

Abstract
ATP-dependent cob(I)alamin adenosyltransferase (ATR) is a bifunctional protein; an enzyme that catalyzes the adenosylation of cob(I)alamin and an escort that delivers the product, adenosylcobalamin (AdoCbl or coenzyme B12), to methylmalonyl-CoA mutase (MCM), resulting in holoenzyme formation. Failure to assemble holo-MCM leads to methylmalonic aciduria. We have previously demonstrated that only two equivalents of AdoCbl bind per homotrimer of ATR and that binding of ATP to the vacant active site triggers ejection of one equivalent of AdoCbl from an adjacent site. In this study, we have mimicked in the Methylobacterium extorquens ATR, a C-terminal truncation mutation, D180X, described in a patient with methylmalonic aciduria, and characterized the associated biochemical penalties. We demonstrate that while kcat and KMCob(I) for D180X ATR are only modestly decreased (by 3- and 2-fold, respectively), affinity for the product, AdoCbl, is significantly diminished (400-fold) and the negative cooperativity associated with its binding is lost. We also demonstrate that the D180X mutation corrupts ATP-dependent cofactor ejection, which leads to transfer of AdoCbl from wild-type ATR to MCM. These results suggest that the pathogenicity of the corresponding human truncation mutant results from its inability to sequester AdoCbl for direct transfer to MCM. Instead, cofactor release into solution is predicted to reduce the capacity for holo-MCM formation, leading to disease.
Coenzyme B12 or 5′-deoxyadenoslycobalamin (AdoCbl) is a complex organometallic cofactor that is used in all domains of life by enzymes catalyzing diverse reactions including: dehydrations, intramolecular carbon skeleton rearrangements, reductive eliminations, and deaminations (1–3). AdoCbl comprises a central cobalt ion coordinated equatorially by four nitrogen atoms donated by the corrin macrocycle. A novel nucleotide base, dimethylbenzimidazole (DMB), occupies the lower axial coordination position while the upper axial position is occupied by the 5′-deoxyadenosyl group, via the organometallic cobalt-carbon bond.
AdoCbl is a “high value” product that is either biosynthesized de novo by some bacteria or generated via adenosylation of cob(I)alamin in organisms such as mammals, which lack a de novo pathway for its synthesis (4). There are at least three families of ATP-dependent cob(I)alamin adenosyltransferases (ATR): EutT, PduO, and CobA, which catalyze the addition of the 5′-deoxyadenosyl group from ATP to cob(I)alamin; however, their roles in metabolism are distinct. Members of the three ATR subfamilies do not share sequence or structural homology and represent examples of convergent evolution. In some organisms, such as Salmonella enterica, members of all three ATR subfamilies are used to meet metabolic requirements under distinct physiological conditions (4). The EutT- and PduO-type ATRs of S. enterica furnish AdoCbl for the B12-dependent enzymes, diol dehydratase and ethanolamine ammonia lyase, for catabolism of alkane diols and growth on ethanolamine, respectively (2, 5). In contrast, CobA is involved in the de novo synthesis of AdoCbl (6). Humans use a PduO-type ATR for supplying AdoCbl to the only enzyme that uses this cofactor, i.e. MCM, for catabolism of cholesterol, branched chain amino acids, and odd-chain fatty acids (7, 8).
To overcome the twin challenges posed by the lability of the cofactor in the three biologically relevant cobalt oxidation states and its low tissue concentration in humans, it has been proposed that following intracellular delivery, B12 remains protein-bound as it is processed and delivered to its target enzymes (9, 10). In support of this model, it has been demonstrated that following AdoCbl synthesis by ATR, the cofactor is directly transferred to the active site of MCM via a transient protein-protein interaction (11–13). Mutations in ATR lead to deficiencies in the enzymatic activity of MCM resulting in methylmalonic aciduria (7, 14, 15).
Structure-function studies on PduO-type ATRs from Lactobacillus reuteri (16–19) and Methylbacterium extorquens (13, 20, 21) have provided mechanistic insights into these enzymes. Cob(II)alamin binds to ATR in a “base-off” conformation in which the lower axial DMB base is displaced from the cobalt ion (22, 23). A yet unknown mitochondrial oxidoreductase catalyzes the one electron reduction of mammalian ATR-bound cob(II)alamin, yielding cob(I)alamin (24) (equation 1). Cob(I)alamin is a potent nucleophile and attacks the C-5′ribosyl carbon of ATP forming PPPi and AdoCbl, which is “base-off” and five-coordinate (16, 25, 26).
| [1] |
In contrast, AdoCbl bound to MCM is in a six-coordinate “base-off/His-on” conformation in which a histidine residue donated by MCM, occupies the lower axial position (1). This difference in coordination number results in large differences in the absorption spectra and is experimentally useful for locating the cofactor (13). Thus, “base-off” AdoCbl bound to ATR exhibits an absorbance maximum at 458 nm while “base-off/His-on” AdoCbl bound to MCM or free in solution has a maximum at 525 nm (13).
PduO-type ATR is a homotrimer that appears to use only two of its three active sites at a time and exhibits negative cooperativity for substrate and product binding (13, 20, 27). In the crystal structure of human PduO-type ATR, two equivalents of ATP are bound per homotrimer (28). ATP binding to the vacant site in the M. extorquens ATR, triggers ejection of only one of two equivalents of AdoCbl providing a mechanism for driving transfer to MCM in what has been described as a “rotary mechanism” (Fig. 1) (27). However, the crystal structures of the L. reuteri ATR show occupancy of all three active sites with either ATP or cob(II)alamin (18). This difference may arise from altered behavior in the crystalline versus solution state or differences between PduO-type ATRs from different organisms. The apparent conservation of function between human and M. extorquens PduO-type ATR suggest that the M. extorquens ATR is a viable model for the human enzyme.
Figure 1.
