

Abstract
An abnormal neutrophil subset has been identified in the PBMC fractions from lupus patients. We have proposed that these “low density granulocytes” (LDGs) play an important role in lupus pathogenesis by damaging endothelial cells and synthesizing increased levels of proinflammatory cytokines and type I interferons. To directly establish LDGs as a distinct neutrophil subset, their gene array profiles were compared to those of autologous normal density neutrophils and control neutrophils. LDGs significantly overexpress mRNA of various immunostimulatory bactericidal proteins and alarmins, relative to lupus and control neutrophils. In contrast, gene profiles of lupus normal density neutrophils do not differ from those of controls. LDGs have heightened capacity to synthesize extracellular traps (NETs) which display increased externalization of bactericidal, immunostimulatory proteins and autoantigens, including LL-37, IL-17, and double-stranded DNA (dsDNA). Through NETosis, LDGs have increased capacity to kill endothelial cells and to stimulate IFN-α synthesis by pDCs. Affected skin and kidneys from lupus patients are infiltrated by netting neutrophils, which expose LL-37 and ds-DNA. Tissue NETosis is associated with increased anti-dsDNA in sera. These results expand the potential pathogenic roles of aberrant lupus neutrophils and suggest that dysregulation of NET formation and its subsequent responses may play a prominent deleterious role.
INTRODUCTION
Recent evidence from our group and others indicates that neutrophils may play an important role in the induction of autoimmune responses and organ damage in systemic lupus erythematosus (SLE) (1–3). Furthermore, microarray data indicates that neutrophil-specific genes are highly expressed in PBMCs from lupus patients because of the co-segregation of low-density granulocytes (LDGs) in mononuclear cell fractions (3, 4). These LDGs represent a distinct neutrophil subset which is present in the peripheral blood of all adult SLE patients analyzed. Lupus LDGs are likely to be pathogenic, given their heightened capacity to induce vascular damage and synthesize type I IFNs upon exposure to specific stimulants, such as G-CSF and poly (I:C), when compared to autologous lupus normal-density neutrophils and healthy control neutrophils (1). Furthermore, lupus patients with higher circulating LDG numbers have increased prevalence of skin involvement and/or vasculitis (1). While currently no specific LDG surface markers have been identified that would allow them to be distinguished from normal-density granulocytes, their nuclear morphology suggests that these cells present a more immature phenotype (1, 3). However, it is still unknown whether lupus LDGs play prominent roles, when compared to normal density lupus neutrophils, in other aspects of disease pathogenesis, including providing a supply of potential autoantigens or inducing or perpetuating autoimmune responses and tissue damage.
Neutrophils immobilize and kill invading microbes extracellularly through the formation of extracellular traps (NETs). As a unique type of neutrophil cell death, recently described as NETosis, this response is distinct from apoptosis and necrosis and is characterized by the active release of nuclear chromatin fibers (5, 6). NETosis is triggered by a variety of stimuli including microorganisms, proinflammatory cytokines, activated platelets and endothelial cells (6, 7). A variety of putative autoantigens are present within and attached to NET chromatin fibers, including citrullinated histones (8) and various bactericidal proteins and/or enzymes such as the cathelicidin LL-37, neutrophil elastase and myeloperoxidase (MPO)(9). Upon neutrophil activation, elastase migrates from azurophilic granules to the nucleus, where it partially degrades specific histones and promotes chromatin decondensation. MPO synergizes with elastase in driving this decondensation (9), a phenomenon that is considered key in NET formation.
Due to the potential role of netting neutrophils in externalizing autoantigens and DNA-modifying factors, thereby making these molecules more exposed to the adaptive and innate immune systems, a putative link between NETosis and autoimmunity has been recently proposed. It has been shown that neutrophils from patients with ANCA-positive vasculitis release NETs enriched in MPO and LL-37 (10). NETs are also present in the kidneys from patients with this disease, where they may provide a source of antigenic nucleosomes and promote immune complex formation(10). Further, impaired NET degradation has been identified in a subset of SLE patients, secondary to DNase1 inhibitors and anti-NET antibodies that prevent DNase1 access to NETs (2). LDGs may represent an additional source of NETs, leading to heightened autoantigen exposure and modification of tissue damage. Recent evidence indicates that NETosis may be enhanced in IFN-α-primed lupus neutrophils upon exposure to anti-RNP antibodies. This is accompanied by the release of LL-37 and high-mobility group protein B1, which facilitate uptake and recognition of mammalian DNA by plasmacytoid dendritic cells (pDCs)(11). In addition, recent evidence indicates that NETs may be harmful to the endothelium and promote thrombosis (7, 12).
To further clarify the origin of LDGs and to assess their pathogenic potential and role in the induction of autoimmune responses, we compared the gene array expression profiles of purified LDGs with those of autologous normal-density lupus neutrophils (referred in the text as lupus neutrophils) or healthy control normal density neutrophils (referred in the text as control neutrophils). We also compared the capacity of isolated LDGs to form NETs, externalize autoantigens and immunostimulatory molecules, stimulate pDCs and induce endothelial cytotoxicity through NETosis. Finally, we assessed whether netting-neutrophils are present in vivo in involved lupus tissue from various organs, as an additional indication of their putative pathogenic role.
MATERIALS AND METHODS
Patient selection
The University of Michigan institutional review board approved this study. Subjects gave informed consent in accordance with the Declaration of Helsinki. Lupus patients fulfilled the revised American College of Rheumatology criteria for SLE (13) and were enrolled from the University of Michigan outpatient rheumatology clinic. Disease activity was assessed by the SLE disease activity index (14). Gender-matched healthy controls were recruited by advertisement. Demographic and clinical information on the lupus patients enrolled in the study (including medications) is included in Table II.
Table II.
Clinical characteristics of lupus patients included in the study
| PERIPHERAL BLOOD | |||||||
|---|---|---|---|---|---|---|---|
| Patient | SLEDAI | ds-DNA-Ab | C3, C4 | ENA | ANA | Clinical Manifestations | Medications |
| 1B | 2 | + | Normal | Negative | 1:160 | Skin, joints, CNS | MMF, antimalarials |
| 2B | 10 | + | Normal | Negative | 1:320 | Nephritis, arthritis | None |
| 3B | 8 | + | Low | Sm, RNP | 1:2560 | Cytopenias, nephritis | PDN, antimalarials |
| 4B | 0 | − | Normal | Ro | 1:2560 | Neuropathy, Raynaud’s | Antimalarials |
| 5B | 0 | − | Normal | Negative | 1:320 | Arthritis, skin, mucositis | Antimalarials |
| 6B | 2 | + | Normal | Sm, RNP | 1:320 | Skin, cytopenias, fever | PDN, antimalarials |
| 7B | 20 | + | Low | Sm, RNP | 1:2560 | Arthritis, CNS, cytopenias, skin | PDN, antimalarials, MMF |
| 8B | 2 | − | Low | Sm, RNP | 1:320 | Nephritis, synovitis, serositis | PDN, antimalarials, MMF |
| 9B | 4 | + | Normal | Negative | 1:160 | Serositis, synovitis, mucositis, skin | Antimalarials, MMF |
| 10B | 2 | − | Normal | Negative | 1:160 | Skin, nephritis, arthritis | PDN, antimalarials, MMF |
| 11B | 4 | + | Normal | Ro | 1:640 | Arthritis, cytopenias | Antimalarials |
| 12B | 0 | − | Normal | Negative | 1:320 | Arthritis, eye | Antimalarials |
| 13B | 10 | + | Low | Ro, La | 1:2560 | Arthritis, nephritis, Raynaud’s | MMF, PDN |
| 14B | 12 | + | Low | Ro, Sm, RNP | 1:2560 | Arthritis, skin, serositis, kidney, CNS, | MMF, MTX, PDN |
| 15B | 4 | + | Low | Sm, RNP | 1:320 | Skin, arthritis, serositis, leukopenia | Antimalarials |
| 16B | 6 | − | Normal | Sm, RNP, Ro | 1:2560 | Nephritis, cytopenias, arthritis | PDN, antimalarials, AZA |
| 17B | 4 | − | Normal | Negative | 1:2560 | Serositis, arthritis, skin | Antimalarials |
| 18B | 0 | − | Normal | Sm, RNP | Negative | Arthritis | PDN, antimalarials |
| 19B | 10 | + | Low | Ro | 1:320 | Nephritis, arthritis | PDN, MMF, antimalarials |
| 20B | 6 | − | Normal | Negative | 1:320 | Arthritis, skin | PDN, antimalarials |
| SKIN | |||||||
|---|---|---|---|---|---|---|---|
| Patient | Diagnosis | ds-DNA-Ab | C3, C4 | ENA | ANA | Other Clinical Manifestations | Medications |
| 1S | DL | + | Low | Negative | 1:160 | CNS, nephritis | MMF, PDN |
| 2S | DL | + | Low | RNP, Ro | 1:2560 | CNS, Raynaud’s | PDN, antimalarials |
| 3S | Interface dermatitis | + | Low | SmRNP | 1:2560 | Nephritis | PDN, antimalarials |
| 4S | Lupus panniculitis | + | Low | Negative | 1:640 | - | None |
| 5S | Lupus panniculitis | + | Low | SmRNP, RNP | 1:2560 | Arthritis | PDN, antimalarials |
| 6S | SCLE | - | Normal | Ro | Negative | Sicca | Antimalarials |
| 7S | DL | N/A | N/A | Negative | Negative | - | None |
| 8S | DL | N/A | N/A | N/A | Negative | - | None |
| 9S | DL | N/A | Normal | N/A | Negative | - | None |
| 10S | DL | N/A | Normal | Negative | 1:640 | Arthritis | None |
| 11S | Interface dermatitis | − | N/A | Ro, La | Negative | - | None |
| KIDNEY | |||||||
|---|---|---|---|---|---|---|---|
| Patient | Kidney Diagnosis Activity/Chronicity | ds-DNA-Ab | C3, C4 | ENA | ANA | Other Clinical Manifestations | Medications at Time of Biopsy |
| 1N | Class IV/S 12/8 |
+ | Low | Sm, RNP | 1:160 | Serositis, arthritis, | PDN, AZA |
| 2N | Class IV/S 11/3 |
+ | Low | Sm | 1:320 | Skin, serositis, arthritis, cytopenia | PDN, antimalarials |
| 3N | Class IV/S 13/2 |
N/A | Low | N/A | 1:640 | Cytopenia | None |
| 4N | ClassIII/V 13/2 |
+ | Low | Ro, La, | 1:1280 | Skin, joints, serositis | PDN, MMF |
| 5N | Class IV 17/11 |
+ | Normal | Sm, RNP Sm | 1:2560 | CNS | PDN |
| 6N | Class III/V 6/8 |
+ | Low | Ro, Sm, RNP | 1:1280 | - | PDN, AZA |
| 7N | Class III 6/2 |
+ | Low | Negative | 1:160 | Cytopenias, arthritis | PDN, MMF, antimalarials |
| 8N | Class III 8/1/ |
+ | Low | Negative | 1:160 | Arthritis, cytopenias | PDN, AZA, antimalarials |
| 9N | Class IV 17/4 |
+ | Low | Ro, Sm, RNP | 1:2560 | Arthritis, lung | PDN |
| 10N | Henoch-Schonlein crescentic glomerulonephritis | N/A | Low | N/A | N/A | Skin, arthritis | Steroids |
| MICROARRAY ANALYSIS | |||||||
|---|---|---|---|---|---|---|---|
| Patient | SLEDAI | ds-DNA-Ab | C3, C4 | ENA | ANA | Clinical Manifestations | Medications |
| 1M | 4 | − | Normal | RNP, Sm | Negative | Skin | Antimalarials |
| 2M | 2 | − | Low | Negative | 1:160 | Skin | Antimalarials |
| 3M | 4 | + | Low | Sm, RNP | 1:2560 | Skin, arthritis | PDN, antimalarials |
| 4M | 2 | + | Normal | Negative | 1:160 | Skin, arthritis, pleuritis | Antimalarials |
| 5M | 10 | − | Low | Ro | 1:160 | Arthritis, Skin, Fever | PDN, antimalarials |
| 6M | 4 | − | Normal | Negative | 1:2560 | Cytopenias, arthritis | None |
| 7M | 14 | + | Low | La, RNP | 1:2560 | Skin, arthritis, serositis | PDN, antimalarials |
| 8M | 4 | − | Normal | Negative | 1:2560 | Arthritis, CNS | Antimalarials |
| 9M | 8 | + | Low | Negative | 1:640 | Arthritis | None |
| 10M | 10 | + | Low | Sm, RNP | 1:2560 | Nephritis, skin, arthritis | Antimalarials |
Reagents
For LDG purification, biotinylated Abs recognizing CD3, CD7, CD19, CD79b, CD56, MHC class II, CD86, and CD235a were obtained from Ancell (Bayport, MN). For immunohistochemical staining, rabbit Abs recognizing human neutrophil elastase (Abcam, Cambridge, MA), human Ro/SSA and human La/SSB (Santa Cruz Biotechnology, Santa Cruz, CA), mouse Abs recognizing human Smith (Abcam), human Cathelicidin/OSX12 (Abcam), and human ds-DNA (Millipore, Temecula, California) and goat-anti-human IL-17 (R&D systems, Minneapolis, MN) were used. Secondary detection was performed using goat anti-rabbit-FITC (SouthernBiotech, Birmingham, AL), donkey anti-goat Alexa 568 (Invitrogen, Carlsbad, CA) and/or anti-mouse Cy3 (Sigma, St. Louis, MO). Prolong Gold Antifade and Hoechst 33342 were from Invitrogen. Micrococcal nuclease was from Thermo Scientific (Rockford, IL).
