

Abstract
Background
Our previous studies demonstrated that simvastatin reduced neuronal death, increased neurogenesis, and promoted functional recovery after TBI. Objective: To investigate the effect of simvastatin on angiogenesis after TBI, and the related signaling pathways.
Methods
Saline or simvastatin (1 mg/kg) was administered orally to rats starting at day 1 after TBI or sham surgery and then daily for 14 days. Rats were sacrificed at 3 and 14 days after treatment. Brain sections and tissues were prepared for immunohistochemical staining, ELISA, and Western blot analysis, respectively. Cultured rat brain microvascular endothelial cells (RBMVECs) were subjected to oxygen-glucose deprivation (OGD) followed by immunocytochemical staining with phallotoxins and vascular endothelial growth factor receptor-2 (VEGFR-2). Western blot analysis was carried out to examine the simvastatin-induced activation of the v-akt murine thymoma viral oncogene homolog (Akt) signaling pathway. The expression of VEGFR-2 was detected by ELISA.
Results
Simvastatin significantly increased the length of vascular perimeter, promoted the proliferation of endothelial cells, and improved the sensorimotor function after TBI. Simvastatin stimulated endothelial cell tube formation after OGD in vitro. VEGFR-2 expression in both brain tissues and cultured RBMVECs was enhanced after simvastatin treatment, which may be modulated by activation of Akt. Akt-dependent endothelial nitric oxide synthase (eNOS) phosphorylation was also induced by simvastatin in vivo and in vitro.
Conclusion
Simvastatin augments TBI-induced angiogenesis in the lesion boundary zone and hippocampus and improves functional recovery. Simvastatin also promotes angiogenesis in vitro. These beneficial effects on angiogenesis may be related to simvastatin-induced activation of the VEGFR-2/Akt/eNOS signaling pathway.
Keywords: Angiogenesis, Simvastatin, Traumatic brain injury, VEGFR-2
Traumatic brain injury (TBI) remains a major cause of serious, long-term disability worldwide. Despite the extensive research being conducted in this field, there still is no effective clinical treatment for TBI. Regional ischemia early after TBI and a relationship between the volume of ischemic tissue and neurological outcomes has been reported.1
Following TBI there is a substantial increase in angiogenesis, and focal angiogenesis may provide oxygen and nutrition for cerebral reconstruction.2 Solely targeting neuroprotection following TBI has not proven sufficient for promoting functional recovery.3 To reduce brain tissue damage and restore neural function, one may have to rescue all the complex signals and interactions between a network of multiple cell types, including neurons, astrocytes, and microvascular endothelial cells.4 Therefore, cerebral vasculature-based therapy combined with neuroprotection may be a reasonable approach for TBI therapy.
The 3-hydroxy-3-methylglutaryl co-enzyme A (HMGCoA) reductase inhibitors referred to as statins have pleiotropic effects that are independent of cholesterol metabolism. A low dose of statins activates angiogenesis in a number of animal models including ischemic stroke5–7 and TBI.8 Simvastatin at the same dose is superior to atorvastatin in treating TBI because of its ability to cross the blood brain barrier.9 In previous studies, we demonstrated that simvastatin modulates neuronal death, increases neurogenesis, and promotes neurological functional recovery after TBI.10 Angiogenesis and neurogenesis are coupled in the brain,11 and simvastatin treatment also promotes angiogenesis.9 However, the present study is the first endeavor to seek the molecular basis of induction of angiogenesis by simvastatin after TBI.
Simvastatin upregulates vascular endothelial growth factor (VEGF) expression after TBI,12 and VEGF is an important mitogen in the process of angiogenesis. The binding of VEGF to its receptors, VEGFR-2 (flk-1), on the surface of endothelial cells activates intracellular tyrosine kinases, triggering multiple downstream signals that promote angiogenesis. Among these signals, Akt-dependent eNOS phosphorylation is crucial for angiogenesis.13
In this study, we investigate the effect of simvastatin on angiogenesis after TBI. To examine a possible mechanism for the angiogenic effects of simvastatin that subsequently enhanced the functional recovery we focused on the VEGFR-2/Akt/eNOS pathway, which has been implicated as a key signaling pathway for angiogenesis.
Materials and Methods
Materials
Simvastatin for in vivo study was purchased from Merck (Whitehouse Station, NJ). Five mg of simvastatin for in vitro study was obtained from Sigma Aldrich (St Louis, MO) and dissolved in 0.2 ml of 100% ethanol, then diluted to a volume of 1 ml with DSMO. The stock solution of simvastatin (12 mM) was further diluted to appropriate concentrations with cell culture medium immediately before use. The control experiments used DMSO only. RBMVECs were purchased from American Type Culture Collection (ATCC Cat# R840-05, Manassas, VA). Von Willebrand factor (vWF) antibody was obtained from DakoCytomation (Carpinteria, CA). Glycogen synthase kinase-3 (GSK-3) inhibitor (lithium chloride, LiCl) and bromodeoxyuridine (BrdU) were purchased from Sigma Aldrich (St Louis, MO). A specific PI3K inhibitor LY294002 [2-(4-Morpholinyl)-8-phenyl-4H-1-benzopyran-4-one] was obtained from Calbiochem (La Jolla, CA). A selective VEGFR-2 antagonist SU1498 was obtained from LC Laboratories (Woburn, MA). DC101, a neutralizing antibody against VEGFR-2, was obtained from ImClone Systems, Inc. (New York, NY). Antibodies against VEGFR-2, phospho-Akt (ser473), Akt, phospho-GSK-3β (Ser9), and phospho-eNOS (Ser1177) were purchased from Cell Signaling Technology (Beverly, MA). Antibodies against actin (I-19) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA).
