

Abstract
During maturation, procaspase-3 is cleaved at D175, which resides in a linker that connects the large and small subunits. The intersubunit linker also connects two active site loops that rearrange following cleavage and, in part, form the so-called loop bundle. As a result of chain cleavage, new hydrogen bonds and van der Waals contacts form among three active site loops. The new interactions are predicted to stabilize the active site. One unresolved issue is the extent to which the loop bundle residues also stabilize the procaspase active site. We examined the effects of replacing four loop bundle residues (E167, D169, E173, and Y203) on the biochemical and structural properties of the (pro)caspase. We show that replacing the residues affects the activity of the procaspase as well as the mature caspase, with D169A and E167A replacements having the largest effects. Replacement of D169 prevents caspase-3 autoactivation, and its cleavage at D175 no longer leads to an active enzyme. In addition, the E173A mutation, when coupled to a second mutation in the procaspase, D175A, may alter the substrate specificity of the procaspase. The mutations affected the active site environment as assessed by changes in fluorescence emission, accessibility to quencher, and cleavage by either trypsin or V8 proteases. High-resolution X-ray crystallographic structures of E167A, D173A, and Y203F caspases show that changes in the active site environment may be due to the increased flexibility of several residues in the N-terminus of the small subunit. Overall, the results show that these residues are important for stabilizing the procaspase active site as well as that of the mature caspase.
Two subfamilies of caspases, the initiators and the effectors, are involved in apoptosis. Effector caspases, such as caspase-3, exist as stable dimers both before and after activation of the zymogen (1, 2). In contrast, procaspase-9, an initiator caspase, exists largely as a monomer, while procaspase-8, also an initiator, exists in equilibrium between monomers and dimers (3, 4). Dimerization of initiator caspases results from their interactions with multisubunit complexes, such as the apoptosome (5) or the DISC (death-inducing signaling complex) (6), and is a fundamental mechanism for autolytic activation of initiator caspases (3, 7, 8). For example, the association of procaspase-9 with the apoptosome results in an increase in activity of 3 orders of magnitude (7).
By contrast, activation of effector procaspases is fundamentally different since it requires cleavage of the preexisting procaspase dimer by initiator caspases. Cleavage occurs in a flexible loop, called the intersubunit linker, and separates the large and small subunits. While the core structure is similar between the zymogen and the activated caspase, cleavage allows for the rearrangement of several active site loops, resulting in an increase in activity of at least 2 orders of magnitude (9). Subsequent cleavages remove the pro-domain, although there is no further increase in enzymatic activity upon prodomain removal (10).
While structures have been determined for many mature caspases (11–16), only one procaspase structure has been determined, that of procaspase-7 (17, 18), which is an effector procaspase. The structures show that the caspases are dimers of heterodimers arranged in an αββα configuration, where α represents a large subunit and β represents a small subunit. The heterodimers (αβ) originate from a cleaved procaspase dimer. Caspases have four active site loops, L1–L4, which originate from one heterodimer and one loop, L2′, which originates from the second heterodimer (Figure 1). L1 (residues 52–66) and L4 (residues 247–263) comprise the sides of the active site; L3 (residues 198–213) comprises the bottom, and L2 (residues 163–175) contains the catalytic cysteine (C163). In addition, interactions among L2′ (residues 176′–192′), from the other heterodimer, and L2 and L4 appear to stabilize the active conformation. These interactions comprise the so-called loop bundle (17). In the zymogen, L2 and L2′ are covalently connected in the intersubunit linker, and cleavage at D175 separates the two loops (Figure 1A,B).
Figure 1.

Structure of caspase-3 and loop bundle interactions. (A) Primary sequence of human procaspase-3. Amino acid residues in L1 (52–66), L2 (163–175), L3 (198–213), L4 (247–263), and L2′ (176′–192′) are underlined, and the colors correspond to those in panel B. Cleavage sites for trypsin (white arrows) and V8 proteases (black arrows) are shown. The catalytic cysteine (C163), two tryptophans (W206 and W214), and amino acids in the loop bundle (E167, E169, E173, and Y203) are shown in bold. Boundaries observed in the crystal structures for the large subunit (S29–D175) and the small subunit (G177–H277) are indicated by angle brackets. Processing sites in the procaspase (D9, D28, and D175) are denoted with triangles. (B) Mature caspase-3 was generated with PyMOL (Delano Scientific, San Carlos, CA). The positions of L1–L4 and the active site tryptophan residues (W206 and W214) are indicated. (C) The position of E167 is highlighted relative to the active site and residues in L3. (D) Hydrogen bonds contributed by D169 to residues in L4 (brown) and L2′ (cyan) are highlighted. (E) The position of Y203 relative to the second heterodimer (R144′, E190′, and K137′) and L2 (C163 and R164) is highlighted. (F) Contacts between L2′ (cyan) and L4 (brown) are shown for residues near E173. For panels C–F, colors for L1–L4 are the same as those for panel B except for L2. For clarity, residues in this loop are colored gray in panels C–F. Red spheres represent water molecules. The small numbers represent distances (in angstroms) between two atoms connected by the dashed lines, and the primes denote residues in the second monomer.
Because procaspase-7 is a close homologue of pro-caspase-3 and indeed resides in the same subfamily (19), it is commonly used as a model for the effector caspases. Structural studies show that procaspase-7 is not catalytically competent because of the covalent connection between L2 and L2′ (17, 18). The intersubunit linker extends across the body of the protein to connect the large and small subunits. As a result, the catalytic cysteine is rotated away from the S1 binding site, and L2′ occupies the central cavity of the protein so that the active site is in a misaligned conformation. Consequently, most of the interactions that stabilize the loop bundle do not form. In addition, the misaligned configuration results in an increased flexibility of L3 and L4 so that the active site is incompatible with efficient catalysis. Upon cleavage of the intersubunit linker, L2′ changes orientation by almost 180° and forms 11 hydrogen bonds and numerous van der Waals interactions with L2 and L4 of the second heterodimer (17). Thus, interactions between the two heterodimers stabilize the active sites.
The structural data suggest that the constraints imposed upon L2 and L2′ result in the low activity of the zymogen. In the case of the effector caspases, cleavage of the intersubunit linker releases the constraints. For procaspase-9, however, the intersubunit linker is several residues longer than for procaspase-3 or -7. Consequently, cleavage of the polypeptide chain results in modest increases in activity. We note that a mutation in the dimer interface of procaspase-3 also resulted in activation of the zymogen without cleavage of the intersubunit linker, and it was suggested that the mutation increased the flexibility of the intersubunit linker, allowing the loop bundle to form, at least partially (20). Increasing the flexibility of the intersubunit linker, either by increasing the length of the polypeptide chain or by other mechanisms, may be sufficient to activate the procaspase if the loop bundle can form.
In the studies presented here, we mutated several acidic residues in L2 of caspase-3 to examine interactions that occur in the loop bundle as well as potential interactions that occur in procaspase-3. In addition, we mutated one residue in L3, Y203, that interacts with residues across the dimer interface. We show that the mutations affected the conformations and activities of both the mature caspases and the procaspases, demonstrating that some interactions in the loop bundle of caspase-3 are important for stabilizing the procaspase as well.
EXPERIMENTAL PROCEDURES
Materials
Acrylamide, ampicillin, citric acid, DEAE-Sepharose, DFP,1 DMSO, DTT, glycerol, IPTG, kanamycin, nickel sulfate, PMSF, potassium iodide, monobasic and dibasic potassium phosphate, TLCK, and TPCK were from Sigma. Imidazole was from ICN. Sodium chloride, sodium azide, hydrochloric acid, sodium hydroxide, Tris base, tryptone, and yeast extract were from Fisher. Nickel–Sepharose resin was from Novagen. Granzyme B, Ac-DEVD-AFC, and Ac-DEVD-CMK were from Calbiochem. V8 protease, trypsin, MgCl2, and dNTPs were from Roche Biochemicals. Polyclonal anti-caspase-3 antibody (9662) was from Cell Signaling Technology. Alkaline phosphatase-coupled secondary antibody (goat anti-rabbit), BCIP, NTB, and nonfat dry milk were from Bio-Rad. The ECL kit was from Amersham. X-OMAT-AR film was from Kodak. Restriction enzymes, deep vent polymerase, and DpnI were from New England Biolabs. PEG 200, PEG 400, PEG 600, PEG 4000, PEG 6000, propanol, 2-propanol, glucose, inositol, MPD, and paraffin oil were from Hampton.
Mutagenesis
All mutations were made in the backgrounds of wild-type caspase-3 using plasmid pHC332 (2) or the uncleavable procaspase-3(D9A/D28A/D175A) [called pro-caspase-3(D3A)] using plasmid pHC33209 (9). Mutations were confirmed by sequencing both DNA strands (University of Maine DNA Sequencing Facility, Orono, ME). In the mutations described below, changes to the DNA sequence are shown in bold.