Purification of LDGs
Lupus LDGs were purified as described by our group (1). In brief, PBMCs were isolated by Ficoll/Hypaque gradient and cell pellets were incubated with LDG isolation mixture (equal volumes of biotinylated Abs recognizing human CD3, CD7, CD19, CD79b, CD56, MHC class II, CD86, and CD235a), followed by anti-biotin MACS beads (Miltenyi Biotech, Auburn, CA). Cells were applied to a MACS-LS column and nonimmobilized cells were recovered by negative selection. The purity of the LDG fraction was >95% and cells were identified as CD15+/CD14lo or CD10+/CD14lo.
Isolation of neutrophils
Normal-density neutrophils were isolated by dextran sedimentation of RBC pellets, as described (15). Purity was at least 95%. In previous studies, we had compared the activation status of neutrophils obtained by dextran sedimentation versus double gradient and found no differences in their phenotype or function (1).
NETs Immunohistochemical Staining and Quantification
Neutrophils or LDGs were isolated as above and 1–2 × 105 cells/mL were seeded in poly-L-lysine coverslips and incubated at 37°C, 5% CO2 for 15 minutes. Cells were washed with ice-cold PBS and either fixed right away with 4% paraformaldehyde and then blocked overnight at 4°C with 10% FBS/1% BSA/0.05% Tween 20 and 2mM EDTA/PBS, or incubated for 2 hours in RPMI/glutamine/2% BSA in the presence or absence of 20 nM PMA to induce NET formation, followed by fixation and overnight blocking at 4 C. NETs were detected by washing the fixed cells with ice-cold 10% FBS/PBS and incubating with anti-human elastase (1:100) or isotype control for 45 minutes at 4°C, followed by incubation with secondary fluorochrome-conjugated antibodies for 45 minutes at 4°C. Nuclear DNA was detected by incubating cells with Hoechst 33342 (1:100) for 10 minutes at room temperature. Coverslips were mounted in Prolong Antifade Reagent and analyzed using an Olympus microscope (IX70, Center Valley, PA). Statistical background and shading subtraction and image overlay were performed with Metamorph v7.7 software (Molecular Devices, Inc., Sunnyvale, CA). The recorded images were loaded onto Adobe Photoshop for further analysis, where NETs were manually quantified by two independent observers. The number of cells positive for both neutrophil elastase and nuclear staining (Hoechst) were considered a NET and digitally recorded to prevent multiple counts. The percentage of NETs was calculated as the average of five to six fields (40X) normalized to the total number of cells. For LL-37 (1:100), ds-DNA (1:10), Ro (1:20), La (1:20), Smith (1:20) and IL-17 (1:10) quantification, the number of cells positive for either of these markers colocalized with elastase and Hoechst were counted as part of the overlay (RGB) image and recorded digitally to prevent multiple counts.
RNA isolation
Total RNA was isolated with Tripure (Roche, Indianapolis, IN), following manufacturer’s recommendations. For microarray analysis, RNA was further purified and concentrated using an RNeasy micro kit (Qiagen, Valencia, CA). RNA samples were processed on an Agilent 2100 BioAnalyzer (Agilent Technologies, Santa Clara, CA) to assess integrity.
Microarray data processing, analysis, and pathway mapping
Affymetrix Human U133 Plus 2.0 Genechips (Affymetrix, Santa Clara, CA) were processed at the University of Michigan Microarray Core Facility. The samples analyzed and compared were: normal density neutrophils from healthy individuals (n=9), normal density neutrophils and autologous LDGs from lupus patients (n=10 for each group). CEL files were normalized in GenePattern pipeline (http://www.GenePattern.org) using the RMA (Robust MultiChip Average) method and the Human Entrez Gene custom CDF annotation version 10 (http://brainarray.mbni.med.umich.edu/Brainarray/default.asp). As samples were part of two hybridization rounds, the two resulting normalized files were corrected for batch effect (16) within GenePattern. Of the 17,527 gene IDs (corresponding to the 54,675 Affymetrix probesets), the number of genes expressed above the Poly-A Affymetrix control expression baseline (negative controls) and used for further analyses were 15,929. The GEO access number for these arrays is GSE26975 (http://www.ncbi.nlm.nih.gov/geo/).
Statistical paired analyses were performed using Significance Analysis of Microarrays (SAM) method implemented in MultiExperiment Viewer (MeV) application (17, 18), for comparing lupus neutrophils with lupus LDGs; unpaired analyses were performed in the comparison of healthy control neutrophils with lupus neutrophils and healthy control neutrophils with lupus LDGs. The canonical pathways derived from the significantly regulated genes between the groups (q < 0.05 depicting the false discovery rate) were analyzed using the Ingenuity Pathway Analysis Software (http://www.ingenuity.com).
Real-time quantitative PCR
Real time-PCR reactions were run on an ABI Prism 7900HT in duplicate using 2×SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA). Oligo nucleotide primers (IDT, Coralville, IA) used in the reactions were:
Lipocalin: 5′CCCAGCCCCACCTCTGA3′ (forward), 5′CTTCCCCTGGAATTGGTTGTC3′ (reverse);
MMP8:5′GCTGAGGTAGAAAGAGCTATCAAGGA3′(forward), 5′AGCAATGTTGATATCTGCCTCTCC3′ (reverse);
MPO:5′TTTGACAACCTGCACGATGAC3′(forward), 5′CGGTTGTGCTCCCGAAGTAA3′(reverse);
Cathepsin-G:5′AGAAGAGTCAGACGGAATCGA3′(forward), 5′CCCTGACGACTTTCCATAGGA 3′(reverse);
LL-37: 5′-GCAGTCACCAGAGGATTGTGAC-3′(forward), 5′CACCGCTTCACCAGCCC3′ (reverse).
IL-17A: 5′-ACTCCTGGGAAGACCTCATTGG-3′(forward), 5′-GGCCACATGGTGGACAAT CG-3′ (reverse)
GAPDH:5′-TTGCCATCAATGACCCCTTCA-3′(forward), 5′CGCCCCACTTGATTTTGGA-3′ (reverse).
Endothelial cell cytotoxicity assay
Human umbilical vein endothelial cells (HUVECs) were cultured in MCDB131 basal media (Gibco-Carlsbad, CA) supplemented with EGM-2MV (without hydrocortisone) bullet kit (Lonza-Walkersville, MD), on 0.2. % gelatin coated 24- well plates (Fisher, Pittsburgh, PA). Lupus LDGs, their autologous lupus neutrophils and healthy control neutrophils were incubated with HUVECs at 2:1 effector (neutrophil): target (HUVEC) ratio in MCDB131/EGM-2MV and 0.1μg/ml PMA in the presence or absence of MNase (10U/mL) for 16–20 hours. Alternatively, cells were also co-cultured with 0.1ug/ml PMA, with or without MNase for 1 to 2 hours, followed by replacement with fresh media with or without nuclease. Following the incubation, nuclease reaction was stopped by adding EDTA to final concentration of 2mM. Adherent LDGs and neutrophils were collected by gentle pipetting, and HUVECs were harvested with 0.05% trypsin-EDTA (Gibco), followed by centrifugation at 1600 RPM for 5 minutes. HUVECs were resuspended in 1% horse serum/1% BSA in PBS and 104–105 cells were incubated with PE-anti-human CD146, APC-Annexin-V, PE/Cy5-anti-human CD45 (BD Pharmingen, San Diego, CA) and/or PE/Cy5-anti-human CD10 (Biolegend), followed by fixation with 4% paraformaldehyde. The percentage of HUVEC cytotoxicity was measured in cells double-positive for CD146 and Annexin-V in the CD45 or CD10 negative gate.
PDC lines and IFN-α quantification
The previously described human pDC cell line GEN 2.2 (19) was cultured in RPMI1640 Glutamax/1 mM sodium pyruvate/4mM glutamine/nonessential amino acids/10% heat-inactivated fetal calf serum. Cells were stimulated for 16 hours with either CpG (1 μg/ml) or supernatants collected from either control or lupus neutrophils or autologous lupus LDGs that had been cultured for 1 hr and supernatants were treated in the presence or absence of MNase (10 U/mL, 10min). IFN-α mRNA from pDCs was quantified by real-time PCR as previously described by our group(1).