TBI model
A modified controlled cortical injury (CCI) model of TBI in rats was used in the present study.14 Adult male Wistar rats weighing 300-400 g were anesthetized intraperitoneally with chloral hydrate (350 mg/kg body weight). Rectal temperature was maintained at 37°C by using a feedback-regulated water heating pad. A CCI device was used to induce injury. Rats were placed in a stereotactic frame. Two 10-mm diameter craniotomies were performed adjacent to the central suture, midway between the lambda and the bregma. The second craniotomy allowed for lateral movement of cortical tissue. The dura mater was kept intact over the cortex. Injury was induced by striking the left (ipsilateral) cortex with a pneumatic piston with a 6-mm diameter tip at a rate of 4 m/sec and 2.5 mm of compression. Velocity was measured using a linear velocity displacement transducer. Sham-injured animals were similarly anesthetized and craniectomy performed without cortical injury.
Experimental Groups
Male Wistar rats were randomly divided into three groups (n = 12/group). Rats in the first group were subjected to TBI and given saline orally 1 day later and then consecutively for 14 days. Rats in the second group were subjected to TBI, and 1 day later simvastatin was administered orally at a dose of 1 mg/kg/day for 14 consecutive days. Rats in the third group were subjected to sham surgery. Four rats in each group were sacrificed at 3 days after administration of simvastatin and cortical tissues in lesion boundary zone were harvested for ELISA of VEGFR-2. The other eight rats were sacrificed at 14 days after administration of simvastatin.
Foot-Fault Functional Test
The foot-fault test15 was performed before TBI and at 1, 3, 7, and 14 days after treatment by an investigator who was blinded to the experimental groups. A total of 50 steps were measured for each right hind limb. The percentage of foot-faults of the right hind limb to total steps was determined.
vWF staining
To identify vascular structure, coronal sections were immunohistochemically stained with von Willebrand factor (vWF) antibody.16 Brain sections were deparaffinized and then incubated with 0.4% Pepsin solution at 37°C for 1 hour. After washing, the sections were blocked with 1% BSA at room temperature for 1 hour, and then incubated with rabbit anti-human vWF (1:200; DakoCytomation, Carpinteria, CA) at 4°C overnight. After washing, sections were incubated with biotinylated anti-rabbit antibody (1:200; Vector Laboratories, Inc., Burlingame, CA) and then with an avidin-biotin-peroxidase system, visualized with diaminobenzidine and counterstained with hematoxylin.
Immunofluorescent staining
Newly generated endothelial cells were identified by double labeling for BrdU and vWF. After dehydration, tissue sections were boiled in 10-mM citric acid buffer (pH 6) for 10 minutes. Sections were incubated with 1% BSA containing 0.3% Triton-X-100 in PBS. Sections were then incubated with rabbit anti-vWF antibody (1:200; Chemicon, Temecula, CA) at 4°C overnight. Fluorescein isothiocyanate (FITC) conjugated anti-rabbit antibody (1:400; Jackson ImmunoResearch, West Grove, PA) was added to sections at room temperature for 2 hours. The sections were then treated with 50% formamide in 2 × SSc at 65°C for 30 minutes, followed by 10 minutes at 37°C in 2 N HCl and 3 minutes at room temperature in 0.1 M boric acid. After PBS wash, sections were then incubated with mouse anti-BrdU antibody (1:200; Dako, Glostrup, Denmark) at 4°C overnight. Sections were then incubated with Cy3-conjugated anti-mouse antibody (1:400; Jackson ImmunoResearch, West Grove, PA) at room temperature for 2 hours. Each of the steps was followed by three 5-minute rinses in PBS. Tissue sections were mounted with Vectashield mounting medium (Vector laboratories, Burlingame, CA). Sections were observed with the aid of a fluorescent microscope.
Stereology
The unbiased stereology method was employed in cell counting using MCIDTM stereology software (v7.0, Linton, Cambridge, UK). A series of −2.12 mm to -6.04 mm coronal sections were cut from the bregma of each rat.17 A pilot study was performed to determine Gunderson coefficients and counting space.18 Based on the pilot study, we determined the appropriate grid size (distance between sampling sites on each section), counting frame size, dissector height, top and bottom guard zone sizes, and target Coefficient of Error (CE) for the sampling parameters to achieve an acceptable level of sampling error. Using these parameters, we then implemented the 3D Fractionator (MCID). Using the automated optical fractionator method, we drew the lesion boundary zone and hippocampus areas on coronal sections at low magnification. A higher power was selected, and the system used random systematic sampling to sample a 5-10% defined region of interest depending on cellular density within the region.