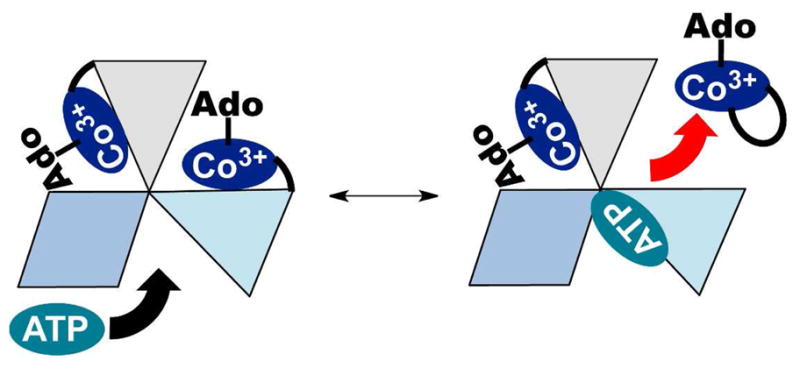
ATP binding triggers AdoCbl release. The ATR•product complex has two of three active sites occupied by AdoCbl. The three subunits of ATR are depicted in shades of blue an grey. ATP binds to the ATR•2AdoCbl complex and triggers the release of one equivalent of AdoCbl. In the presence of MCM, AdoCbl is transferred directly in the “base-off” conformation.
The ATR monomer comprises a five-helix bundle arranged in a head-to-tail fashion relative to the neighboring subunit. The active sites are located at the subunit interfaces (Fig. 2) (18, 28). Residues at the N-terminus become ordered in response to ATP-binding and residues from the adjacent monomer contribute to the ATP binding site. Cobalamin binds in a C-terminal crevice and backbone amides from the adjacent subunit participate in the majority of hydrogen bonding interactions with the amide substituents on the corrin ring (18). Two conformational changes accompany binding of B12: the ordering of the C-terminus and positioning of a mobile loop proximal to the lower axial face of B12. This loop is hydrogen bonded to B12 and its movement facilitates generation of four-coordinate cob(II)alamin (17). In contrast, the significance of the C-terminal ordering for ATR function is less well understood. Truncation of the final 16 residues in human ATR due to the nonsense Q234X mutation, results in early onset methylmalonic aciduria (15). Structural studies on LrPduO-Δ183–188, an L. reuteri ATR missing the terminal six amino acids, revealed decreased conformational rigidity of the mobile loop, although cob(II)alamin was nonetheless bound in a four-coordinate “base-off” state (17). The LrPduO-Δ183–188 mutant exhibited a modest decrease in the catalytic efficiency relative to wild-type ATR, providing limited insight into the role of the C-terminal tail and the biochemical basis of pathogenicity of the Q234X patient mutation (17).
Figure 2.

Structure of PduO-type ATR from L. reuteri with AdoCbl and ATP bound (PDB entry 3CI1). The ATR holoenzyme is shown in ribbon representation with the individual subunits of the ATR trimer are colored as in Fig. 1. ATP and cobalamin are shown in stick representation in cyan and dark blue, respectively. The C-terminal loop (red) in one subunit corresponds to the boundaries of the truncation in M. extorquens D180X ATR (D180–R190).
In this study, we have mimicked the Q234X mutation in the M. extorquens ATR by introducing the D180X mutation, which corresponds to the site of the nonsense mutation in the human sequence (Fig. 3). We demonstrate that while this C-terminal truncation has modest effects on the steady-state kinetic parameters, the affinity for the product, AdoCbl, and allosteric communication between active sites leading to negative cooperativity, are adversely affected. These changes lead to corruption of the direct transfer mechanism of AdoCbl from ATR to MCM, and provide insights into the importance of the C-terminal loop in gating cofactor transfer and for communication between adjoining active sites.
Figure 3.

Sequence comparison of select PduO-type ATRs. Multiple sequence alignment of PduO-type ATRs in which amino acids that are identical (black) or conserved (grey) are highlighted. The sequence of the human ATR includes an N-terminal 32 amino acid mitochondrial leader sequence. An asterisk indicates the position of residue D180.
Experimental Procedures
Materials
MANT-ATP (2′ (3′)-O-(N-methylanthraniloyl)-ATP) was purchased from Jen Biosciences (Germany). ATP, AdoCbl, phenylmethylsulfonyl fluoride, lysozyme, and all other chemical reagents were purchased from Sigma. The Stratagene Quikchange® II XL Site-Directed Mutagenesis Kit was purchased from Agilent.
Construction of the D180X ATR Mutant
The mutation was created using the Quikchange kit and the following primers: Sense: 5′-CAACCGCGACGGCGCCTAGGACGTGCTCTGGGTG-3′ and an antisense primary containing the complimentary sequence. The 6xHis-wild-type ATR expression plasmid described previously (20), was used as template. The mutation was confirmed by nucleotide sequence determination at the University of Michigan DNA Sequencing Core.
Expression and Purification of ATR
Escherichia coli (BL21 (DE3)) was transformed with plasmids encoding recombinant D180X ATR or 6xHis-wild-type ATR and grown at 37 °C in 1 L of Luria-Bertani media containing kanamycyin (50 μg/mL). Cultures were grown to an optical density at 600 nm of 0.5–0.6 and induced with either 0.1 mM (for D180X ATR) or 1 mM (for wild-type ATR) isopropyl-β-D-thiogalactopyranoside. Cultures were then grown at 20 °C for 14–16 h (D180X ATR) or at 37 °C for 6–10 h (wild-type ATR) prior to harvesting.
ATR (wild-type and D180X) was purified as follows. Bacterial pellets were suspended in 50 mM sodium phosphate buffer, pH 8.0, containing 0.3 M KCl, 20 mM imidazole, and 1 mM phenylmethylsulfonyl fluoride (Buffer A). Cells were lysed first by treatment with lysozyme (25 mg/L culture; 40,000 U/mg) for 1 h with stirring at 4 °C, and then disrupted by sonication as described previously (20). The pellet was separated by centrifugation at 19,000 × g at 4 °C. The supernatant was filtered through a 0.4 μm syringe filter, and diluted to 4–5 mg protein/mL before loading onto a 50-mL nickel-nitrilotriacetic acid-Sepharose column pre-equilibrated with 10–20 column volumes of Buffer A. The column was washed with 10 column-volumes of Buffer A and eluted with a linear gradient of 20–300 mM imidazole in Buffer A. Fractions containing the target protein were pooled, concentrated, dialyzed extensively against 50 mM HEPES, pH 8, 0.3 M KCl, 5 % glycerol (Buffer B) at 4 °C, and then dialyzed extensively against Buffer B containing 10 mM MgCl2 at 4 °C. This purification protocol typically yielded 15–20 mg of ≥ 95% pure protein per liter of culture.