Human kidney tissue and immunofluorescence
Kidney tissue was obtained from 9 renal biopsies from subjects with clinical and histological diagnosis of lupus nephritis according to the new SLE nephritis classification (20) and from one patient with fulminant Henoch-Schönlein purpura with renal involvement. Relevant clinicohistological parameters are in Table II. Immunohistochemical analyses were carried out at the University of Michigan Histology and Immunohistochemistry Core. Three-micrometer sections of formalin-fixed, paraffin-embedded tissues were mounted on plus slides, deparaffinized in xylene, and then rehydrated with distilled H2O through graded alcohols. Antigen retrieval was enhanced by microwaving the slides in citrate buffer (pH 6.0; Biogenex) for 10 min. Sections were blocked for 30 minutes in 10% horse serum prior to one hour incubation of primary antibodies at 4 °C. Simultaneous staining was performed with mouse monoclonal anti-human MPO (1:1000; Abcam) and rabbit polyclonal anti-human histone H2A (1:500; Abcam) followed by 30 minute incubation with matched secondary antibodies: anti-mouse IgG Alexa-Fluor 488 (1:300; Invitrogen), or anti-rabbit IgG Alexa-Fluor 555 (1:300; Invitrogen). DAPI was added to the slides prior to mounting with cover slips (Invitrogen). For NETosis detection images were acquired by a Leica DMIRB inverted microscope and an RT slider digital camera (model 2.3.1; Diagnostic Instruments) using DP Controller, DP Manager (Olympus) software, and NIH ImageJ software. RBCs were excluded. NETs were considered as extracellular structures that costained for MPO, histone H2A and DAPI.
Skin tissue immunohistochemistry and quantification of NETosis
Skin biopsies from 11 patients with cutaneous lupus involvement and 10 gender-matched healthy controls were analyzed for neutrophil infiltration and NETosis. In addition, skin biopsies from 10 patients with psoriasis were included as positive controls of IL-17+ cells in the skin. Control donors were identified from respondents to advertisements, had no personal or family history of lupus, and were free of inflammatory skin disease at the time of biopsy. Five-micrometer sections of skin were deparaffinized on a hot plate at 65°C for 1 hr. They were rehydrated by incubation and heat retrieval in Cell Conditioning Solution (CC1, Ventana Med, Tucson, AZ). The sections were blocked with 0.2% horse serum (Invitrogen) for 30 minutes. Simultaneous staining was first performed for 30 minutes with goat anti–IL-17 (100 μg/ml; R&D Systems), mouse anti-human Cathelicidin/OSX12 (Abcam) or anti-ds-DNA (Millipore) and another primary antibody, either rabbit anti-myeloperoxidase (MPO) (1:1500; Dako USA) or rabbit anti-human neutrophil elastase (10 μg/mL; Abcam). This was followed by 30 min incubation with matched secondary antibodies: chicken anti-goat IgG Alexa-Fluor 488 (1:300; Invitrogen), or chicken anti-rabbit IgG Alexa-Fluor 594 (1:300; Invitrogen). ProLong Gold antifade reagent with DAPI was added to the slides prior to mounting with cover slips (Invitrogen). Images were captured with a fluorescent imaging microscope (BX50; Olympus, Essex, U.K.) using DP Controller and DP Manager (Olympus) software. The recorded images were loaded onto Adobe Photoshop. The number of cells expressing one or both markers of interest in each 200x field was manually counted by two independent blinded observers.
Statistical analysis
For NET quantification, immunofluorescence was performed in neutrophils isolated from individual patients (n ≥ 4 for both SLE and control patients) and counts were averaged, represented as mean ± SEM, and analyzed by two-tailed Student’s t-test, where a p ≤ 0.05 was considered significant. Comparison of NETOsis within different areas of the skin was performed by repeated measures one way ANOVA with a Tukey’s multiple comparison post test. For all other studies, paired or unpaired Student’s t tests were performed. Microarray statistical analysis is detailed above.
RESULTS
Lupus LDGs have a distinct gene expression profile from autologous lupus neutrophils and control neutrophils
To determine whether LDGs represent a distinct pool of neutrophils, gene expression profiling of purified neutrophil fractions of lupus LDGs and autologous lupus neutrophils and gender-matched control neutrophils was assessed using Affymetrix genechip microarrays. Samples were matched-paired in comparing lupus LDGs with autologous lupus neutrophils. While there were no genes significantly differentially regulated when comparing normal density lupus neutrophils with healthy control neutrophils, several genes were differentially expressed in LDGs relative to either autologous lupus neutrophils or healthy control neutrophils. In all, 302 genes were differentially expressed in lupus LDGs when compared to control neutrophils and 281 genes were identified as altered by pair-wise comparison of each patient’s LDGs to their autologous lupus neutrophils (q-value < 0.01, fold-change ≥ 1.5 and ≤ 0.7 for the up-regulated and down-regulated genes respectively) (Figure 1A and Supplementary Tables I and II). A total of 224 genes were identified as upregulated in both comparisons (Figure 1A). Conversely, a total of 57 genes were found to be selectively regulated in lupus LDGs when compared to autologous lupus neutrophils, and 78 genes were restricted to LDGs alone when compared to control neutrophils (Figure 1A and Supplementary Tables I and II).
Figure 1. LDGs overexpress defensins and proinflammatory molecules.

A. Schematic representation of the sample group and gene list comparisons made using the gene microarray data (q-value <0.01, fold-change ≥1.5 and ≤ 0.7 for the up-regulated and down-regulated genes, respectively). Samples were matched-paired when comparing lupus neutrophils with autologous lupus LDGs (n=10 for each group). B. Lupus LDGs express elevated levels of azurophilic granule genes. Log base 2 mRNA mean expression values of five azurophil genes in control neutrophils (n=9), lupus neutrophils (n=10) and lupus LDGs (n=10): Myeloperoxidase (MPO), Elastase (ELANE), Defensin alpha 4 (DEFA4), Cathepsin G (CTSG) and azurocidin 1 (AZU1). Data are presented as mean ± SD. ***p<0.0001. C. Confirmation of enhanced mRNA expression by real-time PCR of various neutrophils genes in lupus LDGs when compared to autologous lupus neutrophils (n=7–12) and control neutrophils (n=7). Bar graph represents fold mRNA expression (mean ± SEM) after adjusting for housekeeping gene (GAPDH). *p<0.05 LDGs compared to control and/or autologous lupus neutrophils.
The defined transcripts were associated to canonical pathways using Ingenuity Pathway Analysis (IPA). The pathways significantly regulated in LDGs compared with control neutrophils and autologous lupus neutrophils are listed in Supplementary Table III. Actin cytoskeleton, macropinocytosis, clathrin-mediated endocytosis and integrin signaling pathways were among the most significantly regulated pathways in lupus LDGs compared to control and lupus neutrophils. Inhibition of angiogenesis by Tsp1 was a pathway significantly regulated only in lupus LDGs compared to control neutrophils; whereas Ephrin receptor signaling was only significantly regulated in lupus LDGs compared to lupus neutrophils. The top functions identified using IPA included inflammatory response, hematological and cardiovascular diseases.
The top up-regulated genes in lupus LDGs in both comparisons included a number of serine proteases, bactericidal proteins and other molecules involved in neutrophil regulation of the inflammatory response (3, 21–25), when compared with normal density lupus and control neutrophils (Table I). Among them, mRNA of the cathelicidin gene CAMP (LL-37) was 3.5 and 4.6-fold higher in lupus LDGs when compared to control and lupus neutrophils, respectively (q-value <0.01). In the same group, defensin α-4 (DEFA4) was highly up-regulated in LDGs compared to control and lupus neutrophils (22.4 and 25.6 fold, respectively). Lactotransferrin (LTF), lipocalin-2 (LCN2), MPO, elastase-2 (ELANE), matrix metalloproteinase-8 (MMP-8) and cathepsin-G (CTSG) were also significantly upregulated in the lupus LDGs when compared to control and lupus neutrophils (fold-change range from 10.9 to 41.5). These results indicate that lupus LDGs show increased expression of azurophilic granule genes which is not secondary to a general induction of these genes in SLE, as the autologous lupus neutrophils expressed levels comparable to control neutrophils (Figure 1B). These findings were confirmed by real time PCR for several of the molecules mentioned above (Figure 1C). Thus, microarray analysis of lupus LDGs confirmed an enhanced bactericidal and activated signature compared to control or normal density lupus neutrophils.
Table I.
Affymetrix microarray expression data in lupus LDGs of genes corresponding to enzymes, bactericidal and other molecules known to be implicated in ROS generation and/or NET formation.
| Lupus LDGs compared to control neutrophils | Lupus LDGs compared to lupus neutrophils | |||||
|---|---|---|---|---|---|---|
| Entrez gene ID | Gene symbol | Gene name | Fold-change | q-valuea | Fold-change | q-valuea |
| Enzyme and enzyme inhibitor genes | ||||||
| 6036 | RNASE2 | Ribonuclease, RNaseA family, 2 (liver, eosinophil-derived neurotoxin) | 4.24 | 0.004* | 3.06 | 0.066 |
| 4317 | MMP8 | Matrixmetallopeptidase 8 (neutrophil collagenase) | 38.53 | 0.000* | 41.53 | 0.000* |
| 4318 | MMP9 | Matrix metallopeptidase 9 (gelatinase B,92 kDa gelatinase,92 kDa type IV collagenase) | 1.08 | 0.999 | 1.37 | 0.047* |
| 6037 | RNASE3 | Ribonuclease, RNase A family, 3 (eosinophil cationic protein) | 12.34 | 0.000* | 12.07 | 0.000* |
| 1991 | ELANE (Elastase 2) | Elastase, neutrophil expressed | 11.64 | 0.000* | 10.92 | 0.000* |
| 3934 | LCN2 | Lipocalin2 | 24.49 | 0.000* | 19.02 | 0.000* |
| 5476 | CTSA | Cathepsin A | 1.69 | 0.004* | 1.54 | 0.043* |
| 1511 | CTSG | CathepsinG | 13.53 | 0.000* | 17.73 | 0.000* |
| 4353 | MPO | Myeloperoxidase | 12.02 | 0.000* | 14.66 | 0.000* |
| Bactericidal molecule genes | ||||||
| 1669 | DEFA4 | Defensin, alpha 4, corticostatin | 22.41 | 0.000* | 25.57 | 0.000* |
| 820 | CAMP (LL-37) | Cathelicidin antimicrobial peptide | 3.44 | 0.003* | 4.62 | 0.000* |
| 671 | BPI | Bactericidal/permeability-increasing protein | 13.80 | 0.000* | 16.59 | 0.000* |
| 566 | AZU1 | Azurocidin 1 | 11.23 | 0.000* | 12.22 | 0.000* |
| Other genes | ||||||
| 1088 | CEACAM 8 (CD66b) | Carcino embryonic antigen-related cell adhesion molecule8 | 16.63 | 0.000* | 18.27 | 0.000* |
| 10321 | CRISP3 | Cysteine-rich secretory protein3 | 18.39 | 0.000* | 12.18 | 0.000* |
| 634 | CEACAM 1 (CD66a) | Carcino embryonic antigen-related cell adhesion molecule 1 (biliary glycoprotein) | 1.50 | 0.099 | 1.30 | 0.017* |
| 4057 | LTF | Lactotransferrin | 16.91 | 0.000* | 12.01 | 0.000 |
| 1191 | CLU | Clusterin | 21.24 | 0.000* | 17.39 | 0.000* |
A q-value below 0.05 was considered as significant (in bold and with asterisk); N/A: not expressed above the Affymetrix control baseline.