Enzyme-Linked ImmunoSorbent Assay (ELISA)
Using ELISA kits (R&D Systems, Minneapolis, MN Cat # MVR200B), VEGFR-2 expression was measured using equal amounts of lysate from brain tissue samples in lesion boundary zone or from cultured RBMVECs.
Cell Culture
RBMVECs were cultured in RBMVECs growth medium (ATCC, Cat# R819-500) at 37°C in a humidified incubator containing 5% CO2. To explore the signaling pathways and proteins mediating the pro-angiogenetic effects of simvastatin on endothelial cells, phosphatidylinositol 3-kinase (PI3K) inhibitor LY294002 (10μM), which subsequently inhibits phosphorylation of Akt, GSK-3 activation inhibitor LiCl (3mM), and VEGFR-2 antagonist SU1498 (5μM) were added in the culture medium along with simvastatin treatment to block the corresponding signaling targets.
In Vitro Oxygen Glucose Deprivation/reoxygenation model (OGD)
RBMVEC were seeded in tissue culture plates containing normal medium until they attached to the plates. Normal growth medium was then replaced with DMEM without glucose and the cells were incubated in an anaerobic chamber (model 1025; Forma Scientific) for 3 hours. The oxygen level within the anaerobic chamber was routinely measured with a BD Disposable Anaerobic Indicator (Becton, Dickinson and Company, Sparks, MD), which confirmed that the oxygen level remained below 0.2%. After OGD, glucose-free DMEM was replaced with normal growth medium and the cells were incubated under normal culture conditions. Simvastatin was added at the start of OGD and maintained during the reoxygenation process. After 24 hours, cells were detached and collected for further study.
Immunocytochemistry
Cells were fixed with a fixing solution (i.e., 4% paraformaldehyde in 0.01 M PBS, pH 7.5) at 25°C for 15 minutes. Cells were then washed three times for 5 minutes with PBS and permeabilized with a solution containing 1% bovine serum albumin (BSA) and 0.3% Triton X-100 in PBS for 30 minutes. Cells were incubated with fluorescent phallotoxins (200 units/ml; Invitrogen, Carlsbad, CA) for 20 minutes in the dark. After PBS washing, cells were then incubated with a monoclonal rabbit anti-VEGFR-2 antibody (diluted 1:100, Cell Signaling Technology) in 1% BSA overnight at 4°C. Cells were then washed three times for 5 minutes with PBS. VEGFR-2 immunoreactivity was visualized with a goat anti-rabbit secondary antibody conjugated to CY-3 (Jackson ImmunoResearch). Cell nucleus was counter staining with 4'-6-Diamidino-2-phenylindole (DAPI). Slides were mounted with Fluoromount-GR mounting media on glass and analyzed under a fluorescence microscope (Nikon, Tokyo, Japan).
Western Blot Analysis
After various treatments, cells were collected and washed once with 1 × PBS, lysed in lysis buffer on ice for 30 minutes, and briefly sonicated. Protein concentrations were determined. Equal amounts of cell lysate were subjected to SDS-polyacrylamide electrophoresis on Novex tris-glycine gels (Invitrogen) and separated proteins were then electrotransferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Bedford, MA). After exposure to various antibodies, specific proteins were detected using the SuperSignal West Pico chemiluminescence substrate system (Pierce, Rockford, IL). The band intensity was analyzed using Scion image software (Scion, Frederick, MD).
Matrigel–based capillary tube formation assay
Matrigel was thawed on ice overnight and diluted 1:3 in serum free cell culture medium. One hundred microliters of diluted Matrigel was put onto a pre-chilled well of a 96-well plate and then placed in an incubator at 37°C for 30 minutes until solidified. The cells to be tested (2 × 104 cells/well) were resuspended in growth medium and carefully placed onto the solidified matrigel and incubated at 37°C with 5% CO2. Development of capillary-like networks was evaluated between 2 and 4 hours after plating. The plates were photographed using an inverted microscope (Leica, Bensheim, Germany). The length of completed tube-like structures in 5 random fields (10-fold magnification) was quantified in a blinded fashion using the MCID image analysis software (Linton, Cambridge, UK).
Statistical Analyses
All data are presented as the means with standard deviations (SD). For cell counting and vWF-stained vascular density, a one-way analysis of variance (ANOVA) test followed by a post hoc Student-Newman-Keuls (SNK) test was used to compare the difference between different groups. Data were analyzed by repeated measurements ANOVA for sensorimotor function. Statistical significance was taken when p-values were less than 0.05.
Results
Simvastatin improves sensorimotor function after TBI
The incidence of right forelimb (contralateral to TBI) foot-faults before TBI was approximately 3%. Animals in the sham group showed only a slight increase in the incidence of foot-fault one day after surgery.
TBI significantly increased the incidence of right hind limb foot-faults at 1 to 14 days post-injury as compared to the sham rats _(P = 0.001 vs. sham ) (Fig. 1). Simvastatin treatment significantly reduced the incidence of foot-faults from four days after injury (P = 0.003 vs saline) and this effect persisted to the last observation timepoint (P = 0.01 vs saline) (Fig. 1). There is no significant difference on the incidence of foot-faults between the simvastatin-treated group and the sham group on day 14 after injury (P = 0.055 vs sham), which suggests that simvastatin promotes the sensorimotor functional recovery after TBI.