The E173A mutant in the background of caspase-3 was made using forward primer 5′-GAACTGGACTGTGGCA-TCGCGACAGACAGTGGTG-3′ and reverse primer 5′-CACCACTGTCTGTCGCGATGCCACAGTCCAGTTC-3′. The resulting plasmid is called pHC33233. The E173A mutant in the background of procaspase-3(D3A) was made using forward primer 5′-GGACTGTGGCATTGCGACAG-CTAGCGGTG-3′ and reverse primer 5′-CACCGCTAGCT-GTCGCAATGCCACAGTCC-3′. The resulting plasmid is called pHC33246. The D169A mutant in the background of caspase-3 was made using forward primer 5′-CCTGCC-GTGGTACCGAACTGGCCTGTGGCATTGAGAC-3′ and reverse primer 5′-GTCTCAATGCCACAGGCCAGTTCG-GTACCACGGCAGG-3′. A unique restriction site for KpnI was introduced (underlined). The resulting plasmid is called pHC33232. Note that the D169A mutation was not made in the background of procaspase-3(D3A). The E167A mutants were made in both backgrounds using forward primer 5′-GCCTGCCGTGGTACCGCACTGGACTGTGGCATTGAG-3′ and reverse primer 5′-CTCAATGCCACAGTCCAGT-GCGGTACCACGGCAGGC-3′. A unique KpnI site was introduced (underlined). The resulting plasmids are called pHC33231 (background of caspase-3) or pHC33241 [back-ground of procaspase-3(D3A)]. The Y203F mutant was made using forward primer 5′-CATACTCCACAGCACCCGGG-TTTTATTCTTGGCGAAATTC-3′ and reverse primer 5′-GAATTTCGCCAAGAATAAAACCCGGGTGCTGTGG-AGTATG-3′. A SmaI site was introduced (underlined). The resulting plasmids are called pHC33250 (background of caspase-3) or pHC33252 [background of procaspase-3(D3A)].
Protein Purification
Escherichia coli BL21(DE3) pLys S cells were transformed with the plasmids described above, and proteins were expressed and purified as described previously (9). In addition, immunoblots were performed as described previously (20).
Enzymatic Assays
The activities of the proteins were determined as described previously (9, 21) using the fluorescent substrate Ac-DEVD-AFC. The data were fit as described previously (22) to determine the steady-state parameters, kcat and Km, as well as the specificity constant, kcat/Km.
Tryptophan Quenching
Proteins were examined in citrate [20 mM citrate (pH 3.0–6.0)], potassium phosphate [50 mM KH2PO4/K2HPO4 (pH 6.0–8.0)], or Tris [50 mM Tris-HCl (pH 7.2–9.5)] buffers. All buffers contained 1 mM DTT, and the final protein concentrations were 2 μM. Stock solutions of KI, CsCl, or acrylamide were prepared in the respective buffers and then added to the protein samples to give the final concentrations shown in the figures. Following addition of quencher, each sample was incubated for 5 min at 25 °C. Samples were excited at 295 nm, and fluorescence emission was measured at 345 nm (PTI C61 spectrofluorimeter, Photon Technology International). All data were corrected for the background signal. As described previously (9), the percent quenching, (ΔF/F0) × 100, was plotted versus the concentration of KI, and the data were fit to eq 1:
| (1) |
where ΔF is the change in fluorescence emission at each concentration of quencher (KI), F0 is the fluorescence intensity of the protein in the absence of quencher, KSV is the Stern–Volmer constant, [Q] is the quencher concentration, and fa is the fraction of the initial fluorescence emission that is accessible to the quencher.
The Stern–Volmer constants (KSV), determined from the fits described by eq 1, were plotted versus pH. The resulting data were fit to eq 2:
| (2) |
where A is the value of KSV at the lowest pH, ΔKSVn represents the changes in the Stern–Volmer constant for each transition, yn is the number of protons titrated in each transition, and pKan is the apparent pKa of the transitions. The data were fit to one, two, or three transitions (n = 1–3) as shown in the figures.
Fluorescence Emission as a Function of pH
Protein stocks were initially dialyzed against a buffer of 20 mM citrate (pH 6.2) (for experiments between pH 2.5 and 6.2) or a buffer of 20 mM potassium phosphate (pH 9.0) (for experiments between pH 6.0 and 9.0). All buffers contained 1 mM DTT. Following dilution into each buffer, the samples (2 μM) were incubated for at least 1 h at 25 °C. In separate experiments, the equilibration time was determined to be between 15 and 30 min. Samples were excited at 280 nm, and the fluorescence emission was monitored from 305 to 400 nm. All data were corrected for the background signal. The average emission wavelength, 〈λ〉, was calculated as described previously (23), and plots of 〈λ〉 versus pH were fit as described previously (9) to determine the apparent pKa of each transition.
Limited Proteolysis with Trypsin
Proteins were digested with 1/15 of their concentration (w/w) of trypsin in a buffer of 20 mM potassium phosphate (pH 7.5) and 1.0 mM DTT at 25 °C. After trypsin was added to a stock protein solution, aliquots (25 μL) were taken at time points between 2 min and 24 h. Reactions were inhibited by adding TLCK in 3-fold excess to trypsin (w/w). The samples were frozen at −20 °C until they were visualized on a SDS–PAGE gel using either 10 to 25% (procaspase-3 mutants) or 15 to 25% (caspase-3 and processed caspase-3 mutants) polyacrylamide gradients. The (pro)caspase-3 trypsin cleavage sites have been identified (9).
Limited Proteolysis with V8 Protease
Proteins were digested with V8 protease as described above for trypsin digests except that the reactions were inhibited with DFP at 3 times (w/w) the concentration of protease. The (pro)-caspase-3 V8 protease cleavage sites have been identified (9).
Procaspase-3(D3A/E173A) Autoproteolyis
To examine the proteolysis of procaspase-3(D3A/E173A), 30 μM protein was incubated at 25 °C for 8 days in a buffer of 50 mM Tris-HCl (pH 7.5), 50 mM NaCl, and 1 mM DTT. Aliquots were removed at 24 h intervals and stored at −20 °C until they were visualized on a 10–25% SDS–PAGE gel stained with Coomassie brilliant blue dye. One reaction mixture contained the protease inhibitors TLCK, TPCK, and PMSF (0.66, 1.0, and 1.14 μg/mL, respectively). The second reaction mixture contained the caspase-3 inhibitor Ac-DEVD-CMK at a 1:5 protein:inhibitor ratio (w/w).
Crystallography
Proteins were dialyzed in a buffer of 10 mM Tris-HCl (pH 8.5) and 1 mM DTT. The protein was concentrated to ~10 mg/mL using Amicon ultrafree centrifugal filter devices, and inhibitor, Ac-DEVD-CMK reconstituted in DMSO, was then added at a 5:1 inhibitor:peptide ratio (w/w). The protein was diluted to a concentration of 8 mg/mL by adding 10 mM Tris-HCl (pH 8.5), concentrated DTT, and concentrated NaN3 so that the final buffer consisted of 10 mM Tris-HCl (pH 8.5), 10 mM DTT, and 3 mM NaN3. Crystals were obtained by the hanging drop vapor diffusion method. Concentrated protein (2 μL) was mixed 1:1 with a reservoir solution that contained 100 mM sodium citrate (pH 5), 3 mM NaN3, 10 mM DTT, and 17% PEG 6000 (w/v). The crystallization plates were incubated at 18 °C. Crystals grew within 3 days for wild-type caspase-3 and within 2 weeks for the mutants.
Cryogenic conditions were screened using the wild-type caspase-3 crystals and were subsequently applied to the mutants. Crystals were flash-frozen using a variety of cryoprotectants, including glycerol, ethylene glycol, PEG 200, 400, 600, 4000, and 6000, paraffin oil, propanol, 2-propanol, glucose, inositol, and MPD at various concentrations. The ideal cryoprotectant solution was found to be 80% reservoir solution and 20% PEG 400. Data sets were then collected at 100 K at the SER-CAT synchrotron beamline (Advance Photon Source, Argonne National Laboratory, Argonne, IL). The X-rays had a wavelength of 1 Å. Ninety degrees of data at 1° intervals were collected for each protein. The caspase-3 wild-type structure was determined using molecular replacement with the previously published caspase-3 structure for intial phasing (PDB entry 1CP3) (11). Refinement of the model was performed using CNS (24) and O (25). Structures of the mutants were phased with our caspase-3 structure, and the refinement was performed as with the wild-type structure. The data collection and refinement statistics are listed in Table 3.
Table 3.
Summary of Statistics for Data Collection and Refinement for Caspase-3 and Loop Bundle Mutants
| wild type | E167A | E173A | Y203F | |
|---|---|---|---|---|
| temperature (K) | 100 | 100 | 100 | 100 |
| resolution (Å) | 35.0–1.4 | 35–1.5 | 35–1.3 | 35–2.0 |
| space group | I222 | I222 | I222 | I222 |
| unit cell | ||||
| a (Å) | 68.73 | 67.33 | 68.80 | 67.38 |
| b (Å) | 84.40 | 83.93 | 84.23 | 83.30 |
| c (Å) | 96.35 | 95.50 | 96.14 | 95.92 |
| α, β, γ (deg) | 90 | 90 | 90 | 90 |
| no. of reflections | 54279 | 49333 | 66657 | 58178 |
| completeness (%) | 97.9 (83.8)d | 76.0 (92.9)d | 98.5 (88.3)d | 86.9 (37.2)d |
| I/σ | 46.23 (1.34)d | 34.6 (2.52)d | 50.7 (2.04)d | 45.5 (1.7)d |
| Rmerge (%)a | 5.9 | 9.3 | 11.5 | 14.7 |
| redundancy | 5.8 (3.5)d | 5.1 (2.5)d | 6.5 (3.4)d | 6.5 (6.9)d |
| Rwork (%)b | 19.6 | 20.0 | 19.4 | 17.6 |
| Rfree (%)c | 20.7 | 21.6 | 20.2 | 20.5 |
| rmsd for bond lengths (Å) | 0.005 | 0.005 | 0.005 | 0.006 |
| rmsd for bond angles (deg) | 1.30 | 1.26 | 1.28 | 1.26 |
| rmsd for dihedral angles (deg) | 24.39 | 24.26 | 24.37 | 24.07 |
| rmsd for improper angles (deg) | 1.08 | 1.00 | 1.06 | 0.98 |
| average B factor (Å 2) | 24.84 | 23.8 | 19.71 | 21.83 |
| no. of protein atoms | 1974 | 1935 | 1935 | 1894 |
| no. of water molecules | 290 | 204 | 305 | 251 |
| rmsd | 0.40e | 0.19f | 0.09f | 0.18f |
Rmerge = ΣhΣi|I(h,i) − I(h)|/ΣhΣiI(h,i), where I(h,i) values are symmetry-related intensities and I(h) is the mean intensity of the reflection with unique index h.