ATR Activity Assays
ATR activity was monitored in a steady-state assay as described previously (20) and data were fit to the Michaelis-Menten equation to obtain the kinetic parameters. The KM for ATP was determined at saturating HOCbl (55 μM) and varying ATP (from 0.5–300 μM). The KM for HOCbl was determined at saturating ATP (1 mM) and varying HOCbl (from 0.5–60 μM) concentrations.
Thermal Denaturation Assays
Thermal denaturation of ATR was performed to assess the relative stabilities of the wild-type and mutant proteins. For this, 0.5 mg/mL of purified ATR in Buffer B containing 10 mM MgCl2 was placed in a cuvette and the temperature was increased from 4–68 °C in a Cary 100 Bio UV/Vis spectrophotometer with a heating block connected to a Fisher Scientific Isotemp 3016S water bath. Unfolding was measured by increased light scattering at 600 nm in 5 °C increments until no further change in the optical density was observed. Data were analyzed as described previously (29).
Isothermal Titration Calorimetry (ITC)
ITC experiments were performed as described previously (13, 30) with the exception that the temperature was maintained at 4 °C due to the instability of D180X ATR at ambient temperature for prolonged periods. ITC experiments, conducted in triplicate, were analyzed with MicroCal ORIGIN software to determine the bimolecular association constant (KA), binding enthalpy (ΔH°), and binding entropy (ΔS°). Briefly, 40–45 μM D180X ATR in 50 mM HEPES, 0.3 M KCl, 10 mM MgCl2, 5 % glycerol at pH 8 was titrated with forty five 6 μL aliquots of 1–2 mM ATP or AdoCbl. Following integration of the calorimetric signals in MicroCal ORIGIN software, ITC data for the titration of D180X ATR with ATP were fitted to a two-sites model that describes two populations of active sites that bind ATP with distinct thermodynamic parameters. When D180X ATR was titrated with AdoCbl, a single-site model was used to fit the data. The Gibbs free energy of binding either ligand was calculated using equations 2 and 3.
| [2] |
| [3] |
Fluorescence and UV/visible Titrations
For the UV/visible titration of AdoCbl with D180X ATR, experiments were performed in quadruplicate and the large hypsochromic shift from 525 nm to 458 nm associated with the “base-on” to “base-off” transition for AdoCbl was used to monitor binding. Titrations were performed at 4 °C, and all stock solutions were kept chilled throughout the experiment. Briefly, 1.5–10 μM AdoCbl in 50 mM HEPES, pH 8 containing 0.3 M KCl, 10 mM MgCl2 and 5 % glycerol was titrated with 1–100 μM D180X ATR in the same buffer. Spectra were recorded 8–10 min after each addition of enzyme to allow binding to reach equilibrium. Data were fit to a hyperbolic binding isotherm to determine the dissociation constant, KD, for binding of AdoCbl to D180X ATR. For wild-type ATR, titrations were performed using 4 μM enzyme + 8 μM AdoCbl and aliquots of a 1 mM ATP stock solution. The corresponding titration with D180X ATR was performed using 130 μM enzyme, 2 μM AdoCbl, and aliquots of a 20 mM ATP stock solution. Titrations of MANT-ATP with D180X were monitored via the quenching of MANT fluorescence upon binding. Briefly, 10–15 μM of MANT-ATP in 50 mM HEPES, 0.3 M KCl, 10 mM MgCl2, 5 % glycerol at pH 8 was titrated with 100 μM D180X ATR in the same buffer to a final concentration of 35 μM protein at 4 °C. Fluorescence quenching was monitored at 444 nm (360 nm excitation) and its dependence on D180X ATR concentration was fitted to a hyperbolic binding isotherm. The MANT-ATP titrations were performed in triplicate.
Results
Stability of Wild-type and D180X ATR
During purification, the stability of the D180X mutant was noted to be lower than for wild-type ATR, as evidenced by precipitation especially during concentration. Comparison of the thermal denaturation profiles confirmed that the D180X mutant is indeed less stable relative to wild-type ATR with a ΔTm = 11.1 ± 0.4 °C (Fig. 4).
Figure 4.

Thermal melting curves for wild-type ATR (circles) and D180X ATR (squares). Plots were normalized to the maximum turbidity observed at 600 nm. Protein unfolding was monitored as described under Experimental Procedures.
Kinetic Characterization of Wild-type and D180X ATR
The initial velocity of the adenosylation reaction was monitored at 390 nm (corresponding to conversion of cob(I)alamin to AdoCbl) by varying the concentration of cob(I)alamin at saturating concentrations of ATP, or vice versa (Table 1). The apparent KMATP (8.4 ± 1.4 μM) for wild-type ATR is similar to previously reported values for the N-terminally His8-tagged human (KM, 7.2 μM) and L. reuteri (KM, 2 μM) ATRs. Similarly, the KMCob(I) for wild-type ATR is similar to values reported for other PduO-type ATRs (KM, 0.2–5 μM (8, 31–33)) but 5-fold lower than the value reported for His-tagged human ATR (31). The kcat for wild-type M. extorquens ATR is 4-fold larger than any value previously reported for a PduO-type ATR.
Table 1.