Fig. 1.
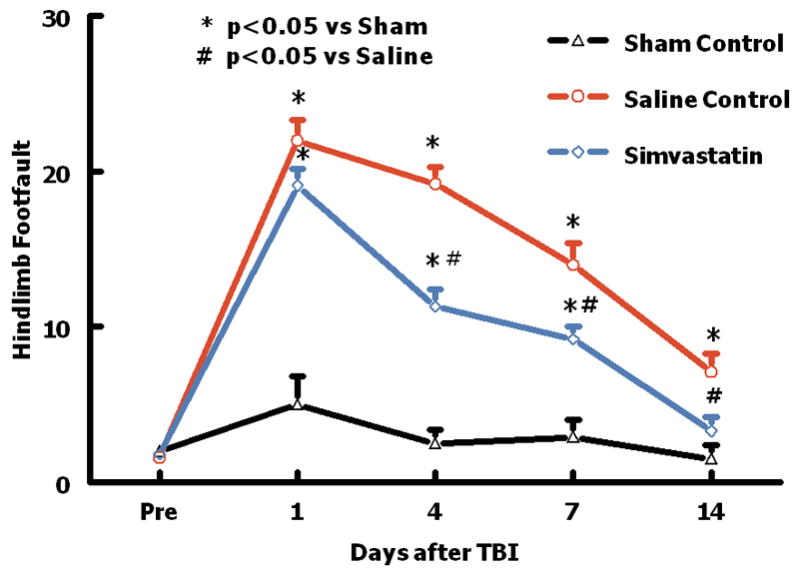
Plot showing the temporal profile of sensorimotor function (foot-fault) before and after TBI or sham. “Pre” represents pre-injury level. TBI impaired sensorimotor performance in the contralateral hind limb. Simvastatin enhanced the recovery of sensorimotor function. Data are represented as mean ± SD. n = 8/group.
Simvastatin treatment of TBI increases angiogenesis in the injured brain
To evaluate the angiogenesis after TBI, vascular perimeter was measured with vWF staining. Figures 2A-G show that vascular perimeter (P = 0.008 vs. saline in cortex and P = 0.006 vs. saline in hippocampus, Fig. 2G) significantly increased in the simvastatin treatment group compared with the saline control group after TBI. Figures 2H-K show that BrdU-positive endothelial cells were significantly increased in the TBI rats (P = 0.003 vs. sham, Fig. 2K) compared to sham rats. Furthermore, simvastatin increased the BrdU-positive endothelial cells (P = 0.001 vs. saline, Fig. 2K) compared with saline treatment after TBI, suggesting that simvastatin enhanced the proliferation of endothelial cells after TBI.
Fig. 2.

Simvastatin treatment of TBI increases angiogenesis in the cortex and hippocampus. A-F - vWF immunostaining in the ipsilateral cortex and hippocampus after TBI. G - quantitative data of cerebral vascular perimeters (vWF). H-J - Immuno-fluorescent staining of vWF (green) and BrdU (red). Nuclei were counterstained with DAPI (blue). Arrows show BrdU-reactive cells co-localized with vWF in the vessel. N = 4/group. “Sim” represents simvastatin. Scale bar = 50 μm in F, J.
Simvastatin stimulates endothelial cell tube formation after oxygen glucose deprivation (OGD)
Capillary-like tube formation was performed to examine the effects of simvastatin on endothelial cells after OGD. Simvastatin significantly (P = 0.001) increased RBMVECs tube formation (5.6 ± 0.6 mm/mm2; Fig. 3C) compared with OGD (2.4 ± 0.3 mm/mm2; Fig. 3B). However, treatment of simvastatin did not increase the tube formation in the presence of the PI3K/Akt inhibitor (LY29402) (2.8 ± 0.2 mm/mm2; Fig. 3D). Inhibition of GSK-3 with lithium chloride (LiCl) (6.1 ± 0.6 mm/mm2; Fig. 3E) enhanced the effect of simvastatin after OGD. Blockage of VEGFR-2 with SU1498, a specific antagonist of VEGFR-2, suppressed capillary-like tube formation induced by the simvastatin (P = 0.001, Fig. 3F). These data suggest that simvastatin induces in vitro angiogenesis of RBMVECs after ischemic injury.
Fig. 3.

Simvastatin regulates capillary like tube formation. A-F - tube formation of rat endothelial cells under various conditions. G - quantitative data of capillary like tube formation. O + S: simvastatin treatment post OGD; O + S + LY: simvastatin plus LY294002 post OGD; O + S + LiCl: simvastatin plus lithium chloride after OGD; O + S + SU: simvastatin plus SU1498 after OGD. n=3/group, Scale bar = 0.5 mm in A.