Rwork = Σ ||Fobs| − |Fcalc||/Σ |Fobs|, where Fobs and Fcalc are observed and calculated structure factors, respectively.
Rfree = ΣT||Fobs| − |Fcalc||/ΣT|Fobs|, where T is a test data set of 10% of the total reflections randomly chosen and set aside prior to refinement.
Data for the highest-resolution shell in parentheses.
Compared to the 1CP3 structure as described in the text.
Compared to our wild-type caspase-3 structure.
Accession Numbers
The atomic coordinates and structure factors of the four structures have been deposited in the Protein Data Bank as entries 2J30 (wild-type caspase-3), 2J31 [caspase-3(E167A)], 2J32 [caspase-3(E173A)], and 2J33 [caspase-3(Y203F)].
RESULTS
E167, from L2, forms three hydrogen bonds with residues from other loops (Figure 1C). The backbone amide of E167 hydrogen bonds with the carbonyl of G202, from L3, and the backbone carbonyl hydrogen bonds with the backbone amide of Y204 (Figure 1C). Y204 is important because the side chain forms part of the S2 subsite. The side chain of E167 hydrogen bonds through a water molecule with the backbone carbonyl of P201, which is located in L3. P201 resides in the so-called elbow loop region of L3 (18) and stacks with the side chains of R164 and Y197 at the bottom of the active site. In procaspase-7, extrusion of the elbow loop correlates with unstructured S1–S3 binding sites (18).
The side chain of D169 (in L2) forms hydrogen bonds across the dimer interface with the backbone amides of V189′ and E190′ (both in L2′) (Figure 1D). In addition, the backbone carbonyl of D169 hydrogen bonds to the backbone amide of K260, from L4 (Figure 1D), and the backbone amide of D169 interacts with the carbonyl of K260 and the side chain of W206 through a water molecule. The aromatic ring of W206 is important because it stacks with W214 (see Figure 1B), which hydrogen bonds to the substrate in the P4 site. Thus, D169 forms three hydrogen bonding interactions that would appear to stabilize three active site loops, L2–L2′–L4. The side chain also is within hydrogen bonding distance of the backbone amide of G171 (in L2), although it is not known whether this in an important interaction.
The side chain of Y203 is within hydrogen bonding distance of the side chain of E190′ across the dimer interface (Figure 1E). In addition, it may interact with the side chain of R144′ through at least one water molecule. R144′ is located between helices 2′ and 3′ on the surface of the second heterodimer. The backbone carbonyl of Y203 hydrogen bonds with the backbone amide of R164 (in L2) (Figure 1E). Thus, Y203 interacts across the dimer interface, like D169 discussed above, but also appears to stabilize L2 near the catalytic cysteine, C163.
In the crystal structure, T174 is the final residue with electron density at the C-terminus of the large subunit (see Figure 1A). While the E173 side chain does not interact with other residues in the loop bundle, the region encompassing E173 does appear to be important in stabilizing the loop bundle. The backbone carbonyl and amide groups of I172 form two hydrogen bonds with I187′ from L2′ (Figure 1F). At the N-terminal end of L2′, the side chain of H185′ interacts with the carbonyl of T245, from L4, and K186′ forms two hydrogen bonds with K260 and A258, also in L4. These interactions likely stabilize L4 since T245 and K260 reside at apposing sides of the base of L4.
We investigated the effects of removing hydrogen bonds contributed by several side chains in the loop bundle by substituting E167, D169, and E173 with alanine. In addition, Y203 was replaced with phenylalanine. With the exception of D169, the mutations were made in the context of wild-type procaspase-3 as well as the uncleavable procaspase-3(D3A) in an attempt to examine the effects in the mature caspase as well as the procaspase.
The proteins were purified after overexpression in E. coli, and we noticed a number of surprising results from the purification. First, it has been well-established that the wild-type procaspase-3 undergoes automaturation when overexpressed in E. coli so that the mature caspase, rather than the procaspase, is obtained following expression and purification. The mutants, with the exception of D169A, also underwent automaturation. The lack of automaturation of procaspase-3(D169A) can be observed experimentally in the Western blot shown in Figure 2A. We found no evidence of processing in this variant using an antibody to the large subunit of caspase-3, and the results were compared to those for several uncleavable procaspase variants. In addition, we found that D175 was accessible to cleavage since the mutant could be processed efficiently with wild-type caspase-3 (Figure 2B) or granzyme B (data not shown), which cleaves the polypeptide chain following D175. In Figure 2B, bands 2 and 3 represent cleavage in the prodomain at D9 and D28, respectively; band 4 is the large subunit with a full-length prodomain, and band 5 is the large subunit cleaved at D9. Band 6 is the processed large subunit, and band 7 is the processed small subunit (17 and 12 kDa, respectively). The cleaved caspase-3(D169A) mutant had extremely low activity, as described below.
Figure 2.
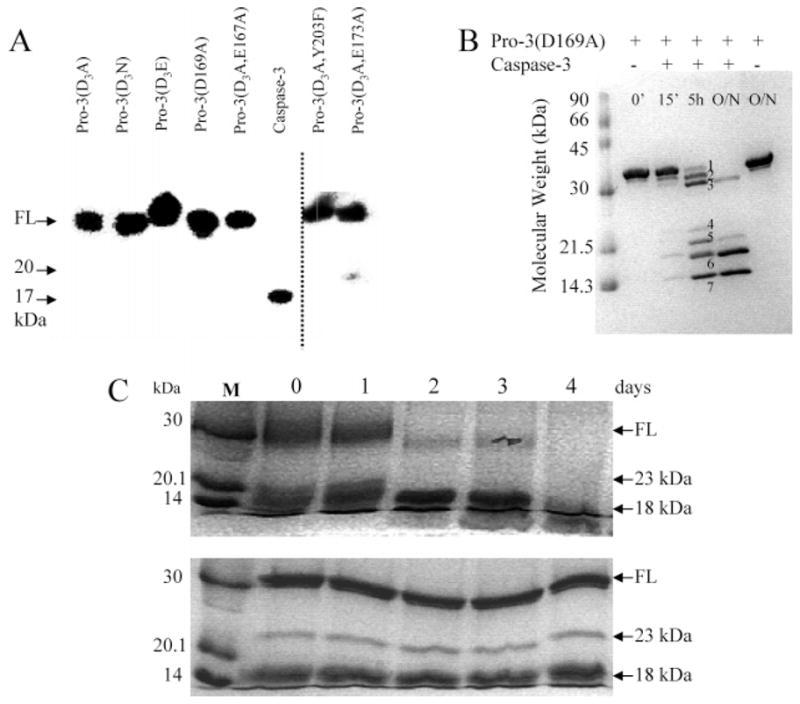
Properties of caspase variants. (A) Anti-caspase-3 immunoblot of procaspase-3(D3A), procaspase-3(D3N), procaspase-3(D3E), procaspase-3(D169A), procaspase-3(D3A/E167A), mature caspase-3, procaspase-3(D3A/Y203F), or procaspase-3(D3A/E173A). The dashed line signifies the use of two gels. (B) Procaspase-3(D169A) incubated with mature caspase-3. Bands 1–7 represent cleavage products as described in the text. O/N refers to overnight incubation (>16 h). (C) In the top panel, procaspase-3(D3A/E173A) was incubated with serine protease inhibitors for 4 days. In the bottom panel, procaspase-3(D3A/E173A) was incubated with the irreversible inhibitor Ac-DAVD-CMK for 4 days. Aliquots were removed each 24 h, and the samples were examined as described in Experimental Procedures. FL refers to full-length protein (32 kDa), and M refers to molecular mass markers.
The second surprising result was that procaspase-3(D3A/E173A) was partially processed (Figure 2A,C), whereas E173A in the context of wild-type caspase-3 was processed normally (see Figure 1 of the Supporting Information). The three processing sites (D9, D28, and D175) were removed previously and replaced with alanine to generate the triple mutant called procaspase-3(D3A) (9). We showed (9) that procaspase-3(D3A) does not autoprocess when overexpressed in E. coli (see also Figure 2A). Other mutations at the three processing sites also abrogated autoprocessing (Figure 2A). For procaspase-3(D3A/E173A), however, the predominant 32 kDa band, representing the full-length protein, was cleaved initially into ~23 and ~9 kDa species (see Figure 2C). The large band was then cleaved more slowly to an ~18 kDa fragment (see Figure 2C and Figure 1 of the Supporting Information). We used N-terminal sequencing to determine that the protein was cleaved initially at N208, in L3. In addition, the protein was degraded over a period of several days following this initial cleavage (Figure 2C, top panel). To examine whether the alternate cleavage occurred as a result of autoproteolysis, the time course for degradation of the protein was monitored by incubating procaspase-3(D3A/E173A) with several serine protease inhibitors (Figure 2C, top panel) or with the caspase-3 inhibitor Ac-DEVD-CMK (Figure 2C, bottom panel). The sample incubated in the presence of serine protease inhibitors was completely processed after ~3 days, whereas most of the procaspase was degraded after 2 days. Alternatively, the sample incubated in the presence of the caspase-3 inhibitor remained intact after 8 days, with the exception of the processing that occurred during purification. Note that the data for the first 4 days are shown in Figure 2C. Because caspase-3(E173A) was processed normally, the results showed that the alternate processing of procaspase-3(D3A/E173A) at N208 resulted from mutating both E173 and D175 to alanine. It is interesting to note that this mutant cleaves itself after the sequence SWRN208/S, although at this point it is not clear if the E173A/D175A double mutation results in a change in the substrate specificity. In addition, it is not yet clear why mutating both residues leads to the cleavage at N208 in L3. Finally, we note that the Y203F variant was not fully processed following overexpression in E. coli. The protein was cleaved at D175, but the prodomain was not removed efficiently. As shown in Figure 1 of the Supporting Information, two bands were observed for the large subunit, the larger of which represents the large subunit containing the pro-domain. It has been shown, however, that the prodomain does not bind to the active site or affect activity (2). As noted below, the catalytic parameters for this variant on the artificial substrate DEVD-AFC were unchanged versus wild-type caspase-3.
