

Abstract
Periodontal disease (PD) is a chronic inflammation of the tooth supporting soft tissue and alveolar bone due to infection by a select group of gram negative microbes, and leads to tooth loss if untreated. Since mice deficient in CD4+ cells are resistant to infection-induced alveolar bone loss, Th cells have been implicated in bone destructive processes during PD. However, the extent to which different Th-cell subtypes play roles in pathogenesis or host protection remains to be defined, and is likely to vary depending on the dominant microorganism involved. By far the best studied periodontal microbe in PD is Porphyromonas gingivalis. Even though the gram negative anaerobe Tannerella forsythia is also a vital contributor to periodontal bone loss, almost nothing is known about immune responses to this organism. Previous studies from our laboratory have revealed that T. forsythia induces periodontal bone loss in mice, and that this bone loss depends on the bacterially-expressed BspA protein. In this study, we show that T. forsythia activates murine APCs primarily through TLR2-dependent signaling via BspA. Furthermore, T. forsythia infection causes a pronounced Th2 bias, evidenced by T cell expression of IL-5 but not IFN-γ or IL-17 in draining LN. Consistently, deficiencies in TLR2 or STAT6 result in resistance to T. forsythia-induced alveolar bone loss. Thus, TLR2 signaling and Th2 cells play pathogenic roles in T. forsythia-induced alveolar bone destruction.
INTRODUCTION
Periodontal disease (PD) is an inflammatory disease of the tooth-supporting tissue (periodontium) that frequently leads to tooth loss (1), and is the most common cause of inflammation-induced bone loss in humans. PD is caused by a select group of anaerobic gram-negative bacteria that colonize subgingival spaces as biofilms. Periodontal bone destruction results mainly from the effects of immune response to these biofilm bacteria (2, 3). The pathogenesis of PD is mediated by a polymicrobial consortium consisting of gram negative pathogens Porphyromonas gingivalis, Treponema denticola and Tannerella forsythia, collectively known as the ‘red complex’ (4–6). Although T. forsythia has been increasingly implicated in the development and severity of PD, it remains a highly understudied pathogen (7, 8). Its etiological role in PD has only been recognized relatively recently, and the virulence mechanisms of T. forsythia are only just beginning to be defined (7). Unlike P. gingivalis, T. forsythia is a fastidious microbe with stringent growth requirements. We were the first to document the virulence potential of T. forsythia in a murine model of PD (9). In so doing, we found that the alveolar bone loss is dependent on the bacterially-expressed virulence protein BspA (9). However, the immune response to T. forsythia remains almost entirely undefined.
Alveolar bone loss in response to oral infection by P. gingivalis is dependent on host response. For example, SCID mice or mice specifically deficient in CD4+ T cells are resistant to alveolar bone loss to P. gingivalis infection (10–12). Moreover, Th1 responses are associated with P. gingivalis-stimulated alveolar bone loss in mice (13). In humans, the role of T cells in PD pathobiology is complex, and data support a role for T cells in both protection and pathogenesis (6, 14–16). The existence of two distinct effector CD4 T cell subsets has been recognized since 1986 (17), with the description of the Th1 (IFN-γ producing) and Th2 (IL-4-, IL-5- and IL-13-producing) cells. More recently, the Th17 lineage of IL-17-producing T cells was recognized in 2005 (18). In P. gingivalis infection, both Th1 and Th2 cytokines are found in the periodontal lesion (6, 14–16). Prior to the discovery of Th17 cells, it was suggested that Th1 cells are characteristic of a ‘stable’ lesions, whereas Th2 cells are associated with disease (14). However, IL-17 has also been documented in periodontitis patients with severe disease (19–22), and a significant number of CD4+ T cells isolated from gingival tissue of periodontitis patients express IL-17 (23). In P. gingivalis murine infections, IL-17 signaling is host-protective by virtue of limiting infection via neutrophil mobilization (24).
The Th effector responses to T. forsythia are still poorly defined, and conceivably may be different from responses to P. gingivalis. This could arise from differences in the nature of the pathogen-associated molecular patterns (PAMPs) expressed on different periodontal pathogens, which ultimately shape the adaptive response. In support of this notion, T. forsythia, unlike P. gingivalis, is unable to block neutrophil recruitment during infection in a mouse model (25), and recent studies demonstrated that T. forsythia, but not P. gingivalis, preferentially activates TLR2. For instance, although P. gingivalis through its fimbriae and LPS predominantly activate TLR2 (26), whole bacteria (27) and both minor and major fimbrial proteins activate TLR4/CD14/MD2 (28, 29). Thus, the role of CD4 effector responses in PD bone loss remain poorly defined (14, 30), particularly specific responses to T. forsythia.
TLRs can play protective or destructive roles depending on the nature of the invading pathogen and its associated virulence determinants (31). T. forsythia as well as its virulence factor BspA induce proinflammatory cytokine and chemokine secretion through TLR2 (32–34). Signaling by TLR2 in APCs, and expression of specific cytokines, has been suggested to favor Th2 responses (35–38). Moreover, the Th2-specific transcription factor STAT6 has been linked to susceptibility to PD in mice (39). Accordingly, it was compelling to determine the role of TLR2 and STAT6-mediated responses in T. forsythia-induced bone destruction. We predicted that T. forsythia would favor a Th2 inflammatory response by activating TLR2. We further hypothesized that Th2 response would exert a destructive role, based on our prior observation that T. forsythia causes alveolar bone loss in mice (9). As shown herein, we found that TLR2−/− or STAT6−/− mice indeed showed markedly decreased susceptibility to T. forsythia-induced PD bone loss, associated with decreased Th2 responses. These results imply a critical involvement of a bone-destructive Th2 response following oral infection with T. forsythia, mediated via TLR2 signaling.
MATERIALS AND METHODS
Mice
Specific-pathogen free BALB/cJ mice (WT) and STAT6−/− mice (BALB/cJ background) were from The Jackson Laboratory (Bar Harbor, ME). TLR2−/− mice on a BALB/cJ background used in this study represent progeny obtained after backcrossing the tlr2 deletion (tlr2−/−) originally on C57BL/6 to BALB/cJ background for 6 generations (TDC, unpublished data). These mice were healthy and showed no signs of gross abnormalities. Mice were maintained in HEPA-filtered cages with autoclaved food, water, and bedding. All procedures were performed in accordance with protocols approved by the University at Buffalo Institutional Animal Care and Use Committee (IACUC).
Bacterial strains and culture Conditions
T. forsythia was cultured in TF broth or on TF-agar plates (1.5 % agar in TF-Broth) under anaerobic conditions as described previously (40).
Oral infection and alveolar bone loss assessment
Animals within groups were age-and sex-matched (6–7 weeks at the start of the experiment; n=8–10 per group) and quarantined for 1 week prior to the experiment. Mice were infected with T. forsythia as previously described with the following modifications (9): mice were treated with kanamycin (1 mg/mL) for 7-days ad libitum, followed by a 3-day antibiotic-free period. This was followed by infection with live bacteria (T. forsythia ATCC43037) via oral gavage. Infection was given as 100 μL bacterial suspensions (109 cfu/mL) in 2% carboxymethyl cellulose (CMC) 3 times at 48 h intervals for 2 weeks. The control (sham-infected) mice received antibiotic pre-treatment and 100 μL 2% CMC. Mice were sacrificed after 6 weeks, and serum was collected by cardiac puncture. Jaws were autoclaved, defleshed, immersed overnight in 3% hydrogen peroxide, and stained with 1% methylene blue. Horizontal bone loss was assessed morphometrically by measuring the distance between the cementoenamel junction (CEJ) and the alveolar bone crest (ABC). Measurements at 14 buccal sites per mouse (7 on the left and right maxillary molars) were made under a dissecting microscope (Brook-Anco, Rochester, NY) fitted with an Aquinto imaging measurement system (a4i America). Random, blinded bone measurements were taken by 2 independent evaluators. Data were analyzed on GraphPad Prism 5 (GraphPad, San Diego, CA).