All together, the transcriptional analysis results indicate that LDGs, as a distinct subset of lupus granulocytes, have a specific molecular pattern that differs from autologous lupus neutrophils and from control neutrophils, while lupus neutrophils display no significant differences in gene expression compared to control neutrophils. These results support the hypothesis that LDGs are a specific subset of granulocytes with distinct phenotype and functional capabilities.
NET formation is increased in lupus LDGs leading to enhanced externalization of autoantigens and immunostimulatory molecules
Serine proteases and cathelicidins released from neutrophils are incorporated into NETs, allowing the delivery of high concentrations of molecules that kill bacteria and degrade their virulence factors (8). We tested if NET formation differed in lupus LDGs relative to control neutrophils or autologous neutrophils. At baseline, peripheral blood LDGs demonstrated significantly enhanced NET formation right after isolation when compared to healthy control neutrophils or autologous lupus neutrophils (Figures 2A–B). Both control and normal density lupus neutrophils displayed significantly higher NET formation following a 2 hour in vitro stimulation with PMA, when compared to baseline levels (Figure 2A–B). In contrast, the percentage of NETs in lupus LDGs remained unchanged from baseline (Figure 2A–B), suggesting that LDGs may be pre-stimulated in vivo and resistant to further NET induction. When comparing isolation methods, there were no differences in NET formation when neutrophils were isolated through gradient separation or through negative selection with magnetic beads (data not shown). This excluded a potential effect of the isolation technique in the differences in induction of NETs in vitro.
Figure 2. Circulating lupus LDGs undergo increased NETosis.
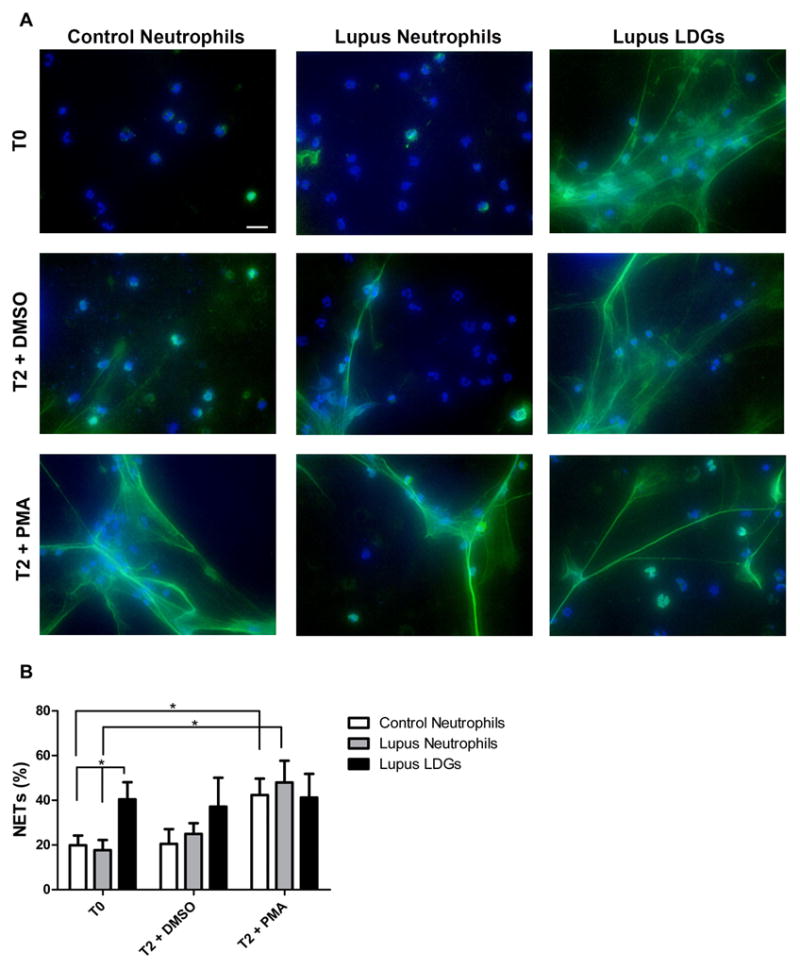
A. Representative images of control neutrophils, lupus neutrophils and lupus LDGs isolated from peripheral blood and analyzed at baseline (T0) or after stimulation for 2 hours with DMSO or PMA. Top panels show immunofluorescent merged images of neutrophil extracellular traps (NETs) which were detected by neutrophil elastase (green) and DNA was labeled with Hoechst 33342 (blue); 40x images, bar graph: 20μm. B. Quantification of the percentage of NETs (elastase-labeled cells over total number of cells) are plotted as mean ± SEM (n = 6 patients/group; * p ≤ 0.05).
There were no significant correlations between the percentage of LDGs undergoing NETosis at baseline and lupus disease activity (as determined by SLE disease activity index (SLEDAI))(14) or presence or titers of autoantibodies. Furthermore, there were no associations between use of various lupus medications (antimalarials, corticosteroids and/or immunosuppressive drugs) and percentage of LDGs undergoing NETosis (data not shown). These results indicate that enhanced NETosis occurs in lupus LDGs independent of the patients’ disease activity.
In order to further characterize the NET composition and functional relevance of extracellular trap formation, the localization and expression of immunostimulatory and bactericidal proteins was investigated. Control and lupus neutrophil and LDG NETs express the bactericidal proteins elastase and LL-37. The latter was detected intracellularly in all neutrophils but was predominantly found colocalized within the extracellular NETs. Since a higher percentage of LDGs undergo NETosis, there was significantly enhanced externalization of LL-37 by these cells when compared to control and lupus neutrophils (Figure 3A–B).
Figure 3. LL-37 externalization in NETs is increased in lupus LDGs.

A. Representative images of control neutrophils, lupus neutrophils and lupus LDGs after isolation from peripheral blood. Cells were stained for detection of LL-37 (red), neutrophil elastase (green) and DNA was labeled with Hoechst 33342 (blue). Top panels show images of LL-37 and Hoechst (left), elastase and Hoechst (middle) and merged LL-37, elastase and Hoechst (right); 40x images, bar graph = 20μm. Arrows represent areas of LL-37 localization within the NETs. B. Quantification of the percentage of cells containing LL-37 colocalized with elastase over total number of cells are plotted as mean ± SEM (n ≥ 3 patients; * p ≤ 0.05).
Because NETs may be sources of autoantigens in other conditions(10), we assessed if various targeted autoantigens were present in LDG NETs. During quantification, it became evident that all NETs expose ds-DNA regardless of the neutrophil source. However, as a higher percentage of LDGs undergo NETosis than healthy controls or lupus neutrophils, they lead to overall enhancement in externalization of ds-DNA (Figure 4A and 4C). In contrast, there was no evidence of expression of other common lupus autoantigens (Ro, La or Smith) within the NET’s structure. Indeed, the expression of Ro, La and Smith was intracellular and equally detected in LDGs, lupus and control neutrophils (data not shown). These observations were independent of whether the serum of the patients was positive or negative for antibodies against ds-DNA, Smith, Ro or La (Table II and data not shown). These results indicate that netting neutrophils externalize ds-DNA and that LDGs may represent an enhanced source of extracellular ds-DNA through heightened NET formation.
Figure 4. Lupus LDGs externalize ds-DNA and IL-17 through NETosis.
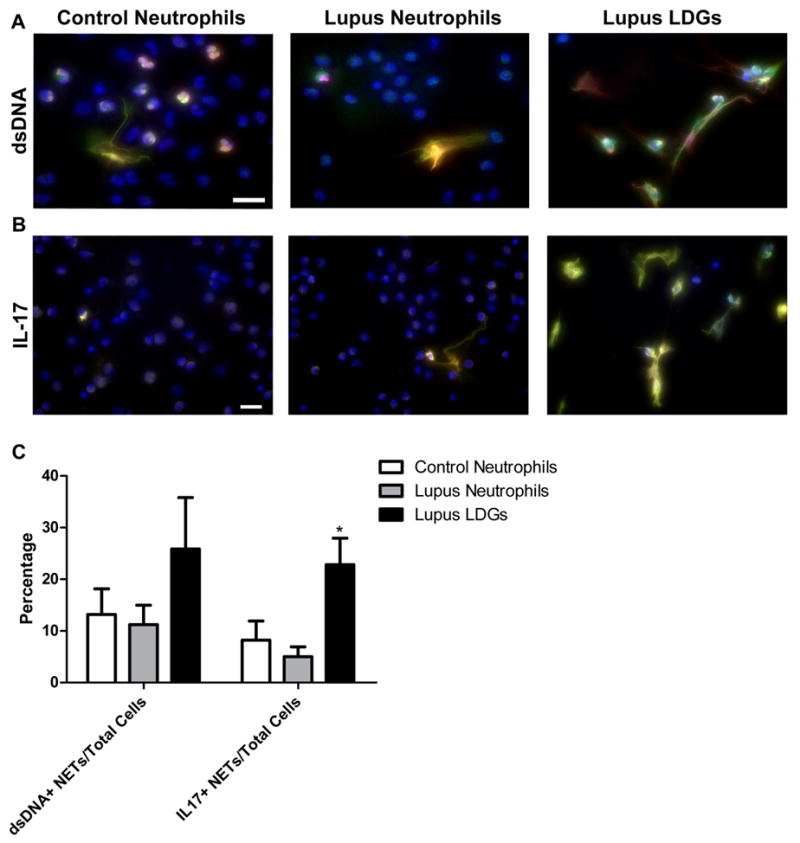
Representative images of control neutrophils, lupus neutrophils and lupus LDGs after isolation from peripheral blood. Cells were stained for detection of neutrophil elastase (green), DNA (Hoechst 33342, blue) and either ds-DNA (red) or IL-17 (red). A. Merged images of ds-DNA, elastase and Hoechst and B. Merged images of IL-17, elastase and Hoechst. C. Quantification of the percentage of cells containing ds-DNA or IL-17 colocalized with elastase over total number of cells are plotted as mean ± SEM (n ≥ 5 patients; * p ≤ 0.05).
NET formation leads to enhanced externalization of IL-17 in lupus LDGs
Innate IL-17-producing cells are considered an integral part of IL-17-mediated immune responses (26). Neutrophils have previously been identified as a source of IL-17, particularly in the context of autoimmunity (27), although the signaling pathways by which these cells synthesize and release this specific cytokine remain uncharacterized. Neutrophils expressing IL-17 have been reported in diseased tissues such as atherosclerotic plaques (28). Furthermore, neutrophils can externalize IL-17 through NETosis in skin and peripheral blood from patients with psoriasis (Lin et al. manuscript under review).