Kinetic Parameters of wild-type and D180X ATRa
| Enzyme | kcat (s−1) | ATP-dependence
|
Cob(I)alamin-dependence
|
||
|---|---|---|---|---|---|
| KM (μM) | kcat/KM (M−1s−1) | KM (μM) | kcat/KM (M−1s−1) | ||
| wild type | (8.9 ± 0.1) × 10−1 | 8.4 ± 1.4 | (5.0 ± 0.3) × 104 | 0.4 ± 0.1 | (2.5 ± 0.2) × 106 |
| D180X | (2.5 ± 0.1) × 10−1 | 9.5 ± 1.2 | (2.7 ± 0.1) × 104 | 0.8 ± 0.1 | (3.5 ± 0.1) × 105 |
The values represent the mean ± SD from at least three independent experiments in the adenosylation reaction.
The D180X mutation has modest effects on the steady-state kinetic parameters: KMcob(I) and kcat are 2- and 3.5-fold smaller, respectively, resulting in a 7-fold lower catalytic efficiency with respect to the cob(I)alamin substrate (Table 1). The KM for ATP is unaffected by the D180X mutation.
Thermodynamic Characterization of ATP and AdoCbl Binding
Binding of ATP to D180X ATR was characterized by ITC and by fluorescence spectroscopy using MANT-ATP (Fig. 5A). A monophasic binding isotherm was obtained from the fluorescence titration and yielded a KD value of 5.4 ± 1.1 μM for binding of MANT-ATP to D180X ATR. This value is comparable to the KD value of 5.9 ± 1.4 μM reported for binding of the second equivalent of ATP to wild-type ATR (27). In contrast, ITC data for ATP binding to D180X ATR exhibits biphasic behavior (Fig. 5B). The discrepancy between the fluorescence and ITC data may be derived from the low amplitude of fluorescence quenching upon binding of MANT-ATP to site 1 and is consistent with prior data on MANT-ATP binding to ATR by stopped-flow fluorescence spectroscopy (27).
Figure 5.

Binding of ATP to D180X ATR. (A) The inset shows a representative fluorescence titration of MANT-ATP (15 μM) with D180X ATR (0–40 μM) at 10 °C. (B) Representative calorimetric titration data for binding of ATP to ATR (50 μM) in Buffer B at 4 °C. The top panel depicts the raw data for ATP-binding in power versus time. The lower panel shows integration of data in the top panel, which is proportional to the heat release that accompanies binding as a function of time. The data in the lower panel were best fit to a two-sites binding model and yielded the thermodynamic parameters that govern ATP binding to D180X ATR at 4 °C (shown in Table 2).
Analysis of the ITC data yielded ΔG1o and ΔG2o values of −8.1 kcal/mol and −6.9 kcal/mol for binding of ATP to sites 1 and 2 in D180X ATR (Table 2), which are virtually identical to those reported for wild-type ATR (−8.3 kcal/mol and −6.6 kcal/mol, respectively). However, the relative contributions of the entropic and enthalpic terms to the ΔGo values differ between the D180X mutant and wild-type ATR. Thus, binding of ATP to sites 1 and 2 is enthalpically dominated (ΔH1o = −8.1 kcal/mol and ΔH2o = −6.0 kcal/mol) in wild-type ATR, while it is entropically driven in the D180X mutant (TΔS1o = 7.9 kcal/mol and TΔS2o = 3.4 kcal/mol).
Table 2.
Thermodynamic Parameters for Binding of ATP and AdoCbl to D180X ATR
| ATPa |
AdoCblb |
||
|---|---|---|---|
| Site 1 | Site 2 | Sites 1 and 2 | |
| KD (μM) | 0.48 ± 0.02 | 4.01 ± 0.61 | 44.1 ± 3.2 |
| ΔHo (kcal/mol) | −0.27 ± 0.02 | −3.40 ± 0.04 | 5.61 ± 0.26 |
| TΔSo (kcal/mol) | 7.86 ± 0.91 | 3.41 ± 0.04 | 11.10 ± 0.63 |
| ΔGo | −8.12 ± 0.90 | −6.87 ± 0.61 | −5.49 ± 0.89 |
| Nc (sites) | 0.38 ± 0.08 | 1.43 ± 0.07 | 2.07 ± 0.07 |
ITC data for ATP binding were fitted to a 2-site model.
The isotherms for binding of AdoCbl to D180X ATR were best fit to a single-binding-site model.
N represents the number of unique sites described by the fitting model.
Binding of AdoCbl is significantly impacted in the D180X mutant, which exhibits a KDAdoCbl of 71 ± 24 μM (Fig. 6A) in comparison to the 0.1 μM and 2.0 μM values for sites 1 and 2 reported for the wild-type enzyme (27). The relatively low affinity of the D180X mutant for AdoCbl necessitated high protein concentrations during spectrophotometric titrations and was limited by the instability of the mutant protein. Hence only ~83% saturation with AdoCbl could be observed in the spectrophotometric binding assay (Fig. 6A). In principle, the inability to observe binding of two equivalents of AdoCbl in the “base-off” state per trimer could arise from two possibilities: (i) only one equivalent of AdoCbl binds to the D180X mutant in the “base-off” conformation while the second equivalent binds in the “base-on” conformation and (ii) the dissociation constant for AdoCbl binding at site 2 is »70 μM measured by spectral titration. To distinguish between these possibilities, AdoCbl-binding was also monitored by ITC (Fig. 6B). The calorimetric titrations showed monophasic behavior for AdoCbl binding and the data were best fit to a single-binding site model with N = 2 per ATR homotrimer (Table 2). The ITC data confirm that AdoCbl binds to D180X ATR with an ~700- and 35-fold weaker binding affinity at sites 1 and 2 compared to wild-type enzyme. Hence, the C-terminal deletion leads to loss of negative cooperativity associated with AdoCbl binding to wild-type ATR.
Figure 6.

Binding of AdoCbl to D180X ATR. (A) The UV/visible spectra were obtained by titrating a fixed concentration of AdoCbl (7.5 μM) with increasing concentrations of D180X ATR (0–150 μM). Approximately 83% saturation of the two AdoCbl binding sites per trimer was achieved based on Δε525 = 6.69 M−1cm−1. (B) Representative ITC data for binding of AdoCbl to D180X ATR (50 μM) in Buffer B at 4 °C. The top panel depicts the raw data for AdoCbl-binding in power versus time. The lower panel shows integration of data in the top panel. The data in the lower panel were best fit to a single-site binding model and gave the thermodynamic parameters associated with binding of AdoCbl to D180X ATR at 4 °C.