Simvastatin enhances VEGFR-2 expression in brain tissues and in the cultured RBMVECs
To elucidate the mechanism of simvastatin on angiogenesis, we examined the VEGFR-2 expression in the injured brain tissues by ELISA (Fig. 4C). The lesion boundary zone is a special area where active angiogenesis takes place after TBI and ischemic stroke.19, 20 Therefore, tissues from the lesion boundary zone were sampled to study the expression of proteins related to angiogenesis. TBI alone increased the expression of VEGFR-2 in the lesion boundary zone in cortex (1298.3 ± 64 pg/mg) compared with the sham rats (856.2 ± 89 pg/mg) (P = 0.003). Simvastatin further enhanced the expression of VEGFR-2 (1634.1 ± 151 pg/mg) in the injured cortex compared with the saline-treated rats (P = 0.02 versus saline). In addition, the expression of VEGFR-2 in cultured RBMVECs was tested by Immunocytochemistry and ELISA (Fig. 4A, B). VEGFR-2 expression in hypoxic cells was significantly upregulated compared with normoxic cells. Simvastatin increased the expression of VEGFR-2 in both the cytoplasm and nucleus of the RBMVECs compared to the OGD-alone cells. The effects of simvastatin on VEGFR-2 were enhanced by the GSK-3 inhibitor, lithium chloride, and were reversed by the PI3K/Akt inhibitor LY29402 and the VEGFR-2 neutralizing protein DC101. A similar pattern of VEGFR-2 expression in cultured RBMVECs was detected by ELISA (Fig. 4B). OGD-alone induced the VEGFR-2 expression (P = 0.03 versus control) compared to the control cells. Simvastatin further increased the VEGFR-2 level compared to OGD (P = 0.01 vs. OGD). LY29402 (P = 0.006 vs. simvastatin) and DC101 (P = 0.008 vs. simvastatin) suppressed the effects of simvastatin on VEGFR-2 expression, whereas lithium chloride (P = 0.02 versus simvastatin) did not alter the effect of simvastatin.
Fig. 4.

Effect of simvastatin on VEGFR-2 expression in brain tissue after TBI and in cultured rat endothelial cells. A - double fluorescent immunostaining of F-Actin (green) and VEGFR-2 (red) in rat endothelial cells. Nuclei were counterstained with DAPI (blue). Arrows: VEGFR-2 expression in nucleus and cytoplasm. Arrowheads: VEGFR-2 expression in cell membrane. B - ELISA on expression of VEGFR-2 in rat endothelial cells. C - ELISA on expression of VEGFR-2 in rat brain tissue after TBI. n = 3 (B), 4 (C). Scale bar = 25 μm.
Simvastatin induces phosphorylation of eNOS via Akt pathway in vivo and in vitro
eNOS plays a key role in the process of angiogenesis after brain insult.21 To explore the signal transduction pathway related to the simvastatin-induced angiogenesis, phosphorylation of eNOS and Akt were examined by Western Blot analyses. The cortical tissues from the lesion boundary zone were harvested and homogenized for immunoblotting (Fig. 5A, C). Simvastatin induced the phosphorylation of eNOS in the injured cortex as compared to the saline-treated animals (P = 0.002 versus saline, Fig. 5C). The phosphorylation of Akt and GSK-3 were also increased in the simvastatin-treated rats compared with that in the saline-treated rats (Fig. 5A). The effect of simvastatin on phosphorylation of eNOS was also tested in the cultured RBMVECs (Fig. 5B, D). Simvastatin promoted the phosphorylation of eNOS in the RBMVECs after OGD, whereas LY29402 and SU1498 suppressed this simvastatin-induced phosphorylation. These results suggest that simvastatin-induced phosphorylation of eNOS may be related to the activation of VEGFR-2/PI3K/Akt pathway.
Fig. 5.

Simvastatin enhanced Akt-dependent phosphorylation of eNOS in brain after TBI and in cultured rat endothelial cells. Western blot analyses show the effects of simvastatin on phosphorylation of Akt and eNOS in brain tissue after TBI (A) and the quantitative data of protein band density (C), n = 4. Western blot analysis shows the effects of simvastatin on phosphorylation of eNOS in cultured rat endothelial cells (B) and the quantitative data of protein band density (D), n = 3.
Discussion
The primary findings in the present study are: 1) Treatment of simvastatin promotes angiogenesis and evokes the functional recovery after TBI; 2) Simvastatin stimulates in vitro angiogenesis after OGD, which could be suppressed by inhibition of Akt or blockage of VEGFR-2; and 3) Simvastatin-induced pro-angiogenetic effects may be related to the activation of VEGFR-2/PI3K/Akt/eNOS pathway in endothelial cells.
One of the most prominent pathophysiological changes after TBI is the ischemia and hypoxia in the lesion boundary area.22 Early after the injury, the direct trauma causes necrotic neuronal cell death, followed by increased neuronal apoptosis hours and days later as a result of secondary injury.23 The post-traumatic ischemia plays an important role in secondary injury after TBI. Regional ischemia early after TBI has been shown to correlate with neurological outcome,1 which suggests that TBI and ischemic stroke share fundamental mechanisms of cell damage.22 To date, drugs that solely target neuroprotection have not been proven efficacious for TBI amelioration.3 The failure of therapies directed only to neuronal protection is, in part, attributable to the lack of concomitant effects on cerebral blood vessels after injury. The ‘neurovascular unit’ has emerged as a new paradigm for understanding the pathology of CNS disease24 and injury.25 To improve neurological function after TBI, one may need not only to promote neuroprotection but also to enhance angiogenesis.