Recent evidence indicates that IL-17 may be involved in the pathogenesis of SLE through its capacity to amplify local inflammation by recruiting cells from the innate immune system and its ability to stimulate B cell adaptive immune responses (29). Indeed, elevated levels of IL-17 and increased numbers of Th17 cells have been reported in human and murine lupus and there is evidence that this cytokine is synthesized in target organs from patients with this disease including skin and kidney (29–31). However, it is unclear if, in addition to T cells, innate cells including neutrophils could represent an enhanced source of IL-17 production in blood and the periphery in SLE. Indeed, IL-17A was detected at the mRNA level in control and lupus neutrophils and in LDGs and there were no significant differences in expression between these three cell subsets (data now shown). In order to determine whether NETosis could be a source of enhanced IL-17 externalization in SLE, the expression of this cytokine in the NETs of peripheral blood neutrophils and LDGs was compared. Overall, between 8 ± 4 % to 23 ± 5 % of all neutrophils isolated from peripheral blood externalize IL-17 in vitro during NETosis and all NETS from peripheral blood neutrophils express IL-17. Both control and lupus neutrophils externalize IL-17 with similar frequency. As NETosis was significantly enhanced in lupus LDGs, a higher proportion of these cells externalized IL-17 (Figure 4B–C). These results suggest that NET formation by LDGs in situ may potentiate IL-17 dependent responses.
Overall, these results indicate that, through enhanced NETosis, LDGs externalize various immunostimulatory molecules and autoantigens that may be crucial in stimulation of adaptive and innate immunity.
Netting neutrophils infiltrate lupus kidneys affected by glomerulonephritis
Previous studies have indicated that impaired NET degradation in SLE is associated with the development of lupus nephritis (2). Furthermore, patients with ANCA+ vasculitis have evidence of netting neutrophils in their kidneys (10). While a potential role for neutrophils in lupus nephritis was proposed decades ago (32), it is unclear if NETosis is observed in lupus nephritis and if NETs play a pathogenic role in associated renal damage. We analyzed kidney biopsies from 9 lupus patients with WHO class III or IV glomerulonephritis (Table II). NETs visualized as web-like or granular structures co-staining with MPO, histone H2A, and DAPI were observed in the majority of lupus nephritis cases (67%) (Figure 5). Overall, 17% of glomeruli examined displayed netting neutrophils (range 0–50%). Patients with class IV lupus nephritis had a higher percentage of glomeruli infiltrated by netting neutrophils than patients with class III nephritis (27.5 ± 10% versus 9.84 ± 3% respectively, mean±SEM p=0.05). Renal biopsies with higher activity index, as per WHO classification, had higher percentage of glomeruli infiltrated by netting neutrophils (19.5 ± 5 % for biopsies with activity index ≥10 versus 6.6 ± 1.5% for biopsies with activity index <10). In addition, those patients with netting neutrophils in glomeruli had on average higher levels of anti-ds-DNA antibodies in serum that those without evidence of these cells (669 ± 380 IU/mL versus 189±150 IU/mL). In contrast, no NETosis was observed in the kidney biopsy from a patient with fulminant Henoch-Schönlein purpura and no lupus (not shown).
Figure 5. Netting neutrophils are present in glomeruli from patients with lupus nephritis.

Colocalization of histone H2A (green), MPO (red), and DNA (white) by direct immunofluorescence reveals in vivo evidence of NET formation in a glomerulus from a representative kidney microphotograph from a patient with class IV lupus nephritis. Yellow arrow and inset box highlights intraglomerular NET formation. Original magnification 20X.
Netting neutrophils infiltrate lupus skin and expose ds-DNA and LL-37
In previous studies, we identified that SLE patients with high levels of LDGs in the circulation had increased prevalence of skin involvement (1). After identifying increased NETosis in peripheral blood lupus LDGs, we proceeded to assess if lupus skin lesions show evidence of infiltration by netting neutrophils. We analyzed skin biopsies from 11 patients with several forms of cutaneous lupus including discoid lupus, acute cutaneous lupus, subacute cutaneous lupus (SCLE) and lupus panniculitis (Table II). High power examination of dual color immunofluorescence slides of SLE patient and control skin revealed numerous NETs visualized as web-like structures co-staining with MPO and DAPI (Figure 6). Similar distribution was seen when skin biopsies were stained with a neutrophil elastase antibody (not shown). Frequently observed in all cutaneous lupus lesions, these NETS were particularly enriched near the dermal-epidermal junction (DEJ) in the papillary dermis, and around blood vessels and adnexae, including hair follicles and eccrine glands (Figure 6A–C). In the two patients with lupus panniculitis, aggregates of netting neutrophils were frequently observed in the lobular adipose tissue with extension into adjacent reticular dermis. (Figure 6D–G). Overall, in lupus biopsies, 7% of all neutrophils in the epidermis, 24.3% of neutrophils in the papillary dermis and 25.4% of neutrophils in the reticular dermis and subcutis had formed NETs. NETosis was particularly enriched in areas with large aggregates of neutrophils (Figure 6). In contrast, neutrophil infiltration and NETosis were not observed in skin biopsies from 10 healthy control individuals (data not shown).
Figure 6. Netting neutrophils are present in affected lupus dermis and subcutis.

Direct immunofluorescence staining of DNA (blue) and MPO (red) reveal NETs throughout affected lupus skin. A. Low power view of a punch biopsy from lesional lupus skin. Bar = 200 μm. Arrows highlight perifollicular infiltration of NETs (B, bar = 50 μm), and NETs within the papillary dermis (C, bar = 20 μm). D. Low power view of lupus reticular dermis (bar = 200 μm) with arrows highlighting infiltration by NETs (E, bar = 50 μm). F. Low power view of a large blood vessel in affected lupus subcutis (bar = 200 μm), with arrows highlighting NETs in an area of panniculitis (G, bar = 20 μm). Dotted line delineates the dermal-epidermal junction (in A, C, D) and circumscribes the follicle (“f”) in B. Epidermis is designated “e” and dermis by “d”. H. Cathelicidin (LL-37) is present in NETs within inflamed lupus skin lesions. Expression of LL-37 in neutrophils in lupus tissue was examined by dual-color immunofluorescence staining for LL-37 (green) and MPO (red) with DAPI counterstain (blue). Representative images from one of 3 sections stained with LL-37 and MPO are shown at 600x magnification. Bar = 100 μm. I. Frequency of NETosis in cutaneous lupus lesions. NETs and neutrophils were counted after immunofluorescence staining with MPO and DAPI. Percentage of neutrophils undergoing NETosis of all neutrophils were calculated for the epidermis, papillary dermis, reticular dermis and subcutis. *p<0.05.
Interestingly, large aggregates of NETs were seen in the skin from all five lupus patients who had increased levels of anti-ds-DNA antibodies in sera (Table II). Because SLE is frequently associated with the presence of circulating anti-ds-DNA antibodies, we tested if large aggregates of NETs in skin could be a potential antigenic source for formation of these autoantibodies, especially since LL-37-DNA complexes present in the NETs may activate pDCs (33). Confirming the findings from peripheral blood neutrophils, analysis of these cells in affected lupus skin revealed that NETs expressed ds-DNA (data not shown). Furthermore, LL-37 was clearly present in the NETs in lupus skin biopsies (Figure 6H) and was most prominent in netting neutrophils from the deep dermis areas. These results indicate that NETs extrude ds-DNA and LL-37, possibly providing antigenic stimuli for anti-ds-DNA antibody formation.
Skin biopsies from lupus patients display higher numbers of IL-17+ neutrophils
Since IL-17 was detected in the NETs from peripheral blood neutrophils, we also assessed if IL-17-positive (IL-17+) neutrophils were detected in the affected lupus skin. Using dual color immunofluorescence for IL-17 and MPO, we observed that 51.5 ± 6.8% of all IL-17-expressing cells in lupus skin were neutrophils. Further, 19.7 ± 7.5% of intact neutrophils stained brightly with IL-17. These intact neutrophils were predominantly observed in dermal blood vessels and in the dermal interstitium of lupus lesions (Figure 7A–C). Only a small proportion (1 ± 0.8%) of NETs located in lupus skin were IL-17+. While the temporal events surrounding neutrophil extravasation, migration in tissue, and NETosis are unclear, our observations are consistent with a model where intact neutrophils containing IL-17 circulate in blood vessels, and then release IL-17 upon extravasation and migration into tissue, with all IL-17 released by the completion of extracellular trap formation. The percentage of neutrophils expressing IL-17 in lupus skin was significantly higher than in healthy control skin (1.9%, n=8, p<0.001), and similar to that seen in skin from patients with psoriasis (32%, n=12) (Figure 7 and data not shown).
Figure 7. IL-17-positive neutrophils infiltrate SLE skin.

Direct immunofluorescence staining of IL-17 (green) and MPO (red) in affected lupus dermis. A. Arrows highlight intact IL-17+ neutrophils in a blood vessel. Bar = 200 μm. B. IL-17 present in a NET (arrow). Bar = 10μm. C. Frequency of IL-17 expression in intact neutrophils and NETs in SLE skin. Percentages of 1) IL17+ neutrophils of all IL17+ cells, 2) IL17+ neutrophils of all intact neutrophils and 3) IL17+ NETs of all NETs are reported in the graph.
Overall, these results demonstrate that enhanced NETosis and exposure of autoantigens and immunostimulatory proteins occurs in vivo in affected organs of SLE patients.
Netting neutrophils stimulate IFN-α synthesis by pDCs
Recent work by various groups indicates that antimicrobial products, including LL-37, are immunostimulatory when bound to DNA and induce IFN-α synthesis by pDCs (23). Additionally, others have shown that NETosis stimulates pDCs to synthesize increased levels of IFN-α (11). There is also evidence that type I IFNs can induce NET formation(34). Given the important pathogenic role of IFN-α in SLE and the increased NET formation and LL-37 externalization by LDGs, we tested if these cells also induce pDCs to synthesize more IFN-α. Supernatants from control neutrophils did not induce the pDC cell line Gen2.2 (19) to synthesize IFN-α mRNA, when compared to pDCs alone. In contrast, supernatants from both lupus neutrophils and LDGs induced significantly enhanced IFN-α mRNA synthesis in pDCs, when compared to healthy controls (Figure 8A). IFN-α induction by neutrophil supernatants was significantly decreased in lupus neutrophils and LDGs after MNAse treatment, indicating that NETosis is involved in the pDC activation. The capacity to stimulate IFN-α in pDCs did not significantly differ between LDGs and autologous lupus neutrophils. These results confirm that both lupus neutrophils and LDGs have heightened capacity to induce IFN-α synthesis in pDCs, at least in part, through a NET-mediated effect.
Figure 8. Netting lupus LDGs stimulate IFN-α synthesis by PDCs and kill endothelial cells.

A. Gen2.2 pDC cells were stimulated for 16 hours with supernatants from control neutrophils (n=4), lupus neutrophils (n=6) and autologous lupus LDGs (n=6) that had been previously cultured in the presence or absence of micrococcal nuclease (MNase) for 1 hour. Results represent mean ± SEM IFN-α mRNA expression in Gen2.2 cells.*p<0.05, for control neutrophils compared with lupus neutrophils and with LDGs, and for lupus and LDGs compared to lupus or LDGs+MNAse. B. Bar graph represents mean ± SEM percentage of apoptotic HUVECs after exposure to activated LDGs, autologous neutrophils and control neutrophils (n=4–5/group) in the presence or absence of MNase. *p<0.05. C. Representative images of LDGs isolated from peripheral blood, cultured for 2 hours followed by either no MNase treatment (T2) or with MNase (T2+MNase) for 10 min at room temperature and processed for immunofluorescence staining of elastase (green) and DNA (Hoechst 33342, blue); 40X images, Bar= 20μm.