ELISA
T. forsythia-specific ELISAs were performed as described (9). Briefly, 96-well Immuno-Maxisorp plates (Nalgene Nunc International, Rochester, NY) were coated with formalin-fixed T. forsythia (108 cells per well). Sera was added in twofold serial dilutions, and T. forsythia-specific IgG was detected using HRP-conjugated goat anti-mouse IgG (Bethyl Laboratories, TX). Specific serum IgG isotype antibody was detected by addition of biotinylated isotype-specific secondary antibody (rat anti-mouse IgG1 or IgG2a; Southern Biotechnology, AL) followed by streptavidin-conjugated HRP (Southern Biotechnology). ELISA wells were color developed with TMB Microwell enzyme substrate (Kirkgaards and Perry, MD). After stopping the enzyme reaction by adding 0.1N H2SO4, plates were read at 495 nm. The titer was defined as the log2 of the highest dilution with a signal that was 0.1 optical density units above background.
Isolation of dendritic cells and macrophages and stimulation
Bone marrow-derived dendritic cells (BMDCs) were generated as described (41). Briefly, femurs and tibias were flushed with PBS. Erythrocytes were lysed using M-Lyse buffer (R&D Systems, Minneapolis, MN) and cells were suspended in RMPI-1640 supplemented with 10% heat-inactivated fetal calf serum, penicillin (50 U/ml), streptomycin (50 μg/ml), L-glutamine (2 mM), β-mercaptoethanol (50 μM), sodium pyruvate (1 mM), sodium bicarbonate (1.5 mg/ml) and HEPES (25 mM). BMDCs were cultured in 24-well plates at 106 cells/ml/well. GM-CSF (R&D Systems, Minneapolis, MN) was added at 20 ng/ml. Media + GM-CSF was replaced on days 2, 4 and 6. Cells were restimulated on d 7. This protocol yielded BMDCs with dendritic morphology and > 85% pure by CD11c+ staining. Peritoneal macrophages were prepared as described (42).
Cytokine ELISA
Cytokines were measured in triplicate by ELISA kits from eBiosciences. Detection limits were 15 pg/ml for IFN-γ, 4 pg/mlfor IL-10, 10 pg/ml for IL-12 p40, and 8 pg/ml for IL-17, respectively.
Intracellular staining and FACS analysis
Abs and isotype controls were purchased from eBiosciences: phycoerythrin (PE)-conjugated anti-IFN-γ and rat IgG2a; FITC-conjugated anti-IL-4; PECy5 labeled anti-CD4; anti-CD80 (16–10A1); and anti-CD83 (Michel-17); rat IgG1, IgG2a, and IgG2b; PE-conjugated anti-CD11c (N418); hamster IgG; PE-conjugatedanti-IL-17A (eBio17B7) and rat IgG2a. For intracellular staining, cell suspensions from cervical LN were stimulated with anti-CD3 and anti-CD28 Abs (eBioscience) for 48 h followed by with PMA (50 ng/ml), ionomycin (1 μg/ml) and brefeldin A (10 μg/ml) for 6 h. Cells were washed, incubated with FcR block (1 μg/ml; eBiosciences) and stained for CD4. Cells were fixed with 2% paraformaldehyde, permeabilized with 0.1% saponin (Sigma), and stained for IL-4, IL-17 or IFN-γ. Cells were analyzed on a FACScalibur (BD Biosciences) with FCS Express software (DeNovo Software, Inc)
Histological staining
Murine maxillary and mandibular bones (n = 4) were fixed in 10% phosphate-buffered formalin and decalcified in 10% EDTA. Samples were embedded in paraffin, and sections at 4 μm were prepared and stained for tartrate-resistant acid phosphatase (TRAP; Sigma). Slides were digitally scanned with a ScanScope CS system (Aperio) to minimize color fading and viewed with ImageScope viewing software (Aperio). The right maxillary and mandibular interdentalareas (average of 10 higher power fields/slide) of the crestal alveolar bone from the first molar to third molar were used to quantify osteoclasts.
CD45 staining
The right and left halves of the maxillary and mandibular bones were removed, fixed and embedded in paraffin, 4-μm sections were cut and mounted. They were deparafinized in xylene and hydrated in graded ethanol. After antigen retrieval by incubating at 90 °C for 10 min with BD Retrievagen A (BD Pharmingen), specimens were sequentially incubated in: (i) blocking solution containing and 0.1% Triton X-100 in 0.1 M PBS for 1 h at room temperature; (ii) monoclonal rat anti-mouse CD45 (BD Pharmingen) (1:30 dilution in PBS containing 0.1% Triton X-100) for 1 h at room temperature. The slides were incubated with a biotinylated goat anti-rat secondary antibody followed by an avidin-biotin complex developed with 3,3-diaminobenzidine (DAB) (Vector Labs, Burlingame, CA); the counterstain was haematoxylin. After each step, slides were rinsed in PBST (3 × 10 min). Slides were analyzed with a ScanScope CS system (Aperio) and viewed with ImageScope viewing software (Aperio). The interdental areas from the first to third molar were used to quantify inflammatory cells.
Data analysis
Data were analyzed on GraphPad Prism software (GraphPad, San Diego, CA). Comparisons between groups were made using a Student’s t test (between two groups) or ANOVA (multiple group comparisons), as appropriate. Statistical significance was defined as p < 0.05.
RESULTS
T. forsythia-induced cytokine expression in APCs is dependent on TLR2
T. forsythia and its major virulence protein BspA induce cytokine expression in epithelial cells through activation of TLR2 (33, 34). To determine the contribution of TLR2 in APCs in the recognition of and response to T. forsythia, bone marrow-derived dendritic cells (BMDCs) and macrophages were challenged with T. forsythia and cytokines were assayed by ELISA. IL-6, a bone resorptive cytokine, contributes to alveolar bone loss in the context of P. gingivalis infection (11), and IL-10 deficient mice are increasingly susceptible to P. gingivalis-induced bone loss (43). IL12p70, IL-12p40 and IL-23p19 were assayed as T effector polarizing cytokines. BMDCs and peritoneal macrophages were purified by negative selection and confirmed to be 90–95% pure by staining for CD11c, MHC-II, and CD86 (BMDCs) or CD11b (macrophages) by flow cytometry (data not shown). Recombinant BspA (34) and Pam3CSK4 were used as TLR2 agonists, and pure E. coli LPS (eLPS) was used as a TLR4 agonist. T. forsythia whole cells (Tf) induced significantly higher amounts of IL-6 and IL-10 secretion in wild-type (BALB/c) BMDCs and macrophages compared to TLR2−/− cells (Fig 1A). The reduced secretion of IL-6 and IL12p40 in TLR2−/− BMDCs was quite intriguing, considering eLPs is a TLR4 agonist. Future investigations ongoing in our group will address this issue in detail. As expected, BspA and Pam3CSK4 induced low levels of cytokine secretion in TLR2−/− cells, whereas eLPS induced potent production of these cytokines (Fig. 1A). Interestingly, only BMDCs secreted IL-12p40 and IL-12p70 in response to T. forsythia challenge, which was largely independent of TLR2 (Fig. 1B). These results suggest that T. forsythia induces differential responses in APCs with respect to the Th1 cytokine IL-12. This also suggests that other surface receptors or intracellular PRRs (e.g., TLR-9 or NODs) might be involved after the bacteria are taken up by DCs. In contrast, the Th17-related cytokine IL-23p19 was not detected in supernatants from WT or TLR2−/− DCs or macrophages stimulated with T. forsythia (Fig. 1B).
Fig. 1. T. forsythia-induced cytokine expression in macrophages and dendritic cells is TLR2-dependent.