Recent studies show significant benefit of statins in models of TBI26 and related disease processes, including cerebral ischemia,7 intracerebral hemorrhage,27 and subarachnoid hemorrhage.28 We reported earlier that simvastatin increases neurogenesis and reduces neuronal apoptosis in TBI rats. Simvastatin also increases angiogenesis in limb ischemic rabbits13 and stroke rats.7 Therefore, simvastatin possesses the potential to promote angiogenesis after TBI. In the current study, fluorescent double staining with BrdU and vWF was performed to identify the newly generated endothelial cells. Our data show that TBI-alone induces angiogenesis in the lesion boundary zone and ipsilateral hippocampus, which is consistent with the previous study.2 However, simvastatin enhanced the TBI-induced angiogenesis identified by increasing endothelial cell proliferation and vascular length (Fig.2). These data are consistent with the previous findings using atorvastatin.8 In this study, simvastatin induces angiogenesis and promotes functional recovery (Fig. 1), although recovery of neurological function after TBI is mediated by many coupled events, including vascular remodeling, neurogenesis, and synaptogenesis. In the treatment of simvastatin, it is possible that other restorative events, in addition to angiogenesis, contribute to recovery of function.
To investigate the mechanism of the pro-angiogenic effects of simvastatin on TBI, endothelial cell tube formation was used as an in vitro angiogenesis model.29 Since ischemia is a fundamental pathophysiological process after TBI,22 OGD was utilized to induce the injury. Simvastatin significantly increases the RBMVECs capillary-like tube formation compared with OGD. Simvastatin-induced tube formation is inhibited by LY29402, a PI3K/Akt inhibitor; enhanced with LiCl, a GSK-3 inhibitor; and blocked by SU1498, a VEGFR-2 antagonist (Fig. 3). These data suggest that simvastatin-induced angiogenesis is related to the activation of the VEGFR-2/Akt/GSK-3 signaling pathway. Endothelial cell capillary tube formation is enhanced by activating VEGFR-2, and Akt is involved in these activities.13, 30 Akt plays an important role in ischemic and VEGF-mediated angiogenesis.31 It regulates many aspects of endothelial cellular functions including apoptosis, cell motility, and tube formation.32 Our previous studies also demonstrate that simvastatin activates Akt in the rat brain after TBI.10
VEGFR-2 mediates the majority of the downstream angiogenic effects of VEGF, including microvascular permeability, endothelial cell proliferation, migration and survival.33 VEGFR-2 transduces VEGF signals through several intracellular signaling pathways including the Raf-Mek-Erk and the PI3K/Akt pathway.34 In light of the critical role of VEGFR-2 on angiogenesis, VEGFR-2 expression was examined both in the brain tissues post TBI and in the cultured RBMVECs subjected to OGD. Our results show that there is an upregulation of VEGFR-2 following TBI, which is consistent with the previous report about VEGFR-2 expression in brain injury,35 and simvastatin further enhances the level of VEGFR-2 in the injured cortex. A similar pattern is seen in the cultured RBMVECs. Hypoxia induces expression of VEGFR-2 in RBMVECs, and simvastatin promotes this induction both in the cytoplasm and in the nucleus of RBMVECs (Fig. 4A). Recent reports show that VEGFR-2 can internalize and extend its proliferative signaling activity for longer periods within intracellular compartments.36 Simvastatin-induced intracellular distribution of VEGFR-2 may also contribute to the enhanced angiogenesis, as the intracellular receptor has longer signal transduction capacity. Simvastatin-induced upregulation of VEGFR-2 expression may be partially related to Akt activity, as LY29402 attenuates this effect (Fig. 4B). These data indicate that simvastatin modulates VEGFR-2 expression in RBMECs both in vitro and in vivo, which results in an increased endothelial cell proliferation and OGD-resistance that may enhance angiogenesis in the injured brain.
eNOS is a downstream mediator of VEGFR-2 and is critical for angiogenesis.21 Enhanced p-eNOS induces a broad range of effects, including the promotion of angiogenesis and mural cell recruitment to immature angiogenic sprouts.37 Our data show that treatment of simvastatin activates Akt/GSK-3 and promotes phosphorylation of eNOS in the TBI brain. Simvastatin treatment of RBMVECs significantly increases p-eNOS activity after OGD compared with controls, which could be suppressed by inhibition of PI3K/Akt and blockade of VEGFR-2 (Fig. 5). These results suggest that simvastatin evokes phosphorylation of eNOS in a VEGFR-2/ PI3K/Akt dependent manner and that eNOS plays an important role in simvastatin-induced angiogenesis after TBI.
Studies reported here show an association between increased overall expressions of VEGFR-2, elevated Akt/eNOS phosphorylation levels, and higher angiogenesis rates in simvastatin treatment compared with control.
