. 2006;103:7595–7600. doi: 10.1073/pnas.0602571103. [DOI] [PMC free article] [PubMed] [Google Scholar]