Corruption of the ATP-triggered Mechanism for AdoCbl Unloading
A characteristic of AdoCbl transfer from ATR to MCM is the ATP-stimulated ejection of one equivalent of AdoCbl from ATR (Fig. 1) (20). To determine whether ATP triggers product release irrespective of whether one or two AdoCbl molecules are bound, wild-type ATR was loaded under conditions where a vast excess of active sites over AdoCbl (i.e. 100:1) resulted statistically, in the predominance of the 1:1 ATR:AdoCbl complex. Addition of 0–30 mM ATP to this complex (corresponding to a 0–200-fold excess of ATP over ATR) failed to trigger release of AdoCbl into solution, demonstrating that binding of ATP only releases AdoCbl from the low affinity site when two equivalents of AdoCbl are bound (Fig. 7).
Figure 7.
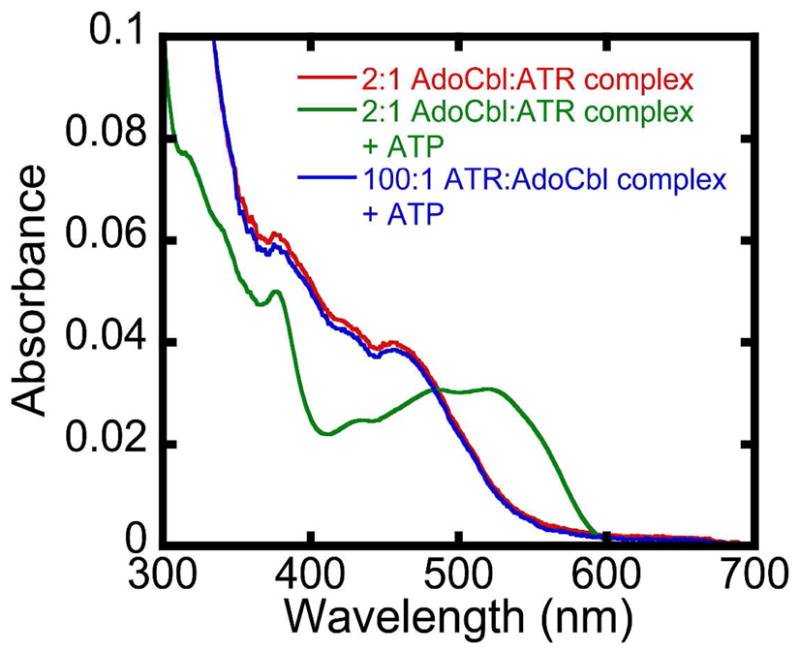
ATP-triggers release of AdoCbl only from the low affinity site in wild-type ATR. The difference in KDs for the two AdoCbl sites on ATR implies that when [ATR]≫[AdoCbl], site 2 is predominantly vacant and site 1 is occupied. Spectrum A is that of “base-off” AdoCbl in the 2:1 (AdoCbl•wild-type ATR) complex where the concentration of AdoCbl is identical to the concentration of AdoCbl-binding sites. Spectrum C is the spectrum after addition of 30 mM ATP to spectrum A (i.e. ~50% bound AdoCbl is released into solution upon addition of ATP). Spectrum B is of “base-off” AdoCbl when AdoCbl•ATR is 1:100 in the presence of 30 mM ATP. ATP is unable to stimulate release of AdoCbl under these conditions when only the high affinity binding site in ATR is occupied by AdoCbl and the low affinity site is vacant. Titrations were performed at 4 °C and recorded after 20 min to allow binding equilibration.
Next, the fidelity of the ATP-dependent AdoCbl release mechanism was assessed in the D180X mutant. The spectrum obtained upon mixing AdoCbl with a 50-fold excess of D180X ATR indicates that at least 85% of the cofactor is bound in the “base-off” conformation (Fig. 8). The low affinity and equivalence of AdoCbl binding to sites 1 and 2 in D180X ATR results in a statistical distribution of populations with 0, 1, or 2 equivalents of AdoCbl bound per ATR trimer. Addition of 5 mM ATP to this sample resulted in full displacement of all bound AdoCbl into solution as evidenced by the appearance of the “base-on” spectrum. The presence of free AdoCbl in solution was confirmed by separation using a Centricon filter.
Figure 8.
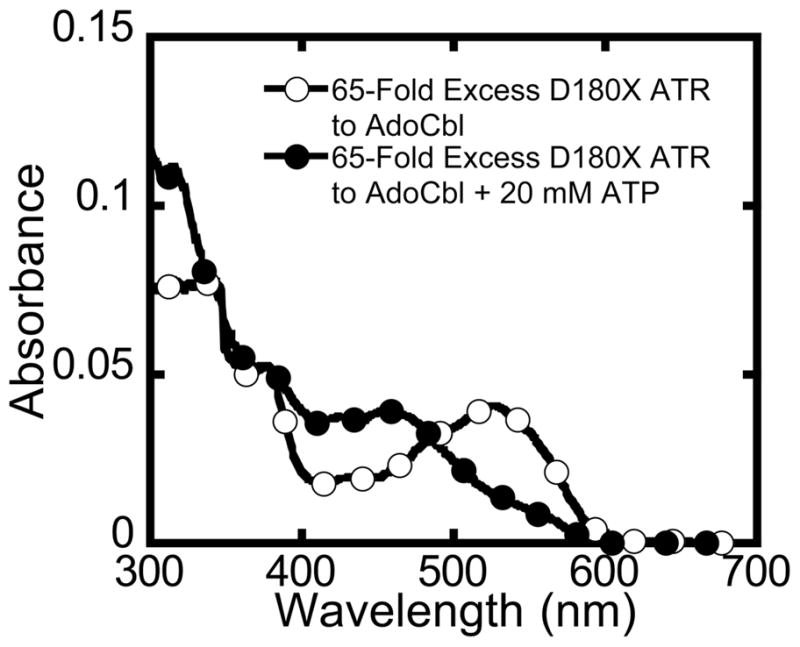
Corruption of the ATP-dependent mechanism for release of a single equivalent of AdoCbl in the D180X mutant. Mixing AdoCbl with a 65-fold excess of D180X ATR generated the spectrum (open circles) with ~90% bound AdoCbl at 4 °C in Buffer B. Due to the equivalence of sites 1 and 2 for AdoCbl binding in D180X ATR, the spectrum represents a statistical distribution of ATR with 0, 1 or 2 equivalents of AdoCbl bound at the active site. Addition of 20 mM ATP resulted in the complete displacement of AdoCbl from D180X ATR. Thus, whereas ATP is only capable of displacing AdoCbl from the low affinity site in wild-type ATR when 2 equivalents of AdoCbl are bound, the D180X mutant binds AdoCbl with equal and low affinity at both binding sites and ejects both equivalents of AdoCbl in response to ATP binding.