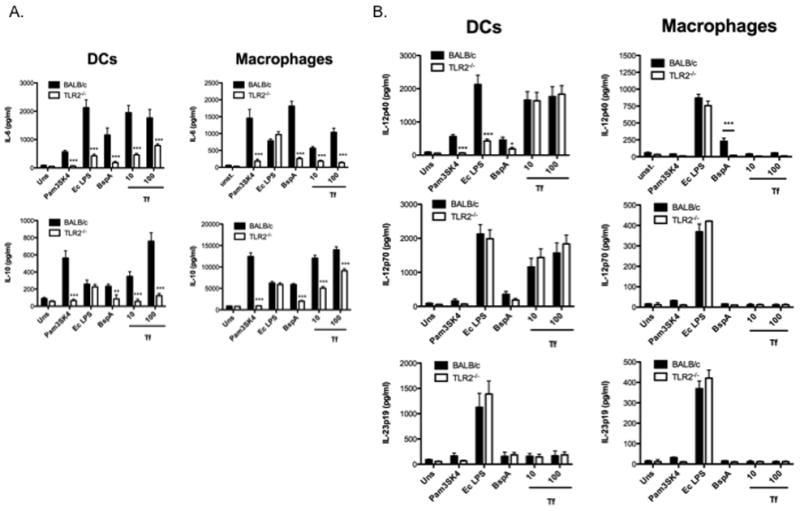
Inflammatory cytokine levels (A. IL-6 and IL-10; B. IL12p40, IL12p70, IL-23p19) were examined by ELISA in the supernatants of mouse DC’s and peritoneal macrophages from BALB/c and TLR2−/− mice following challenge with different agonists. The data show the means ± standard deviations of triplicate determinations in one of 3 independent sets of experiments that yielded similar results; statistically significant differences between the groups are indicated by asterisks (***, p<0.001; **, p<0.01; *, p< 0.05).
TLR2 deficiency attenuates T. forsythia-induced alveolar bone loss
We previously reported that T. forsythia induces alveolar bone loss in BALB/c mice, which is dependent on BspA (9). As a starting point in this study, we confirmed that both purified BspA or intact bacteria activate BMDCs and macrophages through TLR2 (Fig 1).
To determine the contribution of TLR2-mediated responses to alveolar bone loss, we evaluated the susceptibility of TLR2−/− mice to T. forsythia. In this model, mice are pre-treated with antibiotics to reduce normal flora, and then subjected to infection with T. forsythia by oral gavage (9). Analysis of 16S rDNA by PCR indicated that all WT and TLR2−/− (n=6) mice infected with T. forsythia were positive for a 620 bp T. forsythia-specific 16s rDNA as well as 1.4 kb universal eubacteria specific 16s rDNA PCR products, verifying the presence of T. forsythia and common resident bacteria in the oral flora (supplemental figure, S1). As expected, sham-infected mice (WT and TLR2−/−) were positive for the 1.4 kb universal 16s product, but not the 620 bp, product. Currently, reliable methods to quantify periodontal bacteria from the mouse oral cavity are unavailable (44) and estimating bacteria by oral swabbing is not thought to reflect the bacterial load. Periodontal bacteria, and particularly T. forsythia, invade buccal and gingival epithelial cells and form biofilms (45–47), thereby avoiding detection. As another confirmation of infection, T. forsythia-specific serum IgG titers in T. forsythia-infected mice were increased over sham-infected mice. The net Ab response, defined as the titer of sham-infected mice subtracted from T. forsythia-infected mice, increased in both WT and TLR2−/− mice (Fig. 2A). Although there was a low titer even in sham-infected mice, these Abs presumably represent cross-reactive nonspecific Abs against normal resident bacteria. TLR2-deficient mice elicited lower levels of T. forsythia-specific Abs compared to WT mice following infection (Fig. 2A). Thus, both WT and TLR2−/− mice were productively infected with T. forsythia. Conceivably, TLR2 deficiency directly impairs Ab production against T. forsythia by affecting cellular pathways associated with Ab maturation. Alternatively, this occurs indirectly by reducing T. forsythia proliferation and survival, resulting in suboptimal humoral responses. In support of the latter, it has been suggested that P. gingivalis survival is impaired in TLR2−/− mice (48). The lack of suitable methods to quantify bacteria precluded us from determining whether TLR2 deficiency indeed causes lower T. forsythia loads.
Fig. 2. TLR2 promotes T. forsythia-induced alveolar bone loss.

(A) Oral infection with T. forsythia elicits serum IgG response in WT and TLR2−/− mice. Sera from mice after 6 weeks of infection (sham or T. forsythia) in WT and TLR2−/− mice were analyzed for T. forsythia-specific IgG by ELISA. Net antibody response in each group (WT or TLR2−/−) was then determined by subtracting the antibody titers of sham-infected mice from that of T. forsythia-infected mice. Data represents means and standard deviations for each group (n=8–10), and statistical difference between the group means was determined by unpaired t test (*, p<0.05). (B & C) TLR2−/− mice exhibit reduced net alveolar bone loss in response to T. forsythia infection. WT and TLR2−/− mice (n = 8–10) were infected with T. forsythia or sham-infected. Alveolar bone destruction was assessed after 6 weeks by measuring the distance from the ABC to the CEJ at 14 maxillary buccal sites per mouse (R1–R7 = right jaw; L1–L7 = left jaw). (B) Representative maxillary phenotypes of male BALB/c and TLR2−/− mice, sham-infected or T. forsythia-infected. Maxillary jaws were stained with methylene blue and images were acquired with a Nikon SMZ 1000 microscope, magnification ×3. (C) Average alveolar bone loss at 14 buccal sites for BALB/c and TLR2−/− mice either sham- or T. forsythia-infected. Net bone loss shows ABC-CEJ distance of T. forsythia-infected sites minus the mean ABC-CEJ distance of sham-treated sites. Data were analyzed by unpaired t test, Standard deviations are shown, and statistically significant differences of T. forsythia-infected compared with sham-infected for each buccal site are indicated with an asterisk (***, p<0.001; **, p<0.01; *, p< 0.05).
After 6 weeks, alveolar bone loss was measured as the distance between the cementoenamel junction (CEJ) and alveolar bone crest (ABC) at 14 buccal sites per mouse (horizontal bone loss, see Fig. 2B). As expected, significant alveolar bone loss was observed in T. forsythia-infected WT mice compared to controls (Fig. 2, B & C). Strikingly, TLR2−/− mice infected with T. forsythia showed bone loss at fewer sites compared to sham-infected TLR2−/− mice. While the baseline ABC-CEJ distances showed a trend for higher CEJ-ABC distances in TLR2−/− mice compared to WT, this did not reach statistical significance. Nevertheless, the average net alveolar bone loss induced by T. forsythia (measured as total ABC-CEJ for sham-treated mice subtracted from T. forsythia-infected mice) in TLR2−/− was significantly lower than the average net bone loss observed for WT (Fig. 2C). These results imply that TLR2 signaling stimulates bone loss following T. forsythia infection, which is ameliorated in TLR2−/− mice.
Stat6 deletion attenuates T. forsythia induced alveolar bone loss
TLR2 has been suggested to stimulate primarily Th2 responses (35–38), although there are exceptions to this finding. To test the hypothesis that Th2 responses are responsible for inflammatory alveolar bone destruction, mice deficient in STAT6 were assessed. Notably, stat6 gene expression has been linked to alveolar bone loss susceptibility in response to P. gingivalis infection, although its role in T. forsythia infection has never been evaluated (39). One week after the final infection, all mice tested positive for the 620 bp Tf rDNA band by PCR (supplemental figure, S2). Similarly, the presence of increased T. forsythia-specific IgG titers in sera of animals confirmed that all animals were productively infected (Fig. 3A). Moreover, T. forsythia-specific Ab titers in STAT6−/− mice were lower than that in WT mice, indicating that Stat6 deficiency reduces the humoral response to T. forsythia (Fig. 3A). Following infection, average net bone loss in STAT6−/− mice was significantly lower than in WT (Fig. 3B & C), indicating that STAT6 is essential for alveolar bone destruction caused by T. forsythia. Since STAT6 is important in Th2 differentiation, these data imply a role for Th2 cells in driving alveolar bone loss associated with T. forsythia.