Conclusion
Treatment of simvastatin significantly promotes angiogenesis and improves functional outcome after TBI. The pro-angiogenic effect of simvastatin is mediated by activation of VEGFR-2/Akt/eNOS pathway.
Acknowledgments
Financial Support: This work was supported by NIH grants R01NS052280-01A1 (A.M.), and PO1NS23345 (M.C.)
References
- 1.Coles JP, Fryer TD, Smielewski P, et al. Incidence and mechanisms of cerebral ischemia in early clinical head injury. J Cereb Blood Flow Metab. 2004;24(2):202–211. doi: 10.1097/01.WCB.0000103022.98348.24. [DOI] [PubMed] [Google Scholar]
- 2.Morgan R, Kreipke CW, Roberts G, Bagchi M, Rafols JA. Neovascularization following traumatic brain injury: possible evidence for both angiogenesis and vasculogenesis. Neurol Res. 2007;29(4):375–381. doi: 10.1179/016164107X204693. [DOI] [PubMed] [Google Scholar]
- 3.Jain KK. Neuroprotection in traumatic brain injury. Drug Discovery Today. 2008;13(23–24):1082–1089. doi: 10.1016/j.drudis.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 4.del Zoppo GJ. Stroke and neurovascular protection. New Engl J Med. 2006;354(6):553–555. doi: 10.1056/NEJMp058312. [DOI] [PubMed] [Google Scholar]
- 5.Shimamura M, Sato N, Sata M, et al. Delayed postischemic treatment with fluvastatin improved cognitive impairment after stroke in rats. Stroke: J Cereb Circ. 2007;38(12):3251–3258. doi: 10.1161/STROKEAHA.107.485045. [DOI] [PubMed] [Google Scholar]
- 6.Weis M, Heeschen C, Glassford AJ, Cooke JP. Statins have biphasic effects on angiogenesis. Circulation. 2002;105(6):739–745. doi: 10.1161/hc0602.103393. [DOI] [PubMed] [Google Scholar]
- 7.Chen J, Zhang ZG, Li Y, et al. Statins induce angiogenesis, neurogenesis, and synaptogenesis after stroke. Ann Neurol. 2003;53(6):743–751. doi: 10.1002/ana.10555. [DOI] [PubMed] [Google Scholar]
- 8.Lu D, Goussev A, Chen J, et al. Atorvastatin reduces neurological deficit and increases synaptogenesis, angiogenesis, and neuronal survival in rats subjected to traumatic brain injury. J Neurotrauma. 2004;21(1):21–32. doi: 10.1089/089771504772695913. [DOI] [PubMed] [Google Scholar]
- 9.Lu D, Qu C, Goussev A, et al. Statins increase neurogenesis in the dentate gyrus, reduce delayed neuronal death in the hippocampal CA3 region, and improve spatial learning in rat after traumatic brain injury. J Neurotrauma. 2007;24(7):1132–1146. doi: 10.1089/neu.2007.0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu H, Lu D, Jiang H, et al. Increase in phosphorylation of Akt and its downstream signaling targets and suppression of apoptosis by simvastatin after traumatic brain injury. J Neurosurg. 2008;109(4):691–698. doi: 10.3171/JNS/2008/109/10/0691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Palmer TD, Willhoite AR, Gage FH. Vascular niche for adult hippocampal neurogenesis. J Comp Neurol. 2000;425(4):479–494. doi: 10.1002/1096-9861(20001002)425:4<479::aid-cne2>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 12.Wu H, Lu D, Jiang H, et al. Simvastatin-mediated upregulation of VEGF and BDNF, activation of the PI3K/Akt pathway, and increase of neurogenesis are associated with therapeutic improvement after traumatic brain injury. J Neurotrauma. 2008;25(2):130–139. doi: 10.1089/neu.2007.0369. [DOI] [PubMed] [Google Scholar]
- 13.Kureishi Y, Luo Z, Shiojima I, et al. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat Med. 2000;6(9):1004–1010. doi: 10.1038/79510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mahmood A, Lu D, Wang L, Chopp M. Intracerebral transplantation of marrow stromal cells cultured with neurotrophic factors promotes functional recovery in adult rats subjected to traumatic brain injury. J Neurotrauma. 2002;19(12):1609–1617. doi: 10.1089/089771502762300265. [DOI] [PubMed] [Google Scholar]
- 15.Hernandez TD, Schallert T. Seizures and recovery from experimental brain damage. Exp Neurol. 1988;102(3):318–324. doi: 10.1016/0014-4886(88)90226-9. [DOI] [PubMed] [Google Scholar]
- 16.Xiong Y, Mahmood A, Lu D, et al. Histological and functional outcomes after traumatic brain injury in mice null for the erythropoietin receptor in the central nervous system. Brain Res. 2008;1230:247–257. doi: 10.1016/j.brainres.2008.06.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paxinos GWC. The rat brain in stereotaxic coordinates. Brulington: Academic Press; 2007. [Google Scholar]
- 18.Schmitz C, Hof PR. Design-based stereology in neuroscience. Neuroscience. 2005;130(4):813–831. doi: 10.1016/j.neuroscience.2004.08.050. [DOI] [PubMed] [Google Scholar]