Discussion
In this study, we have mimicked the pathogenic Q243X truncation found in human ATR (17) to assess the role of the C-terminal tail in AdoCbl synthesis and transfer to MCM. AdoCbl levels are reduced ~70% in cultured fibroblasts harboring the Q234X deletion, and supplementation with exogenous B12 provides only mild stimulation of MCM activity (15). The corresponding M. extorquens D180X mutant, exhibits lower protein stability (Fig. 4) and the steady-state kinetic parameters for adenosylation of cob(I)alamin are only modestly impacted (Table 1). The steady-state kinetic parameters of the LrPduOΔS183–188 mutant, designed to mimic the Q243X mutation in the human sequence, are similarly mildly affected compared to wild-type ATR. These results suggest that the C-terminus does not play a critical role in synthesis of AdoCbl at least under in vitro conditions (17). However, as indicated by both the primary sequence alignment and the crystal structure of the LrPduO ATR holoenzyme, residue P176 of LrPduO, and not residue S183, corresponds to Q234 in the human sequence. Hence, the LrPduO-ΔS183–188 sequence represents a shorter C-terminal truncation than that resulting from the human Q234X mutation. Importantly, while the wild-type LrPduO gene can rescue the growth of a ΔCobA strain of S. enterica on minimal media, LrPduO-ΔS183–188 is unable to do so. This is consistent with our results on the M. extorquens D180X mutant, which reveal that the C-terminal truncation impairs AdoCbl transfer from ATR to MCM rather than AdoCbl synthesis per se.
In the structure of the L. reuteri ATR {St Maurice, 2008 #155}, cobalamin is bound in a hydrophobic cavity that is capped by an ordered C-terminal tail. Modeling the corresponding Q234X mutation into the LrPduO ATR structure, suggests loss of the C-terminal cap of the B12-binding site (Fig. 9). Thus, a predicted outcome of the Q234X mutation is weaker binding of AdoCbl, which is borne out by the significant reduction in the affinity of the D180X mutant for AdoCbl (Table 2).
Figure 9.

Surface representation of the cobalamin-binding site in ATR. (A) Cob(II)alamin is shown in stick representation, while adjacent subunits colored as in Fig. 2, are shown in surface representation. The C-terminal loop residues deleted in the D180X mutant are shown in red in (A) and are deleted in (B). The figure was created using the PDB file 3CI4.
The C-terminal truncation is also predicted to lead to loss of key hydrogen bonding interactions with AdoCbl and the mobile loop (Fig 10A). In the structure of LrPduO holoenzyme, residues R188, V186, and K184 participate in either direct or water-mediated hydrogen bonds, via backbone amides with the amide substituents of the corrin ring (Fig. 10) (18). The low C-terminal sequence conservation among the PduO-type ATRs from different organisms is likely due to the use of backbone amides rather than side chains for hydrogen bonding. Additionally, ordering of the C-terminus triggers the formation of hydrogen-bonding contacts between B12 and residues I113 and L114 in the mobile loop. Thus, while ordering of the C-terminus may contribute to orienting the mobile loop over the lower face of cobalamin, it is not required for generating “base-off” AdoCbl.
Figure 10.

Map of interactions between cob(II)alamin and LrPduO ATR. (A) Shown are the intermolecular hydrogen bonding interactions formed between the peptide backbone of the C-terminus with cobalamin and the peptide backbone of the mobile loop. All side chains have been excluded for clarity. The C-terminal loop missing in the D180X mutant is shown in red, and the backbone of residues M178, K184, V186, and R188 are shown as sticks. The mobile loop is shown in yellow, and the amide backbone of residues I113 and L114 are indicated. Hydrogen bond interactions are depicted as black dashes. (B) Shown are the proposed allosteric contacts between residues of the C-terminus in one subunit and helix 2 residues on the adjacent subunit. The C-terminus and residues L179, Y180, and R181, shown as sticks, are colored red. For clarity, only the amide backbone of L179 is shown. Residues E61, Q64, and D68 of the adjacent subunit are shown as sticks. Cobalamin is colored dark blue and shown in stick representation.
It is unclear how the C-terminal truncation leads to loss of allosteric communication between adjacent active sites. The binding of ATP to the vacant site in the wild-type ATR•2AdoCbl product complex, triggers ejection of only a single equivalent of AdoCbl, presumably from the low affinity site (Fig. 7). In D180X ATR, the two binding sites are equivalent and both display low affinity for AdoCbl. Binding of ATP leads to release of both equivalents of AdoCbl from the trimer (Fig. 6 and 8). Comparison of the apo- and holo-LrPduO crystal structures suggests a possible mechanism for signal transmission between neighboring subunits (16, 18, 19). B12-binding induces interactions between the C-terminus of one subunit and helix 2 residues in the adjacent subunit (Fig. 10B). These interactions include a salt-bridge between R181 in the C-terminus and E61 and a hydrogen bond network between the side chains of residues D68 and Q64 and peptide backbone of R181 and L179 and the phenolic oxygen of Y180 in the neighboring C-terminus (18). A two amino acid deletion of helix 2 residues in the human sequence, I117-Q118del results in methylmalonic aciduria. Although this deletion mutation has been found in a compound heterozygous state, i.e. the other ATR allele carries a different mutation, it would be of interest to further investigate the importance of this deletion mutation and other helix 2 residues in allosteric communication (14).