Fig. 3. STAT6 promotes T. forsythia-induced alveolar bone loss.

A, Oral infection with T. forsythia elicits serum IgG response in WT and STAT6−/− mice. After 6 wk of infection (sham or T. forsythia) sera from WT and STAT6−/− mice were analyzed for T. forsythia-specific IgG by ELISA. Net antibody response to T. forsythia in WT (males or female) and STAT6−/− (male or female) mice was determined by subtracting the sham-infected animal titers from T. forsythia-infected animal titers (gender-matched). Data represents means and standard deviations for each group (n=8-10). *, p<0.05, unpaired t test. B & C, STAT6−/− mice exhibit reduced alveolar bone loss in response to T. forsythia infection. WT and STAT6−/− mice (n = 8-10) were infected with T. forsythia or sham-infected. Alveolar bone destruction was assessed after 6 wk by measuring the ABC-CEJ distance at 14 maxillary buccal sites per mouse (R1-R7 = right jaw; L1-L7 = left jaw). B, Representative maxillary phenotypes of female BALB/c and STAT6−/− mice. Maxillary jaws were stained with methylene blue and images were acquired with a Nikon SMZ 1000 microscope (original magnification X3). Buccal sites (1-7) are indicated. C, Average alveolar bone loss at 14 buccal sites for BALB/c and STAT6−/− mice (males or females). Net bone loss shows ABC-CEJ distance of T. forsythia-infected sites minus the mean ABC-CEJ distance of sham-treated sites. Data were analyzed by Mann-Whitney unpaired t test. **, p < 0.01, gender matched BALB/c versus STAT6−/− mice.
To assess whether Th1- or Th2-biased immune responses were elicited in response to T. forsythia, we evaluated levels of IgG1 and IgG2a isotypes in serum. In mice, IgG2a and IgG1 immunoglobulin isotypes are considered to represent Th1 and Th2 phenotypes of immune responses, respectively (17). WT mice produced high titers of both T. forsythia-specific IgG1 as well as IgG2a isotypes (Fig. 4). On the other hand, as expected, STAT6−/− mice with a Th1 bias to T. forsythia infection induced significantly higher titers of IgG2a compared to IgG1 (Fig. 4). TLR2−/− mice produced low levels of each isotype, which were not significantly different (Fig. 4). It might be expected to observe increased T. forsythia-specific IgG1 levels in WT mice given the apparent Th2 bias during infection; however in light of the fact that IL-12 is also secreted following T. forsythia challenge in BMDCs (Fig. 1), it is not surprising that the IgG2a response may develop with time. Thus, the isotypes elicited against T. forsythia appeared somewhat linked to Th-responses, and the results further support data in the field that IgG1 and IgG2a subtypes do not functionally play any role in alveolar bone loss.
Fig. 4. T. forsythia infection induces mixed Th1 and Th2 regulated antibodies in wild-type, and selectively induces Th1 regulated antibodies in STAT6−/− mice.

Sera from mice described were analyzed for T. forsythia-specific IgG subclass (IgG1, IgG2a,) antibody levels in WT, TLR2−/− and STAT6−/− mice by ELISA following infection with T. forsythia. Standard deviations are shown. Statistically significant differences in T. forsythia-infected samples compared with sham-infected samples of the same strain, as determined by unpaired t test.
Th2 cell polarization in bone-loss susceptible mice
In order to gain insight into the nature of Th cell responses associated with PD loss, we analyzed in vivo primed Th cells from draining lymph nodes of PD-susceptible (WT) and resistant (TLR2−/− and STAT6−/−) strains. Mice were infected 3 times at 48 h intervals with T. forsythia; 72 h after the last infection, cells from cervical draining lymph nodes (cLN) were stimulatedwith anti-CD3 and -CD28 Abs to induce cytokine production. Cells were treated with PMA-ionomycin, stained for cell surface CD4, and stained intracellularly for IL-5, IFNγ and IL-17 to detect Th2, Th1 and Th17 cells, respectively. As shown (Fig. 5), in WT (susceptible) mice, the number of Th2 cells increased, whereas the number of Th1 and Th17 cells decreased following infection compared to controls (Fig. 5). Conversely, the frequency of Th2 cells was not altered in TLR2−/− or STAT6−/− mice (Fig. 5). Following infection, the number of Th1 cells decreased significantly in TLR2−/− but increased in STAT6−/− mice; conversely, Th17 cells remained unchanged in STAT6−/− but were reduced significantly in TLR2−/− mice. Since T. forsythia-induced net bone loss is reduced in TLR2−/− and STAT6−/− mice, these results imply a destructive capacity for Th2 responses in T. forsythia-induced PD. In contrast, Th1 and Th17 responses might play protective roles, which would be consistent with previous observations in the context of P. gingivalis-induced bone loss and systemic responses (24). (see also Discussion)
Fig. 5. T. forsythia infection in bone-loss susceptible mice causes increased Th2 cell response.
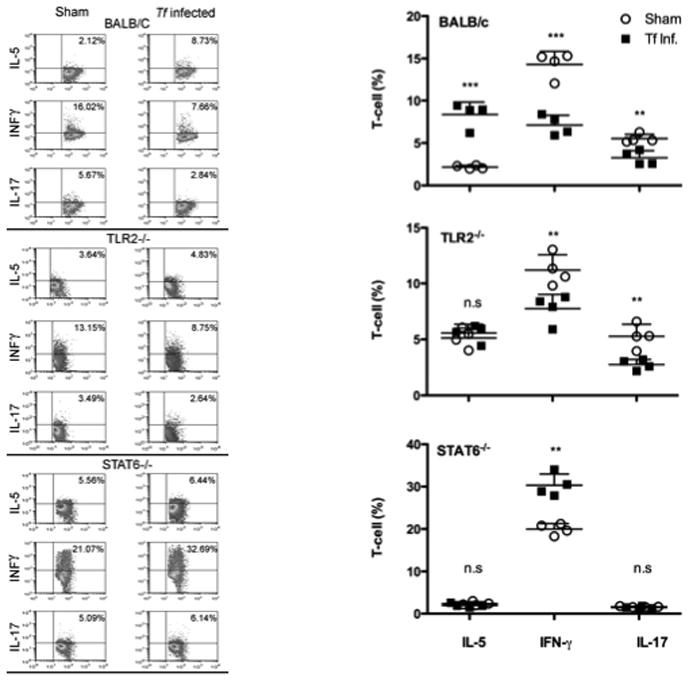
Production of IFN-γ, IL-5 and IL-17 in cervical lymph nodes of WT, TLR2−/− and STAT6−/− mice. Mice (n=4) were infected 3 times at 48 h intervals. 72 h after the final infection, cLN cells were stimulated with anti-CD3 and anti-CD28 Abs for 48 h. Cells were then stimulated with ionomycin and PMA for 4–6 h prior to intracellular staining for IL-5, IFN-γ or IL-17. Left panels show representative FACS profiles of intracellularly stained CD4+ T cells for IL-5, IFN-γ and IL-17 from sham- or T. forsythia-infected mice (a) WT BALB/c, (b) TLR2−/−, and (c) STAT6−/− mice. Right panels show means and standard errors of CD4 T cells positive for IL-5, IFN-γ and IL-17 for each of the sham- or T. forsythia-infected groups (n=4). Statistically significant differences between the T. forsythia-infected and sham-infected groups is indicated by; ***, p<0.001; **, p<0.01; *, p< 0.05, n.s, not significant.