- 19.Beck H, Plate KH. Angiogenesis after cerebral ischemia. Acta Neuropathologica. 2009;117(5):481–496. doi: 10.1007/s00401-009-0483-6. [DOI] [PubMed] [Google Scholar]
- 20.Guo X, Liu L, Zhang M, Angela B, Zhang J. Correlation of CD34+ cells with tissue angiogenesis after traumatic brain injury in a rat model. J Neurotrauma. doi: 10.1089/neu.2008.0733. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murohara T, Asahara T, Silver M, et al. Nitric oxide synthase modulates angiogenesis in response to tissue ischemia. J Clin Invest. 1998;101(11):2567–2578. doi: 10.1172/JCI1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Werner C, Engelhard K. Pathophysiology of traumatic brain injury. Br J Anaesthesia. 2007;99(1):4–9. doi: 10.1093/bja/aem131. [DOI] [PubMed] [Google Scholar]
- 23.Greve MW, Zink BJ. Pathophysiology of traumatic brain injury. Mount Sinai J Med. 2009;76(2):97–104. doi: 10.1002/msj.20104. [DOI] [PubMed] [Google Scholar]
- 24.Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer's disease. Nat Rev. 2004;5(5):347–360. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- 25.Khan M, Im YB, Shunmugavel A, et al. Administration of S-nitrosoglutathione after traumatic brain injury protects the neurovascular unit and reduces secondary injury in a rat model of controlled cortical impact. J Neuroinflammation. 2009;6:32. doi: 10.1186/1742-2094-6-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang H, Lynch JR, Song P, et al. Simvastatin and atorvastatin improve behavioral outcome, reduce hippocampal degeneration, and improve cerebral blood flow after experimental traumatic brain injury. Exp Neurol. 2007;206(1):59–69. doi: 10.1016/j.expneurol.2007.03.031. [DOI] [PubMed] [Google Scholar]
- 27.Karki K, Knight RA, Han Y, et al. Simvastatin and atorvastatin improve neurological outcome after experimental intracerebral hemorrhage. Stroke; J Cereb Circ. 2009;40(10):3384–3389. doi: 10.1161/STROKEAHA.108.544395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sugawara T, Ayer R, Jadhav V, Chen W, Tsubokawa T, Zhang JH. Simvastatin attenuation of cerebral vasospasm after subarachnoid hemorrhage in rats via increased phosphorylation of Akt and endothelial nitric oxide synthase. J Neurosci Res. 2008;86(16):3635–3643. doi: 10.1002/jnr.21807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen J, Cui X, Zacharek A, Roberts C, Chopp M. eNOS mediates TO90317 treatment-induced angiogenesis and functional outcome after stroke in mice. Stroke; J Cereb Circ. 2009;40(7):2532–2538. doi: 10.1161/STROKEAHA.108.545095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morales-Ruiz M, Fulton D, Sowa G, et al. Vascular endothelial growth factor-stimulated actin reorganization and migration of endothelial cells is regulated via the serine/threonine kinase Akt. Circ Res. 2000;86(8):892–896. doi: 10.1161/01.res.86.8.892. [DOI] [PubMed] [Google Scholar]
- 31.Ackah E, Yu J, Zoellner S, et al. Akt1/protein kinase Balpha is critical for ischemic and VEGF-mediated angiogenesis. J Clin Invest. 2005;115(8):2119–2127. doi: 10.1172/JCI24726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shiojima I, Walsh K. Role of Akt signaling in vascular homeostasis and angiogenesis. Circ Res. 2002;90(12):1243–1250. doi: 10.1161/01.res.0000022200.71892.9f. [DOI] [PubMed] [Google Scholar]
- 33.Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol. 2005;23(5):1011–1027. doi: 10.1200/JCO.2005.06.081. [DOI] [PubMed] [Google Scholar]
- 34.Freitas-Andrade M, Carmeliet P, Stanimirovic DB, Moreno M. VEGFR-2-mediated increased proliferation and survival in response to oxygen and glucose deprivation in PlGF knockout astrocytes. J Neurochem. 2008;107(3):756–767. doi: 10.1111/j.1471-4159.2008.05660.x. [DOI] [PubMed] [Google Scholar]
- 35.Lafuente JV, Argandona EG, Mitre B. VEGFR-2 expression in brain injury: its distribution related to brain-blood barrier markers. J Neural Transm. 2006;113(4):487–496. doi: 10.1007/s00702-005-0407-0. [DOI] [PubMed] [Google Scholar]
- 36.Lampugnani MG, Orsenigo F, Gagliani MC, Tacchetti C, Dejana E. Vascular endothelial cadherin controls VEGFR-2 internalization and signaling from intracellular compartments. J Cell Biol. 2006;174(4):593–604. doi: 10.1083/jcb.200602080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kashiwagi S, Izumi Y, Gohongi T, et al. NO mediates mural cell recruitment and vessel morphogenesis in murine melanomas and tissue-engineered blood vessels. J Clin Invest. 2005;115(7):1816–1827. doi: 10.1172/JCI24015. [DOI] [PMC free article] [PubMed] [Google Scholar]