In summary, our studies on the M. extorquens D180X mutation designed to mimic the patient Q243X mutation, provides insights into the functions of ATR not only as an enzyme, but also as an escort responsible for conveying AdoCbl to the client enzyme, MCM. The observations of decreased MCM activity and lower AdoCbl levels in the Q234X patient cell lines this study. We propose that the lower AdoCbl levels in patient fibroblasts likely reflect a combination of instability of the Q234X mutant and weaker affinity for cobalamin. Supplementation of fibroblasts that are homozygous for the Q234X mutation with OHCbl leads to a modest increase in MCM activity and may result from an increased free AdoCbl pool, which facilitates reconstitution of MCM.
Acknowledgments
We thank Tanya Labunska (University of Nebraska) for generating the M. extorquens D180X ATR mutant and Dr. Dominique Padovani for his early guidance in this work.
Abbreviations used
- AdoCbl
5′-deoxyadenosylcobalamin or coenzyme B12
- ATR
adenosyltransferase
- MANT-ATP
(2′(3′)-O-(N-methylanthraniloyl)-adenosine 5′-triphosphate
- ITC
isothermal titration calorimetry
- D180X ATR
ATR from Methylobacterium extorquens AM1 truncated at Asp180
- DMB
dimethylbenzimidazole
- MCM
methylmalonyl-CoA mutase
- HOCbl
hydroxocobalamin
Footnotes
This work was supported in part by grants from the National Institutes of Health (DK45776 and GM007767).
References
- 1.Banerjee R, Ragsdale SW. The many faces of vitamin B12: catalysis by cobalamin-dependent enzymes. Annu Rev Biochem. 2003;72:209–247. doi: 10.1146/annurev.biochem.72.121801.161828. [DOI] [PubMed] [Google Scholar]
- 2.Bandarian VaRGH. Ethanolamine ammonia-lyase. In: Banerjee R, editor. Chemistry and Biochemistry of B12. Wiley; New York: 1999. [Google Scholar]
- 3.Banerjee RaCS. Methylmalonly-CoA mutase. In: Banerjee R, editor. Chemistry and Biochemistry of B12. Wiley; New York: 1999. [Google Scholar]
- 4.Mera PE, Escalante-Semerena JC. Multiple roles of ATP:cob(I)alamin adenosyltransferases in the conversion of B12 to coenzyme B12. Appl Microbiol Biotechnol. 2010;88:41–48. doi: 10.1007/s00253-010-2773-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsang AW, Escalante-Semerena JC. cobB function is required for catabolism of propionate in Salmonella typhimurium LT2: evidence for existence of a substitute function for CobB within the 1,2-propanediol utilization (pdu) operon. J Bacteriol. 1996;178:7016–7019. doi: 10.1128/jb.178.23.7016-7019.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Suh S, Escalante-Semerena JC. Purification and initial characterization of the ATP:corrinoid adenosyltransferase encoded by the cobA gene of Salmonella typhimurium. J Bacteriol. 1995;177:921–925. doi: 10.1128/jb.177.4.921-925.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dobson CM, Wai T, Leclerc D, Kadir H, Narang M, Lerner-Ellis JP, Hudson TJ, Rosenblatt DS, Gravel RA. Identification of the gene responsible for the cblB complementation group of vitamin B12-dependent methylmalonic aciduria. Hum Mol Genet. 2002;11:3361–3369. doi: 10.1093/hmg/11.26.3361. [DOI] [PubMed] [Google Scholar]
- 8.Leal NA, Park SD, Kima PE, Bobik TA. Identification of the human and bovine ATP:Cob(I)alamin adenosyltransferase cDNAs based on complementation of a bacterial mutant. J Biol Chem. 2003;278:9227–9234. doi: 10.1074/jbc.M212739200. [DOI] [PubMed] [Google Scholar]
- 9.Banerjee R. B12 trafficking in mammals: A for coenzyme escort service. ACS Chem Biol. 2006;1:149–159. doi: 10.1021/cb6001174. [DOI] [PubMed] [Google Scholar]
- 10.Banerjee R, Gherasim C, Padovani D. The Tinker, Tailor, Soldier in intracellular B12 Trafficking. Current Op Chem Biol. 2009;13:477–484. doi: 10.1016/j.cbpa.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim J, Hannibal L, Gherasim C, Jacobsen DW, Banerjee R. A human vitamin B12 trafficking protein uses glutathione transferase activity for processing alkylcobalamins. J Biol Chem. 2009;284:33418–33424. doi: 10.1074/jbc.M109.057877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim J, Gherasim C, Banerjee R. Decyanation of vitamin B12 by a trafficking chaperone. Proc Natl Acad Sci U S A. 2008;105:14551–14554. doi: 10.1073/pnas.0805989105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Padovani D, Labunska T, Palfey BA, Ballou DP, Banerjee R. Adenosyltransferase tailors and delivers coenzyme B12. Nat Chem Biol. 2008;4:194–196. doi: 10.1038/nchembio.67. [DOI] [PubMed] [Google Scholar]
- 14.Jorge-Finnigan A, Aguado C, Sanchez-Alcudia R, Abia D, Richard E, Merinero B, Gamez A, Banerjee R, Desviat LR, Ugarte M, Perez B. Functional and structural analysis of five mutations identified in methylmalonic aciduria cblB type. Hum Mutat. 2010;31:1033–1042. doi: 10.1002/humu.21307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lerner-Ellis JP, Gradinger AB, Watkins D, Tirone JC, Villeneuve A, Dobson CM, Montpetit A, Lepage P, Gravel RA, Rosenblatt DS. Mutation and biochemical analysis of patients belonging to the cblB complementation class of vitamin B12-dependent methylmalonic aciduria. Mol Genet Metab. 2006;87:219–225. doi: 10.1016/j.ymgme.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 16.Mera PE, St Maurice M, Rayment I, Escalante-Semerena JC. Structural and functional analyses of the human-type corrinoid adenosyltransferase (PduO) from Lactobacillus reuteri. Biochemistry. 2007;46:13829–13836. doi: 10.1021/bi701622j. [DOI] [PubMed] [Google Scholar]