We examined all of the proteins by circular dichroism (data not shown) and by fluorescence emission (described below) spectroscopies. The results showed that while there were some differences when compared to the controls, all of the proteins were folded and contained well-packed tertiary structures.
Enzymatic Activity
We measured the enzymatic activity of the proteins against the substrate Ac-DEVD-AFC, and the results are summarized in Table 1. Both E167A and Y203F, in the context of the procaspase resulted in higher KM values, and E167A also had a large effect on kcat. As a result, the specificity constant, kcat/KM, was >40-fold lower for this mutant while the effects of Y203F were more modest. E167A also had large effects on the activity of mature caspase-3, where it was observed that both KM and kcat were altered, and the specificity constant was decreased by ~20-fold. In the context of mature caspase-3, neither E173A nor Y203F significantly affected KM or kcat.
Table 1.
Catalytic Parameters of the Caspase-3 Loop Bundle Mutants
| KM (μM) | kcat (s−1) | kcat/KM (M−1 s−1) | |
|---|---|---|---|
| Caspase-3a | 2.2 ± 0.5 | 0.40 ± 0.05 | 1.8 × 105 |
| Caspase-3(E167A)a | 13.2 ± 1.8 | 0.12 ± 0.06 | 9.1 × 103 |
| Caspase-3(E173A)a | 2.4 ± 0.1 | 0.69 ± 0.05 | 2.9 × 105 |
| Caspase-3(Y203F)a | 5.9 ± 0.4 | 0.68 ± 0.10 | 1.2 × 105 |
| Procaspase-3(D3A)b,c | 3.5 ± 0.8 | 0.003 ± (1.4 × 10−4) | 8.6 × 102 |
| Procaspase-3(D3A, E167A)b | 18.2 ± 2.6 | 0.0004 ± (2.0 × 10−4) | 2.2 × 101 |
| Procaspase-3(D3A, Y203F)b | 12.9 ± 1.9 | 0.003 ± (6.0 × 10−4) | 2.3 × 102 |
| Procaspase-3(D3A, E173A)b | no measurable activity | ||
| Caspase-3(D169A)a (unprocessed) |
no measurable activity | ||
| Caspase-3(D169A)a processed with granzyme B |
>75 | NDd | NDd |
pH 7.5.
pH 8.0, which represents the optimal pH for the procaspase.
D3A refers to procaspase-3(D9A, D28A, D175A).
Not determined.
The alternately processed procaspase-3(D3A/E173A) exhibited no activity against the Ac-DEVD-AFC substrate (data not shown), even though it degraded itself over time (Figure 2C). However, E173A had no effect on the activity of mature caspase-3. Likewise, we observed no activity for full-length caspase-3(D169A). In addition, the protein had very little activity following cleavage with granzyme B. Under these conditions, we estimated that the KM was greater than 75 μM. We could not determine the kcat for this mutant since the substrate concentrations were not saturating under these conditions.
Overall, the results showed that E167A and D169A significantly affected the activities of the procaspase and the mature caspase, and the E173A mutation, when coupled to the D175A mutation, resulted in an alternately processed procaspase that had no activity when measured against a synthetic peptide substrate. In contrast, E173A in caspase-3 had no effect on activity. Y203F had little effect on the activity of either the caspase or the procaspase, although, as noted above, it did not efficiently remove its own propeptide.
Limited Proteolysis with V8 Protease
To examine potential conformational changes, the mutants were subjected to limited proteolysis with V8 and trypsin proteases. We determined previously (9) that V8 protease (Endo-Glu C) cleaves procaspase-3 initially at E248/D253, in L4, to generate fragments of ~29 and 4 kDa. The larger band was termed band 2 (Figure 3A). The initial cleavage was followed by cleavage at E25 in the prodomain to generate band 3. Band 3 was then cleaved simultaneously at E98 and E106 and at E173 (L2) and E190 (L2′), giving rise to 16–18 and ~8 kDa bands. These results are shown in Figure 3A and are summarized in Figure 3E and Table 2. Caspase-3 also was cleaved at E248/D253 and at E173, but the cleavages at E25, E98/E106, and E190 did not occur (9) (Figure 3B and Table 2). We suggested previously that hydrogen bonds utilizing the side chains of E98, E106, and E190 are not formed in the procaspase, which affects their accessibility to the protease (9).
Figure 3.

V8 (Endo-Glu C) or trypsin protease digests of (pro)caspases. V8 digest of procaspase-3(D3A) (A) or of wild-type caspase-3 (B). Trypsin digest of procaspase-3(D3A) (C) or of wild-type caspase-3 (D). (E) Trypsin and V8 protease cleavage sites are mapped onto the caspase-3 structure, and the cleavages are summarized in Table 2. The structure was generated using PyMOL (Delano Scientific). The cleavages at K57, R64, and R207 (underlined) are by trypsin, and the cleavages at E98, E106, E173, D190, E248, and D253 (italicized) are by V8 protease. The primes denote residues from the second heterodimer. Results for the loop bundle mutants are provided as Supporting Information and are summarized in Table 2.
Table 2.
Summary of V8 and Trypsin Proteolysis
| V8 Protease
| ||||||
|---|---|---|---|---|---|---|
| protein | cleavage site
|
comments | loops affected by mutation | |||
| E248/D253 | E25 | E98/E106 | E173/E190 | |||
| procaspase-3 | + | + | + | + | bands 2 and 3, 16–18 and 38 kDa bands | − |
| D3A/E167Aa | − | + | − | − | removal of prodomain at E25 is the only cleavage | L2′ and L4 |
| D169A | + | + | + | very little | very slow kinetics of cleavage | L2′ and L4 |
| D3A/E173A | − | + | − | − | cleavage of prodomain at E25; 8 kDa band arises from alternate processing |
L2′ and L4 |
| D3A/Y203F | + | + | + | + | cleavage kinetics similar to those of the control | none |
| caspase-3 | + | − | − | + | cleavage at E173; no cleavage at E190 | – |
| E167A | + | − | − | + | cleavage kinetics similar to those of the control | none |
| E173A | + | − | − | + | cleavage kinetics similar to those of the control | none |
| Y203F | + | − | − | + | cleavage kinetics similar to those of the control | none |
| Trypsin Protease
| ||||||
| cleavage site
|
loops affected | |||||
| protein | K57/R64 | R207 | comments | by mutation | ||
|
| ||||||
| procaspase-3 | + | + | cleavage in L1 and L3 | – | ||
| D3A/E167A | + | + | cleavage kinetics similar to those of the procaspase control | none | ||
| D3A/D169A | + | + | cleavage kinetics similar to those of the procaspase control | none | ||
| D3A/Y203F | + | + | cleavage kinetics similar to those of the procaspase control | none | ||
| caspase-3 | + (large subunit) | + (small subunit) | cleavage is ~10 times faster than for the procaspase | – | ||
| E167A | + | + | cleavage kinetics similar to those of the caspase control | none | ||
| E173A | + | + | cleavages at K57/R64 and R207 much slower than for the control | L1 and L3 | ||
| Y203F | + | + | cleavage at K57/R64 slower than for the control; cleavage at R207 much slower than for the control |
L1 and L3 | ||
D3A refers to procaspase-3(D9A/D28A/D175A).
For the E167A variant, the prodomain was cleaved at E25 in the procaspase, but the other cleavages were not observed (Table 2 and Figure 1 of the Supporting Information). For the full-length D169A variant, cleavages occurred at the same sites that they did for procaspase-3, but the kinetics of cleavage were much slower (Table 2). The data indicate that the sites are less accessible to the protease than the same sites in procaspase-3(D3A). One should note that four large bands were observed in this protein rather than the three bands for procaspase-3(D3A). As described previously (9), the appearance of the fourth band is due to cleavage at D9, which is missing in the procaspase-3(D3A) variants but remains in the D169A variant. In the case of E173A, the prodomain was cleaved at E25 in the alternately processed procaspase variant, but the other sites were not cleaved (Table 2). Interestingly, the alternately processed procaspase was not degraded further, suggesting that the E248/D253, E98/E106, and E173/E190 sites were not accessible to the protease. Finally, the procaspase variant of Y203F was cleaved in a manner similar to that of the control (Table 2). Cleavage of caspase variants E167A, E173A, and Y203F occurred in a manner similar to that of the wild-type protein (Table 2 and Figure 1 of the Supporting Information).
Limited Proteolysis with Trypsin Protease
We showed (9) that trypsin cleaved procaspase-3 in L1 at R64 and K57, which generated fragments of ~25 and ~4 kDa (Figure 3C). Cleavage of the 25 kDa fragment at R207, in L3, led to fragments of 16 and 9.5 kDa. The results of trypsin cleavage also are summarized in Figure 3E and Table 2. These sites are important because R64 forms part of the S1 binding pocket, whereas R207 makes contacts with the substrate in both the S1 and S3 binding pockets (11, 12). For caspase-3 (Figure 3D and ref 9), the same cleavages occurred as in the procaspase except that the protein was cleaved much faster. The large subunit was cleaved rapidly by trypsin at K57/R64 to generate bands of ~14 and ~3.5 kDa, whereas the small subunit was cleaved at R207 to generate fragments of ~9 and ~3 kDa. One should note in Figure 3D that the smaller bands are not observed. When compared to cleavages of the procaspase, the results suggest that the cleavage sites are more accessible in the mature caspase.