Lupus netting neutrophils induce endothelial cytotoxicity
Patients with SLE display evidence of accelerated endothelial cell (EC) apoptosis (35), which strongly correlates with the development of aberrant vascular function and may predispose to the development of atherosclerosis. Our group had previously reported that lupus LDGs, and less so lupus neutrophils, induce enhanced EC cytotoxicity using a co-culture system with human umbilical vein endothelial cells HUVECs (1). However, the mechanisms implicated in this enhanced EC cytotoxicity have not been identified. Because EC activation may induce NET formation, which in turn promotes endothelial cytotoxicity (7), we tested if NET formation by lupus LDGs could enhance EC death. Incubation of HUVEC monolayer with lupus LDGs induces significantly elevated levels of EC cytotoxicity, relative to control and lupus neutrophils (Figure 8B). As previously reported, lupus neutrophils also showed enhanced EC cytotoxicity when compared to healthy control neutrophils, but significantly less when compared to lupus LDGs (1). A component of this enhanced cytotoxicity was mediated by NET formation since disrupting these structures by treating lupus LDGs and neutrophils with micrococcal nuclease (MNase) (Figure 8C), significantly downregulated EC apoptosis (Figure 8B). These results indicate that lupus LDGs mediated more extensive EC killing, at least in part, through their enhanced capacity to form NETs.
DISCUSSION
Recent work from various groups indicates that NET formation may be an important phenomenon in autoantigen modification and exposure to the immune system, as well as in the induction of tissue damage (9, 10). As such, aberrant NET formation may play an important role in the development and perpetuation of autoimmune diseases and organ damage observed in chronic inflammatory disorders. Our work now expands and reinforces this concept by reporting that a distinct subset of neutrophils found in SLE patients (LDGs) have enhanced capacity to form NETs and upregulate expression of various neutrophil proteins and enzymes implicated in NET formation and in autoimmunity induction. These NETs also expose ds-DNA, an autoantigen considered key in lupus pathogenesis. Furthermore, lupus neutrophils and, in particular, LDGs elicit enhanced EC cytotoxicity through NET formation. This phenomenon also appears to play a role in the induction of IFN-α synthesis by pDCs. We have also identified that enhanced NETosis occurs in vivo in SLE in affected skin and kidney. Furthermore, neutrophils from blood and skin from SLE patients frequently externalize IL-17 as part of the NETosis process, which may contribute to tissue damage and immune dysregulation. Overall, these observations further support a pathogenic role for neutrophils in organ damage in SLE.
One of the findings from our study is that no differences in gene expression were found when normal density lupus neutrophils were compared to gender-matched healthy control neutrophils. Thus, previous reports examining alterations in lupus neutrophils may in part reflect responses specifically elicited in the LDG pool. In contrast, lupus LDGs obtained from the same patients from whom the normal density neutrophils were obtained showed significant differences in gene expression when compared to healthy control and lupus neutrophils. The pair-wise comparison of gene expression in LDGs and autologous neutrophils in SLE patients provides a control for many potential sources of variability such as medications, disease activity and clinical manifestations, and exposure to environmental factors. As it is expected that LDGs and autologous lupus neutrophils were exposed to a very similar cytokine milieu, these results indicate that LDGs may indeed represent a distinct subset of proinflammatory and pathogenic cells within the granulocyte spectrum.
It is unclear why LDGs upregulate mRNA of various serine proteases and bactericidal proteins present in azurophilic granules. One possibility is that these findings are indicative of a more immature phenotype of the LDGs, further supported by their immature nuclear morphology (1). Indeed, proteins synthesized at the same time during neutrophil differentiation are co-localized in the same granules. The levels of expression of the mRNA that encode the neutrophil serine proteases are greatest at the pro-myelocytic stage of neutrophil differentiation in the marrow and are downregulated as neutrophils mature (36). This observation could indicate that LDGs are indeed a more immature neutrophil subset, despite their apparent expression of markers of fully matured neutrophils, including CD16 and CD10. Indeed, a previous study has shown that, among the lupus bone marrow up-regulated genes (when compared to controls), the highest overexpression occurs in granulopoiesis-related genes. These genes include several of the “early granulopoiesis genes” upregulated in the LDG microarray in our study, including MPO, ELA2, CTSG, DEFA4 and LTF (24). This is also confirmed by a previous study that showed that the PBMC granulocyte signatures observed in pediatric SLE patients were for genes preferentially transcribed within the earliest granulocytes (myeloblast and promyelocytes) and with the presence of immature neutrophils in their peripheral blood(3). These observations further support that LDGs could represent an aberrant immature subset originating from lupus bone marrow that may persist or expand in the blood and/or other tissues from SLE patients.
The functional consequences of high serine protease expression in LDGs may be varied. It has been proposed that all serine proteases of azurophilic granules (cathepsin G, proteinase 3 and neutrophil elastase), released after encountering immune complexes, may potentiate a positive autocrine feedback on neutrophil activation (37) (38). Further, these molecules have been implicated in the activation of the pro-forms of proinflammatory cytokines including TNF and IL-1β (39). Neutrophil elastase can activate TLR4 eventually resulting in IL-8 production (40). IL-8 levels are elevated in SLE but the exact mechanisms leading to this increase have been unclear (41). One could propose that enhanced exposure to extracellular elastase through NET formation in SLE LDGs could promote enhanced synthesis of IL-8. This cytokine could in turn activate neutrophil recruitment and promote damage in various organs. In addition, we previously showed that LDGs synthesize enhanced levels of IL-8 and TNF upon activation, when compared to control and normal-density lupus neutrophils (1).
The mechanisms by which LDGs are more primed to make NETs are unclear. To date, the exact molecular mechanisms and subcellular events leading to NETosis remain elusive. While a crucial role for elastase, reactive oxygen species and the cytoskeleton has been proposed, recent reports suggest that NETosis is quite complex. The observed higher expression of elastase and MPO in LDGs could play an important role in enhancing extracellular trap synthesis, based on what other groups have recently reported(9). There is also evidence that type I and II IFNs can act as priming factors on mature neutrophils, allowing the formation of NETs upon subsequent stimulation with complement factor 5a (34). One possibility may be that LDGs are more sensitive to the effects of type I IFNs and/or to IFN signaling than normal density lupus neutrophils. However, this would go against the hypothesis that LDGs represent a more immature subset of neutrophils, since previous evidence indicates that granulocyte precursors and less mature cells are fairly insensitive to type I IFN effects, when compared to fully differentiated neutrophils (34). Further, while one possibility is that LDGs represent cells that have been exposed to elevated levels of type I IFNs in the bone marrow, we do not consider this is likely the case since these cells do not display evidence of increased type I IFN gene signature. Future studies need to explore whether the LDGs are derived from normal neutrophils or result from alterations in granulocyte development.
Previous studies have shown that the antimicrobial peptide LL-37 is a key factor that mediates pDC activation in psoriasis (23). LL-37 converts inert self-DNA into a potent trigger of type I IFN production. This occurs by binding the DNA to form condensed structures that are delivered to and retained within early endocytic compartments in pDCs to trigger TLR9 (23). LL-37 also converts self-RNA into a trigger of TLR7 and TLR8 in human DCs (22). In general, LL-37 is involved in a myriad of important immune functions including chemoattraction of immune cells and release of inflammatory mediators from epithelial cells (42). Our findings support that the enhanced release of LL-37 through NETosis could promote enhanced inflammatory responses in SLE organs. Other molecules overexpressed in lupus LDGs, including various defensins, may also be immunostimulatory (43). Our findings are in agreement with a recent study that reported that SLE patients have elevated serum levels of various neutrophil peptides including MPO and defensins (44, 45). It is possible that these are derived from LDGs. Further, increased anti-defensin and cathepsin-G ANCA antibodies are found in lupus patients (46). Given their immunomodulatory role, overproduction of alarmins might activate the adaptive immune and promote autoimmune responses, as is manifested in SLE (47, 48). Another overexpressed molecule in LDGs, neutrophil-gelatinase associated lipocalin (NGAL/lipocalin2), has recently been proposed as a biomarker of and to have a pathogenic role in lupus nephritis(49). Finally, the role of MMP-8 overexpression in LDGs remains to be determined, but this metalloproteinase may play a key role in vascular damage in other conditions (50).
Lupus patients develop accelerated atherosclerosis, leading to premature cardiovascular disease. We had previously found that ECs from lupus patients undergo accelerated apoptosis in vivo and that this phenomenon may be pathogenic, as it correlates with endothelial dysfunction development (35). LDGs are capable of killing ECs but the mechanism involved was unclear (1). The current study shows that, through enhanced NET formation, LDGs acquire a heightened capability to damage the endothelium, as cytotoxicity was abrogated with MNAse treatment. As such, accelerated NETosis may represent an important mechanism of premature vascular damage in SLE.
Death by NETosis may also represent an important immunostimulatory event with regards to activation of innate immunity in SLE, given the enhanced capacity of netting lupus neutrophils to activate pDCs. This could promote chronic activation of the immune system and perpetuation of type I IFN synthesis characteristic of this disease(51). Enhanced NETosis in SLE may be one of the mechanisms explaining why infections may trigger flares in this disease(52), with NET induction by microorganisms which in turn leads to autoantigen externalization and immune system activation. The observation that both lupus neutrophils and LDGs induced comparable induction of IFN-α synthesis by PDCs may indicate that this process cannot be fully explained by pure enhancement of NETosis in LDGs and that other variables are involved in this phenomenon. Future studies are needed to better understand how this process occurs.
The skin is an important target organ of the immune system in SLE and a significant proportion of lupus patients have cutaneous involvement. Although mechanisms of skin damage in SLE are likely multifactorial (53), recent evidence has identified an enhanced type I interferogenic signature and IFN-α producing pDCs in lupus skin(54). The role of pDCs in the inflammation observed in autoimmune skin disease in animal models has recently supported the notion that IFN-α may be crucial in cutaneous involvement in SLE (55). In a lupus murine model, tape stripping led to an influx into skin of neutrophils forming NETs which contain DNA and RNA associated with LL-37 (55). Our data confirm a similar pattern in human lupus skin biopsies, where abundant ds-DNA+, LL-37+ NETs are seen in multiple layers of affected skin. Interestingly, these findings are associated with the presence of elevated anti-ds-DNA antibodies in the circulation, supporting the notion that autoantigen exposure in the NETs could promote autoantibody formation in vivo.
Similar findings were seen in the kidneys of lupus patients affected by class III and IV glomerulonephritis. This supports recent findings that impaired NET degradation in SLE is associated with renal involvement in this disease (2). Indeed, IgG deposition on NETs in tubuli and glomeruli in the kidney of an SLE patient who degraded NETs poorly was reported in that study (2). We have now expanded this observation by studying a larger number of patients with lupus nephritis, where presence of netting neutrophils in glomeruli was observed in a majority of them. Interestingly, patients with higher proportion of glomeruli infiltrated by netting neutrophils had higher levels of circulating anti-ds-DNA antibodies and higher activity index in renal biopsies. It has been previously proposed that anti-NET antibodies and persistent NETs could form NET immune complexes that could be relevant in lupus disease severity (2). It is possible that the presence of infiltrating netting neutrophils in lupus tissue samples represented a combination of enhanced NETosis and impaired NET degradation. We could then propose a model of profound imbalance in NET production and degradation. On one hand, NETosis would be enhanced in lupus LDGs. On the other hand, the activity of DNase1 (an enzyme important in NET degradation) is decreased in a subset of SLE patients (2). This may provide the conditions by which NETs may persist and constitute a prolonged source of autoantigen exposure in an immunostimulatory context, leading to enhanced formation of immune complexes and induction of autoantibodies which could further contribute to tissue damage.