Susceptibility to Alveolar bone loss correlates with osteoclastic activity and lymphocytic infiltration
To evaluate inflammatory and osteoclastic activity, maxillae bones were stained for CD45+ cells and TRAP (tartrate resistance alkaline phosphatase, a marker of osteoclasts). CD45 staining was used as a marker for inflammatory cells in the gingival tissues (49, 50), whereas TRAP staining was used as a marker for osteoclastic activity and a measure of bone resorption in the jaw bones due to infection (51, 52). The results indicated scattered CD45+ inflammatory cells in sham-infected WT mice. Not surprisingly, T. forsythia infection was associated with an increased infiltration of CD45+ cells in gingival tissue of WT mice. However, there was no significant increase in CD45+ infiltrate in TLR2−/− and only a small increase in STAT6−/− animals following infection (Fig. 6A). Mice infected with T. forsythia also showed prominent epithelial hyperplasia, indicative of gingival destruction. T. forsythia-induced bone loss susceptibility correlated with an increased infiltration of CD45+ cells in the gingival connective tissue. There was a ~3-fold increase in CD45+ cells in WT T. forsythia-infected gingival tissue (Fig. 6B). With regard to osteoclastic activity, a 2.5-fold increase in the numbers of TRAP+ cells was observed in infected WT mice compared to controls (Fig. 7A & B). However, only a marginal increase in the number of osteoclasts was observed in TLR2−/− and STAT6−/− mice (Fig. 7). Although the baseline osteoclastic activity in STAT6−/− was marginally higher than WT, these mice nonetheless showed a higher frequency of TRAP+ cells (Fig. 7). Therefore, induction of TLR2/Th2-mediated inflammation by T. forsythia appears to be responsible for increased activation of osteoclasts, ultimately leading to T. forsythia-induced alveolar bone loss.
Fig. 6. Alveolar bone loss correlates with lymphocytic infiltration.
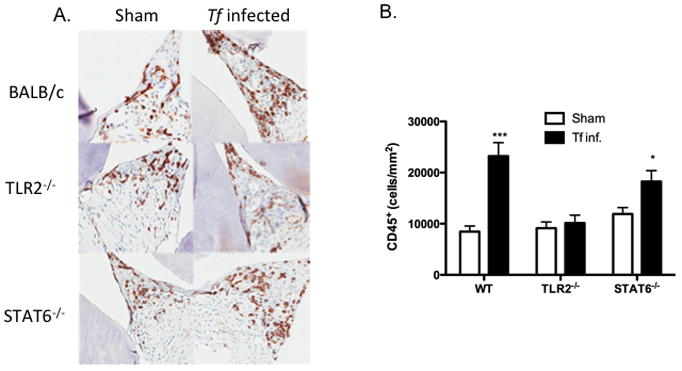
CD45 staining for inflammatory infiltrate cells in gingival tissue 3 weeks following infection of WT, TLR2−/− and STAT6−/− mice. (A) All images are representative of 400x magnification. (B) Inflammatory cells were quantified as number of CD45 positive cells/mm2. Statistically significant differences between the respective T. forsythia-infected and sham-infected groups is indicated by; ***, p<0.001; **, p<0.01; *, p< 0.05, n.s, not significant.
Fig. 7. Alveolar bone loss correlates with osteoclastic activity.
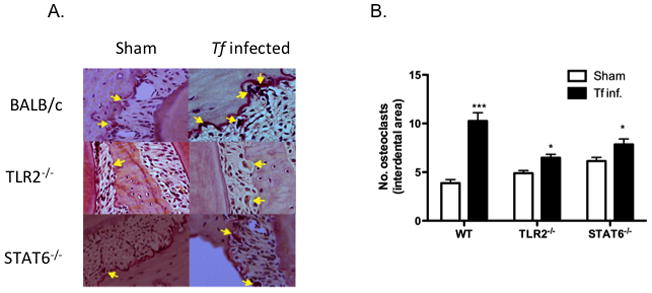
(A) Representative histological sections showing TRAP+ cells (arrows) from T. forsythia-and sham-infected WT, TLR2−/− and STAT6−/− mice. (B) average number of TRAP+ cells in 10 high power magnification fields/slide (4 mice/group). Statistically significant differences between T. forsythia-infected and sham-infected groups is indicated by; ***, p<0.001; **, p<0.01; *, p< 0.05, n.s, not significant.
DISCUSSION
Periodontitis, characterized by alveolar bone destruction of the maxillary jaw, is by far the most common form of bacterially-induced inflammation that affects bone. In this study we have shown for the first time that T. forsythia, a relatively understudied periodontal pathogen, mediates the alveolar bone loss process via the TLR2/Th2 pathway. This is the first detailed analysis of immune responses to T. forsythia.
Although initiated by bacteria, the bone pathology in PD is mediated almost entirely by the host response. No single bacterial species has been implicated as the primary pathogen in PD, and all available evidence is consistent with a polymicrobial disease etiology. A bacterial consortium known as the ‘red-complex’, consisting of P. gingivalis, Treponema denticola and T. forsythia, is strongly implicated in the onset and the severity of PD (4). While there is a plethora of studies on the nature of P. gingivalis-associated host response and its role in disease pathology, similar information regarding T. denticola or T. forsythia is evidently lacking (5). Previous studies have been largely restricted to P. gingivalis as a model periodontal pathogen. Although informative, such studies are limited to the nuances of this organism.
Although initiated by bacteria, bone loss is mediated by the immune response. A destructive role for T- and B cells in PD bone loss is well documented (10–12). On the other hand, impaired neutrophil and macrophage recruitment due to deficiencies in P-selectin and ICAM-1 result in increased susceptibility to P. gingivalis-induced alveolar bone loss (53). Thus, while lymphocytes can drive bone loss, a robust innate immune response controls infection and hence limits overall bone destruction. Despite the potential bone-destructive capacity of IL-17 and Th17 cells seen in the context of rheumatoid arthritis (54), a deficiency in IL-17 signaling in PD results in increased susceptibility to P. gingivalis-induced alveolar bone loss. It has also been demonstrated that TLR2-mediated inflammatory responses are critical to P. gingivalis-induced alveolar bone loss through promoting pathogen survival and burden in the host (48). Taken together, these studies demonstrate that both innate and adaptive arms of immunity play critical roles in PD. However, the contribution of individual T cell responses (namely, Th1, Th2 or Th17) still needs to be further explored in PD (6, 55).
To date, the mechanisms by which T. forsythia induces inflammation and alveolar bone loss are poorly understood. We previously demonstrated that T. forsythia expresses a TLR2-activating molecule, BspA (34), which is required for inducing alveolar bone loss in mice (9). Since both BspA and intact T. forsythia signal through TLR2 (33, 34), the focus of this study was to evaluate the role of TLR2-mediated responses in alveolar bone loss. Moreover, because TLR2-mediated responses have been shown to favor Th2 development (31, 35, 36, 38), we predicted that Th2 responses would be elicited and dictate the alveolar bone loss associated with T. forsythia infection. Indeed, our results show that TLR2 plays a significant role in stimulating DCs and macrophages to elicit cytokine responses via BspA (Fig. 1). DCs or macrophages stimulated with bacteria or BspA did not express IL-23, suggesting that Th17 cells are not induced in response to T. forsythia. Interestingly, DCs but not macrophages produced IL-12 (Th1 cytokine) in response to bacteria or BspA. It is possible that engagement of other pathways, such as through C-type lectin receptors, intracellular NOD-like receptors or TLR9, might be operative in DCs. Indeed, we have shown that T. forsythia DNA is a strong inducer of TLR9 (56). Consistently, Mycobacterium tuberculosis (MTb) induces strong IL-12 expression in DCs through the TLR9 pathway, but weak IL-12 and strong IL-10 and expression in macrophages through engagement of TLR2 (57). TLR2 activation inhibits IL-12 by dampening the Th1 IFN-γ amplification loop in DCs, and promotes induction of Th2 and Th17 responses (38). Furthermore, IL-10, produced primarily via TLR2, inhibits IP-10 and IL12p35, and is therefore considered a Th1-suppressing cytokine. While IL-10 is primarily an inhibitor of Th1, recent studies have also indicated IL-10 suppresses Th2 and Th17 cell responses as well (58). The failure of T. forsythia to induce IL-23 in DCs or macrophages (Fig. 1B) indicated that Th17 responses are likely not induced in this setting, which was confirmed by our findings in T. forsythia-infected mice (Fig. 5). Also consistent with our hypothesis, the net bone loss caused by T. forsythia in TLR2−/− mice was attenuated. This finding indicated that TLR2 activation plays a destructive role. This finding is similar to what has been observed for P. gingivalis infections, where TLR2−/− mice are resistant to P. gingivalis-induced alveolar bone loss (48). It is likely that T. forsythia, which depends on the availability of host factors such as peptides, heme and sialic acid for growth, exploits TLR2-mediated inflammation for its growth and survival.