In the case of the E167A variants, the same cleavages occurred in L1 and L3 and with kinetics similar to those of the respective controls (Table 2 and Figure 2 of the Supporting Information). Likewise, the D169A variant, which is unprocessed, was cleaved in a manner similar to that of the procaspase (Table 2). In contrast, the processed E173A and Y203F caspase variants were cleaved more slowly at K57/R64 in L1 (Table 2). Likewise, cleavage at R207 was very slow for both mutants when compared to the control. The results for E173A were surprising since the side chain of E173 was not predicted to contact other residues in the active site region. Finally, the Y203F procaspase variant was cleaved in a manner similar to the control (Table 2). The data for the mutants are shown in Figure 2 of the Supporting Information and are summarized in Table 2.
Overall, the results of the limited proteolysis with trypsin and V8 proteases suggested that E167A affected L2′ and L4 in the procaspase, but not in the caspase, and did not affect L1 or L3 in either form of the protein. D169A, which is unprocessed, affected L2′ and L4 but not L1 and L3, and E173A affected L1 and L3 in the caspase variant. Because the procaspase is alternately processed, however, it is not clear if E173A also affected the active site loops in the procaspase as well since the sample was a mixture of processed and unprocessed protein. Finally, Y203F had little effect on L4 in either the procaspase or the caspase but affected L1 and L3 in the caspase.
Quenching of Fluorescence Emission
The (pro)caspase-3 dimer has four tryptophans, two on each small subunit, W206 and W214 (see Figure 1). Both tryptophans are located on L3 and stack together in the active site, and W214 hydrogen bonds with the substrate in the S4 subsite (11). We have used a variety of biochemical and biophysical methods, including analytical ultracentrifugation, size exclusion chromatography, and urea denaturation studies (2, 21, 26), to show that the fluorescence emission changes as the pH is decreased from pH 9 to 3 due to dissociation of the dimer (pH ~4–5) and unfolding of the monomer (pH <4). Changes in fluorescence emission versus pH are described in more detail below, but as we show here, the conformational changes also correlate to a change in the accessibility of the active site tryptophans to iodide quencher (9). Thus, a measure of the changes in the Stern–Volmer quenching constant (KSV) versus pH is a good probe of active site conformation, especially when mutations are made in or around the active site. For example, procaspase-3(D3A) underwent two conformational changes between pH 9 and 3, and each conformational change increased the accessibility of the tryptophan residues to quenching by iodide [see Figure 4A (○)]. The first transition occurred between pH 8 and ~6.5, and the second transition occurred between pH 5 and 4. While the exact mechanism for the increase in KSV is not known, the first transition correlates with dissociation of the propeptide from the protease domain, and the second transition correlates with dissociation of the dimer (9). For caspase-3, three transitions were observed as the pH was decreased [see Figure 4B (○)]. The first transition (pH 8 to ~7) resulted in a small decrease in KSV; the second transition (pH ~7 to 4.5) resulted in a small increase in KSV, and the third transition (pH 4.5 to 4) resulted in a sharp increase in KSV. Again, the exact mechanism for the change in KSV is not known, although we have shown that the caspase-3 heterotetramer (equivalent to the procaspase dimer) dissociates between pH ~6 and 4 (9, 21).
Figure 4.

Fluorescence emission quenching by iodide and changes in average emission wavelength, 〈λ〉, vs pH. The Stern–Volmer quenching constants, KSV, are a measure of the accessibility of the active site tryptophan residues to iodide. (A and B) KSV values were determined for the quenching of tryptophan fluorescence emission over the pH range of 3–9 for (A) procaspase-3(D3A) (○) and (B) mature caspase-3 (○) and the (pro)caspase-3 mutants (●). The solid lines represent fits of the data as described in Experimental Procedures. For panels C and D, the average emission wavelength, 〈λ〉, was determined over the pH range of 2.4–9 for procaspase-3(D3A) (○), mature caspase-3 (○), and each (pro)caspase-3 mutant (□). The solid lines represent fits of the data as described in Experimental Procedures. For panels A–D, representative data are shown for E167A, and results for all mutants are provided as Supporting Information.
Overall, the quenching data demonstrate that all of the loop bundle mutations affected the electrostatic environment of the (pro)caspase-3 active site and may have affected the pH-dependent conformational changes of the proteins. For the E167A variants, the first transition (between pH 7.5 and ~6.5) was not observed in the procaspase (Figure 4A), and caspase-3(E167A) demonstrated a large KSV at higher pH and a lower KSV at lower pH (Figure 4B), which was the opposite of that for the wild-type caspase control. Representative data for E167A are shown in Figure 4, while data for all mutants are shown in Figure 3 of the Supporting Information and are summarized here. For the unprocessed D169A variant, KSV did not change as the pH was lowered. The alternately processed E173A procaspase variant demonstrated a higher KSV at higher pH compared to the control. In contrast, the KSV was lower than that of the control for the processed E173A caspase variant at higher pH. Following a single transition between pH ~7.5 and 7, the KSV did not change at lower pH. The procaspase variant of Y203F was most similar to the control as both transitions were observed, although the second transition occurred with a lower pKa of ~4 versus a pKa of 4.5 for the control. In contrast, the caspase variant of Y203F exhibited a lower KSV at both lower and higher pH.
We showed previously (9) that the results are specific to the negatively charged iodide ion since the tryptophan fluorescence quenching was weak in the presence of either cesium or acrylamide. We also observed that quenching of the tryptophanyl fluorescence emission in the mutants was weak in the presence of acrylamide (Figure 3 of the Supporting Information).
Fluorescence Emission versus pH
We showed previously that the fluorescence emission maximum, 〈λ〉, of procaspase-3(D3A) decreased as the pH was lowered from 9 to 2.5 (9, 21). One major transition was observed between pH 5 and 3 in which 〈λ〉 was blue-shifted [see Figure 4C (○)], and we have shown that the transition correlates to dissociation of the dimer (21, 26). A second transition, which was due to unfolding of the monomer (21, 26), was observed at pH <3 and resulted in a red shift in 〈λ〉. For caspase-3, 〈λ〉 was red-shifted at higher pH relative to the procaspase (~345 nm vs 342.5 nm) [see Figure 4D (○)]. We suggested that the higher 〈λ〉 of the caspase is consistent with a more open active site conformation in the caspase in which the tryptophan residues are more solvent accessible relative to their positions in the procaspase (9). As the pH was lowered, 〈λ〉 of caspase-3 was blue-shifted between pH 7 and 4 due to dissociation of the heterotetramer (21, 26). Below pH ~4, 〈λ〉 was red-shifted, and this was due to dissociation of the heterodimer into the individual subunits (21). Thus, the pH dependence of 〈λ〉 is useful in examining the solvent accessibility of the active site tryptophan residues as well as the oligomeric properties of the proteins.
Measures of 〈λ〉 versus pH for the loop bundle mutants showed that all of the mutations affected the active site tryptophans, but the effects were not equivalent. For the E167A procaspase variant, 〈λ〉 was slightly blue-shifted at higher pH compared to the control (342 nm vs 342.5 nm) (Figure 4C). A new transition occurred between pH ~6.5 and 5 that resulted in a blue shift of ~0.5 nm. The major transition then occurred between pH 5 and 4, with a pKa similar to that of the control (pKa ~ 4.3). While it is not clear what the new transition represents in terms of conformational changes in the protein, it has been observed in other (pro)caspase-3 mutants that affect the active site conformation, either in L4 (K242A) (21) or in the dimer interface (V266E) (20). At lower pH, the monomer of procaspase-3(D3A/E167A) may be less stable than that of the control since it unfolded at a higher pH (pKa ~ 3.4 and pKa < 2.5, respectively).
The 〈λ〉 of the caspase variant of E167A also was slightly blue-shifted at higher pH, but the protein underwent the same major transition between pH ~6 and ~4 as the control (Figure 4D). In contrast to the control, however, 〈λ〉 was blue-shifted during the transition below pH ~4. The latter transition was described previously for wild-type caspase-3 in the presence of cations (27), and we suggested that the cations shield negative charges that affect fluorescence emission upon subunit dissociation. The results presented here suggest either that E167 is one of the charges shielded by cations in wild-type caspase-3 or that the mutation affected the conformation of the subunit at low pH, which consequently affected the fluorescence emission in a manner similar to that of the cations.
Representative data for E167A are shown in Figure 4, while data for all mutants are shown in Figure 4 of the Supporting Information and are summarized here. The 〈λ〉 of the D169A variant was similar to the procaspase control at higher pH, but the major transition (from pH ~6 to ~4) occurred with a pKa significantly higher than that of the procaspase control (pKa ~5 and pKa ~4.3, respectively). The increased pKa for the transition of D169A showed that the homodimer was less stable than that of the control, causing the subunits to dissociate at higher pH. The 〈λ〉 of the alternately processed D173A procaspase variant more closely resembled that of caspase-3 rather than the pro-caspase. However, the 〈λ〉 decreased in a broad transition between pH ~6 and 3. Since this variant was alternately processed, it is not clear at present whether the results correlate to the procaspase, the alternately processed caspase, or a combination of the two. In contrast, the correctly processed E173A caspase was very similar to the wild-type control except that its 〈λ〉 was red-shifted at higher pH. The changes in 〈λ〉 show that the active site tryptophanyl residues were somewhat more solvent accessible in E173A than in the control. Finally, the 〈λ〉 values of the Y203F variants were significantly different from either of the controls. In the procaspase variant, 〈λ〉 was blue-shifted at higher pH compared to the control, but the major transition occurred with a similar pKa (4.1 and 4.3, respectively). Like the E167A variant, the Y203F variant also affected the fluorescence emission of the procaspase monomer at low pH. This was true also for the Y203F caspase variant.