Whether the infiltrating neutrophils in the skin and kidney correspond primarily to LDGs is unclear at this point, as no specific cell marker has to this date been identified to distinguish these cells from normal-density neutrophils. A better understanding of the homing characteristics of LDGs versus other neutrophil subsets will be important to assess if, through enhanced NETosis or other yet unidentified mechanism, LDGs could have an increased capacity to migrate to various tissues and induce damage. It is relevant that patients with high levels of LDGs in their circulation have higher prevalence of skin involvement, and this association could also support that this cell subset is pathogenic to cutaneous tissue (1). Future studies in a larger number of patients are required to assess the specific role that these cells play in tissue damage and progression of disease in SLE.
IL-17 has recently been linked to the pathogenesis of SLE(30) by participation in the amplification of autoimmune responses by stimulating autoantibody production by B cells (29). Further, it augments tissue injury and target organ damage in this disease (31). Most studies have focused on Th17+ cells, which are elevated in SLE (56) and infiltrate renal tissue (57, 58). A recent report showed IL-17+ cells infiltrating lupus affected skin, but neutrophils were not specifically studied (59). Similar to what has been shown in other conditions (28) and (Lin et al., manuscript under review), neutrophils expressing IL-17 are seen at significantly enhanced levels in blood and affected skin from lupus patients. This phenomenon could initiate a cycle in which IL-17- secreting cells in lupus skin lesions (including innate and adaptive immune cells) would recruit additional neutrophils. IL-17 can stimulate endothelial cells to produce chemoattractants (IL-8) that selectively drive neutrophil but not lymphocyte chemotaxis. IL-17 increases neutrophil adhesion to the endothelium which may also enhance neutrophil recruitment to organs (60). We had previously been unable to detect IL-17A in LDG supernatants (1). Given that we have now found evidence of IL-17A expression both at the mRNA and protein levels, it is possible that the technique previously used was not sensitive enough or that the NET-bound IL-17 had not been fully released to the supernatants to allow for quantification by ELISA. Overall, these results further support a pathogenic role for IL-17 in organ damage in SLE and that neutrophils represent an important subset of IL-17 producing cells.
Whether enhanced NETosis is a phenomenon present in other autoimmune diseases associated to autoantibody production, interferogenic signatures and/or vascular damage remains to be determined and should be the focus of future investigations. These conditions may include inflammatory myopathies, Sjögren syndrome and rheumatoid arthritis (61–63).
In conclusion, we have identified that LDGs isolated from lupus patients have a higher capacity to synthesize NETs with immunostimulatory and cytotoxic properties. These results further expand the potential pathogenic role of aberrant lupus neutrophils through a NET-mediated effect and indicate that strategies attempting to modulate NET formation with the objective of abrogating autoimmune responses should be investigated.
Supplementary Material
Acknowledgments
The authors thank Dr. Michelle Kahlenberg for critical review of the manuscript.
ABBREVIATIONS
- SLE
systemic lupus erythematosus
- LDGs
low density granulocytes
- NETs
neutrophils extracellular traps
- dsDNA
double-stranded DNA
- ANCA
Anti-Neutrophilic Cytoplasmic Antibodies
- pDCs
plasmacytoid dendritic cells
- SAM
Significance Analysis of Microarrays
- MeV
MultiExperiment Viewer
- IPA
Ingenuity Pathway Analysis
- MNase
micrococcal nuclease
- MPO
myeloperoxidase
- DEFA4
defensin α-4
- LTF
lactotransferrin
- LCN2
lipocalin-2
- ELANE
elastase-2
- MMP-8
matrix metalloproteinase-8
- CTSG
cathepsin-G
- NGAL/Lipocalin2
neutrophil-gelatinase associated lipocalin
- AZU1
azurocidin 1
- SLEDAI
SLE disease activity index
- DEJ
dermal-epidermal junction
Footnotes
This work was supported by the National Institutes of Health (NIH) through Public Health Service Grant HL088419 (to MJK), the Arthritis Foundation (to MJK), the University of Michigan/Centocor Posdoctoral Research Program (to MJK and EV), the Anthony Gramer Fund in Inflammation Research (to MJK); the Babcock Research Endowment (to ATB), and the Training Grant in Cell and Molecular Dermatology 5T32AR007197 (to CR). This work was supported in part by the NIH through the University of Michigan’s Cancer Center Support (Grant P30 CA46592), the Rheumatic Disease Core Center (Grant P30 AR48310) and the Applied Systems Biology Core in the O’Brien Renal Center (Grant P30 DK081943).
References
- 1.Denny MF, Yalavarthi S, Zhao W, Thacker SG, Anderson M, Sandy AR, McCune WJ, Kaplan MJ. A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J Immunol. 184:3284–3297. doi: 10.4049/jimmunol.0902199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hakkim A, Furnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, Herrmann M, Voll RE, Zychlinsky A. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci U S A. 2010;107:9813–9818. doi: 10.1073/pnas.0909927107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, Pascual V. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. 2003;197:711–723. doi: 10.1084/jem.20021553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hacbarth E, Kajdacsy-Balla A. Low density neutrophils in patients with systemic lupus erythematosus, rheumatoid arthritis, and acute rheumatic fever. Arthritis Rheum. 1986;29:1334–1342. doi: 10.1002/art.1780291105. [DOI] [PubMed] [Google Scholar]
- 5.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 6.Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V, Zychlinsky A. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176:231–241. doi: 10.1083/jcb.200606027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gupta AK, Joshi MB, Philippova M, Erne P, Hasler P, Hahn S, Resink TJ. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett. 2010;584:3193–3197. doi: 10.1016/j.febslet.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 8.Neeli I, Khan SN, Radic M. Histone deimination as a response to inflammatory stimuli in neutrophils. J Immunol. 2008;180:1895–1902. doi: 10.4049/jimmunol.180.3.1895. [DOI] [PubMed] [Google Scholar]
- 9.Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. 191:677–691. doi: 10.1083/jcb.201006052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kessenbrock K, Krumbholz M, Schonermarck U, Back W, Gross WL, Werb Z, Grone HJ, Brinkmann V, Jenne DE. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med. 2009 doi: 10.1038/nm.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caielli SG-RG, Vega B, Connolly J, Allantaz F, Xu Z, Punaro M, Baisch J, Guiducci G, Coffman R, Barrat F, Banchereau J, Pascual V. Netting neutrophils are major inducers of type 1 IFN production in SLE (PO1.M.11 Abstract of Poster Session) Lupus. 2010;19(1 Supplement):117–118. [Google Scholar]
- 12.Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD, Jr, Wrobleski SK, Wakefield TW, Hartwig JH, Wagner DD. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A. 2010;107:15880–15885. doi: 10.1073/pnas.1005743107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725. doi: 10.1002/art.1780400928. [DOI] [PubMed] [Google Scholar]
- 14.Bombardier C, Gladman DD, Urowitz MB, Caron D, Chang CH. Derivation of the SLEDAI. A disease activity index for lupus patients. The Committee on Prognosis Studies in SLE. Arthritis Rheum. 1992;35:630–640. doi: 10.1002/art.1780350606. [DOI] [PubMed] [Google Scholar]
- 15.Clark RAaNWM. Isolation and Functional Analysis of neutrophils. 2005 doi: 10.1002/0471142735.im0723s19. [DOI] [PubMed] [Google Scholar]
- 16.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 17.Saeed AI, Bhagabati NK, Braisted JC, Liang W, Sharov V, Howe EA, Li J, Thiagarajan M, White JA, Quackenbush J. TM4 microarray software suite. Methods Enzymol. 2006;411:134–193. doi: 10.1016/S0076-6879(06)11009-5. [DOI] [PubMed] [Google Scholar]
- 18.Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, Sturn A, Snuffin M, Rezantsev A, Popov D, Ryltsov A, Kostukovich E, Borisovsky I, Liu Z, Vinsavich A, Trush V, Quackenbush J. TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003;34:374–378. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]
- 19.Chaperot L, Blum A, Manches O, Lui G, Angel J, Molens JP, Plumas J. Virus or TLR agonists induce TRAIL-mediated cytotoxic activity of plasmacytoid dendritic cells. J Immunol. 2006;176:248–255. doi: 10.4049/jimmunol.176.1.248. [DOI] [PubMed] [Google Scholar]
- 20.Weening JJ, V, D’Agati D, Schwartz MM, Seshan SV, Alpers CE, Appel GB, Balow JE, Bruijn JA, Cook T, Ferrario F, Fogo AB, Ginzler EM, Hebert L, Hill G, Hill P, Jennette JC, Kong NC, Lesavre P, Lockshin M, Looi LM, Makino H, Moura LA, Nagata M. The classification of glomerulonephritis in systemic lupus erythematosus revisited. Kidney Int. 2004;65:521–530. doi: 10.1111/j.1523-1755.2004.00443.x. [DOI] [PubMed] [Google Scholar]
- 21.Papayannopoulos V, Zychlinsky A. NETs: a new strategy for using old weapons. Trends Immunol. 2009;30:513–521. doi: 10.1016/j.it.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 22.Ganguly D, Chamilos G, Lande R, Gregorio J, Meller S, Facchinetti V, Homey B, Barrat FJ, Zal T, Gilliet M. Self-RNA-antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J Exp Med. 2009;206:1983–1994. doi: 10.1084/jem.20090480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lande R, Gregorio J, Facchinetti V, Chatterjee B, Wang YH, Homey B, Cao W, Su B, Nestle FO, Zal T, Mellman I, Schroder JM, Liu YJ, Gilliet M. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature. 2007;449:564–569. doi: 10.1038/nature06116. [DOI] [PubMed] [Google Scholar]
- 24.Nakou M, Knowlton N, Frank MB, Bertsias G, Osban J, Sandel CE, Papadaki H, Raptopoulou A, Sidiropoulos P, Kritikos I, Tassiulas I, Centola M, Boumpas DT. Gene expression in systemic lupus erythematosus: bone marrow analysis differentiates active from inactive disease and reveals apoptosis and granulopoiesis signatures. Arthritis Rheum. 2008;58:3541–3549. doi: 10.1002/art.23961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pham CT. Neutrophil serine proteases: specific regulators of inflammation. Nat Rev Immunol. 2006;6:541–550. doi: 10.1038/nri1841. [DOI] [PubMed] [Google Scholar]