To explore the possibility of polarization of Th2 responses downstream of TLR2, STAT6−/− mice were assessed. In STAT6−/− mice T. forsythia-induced alveolar bone loss was significantly reduced (Fig. 3). Consistent with our findings, the stat6 gene was shown to be associated with increased susceptibility in P. gingivalis-induced PD (39). We observed increased osteoclastic activity following infection only in alveolar bone loss susceptible WT mice. Consistently, WT mice presented increased inflammatory cells in the connective tissue surrounding alveolar bone following infection, which correlated with orthoclastic activity. These results strongly suggest that the TLR2-Th2 inflammatory axis plays a significant role in T. forsythia-induced alveolar bone loss.
Our observations suggest that STAT6 does not play a bone protective role during T. forsythia-induced alveolar bone loss. However, STAT6 is considered important for dampening the activity of osteoclasts via IL-4 and IL-13 in vitro (59, 60), so other mechanisms are likely to be involved whereby Th2 mediates bone loss. In that respect, transgenic expression of IL-4 in mice was shown to suppress osteoblast activity, leading to general osteoporosis (61). Thus, it is possible that Th2 cytokines suppress local osteoblast activity in the jaw. Another possible mechanism is that this occurs via increased levels of RANKL (receptor-activator of nuclear factor-kB ligand) expression, driving osteoclast differentiation. However, RANKL expression is thought to be limited mainly to Th1 (62) and Th17 (63) cells, though this may vary depending on the infection stimulus, and was not assessed in our study. Alternatively, it is tempting to speculate that Th2 responses induce RANKL-mediated osteoclastogenesis through effects on B cells. B cells also express RANKL, and can drive osteoclastogenesis in inflamed peridotium (64, 65).
In summary, we present evidence for the roles of TLR2 signaling and Th2 differentiation in mediating T. forsythia-induced alveolar bone destruction. We show that T. forsythia-induced TLR2 activation results in alveolar bone destruction. Furthermore, Th2 development downstream of TLR2 activation is associated with alveolar bone destruction caused by T. forsythia.
Supplementary Material
Acknowledgments
We thank Raymond J. Kelleher and Rajesh Rao for assistance with flow cytometry and, Moon-Il Cho and Shuying Yang for their advice on processing of jawbones for histological staining.
Abbreviations used in this manuscript
- Tf
Tannerella forsythia
- PD
periodontal disease
Footnotes
This work was supported by U. S. Public Health R01 grants DE1074905 (to A.S. and S.L.G) and DE14749 (to A.S), DE13833 (T. D. K) and AI059775 and AI038985 (to A.D.K.).
References
- 1.Zambon JJ, Grossi S, Dunford R, Harazsthy VI, Preus H, Genco RJ. Epidemiology of subgingival bacterial pathogens in periodontal diseases. In: Genco RJ, Hamada S, Lehrer JR, McGhee JR, Mergenhagen S, editors. Molecular Pathogenesis of Periodontal Disease. Am. Soc. Microbiol; Washington, DC: 1994. pp. 3–12. [Google Scholar]
- 2.Darveau RP. Periodontitis: a polymicrobial disruption of host homeostasis. Nat Rev Microbiol. 2010;8:481–490. doi: 10.1038/nrmicro2337. [DOI] [PubMed] [Google Scholar]
- 3.Graves DT, Li J, Cochran DL. Inflammation and uncoupling as mechanisms of periodontal bone loss. J Dent Res. 2011;90:143–153. doi: 10.1177/0022034510385236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL., Jr Microbial complexes in subgingival plaque. J Clin Periodontol. 1998;25:134–144. doi: 10.1111/j.1600-051x.1998.tb02419.x. [DOI] [PubMed] [Google Scholar]
- 5.Holt SC, Ebersole JL. Porphyromonas gingivalis, Treponema denticola, and Tannerella forsythia: the “red complex”, a prototype polybacterial pathogenic consortium in periodontitis. Periodontol 2000. 2005;38:72–122. doi: 10.1111/j.1600-0757.2005.00113.x. [DOI] [PubMed] [Google Scholar]
- 6.Gaffen SL, Hajishengallis G. A new inflammatory cytokine on the block: re-thinking periodontal disease and the Th1/Th2 paradigm in the context of Th17 cells and IL-17. J Dent Res. 2008;87:817–828. doi: 10.1177/154405910808700908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharma A. Virulence mechanisms of Tannerella forsythia. Periodontol 2000. 2010;54:106–116. doi: 10.1111/j.1600-0757.2009.00332.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tanner AC, Izard J. Tannerella forsythia, a periodontal pathogen entering the genomic era. Periodontol 2000. 2006;42:88–113. doi: 10.1111/j.1600-0757.2006.00184.x. [DOI] [PubMed] [Google Scholar]
- 9.Sharma A, Inagaki S, Honma K, Sfintescu C, Baker PJ, Evans RT. Tannerella forsythia-induced alveolar bone loss in mice involves leucine-rich-repeat BspA protein. J Dent Res. 2005;84:462–467. doi: 10.1177/154405910508400512. [DOI] [PubMed] [Google Scholar]
- 10.Baker PJ. The role of immune responses in bone loss during periodontal disease. Microbes Infect. 2000;2:1181–1192. doi: 10.1016/s1286-4579(00)01272-7. [DOI] [PubMed] [Google Scholar]
- 11.Baker PJ, Dixon M, Evans RT, Dufour L, Johnson E, Roopenian DC. CD4(+) T cells and the proinflammatory cytokines gamma interferon and interleukin-6 contribute to alveolar bone loss in mice. Infect Immun. 1999;67:2804–2809. doi: 10.1128/iai.67.6.2804-2809.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baker PJ, Evans RT, Roopenian DC. Oral infection with Porphyromonas gingivalis and induced alveolar bone loss in immunocompetent and severe combined immunodeficient mice. Arch Oral Biol. 1994;39:1035–1040. doi: 10.1016/0003-9969(94)90055-8. [DOI] [PubMed] [Google Scholar]
- 13.Stashenko P, Goncalves RB, Lipkin B, Ficarelli A, Sasaki H, Campos-Neto A. Th1 Immune Response Promotes Severe Bone Resorption Caused by Porphyromonas gingivalis. Am J Pathol. 2007;170:203–213. doi: 10.2353/ajpath.2007.060597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gemmell E, Yamazaki K, Seymour GJ. The role of T cells in periodontal disease: homeostasis and autoimmunity. Periodontol 2000. 2007;43:14–40. doi: 10.1111/j.1600-0757.2006.00173.x. [DOI] [PubMed] [Google Scholar]
- 15.Teng YT. Protective and Destructive Immunity in the Periodontium: Part 1--Innate and Humoral Immunity and the Periodontium. J Dent Res. 2006;85:198–208. doi: 10.1177/154405910608500301. [DOI] [PubMed] [Google Scholar]
- 16.Teng YT, Nguyen H, Gao X, Kong YY, Gorczynski RM, Singh B, Ellen RP, Penninger JM. Functional human T-cell immunity and osteoprotegerin ligand control alveolar bone destruction in periodontal infection. J Clin Invest. 2000;106:R59–67. doi: 10.1172/jci10763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. 1989;7:145–173. doi: 10.1146/annurev.iy.07.040189.001045. [DOI] [PubMed] [Google Scholar]