Overall, measures of 〈λ〉 versus pH for the loop bundle mutants showed that most of the mutations significantly affected the environment of the tryptophan residues at higher pH. In most cases, except D169A, the mutations did not appear to affect the stability of the dimer; however, E167A decreased the stability of the procaspase monomer. Both E167A and Y203F affected the fluorescence emission of the procaspase monomer and caspase heterodimer at low pH in a manner that suggested a shielding of charges in the active site so that the tryptophan residues were in a more hydrophobic environment.
Crystal Structures of Wild-Type and Three Loop Bundle Mutants
To further examine potential conformational changes in the loop bundle proteins, we crystallized and determined the structures of the E167A, E173A, and Y203F caspase variants with the inhibitor Ac-DEVD-CMK bound. Unfortunately, we were unable to crystallize the D169A mutant. At present, procaspase-3 has been resistant to crystallization; thus, there currently are no structures available for the procaspase. Likewise, there are no structures for apocaspase-3, although we note a report by Ni et al. (28) describing the structure of an “unliganded” caspase-3. In their studies, however, the protein was incubated in the presence of an inhibitor during crystallization, but the inhibitor was not observed in the final structural model. Consequently, it is not clear whether their data are representative of apocaspase-3. So, currently we must use structures of the holoenzyme to infer properties of both the procaspase and the apocaspase.
As part of our studies of the mutants, we also determined the high-resolution structure of wild-type caspase-3. While this structure has been determined (11, 12), data collection occurred at higher temperatures because the crystals were not frozen. As a result, the initial structures of caspase-3 were determined to 2.3–2.5 Å resolution. Since those earlier studies, others have determined higher-resolution structures of caspase-3 by first freezing the crystal in cryoprotectant that usually consisted of 20–30% glycerol (28–32). We screened a variety of solutions and found optimal freezing conditions to consist of 80% reservoir solution and 20% PEG 400 (see Experimental Procedures). These conditions yielded a caspase-3 structure to 1.4 Å resolution and also were used to freeze crystals of the mutant caspases. The space group symmetry for our crystals was I222 (Table 3), the same as that described by Rotonda et al. [PDB entry 1PAU (12)] with one heterodimer per asymmetric unit. The heterotetramer was built from symmetry-related operations and compared to the structure described by Mittl et al. [PDB entry 1CP3 (11)]. In their case, caspase-3 crystallized with symmetry of space group P21, with one heterotetramer per asymmetric unit. Overall, the root-mean-square deviation (rmsd) between our structure of caspase-3 and that of Mittl et al. was ~0.4 Å (Table 3). The primary differences between the two structures were found at the termini of the large and small subunits. In our structure, electron density was observed starting at S29, which is the first residue in the large subunit after maturation, whereas the structure of Mittl et al. began at N35. As a result, we observed some differences in the interactions across the dimer interface. For example, the side chain of R238 was different from that of the 1CP3 structure in that it was clearly interacting with N35′ (the prime indicates residues from the second heterodimer), where the two side chains were 3.2 Å apart (data not shown). In the 1CP3 structure, R238 appeared to interact with E272′ as part of a series of charge–charge interactions across the dimer interface that presumably stabilizes the oligomer. In our structure, E272′ interacted with H234 (3.7 Å) as part of the same series of interactions. At the N-terminus of the small subunit, we observed electron density starting with G177, whereas the earlier structures started either with H185 (11) or with K186 (12). The data showed that residues 177–184 were positioned relatively close to the active site such that D180 was ~4.9 Å from K210 in L3. The consequences of this proximity are not known currently since residues 177–184 also made several contacts with a symmetry-related molecule in the crystal lattice (data not shown). At the C-terminus of the large subunit, we observed electron density for the backbone through T174. In the earlier structures, this electron density was modeled with the side chain of E173. While the consequences of the alternate model for E173 are discussed below, we note that other high-resolution structures of caspase-3 were modeled with the backbone of T174 in this position (30). Finally, we note that residue 190 was a glutamate in our structure of caspase-3, whereas it was an aspartate in the 1CP3 structure.
The structures of E167A, E173A, and Y203F with an inhibitor bound were determined to 1.5, 1.3, and 2.0 Å resolution, respectively (Table 3). Overall, the structures were very similar to that of wild-type caspase-3, and the average rmsd was 0.15 Å (Figure 5A and Table 3). For E167A, the termini were the same as those for caspase-3, except that electron density at the N-terminus of the small subunit was observed beginning with H185 in the mutant rather than G177 as for the wild type. There were no readily observed changes in L1–L4 that might explain the effects of the mutation on kcat or KM or the accessibility of the iodide quencher to the active site tryptophans. We note, however, that L1 and L4 were involved in crystal packing contacts with a symmetry-related molecule, where L1 of the related molecule packed between L1 and L4 (data not shown). As a result, changes in these loops may not be observed in the crystal structure. Nevertheless, the data show that the hydrogen bonds that form between the backbone amide and carbonyl of E167 and the backbone atoms of G202 and Y204 were present also in the A167 variant (Figure 5B), and the distances of the hydrogen bonds were unchanged. Likewise, hydrogen bonds between the backbone atoms of Y203 and R164 and between R164 and G202 were unchanged (Figure 5B). However, the interactions between the side chain of E167 and the carbonyl of P201, mediated by a water molecule, were absent in the mutant. The water molecule was reasonably well-ordered in the wild-type structure, with a temperature factor comparable to that of the E167 side chain oxygen (~34 Å2). This water molecule was not observed in the mutant structure. Instead, a new water molecule was observed in place of the E167 side chain, although it was not well-ordered (temperature factor of 51 Å2) (Figure 5B), and it was too far from the carbonyl of P201 to form a hydrogen bond (5.4 Å). Overall, the data suggest that the lower activity of the mutant resulted from the loss of the hydrogen bond between E167, in L2, and P201, in L3. Destabilizing the elbow loop, which contains P201, could very well affect the substrate binding loop, L3, and thus the ability of the variant to bind substrate. Alternatively, changes in the elbow loop may affect the stacking interactions among P201, R164, and Y197, shown in Figure 5B, which may in turn affect the catalytic cysteine, C163. We note also that the lower activity could be from changes in solution that are not observed in the crystals. That is, the conformation of the protein important for function may not be stabilized in the crystals since loops are flexible.
Figure 5.

X-ray crystal structures of caspase-3 variants. (A) Overlay of structures of wild-type caspase-3 (green), E167A (cyan), E173A (orange), and Y203F (yellow). (B) Stereoview of wild-type (green) and E167A (cyan) structures highlighting residues near E/A167. The red sphere represents a water molecule in the wild-type structure, whereas the yellow sphere represents a water molecule in the mutant structure. (C) Stereoview of wild-type (green) and E173A (orange) structures highlighting contacts in residues near E/A173. (D) Stereoview of wild-type (green) and Y203F (yellow) structures highlighting contacts in residues near Y/F203. The red spheres represent water molecules. For panels B–D, the small numbers represent distances (in angstroms) between two atoms connected by the dashed line, and the primes denote residues from the second monomer.
For the E173A variant, we observed electron density for the small subunit starting at H185 rather than G177, as for the wild-type protein. However, the side chain of H185 still was positioned to hydrogen bond with the backbone carbonyl of T245′ (Figure 5C). In E173A, we clearly observed electron density for the backbone and side chain of T174 of the large subunit. Modeling the backbone of T174 to fill the electron density resulted in a rotation of the E/A173 backbone atoms around the ψ bond when compared to the earlier structure of caspase-3 (11). The rotation of the E/A173 backbone atoms resulted in the relative proximity of the E/A173 carbonyl (in L2) to the side chain of R149′, which is located in a turn between helices 2′ and 3′ of the second heterodimer (Figure 5C). The distance between these atoms was 3.1 Å. In addition, the backbone amide of T174 was within hydrogen bonding distance of the carbonyl oxygen of H185′ from L2′ (2.8 Å). As shown previously (30), these new interactions further confirmed that the electron density in this region represented the backbone and side chain atoms of T174 rather than the side chain of E173. From the solution properties of the E173A variant described above, we found that the mutation affected the conformation of L1 and L3 as well as the environment of the tryptophanyl residues in the active site. Overall, these effects on the conformation were not sufficiently large to affect the activity. While there is little electron density for the side chain atoms of E173 in our data, it was not predicted to interact with other groups on the protein since it appeared to be oriented toward the solvent. Erlanson and co-workers made similar observations in a caspase-3 bound to a tethered inhibitor (30). Overall, the structure of E173A demonstrated little difference from that of wild-type caspase-3, although as noted above, crystal contacts in the regions of L1 and L4 may prevent us from observing changes in these regions that could be correlated to the solution properties of the protein. We note, however, that like E167A, replacing E173 at the C-terminus of the large subunit affected the stability of the N-terminus of the small subunit since there was no electron density for G177–C184 in the mutant. At present, it is not clear how replacing the E173 side chain in L2 could affect the N-terminus of the small subunit since there are no predicted polar contacts for the side chain of E173 except to solvent.