- 26.Cua DJ, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol. 2010;10:479–489. doi: 10.1038/nri2800. [DOI] [PubMed] [Google Scholar]
- 27.Hoshino A, Nagao T, Nagi-Miura N, Ohno N, Yasuhara M, Yamamoto K, Nakayama T, Suzuki K. MPO-ANCA induces IL-17 production by activated neutrophils in vitro via classical complement pathway-dependent manner. J Autoimmun. 2008;31:79–89. doi: 10.1016/j.jaut.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 28.de Boer OJ, van der Meer JJ, Teeling P, van der Loos CM, Idu MM, van Maldegem F, Aten J, van der Wal AC. Differential expression of interleukin-17 family cytokines in intact and complicated human atherosclerotic plaques. J Pathol. 2010;220:499–508. doi: 10.1002/path.2667. [DOI] [PubMed] [Google Scholar]
- 29.Crispin JC, Tsokos GC. IL-17 in systemic lupus erythematosus. J Biomed Biotechnol. 2010;2010:943254. doi: 10.1155/2010/943254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen XQ, Yu YC, Deng HH, Sun JZ, Dai Z, Wu YW, Yang M. Plasma IL-17A is increased in new-onset SLE patients and associated with disease activity. Journal of clinical immunology. 2010;30:221–225. doi: 10.1007/s10875-009-9365-x. [DOI] [PubMed] [Google Scholar]
- 31.Edgerton C, Crispin JC, Moratz CM, Bettelli E, Oukka M, Simovic M, Zacharia A, Egan R, Chen J, Dalle Lucca JJ, Juang YT, Tsokos GC. IL-17 producing CD4+ T cells mediate accelerated ischemia/reperfusion-induced injury in autoimmunity-prone mice. Clin Immunol. 2009;130:313–321. doi: 10.1016/j.clim.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Camussi G, Cappio FC, Messina M, Coppo R, Stratta P, Vercellone A. The polymorphonuclear neutrophil (PMN) immunohistological technique: detection of immune complexes bound to the PMN membrane in acute poststreptococcal and lupus nephritis. Clin Nephrol. 1980;14:280–287. [PubMed] [Google Scholar]
- 33.Gilliet M, Lande R. Antimicrobial peptides and self-DNA in autoimmune skin inflammation. Current opinion in immunology. 2008;20:401–407. doi: 10.1016/j.coi.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 34.Martinelli S, Urosevic M, Daryadel A, Oberholzer PA, Baumann C, Fey MF, Dummer R, Simon HU, Yousefi S. Induction of genes mediating interferon-dependent extracellular trap formation during neutrophil differentiation. J Biol Chem. 2004;279:44123–44132. doi: 10.1074/jbc.M405883200. [DOI] [PubMed] [Google Scholar]
- 35.Rajagopalan S, Somers E, Brook R, Kehrer C, Pfenninger D, Lewis E, Chakrabarti A, Richardson B, Shelden E, McCune WJ, Kaplan M. Endothelial cell apoptosis in systemic lupus erythematosus: a common pathway for abnormal vascular function and thrombosis propensity. Blood. 2004;103:3677–3683. doi: 10.1182/blood-2003-09-3198. [DOI] [PubMed] [Google Scholar]
- 36.Cowland JB, Borregaard N. The individual regulation of granule protein mRNA levels during neutrophil maturation explains the heterogeneity of neutrophil granules. J Leukoc Biol. 1999;66:989–995. doi: 10.1002/jlb.66.6.989. [DOI] [PubMed] [Google Scholar]
- 37.Kessenbrock K, Frohlich L, Sixt M, Lammermann T, Pfister H, Bateman A, Belaaouaj A, Ring J, Ollert M, Fassler R, Jenne DE. Proteinase 3 and neutrophil elastase enhance inflammation in mice by inactivating antiinflammatory progranulin. J Clin Invest. 2008;118:2438–2447. doi: 10.1172/JCI34694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Adkison AM, Raptis SZ, Kelley DG, Pham CT. Dipeptidyl peptidase I activates neutrophil-derived serine proteases and regulates the development of acute experimental arthritis. J Clin Invest. 2002;109:363–371. doi: 10.1172/JCI13462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meyer-Hoffert U. Neutrophil-derived serine proteases modulate innate immune responses. Front Biosci. 2009;14:3409–3418. doi: 10.2741/3462. [DOI] [PubMed] [Google Scholar]
- 40.Devaney JM, Greene CM, Taggart CC, Carroll TP, O’Neill SJ, McElvaney NG. Neutrophil elastase up-regulates interleukin-8 via toll-like receptor 4. FEBS Lett. 2003;544:129–132. doi: 10.1016/s0014-5793(03)00482-4. [DOI] [PubMed] [Google Scholar]
- 41.Ding D, Mehta H, McCune WJ, Kaplan MJ. Aberrant phenotype and function of myeloid dendritic cells in systemic lupus erythematosus. J Immunol. 2006;177:5878–5889. doi: 10.4049/jimmunol.177.9.5878. [DOI] [PubMed] [Google Scholar]
- 42.Bals R, Wilson JM. Cathelicidins--a family of multifunctional antimicrobial peptides. Cell Mol Life Sci. 2003;60:711–720. doi: 10.1007/s00018-003-2186-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.de la Rosa G, Yang D, Tewary P, Varadhachary A, Oppenheim JJ. Lactoferrin acts as an alarmin to promote the recruitment and activation of APCs and antigen-specific immune responses. J Immunol. 2008;180:6868–6876. doi: 10.4049/jimmunol.180.10.6868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vordenbaumen S, Fischer-Betz R, Timm D, Sander O, Chehab G, Richter J, Bleck E, Schneider M. Elevated levels of human beta-defensin 2 and human neutrophil peptides in systemic lupus erythematosus. Lupus. doi: 10.1177/0961203310377089. [DOI] [PubMed] [Google Scholar]
- 45.Telles RW, Ferreira GA, da Silva NP, Sato EI. Increased plasma myeloperoxidase levels in systemic lupus erythematosus. Rheumatology international. 2010;30:779–784. doi: 10.1007/s00296-009-1067-4. [DOI] [PubMed] [Google Scholar]
- 46.Tamiya H, Tani K, Miyata J, Sato K, Urata T, Lkhagvaa B, Otsuka S, Shigekiyo S, Sone S. Defensins- and cathepsin G-ANCA in systemic lupus erythematosus. Rheumatology international. 2006;27:147–152. doi: 10.1007/s00296-006-0173-9. [DOI] [PubMed] [Google Scholar]
- 47.Froy O, Sthoeger ZM. Defensins in systemic lupus erythematosus. Ann N Y Acad Sci. 2009;1173:365–369. doi: 10.1111/j.1749-6632.2009.04622.x. [DOI] [PubMed] [Google Scholar]
- 48.Kumar V, Sharma A. Neutrophils: Cinderella of innate immune system. Int Immunopharmacol. doi: 10.1016/j.intimp.2010.08.012. [DOI] [PubMed] [Google Scholar]
- 49.Viau A, El Karoui K, Laouari D, Burtin M, Nguyen C, Mori K, Pillebout E, Berger T, Mak TW, Knebelmann B, Friedlander G, Barasch J, Terzi F. Lipocalin 2 is essential for chronic kidney disease progression in mice and humans. J Clin Invest. 2010;120:4065–4076. doi: 10.1172/JCI42004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ionita MG, van den Borne P, Catanzariti LM, Moll FL, de Vries JP, Pasterkamp G, Vink A, de Kleijn DP. High neutrophil numbers in human carotid atherosclerotic plaques are associated with characteristics of rupture-prone lesions. Arterioscler Thromb Vasc Biol. 2010;30:1842–1848. doi: 10.1161/ATVBAHA.110.209296. [DOI] [PubMed] [Google Scholar]
- 51.Banchereau J, Pascual V. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity. 2006;25:383–392. doi: 10.1016/j.immuni.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 52.Ruiz-Irastorza G, Khamashta MA, Castellino G, Hughes GR. Systemic lupus erythematosus. Lancet. 2001;357:1027–1032. doi: 10.1016/S0140-6736(00)04239-2. [DOI] [PubMed] [Google Scholar]
- 53.Wenzel J, Zahn S, Tuting T. Pathogenesis of cutaneous lupus erythematosus: common and different features in distinct subsets. Lupus. 2010;19:1020–1028. doi: 10.1177/0961203310370046. [DOI] [PubMed] [Google Scholar]
- 54.Farkas L, Beiske K, Lund-Johansen F, Brandtzaeg P, Jahnsen FL. Plasmacytoid dendritic cells (natural interferon- alpha/beta-producing cells) accumulate in cutaneous lupus erythematosus lesions. Am J Pathol. 2001;159:237–243. doi: 10.1016/s0002-9440(10)61689-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guiducci C, Tripodo C, Gong M, Sangaletti S, Colombo MP, Coffman RL, Barrat FJ. Autoimmune skin inflammation is dependent on plasmacytoid dendritic cell activation by nucleic acids via TLR7 and TLR9. J Exp Med. 2010;207:2931–2942. doi: 10.1084/jem.20101048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shah K, Lee WW, Lee SH, Kim SH, Kang SW, Craft J, Kang I. Dysregulated balance of Th17 and Th1 cells in systemic lupus erythematosus. Arthritis Res Ther. 2010;12:R53. doi: 10.1186/ar2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dolff S, Quandt D, Wilde B, Feldkamp T, Hua F, Cai X, Specker C, Kribben A, Kallenberg CG, Witzke O. Increased expression of costimulatory markers CD134 and CD80 on interleukin-17 producing T cells in patients with systemic lupus erythematosus. Arthritis Res Ther. 2010;12:R150. doi: 10.1186/ar3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang Z, V, Kyttaris C, Tsokos GC. The role of IL-23/IL-17 axis in lupus nephritis. J Immunol. 2009;183:3160–3169. doi: 10.4049/jimmunol.0900385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tanasescu C, Balanescu E, Balanescu P, Olteanu R, Badea C, Grancea C, Vagu C, Bleotu C, Ardeleanu C, Georgescu A. IL-17 in cutaneous lupus erythematosus. Eur J Intern Med. 2010;21:202–207. doi: 10.1016/j.ejim.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 60.Roussel L, Houle F, Chan C, Yao Y, Berube J, Olivenstein R, Martin JG, Huot J, Hamid Q, Ferri L, Rousseau S. IL-17 promotes p38 MAPK-dependent endothelial activation enhancing neutrophil recruitment to sites of inflammation. J Immunol. 2010;184:4531–4537. doi: 10.4049/jimmunol.0903162. [DOI] [PubMed] [Google Scholar]
- 61.Emamian ES, Leon JM, Lessard CJ, Grandits M, Baechler EC, Gaffney PM, Segal B, Rhodus NL, Moser KL. Peripheral blood gene expression profiling in Sjogren’s syndrome. Genes Immun. 2009 doi: 10.1038/gene.2009.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Walsh RJ, Kong SW, Yao Y, Jallal B, Kiener PA, Pinkus JL, Beggs AH, Amato AA, Greenberg SA. Type I interferon-inducible gene expression in blood is present and reflects disease activity in dermatomyositis and polymyositis. Arthritis Rheum. 2007;56:3784–3792. doi: 10.1002/art.22928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang D, Arkfeld D, Fong TL. Development of anti-CCP-positive rheumatoid arthritis following pegylated interferon-alpha2a treatment for chronic hepatitis C infection. J Rheumatol. 2010;37:1777. doi: 10.3899/jrheum.100092. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.