- 18.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 19.Honda T, Aoki Y, Takahashi N, Maekawa T, Nakajima T, Ito H, Tabeta K, Okui T, Kajita K, Domon H, Yamazaki K. Elevated expression of IL-17 and IL-12 genes in chronic inflammatory periodontal disease. Clin Chim Acta. 2008;395:137–141. doi: 10.1016/j.cca.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 20.Johnson RB, Wood N, Serio FG. Interleukin-11 and IL-17 and thepathogenesis of periodontal disease. J Periodontol. 2004;75:37–43. doi: 10.1902/jop.2004.75.1.37. [DOI] [PubMed] [Google Scholar]
- 21.Schenkein HA, Koertge TE, Brooks CN, Sabatini R, Purkall DE, Tew JG. IL-17 in sera from patients with aggressive periodontitis. J Dent Res. 2010;89:943–947. doi: 10.1177/0022034510369297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vernal R, Dutzan N, Chaparro A, Puente J, Antonieta Valenzuela M, Gamonal J. Levels of interleukin-17 in gingival crevicular fluid and in supernatants of cellular cultures of gingival tissue from patients with chronic periodontitis. J Clin Periodontol. 2005;32:383–389. doi: 10.1111/j.1600-051X.2005.00684.x. [DOI] [PubMed] [Google Scholar]
- 23.Ito H, Honda T, Domon H, Oda T, Okui T, Amanuma R, Nakajima T, Yamazaki K. Gene expression analysis of the CD4+ T-cell clones derived from gingival tissues of periodontitis patients. Oral Microbiol Immunol. 2005;20:382–386. doi: 10.1111/j.1399-302X.2005.00241.x. [DOI] [PubMed] [Google Scholar]
- 24.Yu JJ, Ruddy MJ, Wong GC, Sfintescu C, Baker PJ, Smith JB, Evans RT, Gaffen SL. An essential role for IL-17 in preventing pathogen-initiated bone destruction: recruitment of neutrophils to inflamed bone requires IL-17 receptor-dependent signals. Blood. 2007;109:3794–3802. doi: 10.1182/blood-2005-09-010116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gosling PT, Gemmell E, Carter CL, Bird PS, Seymour GJ. Immunohistological analysis of Tannerella forsythia-induced lesions in a murine model. Oral Microbiol Immunol. 2005;20:25–30. doi: 10.1111/j.1399-302X.2004.00188.x. [DOI] [PubMed] [Google Scholar]
- 26.Eskan MA, Hajishengallis G, Kinane DF. Differential activation of human gingival epithelial cells and monocytes by Porphyromonas gingivalis fimbriae. Infect Immun. 2007;75:892–898. doi: 10.1128/IAI.01604-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou Q, Desta T, Fenton M, Graves DT, Amar S. Cytokine Profiling of Macrophages Exposed to Porphyromonas gingivalis, Its Lipopolysaccharide, or Its FimA Protein. Infect Immun. 2005;73:935–943. doi: 10.1128/IAI.73.2.935-943.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davey M, Liu X, Ukai T, Jain V, Gudino C, Gibson FC, 3rd, Golenbock D, Visintin A, Genco CA. Bacterial fimbriae stimulate proinflammatory activation in the endothelium through distinct TLRs. J Immunol. 2008;180:2187–2195. doi: 10.4049/jimmunol.180.4.2187. [DOI] [PubMed] [Google Scholar]
- 29.Gibson FC, 3rd, Ukai T, Genco CA. Engagement of specific innate immune signaling pathways during Porphyromonas gingivalis induced chronic inflammation and atherosclerosis. Frontiers in bioscience a journal and virtual library. 2008;13:2041–2059. doi: 10.2741/2822. [DOI] [PubMed] [Google Scholar]
- 30.Gaffen SL, Kramer JM, Yu JJ, Shen F. The IL-17 cytokine family. Vitam Horm. 2006;74:255–282. doi: 10.1016/S0083-6729(06)74010-9. [DOI] [PubMed] [Google Scholar]
- 31.Netea MG, Van der Meer JWM, Sutmuller RP, Adema GJ, Kullberg BJ. From the Th1/Th2 Paradigm towards a Toll-Like Receptor/T-Helper Bias. Antimicrob Agents Chemother. 2005;49:3991–3996. doi: 10.1128/AAC.49.10.3991-3996.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hasebe A, Yoshimura A, Into T, Kataoka H, Tanaka S, Arakawa S, Ishikura H, Golenbock DT, Sugaya T, Tsuchida N, Kawanami M, Hara Y, Shibata K. Biological activities of Bacteroides forsythus lipoproteins and their possible pathological roles in periodontal disease. Infect Immun. 2004;72:1318–1325. doi: 10.1128/IAI.72.3.1318-1325.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kikkert R, Laine ML, Aarden LA, van Winkelhoff AJ. Activation of toll-like receptors 2 and 4 by gram-negative periodontal bacteria. Oral Microbiol Immunol. 2007;22:145–151. doi: 10.1111/j.1399-302X.2007.00335.x. [DOI] [PubMed] [Google Scholar]
- 34.Onishi S, Honma K, Liang S, Stathopoulou P, Kinane D, Hajishengallis G, Sharma A. Toll-like receptor 2-mediated interleukin-8 expression in gingival epithelial cells by the Tannerella forsythia leucine-rich repeat protein BspA. Infect Immun. 2008;76:198–205. doi: 10.1128/IAI.01139-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dillon S, Agrawal A, Van Dyke T, Landreth G, McCauley L, Koh A, Maliszewski C, Akira S, Pulendran B. A Toll-Like Receptor 2 Ligand Stimulates Th2 Responses In Vivo, via Induction of Extracellular Signal-Regulated Kinase Mitogen-Activated Protein Kinase and c-Fos in Dendritic Cells. J Immunol. 2004;172:4733–4743. doi: 10.4049/jimmunol.172.8.4733. [DOI] [PubMed] [Google Scholar]
- 36.Re F, Strominger JL. IL-10 released by concomitant TLR2 stimulation blocks the induction of a subset of Th1 cytokines that are specifically induced by TLR4 or TLR3 in human dendritic cells. J Immunol. 2004;173:7548–7555. doi: 10.4049/jimmunol.173.12.7548. [DOI] [PubMed] [Google Scholar]
- 37.Redecke V, Hacker H, Datta SK, Fermin A, Pitha PM, Broide DH, Raz E. Cutting edge: activation of Toll-like receptor 2 induces a Th2 immuneresponse and promotes experimental asthma. J Immunol. 2004;172:2739–2743. doi: 10.4049/jimmunol.172.5.2739. [DOI] [PubMed] [Google Scholar]
- 38.Wenink MH, Santegoets KC, Broen JC, van Bon L, Abdollahi-Roodsaz S, Popa C, Huijbens R, Remijn T, Lubberts E, van Riel PL, van den Berg WB, Radstake TR. TLR2 promotes Th2/Th17 responses via TLR4 and TLR7/8 by abrogating the type I IFN amplification loop. J Immunol. 2009;183:6960–6970. doi: 10.4049/jimmunol.0900713. [DOI] [PubMed] [Google Scholar]
- 39.Hart GT, Shaffer DJ, Akilesh S, Brown AC, Moran L, Roopenian DC, Baker PJ. Quantitative gene expression profiling implicates genes for susceptibility and resistance to alveolar bone loss. Infect Immun. 2004;72:4471–4479. doi: 10.1128/IAI.72.8.4471-4479.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sharma A, Sojar HT, Glurich I, Honma K, Kuramitsu HK, Genco RJ. Cloning, expression, and sequencing of a cell surface antigen containing a leucine-rich repeat motif from Bacteroides forsythus ATCC 43037. Infect Immun. 1998;66:5703–5710. doi: 10.1128/iai.66.12.5703-5710.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Inaba WJ, Swiggard RM, Steinman N, Romani N, Schular G, Brinster C. Isolation of dendritic cells. Curr Protoc Immunol. 2009;86:3.7.1–3.7.19. doi: 10.1002/0471142735.im0307s86. [DOI] [PubMed] [Google Scholar]