Like the other mutants, there was little difference between the structures of the Y203F variant and wild-type caspase-3, with a rmsd of 0.18 Å (Figure 5D and Table 3), although this mutation also affected the stability of the N-terminus of the small subunit. For Y203F, we observed electron density for K186 but none for G177–H185. We note that all of the hydrogen bonds formed by E167 in wild-type caspase-3 also formed in the mutant, although for clarity, E167 is not shown in Figure 5D. All of the backbone and side chain interactions involving E167 and D169, described above, were observed in the Y203F mutant as well, including the water-mediated E167–P201 interactions. However, electron density was missing for residues beyond D169, showing that the chain was more flexible for C170–D175 in the mutant. Interestingly, the increased flexibility appeared to be due to loss of hydrogen bonding interactions between F203 and the backbone amide of C170. In wild-type caspase-3, these interactions were mediated by two water molecules (Figure 5D). The first of these was 2.6 Å from the hydroxyl group of Y203, and as shown in Figure 1E, the water molecule was thought to mediate interactions across the dimer interface between Y203 and R144′. However, the data for Y203F showed that the water molecule instead mediated interactions between the hydroxyl of Y203 and the amide nitrogen of C170 through a second water molecule that was 2.9 Å from the NH group of C170 (Figure 5D). The water molecules were absent in the mutant. The lack of interactions between Y203 and C170 appeared to destabilize the chain beyond D169, suggesting that the putative interactions between Y203F and R144′ across the dimer interface were less important structurally than the Y203–C170 interactions. The R144′ side chain was mostly unaffected by the mutation, which lends support to this conclusion. In addition, the data showed that the Y203 hydroxyl likely does not interact with the third water molecule that is 3.4 Å away (Figures 1E and 5D) since the water molecule was present in the mutant as well. Overall, the Y203F mutation had little effect on the enzyme activity, so destabilizing the C-terminus of the large subunit from C170 to D175 did not affect activity. However, it did affect the accessibility of K57/R64, in L1, and R207, in L3, to cleavage by trypsin, and it affected the active site environment of the tryptophan residues as observed in changes in fluorescence emission and accessibility to quenching by iodide.
DISCUSSION
Biochemical and biophysical studies presented here show that all four mutations affected the solution properties of the enzyme. While the enzyme activity was mostly unaffected in the E173A and Y203F mutants, a larger effect was observed in the E167A and D169A mutants. For D169A, the protein did not autoprocess when expressed in E. coli cells, as does wild-type procaspase-3. Treatment of the mutant with granzyme B, which cleaves in the intersubunit linker of procaspase-3 following D175, showed that the processing site was accessible for cleavage, and circular dichroism studies showed that there were no large conformational changes in the protein. The granzyme B-processed D169A variant exhibited little measurable activity, with a KM of > 70 μM. Thus, of the four mutants that were examined, the data showed that the contacts provided by D169 are critical for both producing and maintaining the active site of the mature caspase-3.
The results presented here are surprising for several reasons. First, in caspase-3, the side chain of E173 did not appear to have contacts with other residues. Although the backbone was involved in hydrogen bonding interactions, these should not be affected by replacing the side chain. When E173A was coupled to the D175A variant, the procaspase was alternately processed in L3, and we showed that this occurred by autolysis. At present, it is not clear why the E173A/D175A double mutations resulted in this property since neither of the single mutations resulted in the alternate processing. The ensuing caspase was not catalytically active, which probably was due to cleavage of the substrate binding loop at N208. Second, cleavage of the D169A variant at D175 was not sufficient for activation, as it was for wild-type caspase-3, and this probably was due to the loss of hydrogen bonds among L2, L4, and L2′ centered around D169. The data also showed that replacing D169 abrogated activity in the procaspase and decreased the stability of the procaspase dimer. While it is not known whether the contacts provided by D169 are present in the procaspase as well, the data suggest that this is the case. Third, the Y203F and E167A variants showed the importance of hydrogen bonds mediated by water molecules in stabilizing the loop bundle. E167 interacted with P201 within the elbow loop, whereas Y203 interacted with C170 in L2. In both cases, replacing the side chain resulted in the loss of the water molecules. In addition, removal of the hydroxyl group from the tyrosyl side chain in the Y203F variant and subsequent loss of two water molecules resulted in increased flexibility for all residues C-terminal to D169 in the large subunit.
From the crystal structures of E167A, E173A, and Y203F, we found that the three mutations affected the electron density of the N-terminus of the small subunit (residues 176–185), among other changes. The proteins, in general, demonstrated similar solution properties. Namely, there were changes in fluorescence emission, accessibility of the active site tryptophans to quencher, and accessibility of residues in L1 and L3 to cleavage by trypsin. One interpretation of the results is that the point mutations resulted in conformational changes in the active site. However, the mutations were approximately 20 Å from R64 and R207 in L1 and L3, respectively. Therefore, structural perturbations caused by the mutations would occur over reasonably long distances. With the exception of E167A and D169A, as noted above, the mutations had relatively minor effects on the caspase-3 activity, and the structural data for the holoenzymes showed no changes in the active sites of the mutants. Overall, the results show that the E167A, E173A, and Y203F mutations do not result in large structural perturbations of the active site residues, except for the noted changes in the elbow loop for E167A. We suggest an alternative interpretation in which the effects of the mutations on the observed solution properties are due to the increased flexibility in the N-terminus of the small subunit since this region was affected in the three mutants. The N-terminal region, which includes S176–H185, is located near the base of L4 and extends toward L3 such that D180 is 4.9 Å from K210 in L3. These residues may, in effect, shield the wild-type active site from solvent in a manner that affects tryptophan fluorescence emission and accessibility of the iodide quencher. While the mechanism is not yet clear, the increased flexibility of this region, observed in the mutants, may decrease the accessibility of residues in L1 and L3 to trypsin as well as affect the other solution properties. Since it does not bind in the active site, the N-terminal segment would not be expected to occlude substrate binding. It is important to note that the mutations also affected conformational changes that occur as the pH is lowered. While there are no obvious direct interactions among residues 176–185 and other residues in the protein, there are several acidic amino acids that could be titrated as the pH is lowered. Finally, at pH <4, where the monomer is populated, the results show that E167A affected the stability of the procaspase monomer. At present, it is not clear how this residue is involved in stabilizing the monomer.
One remaining uncertainty is the degree to which the contacts observed in the loop bundle of caspase-3 occur in the procaspase. The loop bundle contacts are not observed in procaspase-7, where L3 is extended away from the active site such that P201 (caspase-3 numbering), from the elbow loop, and Y203 reside in the substrate binding pocket. Because the intersubunit linker occupies the central cavity to connect the large and small subunits, residues E167, D169, and E173, which is a glutamine in procaspase-7, are completely exposed to solvent. Indeed, one observes that E167, D169, E173, and Y203 do not interact with other residues in procaspase-7 (17, 18). So, while mutating these residues may affect the properties of caspase-7, once the loop bundle forms, one predicts that there should be no effect on the procaspase. Clearly, this is not the case for procaspase-3. Our results show that the residues are important for maintaining the active site of the procaspase as well as the caspase. Procaspase-3 may exist in a conformation very similar to that of the cleaved caspase, explaining why the mutations affect the procaspase. However, due to the uncleaved linker and the resulting strain on this conformation, the equilibrium is shifted to favor the inactive state of the protein, as displayed in the structure of procaspase-7. We caution that our results do not imply necessarily that the same contacts occur in the procaspase as in the caspase. Indeed, the results suggest that there are fewer stabilizing interactions in the procaspase active site loops. So, removal of one or two interactions had a much larger effect on the procaspase structure and activity than it did on the mature caspase. Of particular note is the fact that the mutations did not affect the stability of the dimer, with the exception of D169A. The data indicate that the side chain of D169 is involved in interactions across the dimer interface in the procaspase. We conclude that the four residues form contacts in the pro-caspase that are important for stabilizing the active site, the dimer (D169A), and the monomer (E167A). This would not be expected if the sites were solvent exposed as in the model for procaspase-7. In addition to these properties, the residues stabilize the N-terminus of the small subunit (all four residues) and the C-terminus of the large subunit (Y203F) in the caspase.
Acknowledgments
We thank Sean Casey for preliminary studies of iodide quenching and the research agencies of North Carolina State University and the North Carolina Agricultural Research Service for continued technical support. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract W-31-109-ENG-38.
Footnotes
This work was supported by National Institutes of Health Grant GM065970 to A.C.C.
Abbreviations: Ac-DEVD-AFC, N-acetyl-Asp-Glu-Val-Asp-(7-amino-4-trifluoromethylcoumarin); Ac-DEVD-CMK, N-acetyl-Asp-Glu-Val-Asp-chloromethyl ketone; BCIP, 5-bromo-4-chloro-3-indolyl phosphate p-toluidine salt; dNTP, deoxynucleotide triphosphate; DTT, dithiothreitol; DEAE, diethyl amino ethyl; DFP, diisopropyl fluoro-phosphate; DMSO, dimethyl sulfoxide; IPTG, isopropyl β-D-thioga-lactopyranoside; MPD, 2-methyl-2,4-pentanediol; NTB, p-nitro blue tetrazolium chloride; PEG, polyethylene glycol; PMSF, phenylmethane-sulfonyl fluoride; procaspase-3(D3A), procaspase-3(D9A/D28A/D175A); TLCK, N-α-P-tosyl-L-lysine chloromethyl ketone; TPCK, N-tosyl-L-phenylalanine chloromethyl ketone.