- 17.Mera P, St Maurice M, Rayment I, Escalante-Semerena J. Residue Phe112 of the Human-type Corrinoid Adenosyltransferase (PduO) Enzyme of Lactobacillus reuteri is Critical to the Formation of the Four-coordinate Co(II) Corrinoid Substrate and to the Activity of the Enzyme. Biochemistry. 2009;48:3138–3145. doi: 10.1021/bi9000134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.St Maurice M, Mera P, Park K, Brunold TC, Escalante-Semerena JC, Rayment I. Structural characterization of a human-type corrinoid adenosyltransferase confirms that coenzyme B12 is synthesized through a four-coordinate intermediate. Biochemistry. 2008;47:5755–5766. doi: 10.1021/bi800132d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.St Maurice M, Mera PE, Taranto MP, Sesma F, Escalante-Semerena JC, Rayment I. Structural characterization of the active site of the PduO-type ATP:Co(I)rrinoid adenosyltransferase from Lactobacillus reuteri. J Biol Chem. 2007;282:2596–2605. doi: 10.1074/jbc.M609557200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Padovani D, Banerjee R. A Rotary Mechanism for Coenzyme B12 Synthesis by Adenosyltransferase. Biochemistry. 2009;48:5350–5357. doi: 10.1021/bi900454s. [DOI] [PubMed] [Google Scholar]
- 21.Padovani D, Banerjee R. A G-protein editor gates coenzyme B12 loading and is corrupted in methylmalonic aciduria. Proc Natl Acad Sci U S A. 2009;106:21567–21572. doi: 10.1073/pnas.0908106106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stich TA, Buan NR, Escalante-Semerena JC, Brunold TC. Spectroscopic and computational studies of the ATP:corrinoid adenosyltransferase (CobA) from Salmonella enterica: insights into the mechanism of adenosylcobalamin biosynthesis. J Am Chem Soc. 2005;127:8710–8719. doi: 10.1021/ja042142p. [DOI] [PubMed] [Google Scholar]
- 23.Stich TA, Yamanishi M, Banerjee R, Brunold TC. Spectroscopic evidence for the formation of a four-coordinate Co2+ cobalamin species upon binding to the human ATP:cobalamin adenosyltransferase. J Am Chem Soc. 2005;127:7660–7661. doi: 10.1021/ja050546r. [DOI] [PubMed] [Google Scholar]
- 24.Mera PE, Escalante-Semerena JC. Dihydroflavin-driven adenosylation of 4-coordinate Co(II) corrinoids: are cobalamin reductases enzymes or electron transfer proteins? J Biol Chem. 2010;285:2911–2917. doi: 10.1074/jbc.M109.059485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park K, Mera PE, Escalante-Semerena JC, Brunold TC. Kinetic and spectroscopic studies of the ATP:corrinoid adenosyltransferase PduO from Lactobacillus reuteri: substrate specificity and insights into the mechanism of Co(II)corrinoid reduction. Biochemistry. 2008;47:9007–9015. doi: 10.1021/bi800419e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamanishi M, Labunska T, Banerjee R. Mirror “base-off” conformation of coenzyme B12 in human adenosyltransferase and its downstream target, methylmalonyl-CoA mutase. J Am Chem Soc. 2005;127:526–527. doi: 10.1021/ja044365l. [DOI] [PubMed] [Google Scholar]
- 27.Padovani D, Banerjee R. A rotary mechanism for coenzyme B(12) synthesis by adenosyltransferase. Biochemistry. 2009;48:5350–5357. doi: 10.1021/bi900454s. [DOI] [PubMed] [Google Scholar]
- 28.Schubert HL, Hill CP. Structure of ATP-bound human ATP:cobalamin adenosyltransferase. Biochemistry. 2006;45:15188–15196. doi: 10.1021/bi061396f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zoldak G, Zubrik A, Musatov A, Stupak M, Sedlak E. Irreversible thermal denaturation of glucose oxidase from Aspergillus niger is the transition to the denatured state with residual structure. J Biol Chem. 2004;279:47601–47609. doi: 10.1074/jbc.M406883200. [DOI] [PubMed] [Google Scholar]
- 30.Padovani D, Labunska T, Banerjee R. Energetics of interaction between the G-protein chaperone, MeaB, and B12-dependent methylmalonyl-CoA mutase. J Biol Chem. 2006;281:17838–17844. doi: 10.1074/jbc.M600047200. [DOI] [PubMed] [Google Scholar]
- 31.Leal NA, Olteanu H, Banerjee R, Bobik TA. Human ATP: Cob(I)alamin adenosyltransferase and its interaction with methionine synthase reductase. J Biol Chem. 2004;279:47536–47542. doi: 10.1074/jbc.M405449200. [DOI] [PubMed] [Google Scholar]
- 32.Saridakis V, Yakunin A, Xu X, Anandakumar P, Pennycooke M, Gu J, Cheung F, Lew JM, Sanishvili R, Joachimiak A, Arrowsmith CH, Christendat D, Edwards AM. The structural basis for methylmalonic aciduria. The crystal structure of archaeal ATP:cobalamin adenosyltransferase. J Biol Chem. 2004;279:23646–23653. doi: 10.1074/jbc.M401395200. [DOI] [PubMed] [Google Scholar]
- 33.Johnson CL, Buszko ML, Bobik TA. Purification and initial characterization of the Salmonella enterica PduO ATP:Cob(I)alamin adenosyltransferase. J Bacteriol. 2004;186:7881–7887. doi: 10.1128/JB.186.23.7881-7887.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]