- 42.Zheng X, Goncalves R, Mosser DM. The Isolation and Characterization of Murine Macrophages. Curr Protoc Immunol. 2008;83:4.1.1–14.11.14. doi: 10.1002/0471142735.im1401s83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sasaki H, Okamatsu Y, Kawai T, Kent R, Taubman M, Stashenko P. The interleukin-10 knockout mouse is highly susceptible to Porphyromonas gingivalis-induced alveolar bone loss. J Periodontal Res. 2004;39:432–441. doi: 10.1111/j.1600-0765.2004.00760.x. [DOI] [PubMed] [Google Scholar]
- 44.Graves DT, Fine D, Teng YT, Van Dyke TE, Hajishengallis G. The use of rodent models to investigate host-bacteria interactions related to periodontal diseases. J Clin Periodontol. 2008;35:89–105. doi: 10.1111/j.1600-051X.2007.01172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Honma K, Mishima E, Sharma A. Role of Tannerella forsythia NanH sialidase in epithelial cell attachment. Infect Immun. 2010 doi: 10.1128/IAI.00629-10. in press, ePub ahead of print November 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Inagaki S, Onishi S, Kuramitsu HK, Sharma A. Porphyromonas gingivalis Vesicles Enhance Attachment, and the Leucine-Rich Repeat BspA Protein is Required for Invasion of Epithelial Cells by “Tannerella forsythia”. Infect Immun. 2006;74:5023–5028. doi: 10.1128/IAI.00062-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rudney JD, Chen R, Sedgewick GJ. Actinobacillus actinomycetemcomitans, Porphyromonas gingivalis, and Tannerella forsythensis are components of a polymicrobial intracellular flora within human buccal cells. J Dent Res. 2005;84:59–63. doi: 10.1177/154405910508400110. [DOI] [PubMed] [Google Scholar]
- 48.Burns E, Bachrach G, Shapira L, Nussbaum G. Cutting Edge: TLR2 Is Required for the Innate Response to Porphyromonas gingivalis: Activation Leads to Bacterial Persistence and TLR2 Deficiency Attenuates Induced Alveolar Bone Resorption. J Immunol. 2006;177:8296–8300. doi: 10.4049/jimmunol.177.12.8296. [DOI] [PubMed] [Google Scholar]
- 49.Seguier S, Godeau G, Brousse N. Collagen fibers and inflammatory cells in healthy and diseased human gingival tissues: a comparative and quantitative study by immunohistochemistry and automated image analysis. J Periodontol. 2000;71:1079–1085. doi: 10.1902/jop.2000.71.7.1079. [DOI] [PubMed] [Google Scholar]
- 50.Bage T, Kats A, Lopez BS, Morgan G, Nilsson G, Burt I, Korotkova M, Corbett L, Knox AJ, Pino L, Jakobsson PJ, Modeer T, Yucel-Lindberg T. Expression of prostaglandine synthases in periodontitis immunolocalization and cellular regulation. Am J Pathol. 2011;178:1676–1688. doi: 10.1016/j.ajpath.2010.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kawai T, Eisen-Lev R, Seki M, Eastcott JW, Wilson ME, Taubman MA. Requirement of B7 costimulation for Th1-mediated inflammatory bone resorption in experimental periodontal disease. Journal of immunology. 2000;164:2102–2109. doi: 10.4049/jimmunol.164.4.2102. [DOI] [PubMed] [Google Scholar]
- 52.Liu R, Bal HS, Desta T, Krothapalli N, Alyassi M, Luan Q, Graves DT. Diabetes enhances periodontal bone loss through enhanced resorption and diminished bone formation. Journal of Dental Research. 2006;85:510–514. doi: 10.1177/154405910608500606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baker PJ, DuFour L, Dixon M, Roopenian DC. Adhesion molecule deficiencies increase Porphyromonas gingivalis-induced alveolar bone loss in mice. Infect Immun. 2000;68:3103–3107. doi: 10.1128/iai.68.6.3103-3107.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Onishi RM, Gaffen SL. Interleukin-17 and its target genes: mechanisms of interleukin-17 function in disease. Immunology. 2010;129:311–321. doi: 10.1111/j.1365-2567.2009.03240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gemmell E, Yamazaki K, Seymour GJ. Destructive periodontitis lesions are determined by the nature of the lymphocytic response. Crit Rev Oral Biol Med. 2002;13:17–34. doi: 10.1177/154411130201300104. [DOI] [PubMed] [Google Scholar]
- 56.Sahingur SE, Xia XJ, Alamgir S, Honma K, Sharma A, Schenkein HA. DNA from Porphyromonas gingivalis and Tannerella forsythia induce cytokine production in human monocytic cell lines. Mol Oral Microbiol. 2010;25:123–135. doi: 10.1111/j.2041-1014.2009.00551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pompei L, Jang S, Zamlynny B, Ravikumar S, McBride A, Hickman SP, Salgame P. Disparity in IL-12 release in dendritic cells and macrophages in response to Mycobacterium tuberculosis is due to use of distinct TLRs. J Immunol. 2007;178:5192–5199. doi: 10.4049/jimmunol.178.8.5192. [DOI] [PubMed] [Google Scholar]
- 58.Saraiva M, O’Garra A. The regulation of IL-10 production by immune cells. Nat Rev Immunol. 2010;10:170–181. doi: 10.1038/nri2711. [DOI] [PubMed] [Google Scholar]
- 59.Moreno JL, Kaczmarek M, Keegan AD, Tondravi M. IL-4 suppresses osteoclast development and mature osteoclast function by a STAT6-dependent mechanism: irreversible inhibition of the differentiation program activated by RANKL. Blood. 2003;102:1078–1086. doi: 10.1182/blood-2002-11-3437. [DOI] [PubMed] [Google Scholar]
- 60.Palmqvist P, Lundberg P, Persson E, Johansson A, Lundgren I, Lie A, Conaway HH, Lerner UH. Inhibition of hormone and cytokine-stimulated osteoclastogenesis and bone resorption by interleukin-4 and interleukin-13 is associated with increased osteoprotegerin and decreased RANKL and RANK in a STAT6-dependent pathway. J Biol Chem. 2006;281:2414–2429. doi: 10.1074/jbc.M510160200. [DOI] [PubMed] [Google Scholar]
- 61.Lewis DB, Liggitt HD, Effmann EL, Motley ST, Teitelbaum SL, Jepsen KJ, Goldstein SA, Bonadio J, Carpenter J, Perlmutter RM. Osteoporosis induced in mice by overproduction of interleukin 4. Proc Natl Acad Sci U S A. 1993;90:11618–11622. doi: 10.1073/pnas.90.24.11618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kotake S, Nanke Y, Mogi M, Kawamoto M, Furuya T, Yago T, Kobashigawa T, Togari A, Kamatani N. IFN-gamma-producing human T cells directly induce osteoclastogenesis from human monocytes via the expression of RANKL. Eur J Immunol. 2005;35:3353–3363. doi: 10.1002/eji.200526141. [DOI] [PubMed] [Google Scholar]
- 63.Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, Tanaka S, Kodama T, Akira S, Iwakura Y, Cua DJ, Takayanagi H. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med. 2006;203:2673–2682. doi: 10.1084/jem.20061775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lin X, Han X, Kawai T, Taubman MA. Antibody to RANKL Ameliorates T cell-mediated Periodontal Bone Resorption. Infect Immun. 2010 doi: 10.1128/IAI.00944-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Han X, Lin X, Seliger AR, Eastcott J, Kawai T, Taubman MA. Expression of receptor activator of nuclear factor-kappaB ligand by B cells in response to oral bacteria. Oral Microbiol Immunol. 2009;24:190–196. doi: 10.1111/j.1399-302X.2008.00494.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.