SUPPORTING INFORMATION AVAILABLE
V8 protease digests of (pro)caspase variants (Figure 1), trypsin protease digests of (pro)caspase variants (Figure 2), quenching by iodide or acrylamide (Figure 3), and average emission wavelength, 〈λ〉, versus pH (Figure 4). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Cain K, Brown DG, Langlais C, Cohen GM. Caspase activation involves the formation of the aposome, a large (~700 kDa) caspase-activating complex. J Biol Chem. 1999;274:22686–22692. doi: 10.1074/jbc.274.32.22686. [DOI] [PubMed] [Google Scholar]
- 2.Pop C, Chen Y-R, Smith B, Bose K, Bobay B, Tripathy A, Franzen S, Clark AC. Removal of the pro-domain does not affect the conformation of the procaspase-3 dimer. Biochemistry. 2001;40:14224–14235. doi: 10.1021/bi011037e. [DOI] [PubMed] [Google Scholar]
- 3.Boatright KM, Renatus M, Scott FL, Sperandio S, Shin H, Pedersen IM, Ricci JE, Edris WA, Sutherlin DP, Green DR, Salvesen G. A unified model for apical caspase activation. Mol Cell. 2003;11:529–541. doi: 10.1016/s1097-2765(03)00051-0. [DOI] [PubMed] [Google Scholar]
- 4.Donepudi M, Sweeney AM, Briande C, Grutter MG. Insights into the regulatory mechanism for caspase-8 activation. Mol Cell. 2003;11:543–549. doi: 10.1016/s1097-2765(03)00059-5. [DOI] [PubMed] [Google Scholar]
- 5.Zou H, Li Y, Liu X, Wang X. An apaf-1β cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem. 1999;274:11549–11556. doi: 10.1074/jbc.274.17.11549. [DOI] [PubMed] [Google Scholar]
- 6.Medema JP, Scaffidi C, Kischkel FC, Shevchenko A, Mann M, Krammer PH, Peter ME. FLICE is activated by association with the CD95 death-inducing signaling complex (DISC) EMBO J. 1997;16:2794–2804. doi: 10.1093/emboj/16.10.2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stennicke HR, Deveraux QL, Humke EW, Reed JC, Dixit VM, Salvesen GS. Caspase-9 can be activated without proteolytic processing. J Biol Chem. 1999;274:8359–8362. doi: 10.1074/jbc.274.13.8359. [DOI] [PubMed] [Google Scholar]
- 8.Srinivasula SM, Hegde R, Saleh A, Datta P, Shiozaki E, Chai J, Lee R-A, Robbins PD, Fernandes-Alnemri T, Shi Y, Alnemri ES. A conserved XIAP-interaction motif in caspase-9 and Smac/DIABLO regulates caspase activity and apoptosis. Nature. 2001;410:112–116. doi: 10.1038/35065125. [DOI] [PubMed] [Google Scholar]
- 9.Bose K, Pop C, Feeney B, Clark AC. An uncleavable procaspase-3 mutant has a lower catalytic efficiency but an active site similar to that of mature caspase-3. Biochemistry. 2003;42:12298–12310. doi: 10.1021/bi034998x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stennicke HR, Jurgensmeier JM, Shin H, Deveraux Q, Wolf BB, Yang X, Zhou Q, Ellerby M, Ellerby LM, Bredesen D, Green DR, Reed JC, Froelich CJ, Salvesen GS. Pro-caspase-3 is a major physiologic target of caspase-8. J Biol Chem. 1998;273:27084–27090. doi: 10.1074/jbc.273.42.27084. [DOI] [PubMed] [Google Scholar]
- 11.Mittl PRE, DiMarco S, Krebs JF, Bai X, Karanewsky DS, Priestle JP, Tomaselli KJ, Grutter MG. Structure of recombinant human CPP32 in complex with the tetrapeptide acetyl-asp-val-ala-asp fluoromethyl ketone. J Biol Chem. 1997;272:6539–6547. doi: 10.1074/jbc.272.10.6539. [DOI] [PubMed] [Google Scholar]
- 12.Rotonda J, Nicholson DW, Fazil KM, Gallant M, Gareau Y, Labelle M, Peterson EP, Rasper DM, Ruel R, Vaillancourt JP, Thornberry NA, Becker JW. The three-dimensional structure of apopain/CPP32, a key mediator of apoptosis. Nat Struct Biol. 1996;3:619–625. doi: 10.1038/nsb0796-619. [DOI] [PubMed] [Google Scholar]
- 13.Chai J, Shiozaki E, Srinivasula SM, Wu Q, Dataa P, Alnemri ES, Shi Y. Structural basis of caspase-7 inhibition by XIAP. Cell. 2001;104:769–780. doi: 10.1016/s0092-8674(01)00272-0. [DOI] [PubMed] [Google Scholar]
- 14.Blanchard H, Kodandapani L, Mittl PRE, Di Marco S, Krebs JF, Wu JC, Tomaselli KJ, Grutter MG. The three-dimensional structure of caspase-8: An initiator enzyme in apoptosis. Structure. 1999;7:1125–1133. doi: 10.1016/s0969-2126(99)80179-8. [DOI] [PubMed] [Google Scholar]
- 15.Wilson KP, Black J-AF, Thomson JA, Kim EE, Griffith JP, Navia MA, Murcko MA, Chambers SP, Aldape RA, Raybuck SA, Livingston DJ. Structure and mechanism of interleukin-1β converting enzyme. Nature. 1994;370:270–275. doi: 10.1038/370270a0. [DOI] [PubMed] [Google Scholar]
- 16.Schweizer A, Briande C, Grutter MG. Crystal structure of caspase-2, apical initiator of the intrinsic apoptotic pathway. J Biol Chem. 2003;278:42441–42447. doi: 10.1074/jbc.M304895200. [DOI] [PubMed] [Google Scholar]
- 17.Chai J, Wu Q, Shiozaki E, Srinivasula SM, Alnemri ES, Shi Y. Crystal structure of a procaspase-7 zymogen: Mechanisms of activation and substrate binding. Cell. 2001;107:399–407. doi: 10.1016/s0092-8674(01)00544-x. [DOI] [PubMed] [Google Scholar]
- 18.Riedl SJ, Fuentes-Prior P, Renatus M, Kairies N, Krapp S, Huber R, Salvesen GS, Bode W. Structural basis for the activation of human procaspase-7. Proc Natl Acad Sci USA. 2001;98:14790–14795. doi: 10.1073/pnas.221580098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fuentes-Prior P, Salvesen GS. The protein structures that shape caspase activity, specificity activation and inhibition. Biochem J. 2004;384:201–232. doi: 10.1042/BJ20041142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pop C, Feeney B, Tripathy A, Clark AC. Mutations in the procaspase-3 dimer interface affect the activity of the zymogen. Biochemistry. 2003;42:12311–12320. doi: 10.1021/bi034999p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feeney B, Pop C, Tripathy A, Clark AC. Ionic interactions near loop L4 are important for maintaining the active site environment and the dimer stability of (pro)caspase-3. Biochem J. 2004;384:515–525. doi: 10.1042/BJ20040693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stennicke HR, Salvesen GS. Caspases: Preparation and characterization. Methods. 1999;17:313–319. doi: 10.1006/meth.1999.0745. [DOI] [PubMed] [Google Scholar]
- 23.Royer CA, Mann CJ, Matthews CR. Resolution of the fluorescence equilibrium unfolding profile of trp aporepressor using single tryptophan mutants. Protein Sci. 1993;2:1844–1852. doi: 10.1002/pro.5560021106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brunger AT, Adams PD, Clore GM, Delano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Panni NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 25.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 26.Bose K, Clark AC. pH effects on the stability and dimerization of procaspase-3. Protein Sci. 2005;14:24–36. doi: 10.1110/ps.041003305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feeney B, Clark AC. Reassembly of active caspase-3 is facilitated by the propeptide. J Biol Chem. 2005;280:39772–39785. doi: 10.1074/jbc.M505834200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ni CZ, Li C, Wu JC, Spada AP, Ely KR. Conformational restrictions in the active site of unliganded human caspase-3. J Mol Recognit. 2003;16:121–124. doi: 10.1002/jmr.615. [DOI] [PubMed] [Google Scholar]
- 29.Becker J, Rotonda J, Soisson SM, Aspiotis R, Bayly C, Francoeur S, Gallant M, Garcia-Calvo M, Giroux A, Grimm E, Han Y, McKay D, Nicholson DW, Peterson E, Renaud J, Roy S, Thornberry N, Zamboni R. Reducing the peptidyl features of caspase-3 inhibitors: A structural analysis. J Med Chem. 2004;47:2466–2474. doi: 10.1021/jm0305523. [DOI] [PubMed] [Google Scholar]
- 30.Erlanson DA, Lam JW, Wiesmann C, Luong TN, Simmons RL, DeLano WL, Choong IC, Burdett MT, Flanagan WM, Lee D, Gordon EM, O’Brien T. In situ assembly of enzyme inhibitors using extended tethering. Nat Biotechnol. 2003;21:308–314. doi: 10.1038/nbt786. [DOI] [PubMed] [Google Scholar]
- 31.Lee D, Long SA, Adams JM, Chan G, Vaidya KS, Francis TA, Kikly K, Winkler JD, Sung CM, Debouck C, Richardson S, Levy MA, DeWolf WE, Jr, Keller PM, Tomaszek T, Head MS, Ryan DM, Haltiwanger RC, Liang PH, Janson CA, McDevitt PJ, Johanson K, Concha NO, Chan W, Abdel-Meguid SS, Badger AM, Lark MW, Nadeau DP, Suva LJ, Gowen M, Nuttall ME. Potent and selective nonpeptide inhibitors of caspases 3 and 7 inhibit apoptosis and maintain cell functionality. J Biol Chem. 2000;275:16007–16014. doi: 10.1074/jbc.275.21.16007. [DOI] [PubMed] [Google Scholar]
- 32.Riedl SJ, Renatus M, Schwarzenbacher R, Zhou Q, Sun C, Fesik SW, Liddington RC, Salvesen GS. Structural basis for the inhibition of caspase-3 by XIAP. Cell. 2001;104:791–800. doi: 10.1016/s0092-8674(01)00274-4. BI0611964. [DOI] [PubMed] [Google Scholar]