

Abstract
Low-frequency thalamocortical oscillations that underlie drowsiness and slow-wave sleep depend on rhythmic inhibition of relay cells by neurons in the reticular nucleus (RTN) under the influence of corticothalamic fibers that branch to innervate RTN neurons and relay neurons. To generate oscillations, input to RTN predictably should be stronger so disynaptic inhibition of relay cells overcomes direct corticothalamic excitation. Amplitudes of excitatory postsynaptic conductances (EPSCs) evoked in RTN neurons by minimal stimulation of corticothalamic fibers were 2.4 times larger than in relay neurons, and quantal size of RTN EPSCs was 2.6 times greater. GluR4-receptor subunits labeled at corticothalamic synapses on RTN neurons outnumbered those on relay cells by 3.7 times, providing a basis for differences in synaptic strength.
Keywords: reticular nucleus, ventral posterior nucleus, dual whole-cell recordings, minimal stimulation, synaptic strength
Corticothalamic fibers that arise from layer VI cells in the cerebral cortex branch to innervate neurons in the reticular nucleus (RTN) and thalamocortical relay neurons in the underlying thalamus (1, 2) are a powerful influence in the generation of neuronal synchrony in thalamus and cortex (3). In RTN and relay nuclei, they end in excitatory synapses associated with α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), N-methyl-D-aspartate, and metabotropic glutamate receptors (4–8). Corticothalamic fibers strongly excite inhibitory neurons of the RTN and these in turn inhibit relay neurons. Relay neurons fire bursts of action potentials as they recover from inhibition, re-exciting RTN cells so that the sequence continues (9–11). The capacity of the disynaptic RTN-generated inhibitory postsynaptic potential (IPSP) to overcome the monosynaptic excitatory postsynaptic potential (EPSP) generated by the direct corticothalamic input to the relay neurons is a key element in generation of low-frequency oscillations or spindle waves (10, 12), which are the electroencephalographic hallmarks of drowsiness and early stages of slow-wave sleep. In models in which the strength of corticothalamic input to RTN neurons is set equal to that to relay neurons, the disynaptic IPSP is shunted by the monosynaptic EPSP, and the network does not oscillate (13). Here, we demonstrate that excitatory postsynaptic conductances (EPSCs) elicited by minimal stimulation of corticothalamic fibers are stronger quantitatively in RTN than in ventral posterior thalamic nucleus (VP) neurons, and that the number of GluR4 subunits at corticothalamic synapses on RTN neurons is correspondingly greater than at synapses on relay cells, providing a basis for synaptic heterogeneity.
Materials and Methods
Whole-cell recordings were made at 22–25°C from neurons in layer VI of somatosensory cortex, RTN, and VP in brain slices from postnatal day 13 to 24 ICR mice, cut at an angle that preserves corticothalamic and thalamocortical connectivity, and maintained in vitro as described (14). Electrode resistances were 2–5 MΩ. Biocytin 0.5% was present in the internal solution. Holding potential was −70 mV. In paired recordings of layer VI and thalamic neurons, 50–200 current pulses 10 msec in duration and 500 pA to 1 nA in amplitude were injected into the cortical cell to elicit single action potentials or pairs of action potentials separated by 50 or 100 msec at 0.2 Hz.
Corticothalamic synaptic responses were evoked in VP and RTN neurons by electrical stimulation of corticothalamic fibers in layer VI or subcortical white matter using 0.1-msec cathodal stimuli delivered at 0.2 or 0.5 Hz by a monopolar tungsten electrode. Stimulus strength was set 10–15% above absolute threshold to elicit a stable mixture of failures and successes.
EPSC amplitudes were measured as the difference between the average of 0.5-msec regions spanning the baseline immediately before the onset of the EPSC and those straddling the peak of the EPSC. Noise was determined by measuring the difference of means of 0.5-msec regions spanning the baseline region and a region separated by the same interval separating baseline and EPSC-peak regions. All deflections less than 1.5⋅rms of noise were considered failures. rms of noise was 2.33 ± 0.91 pA in VP neurons and 2.75 ± 1.38 pA in RTN neurons and not statistically different.
Slices were fixed in 4% paraformaldehyde in 0.1 M phosphate buffer, incubated in avidin-biotin-horseradish peroxidase complex (ABC kit PK-4000, Vector Laboratories) and reacted with 3,3′-diaminobenzidine-4HCl. Camera lucida drawings of filled cells were made at ×100.
Twelve postnatal day 15 mice were anesthetized and perfused with 4% paraformaldehyde and 0.1–0.15% glutaraldehyde in 0.1 M phosphate buffer. Sections containing VP and RTN were cut on a Vibratome at 500 μm, cryoprotected in glycerol, frozen in −180°C propane, infiltrated with 1.5% uranyl acetate/methanol solution at −90°C, and embedded in Lowicryl HM-20 (Electron Microscopy Sciences, Fort Washington, PA).
Serial 70- to 80-nm sections were mounted on Formvar-coated nickel grids and immersed briefly in a saturated solution of NaOH in absolute ethanol and incubated in 0.1% sodium borohydride and 50 mM glycine in 5 mM Tris buffer containing 0.3–0.6% NaCl and 0.1% Triton X-100, followed by 2% swine albumin in the same solution before immunoreacting with rabbit antisera to GluR2 and GluR3 (GluR2/3) or to GluR4 glutamate-receptor subunits (2 μg/ml; Chemicon) for 4–12 h, followed by incubation in goat anti-rabbit Fab fragments coupled to 10-nm gold particles (1:20; Biocell Laboratories) for 2 h at room temperature. Grids were stained with uranyl acetate and lead citrate and examined in a Philips (Eindhoven, the Netherlands) CM120 electron microscope.
The number of gold particles associated with the postsynaptic density (PSD) was counted in 10 serially sectioned STs (corticothalamic synapses in RT) and 11 serially sectioned RSs (corticothalamic synapses in VP) and in 436 ST and RS synaptic profiles from 279 single sections from RTN and VP. PSDs with no associated gold particles were included. Only gold particles located within 20 nm of the inner leaflet of the postsynaptic membrane and within 20 nm of the lateral edge of the postsynaptic membrane were counted.
Student's t test was used for all tests of statistical significance.
Results
Results are derived from recordings of 100 VP cells, 60 RTN cells, and 60 layer VI cells. At the ages studied, corticothalamic and intrathalamic circuitry is functionally mature (14). To isolate responses of RTN neurons resulting from orthodromic activation of corticothalamic fibers from responses to antidromic activation of thalamocortical collaterals, EPSCs were sorted according to latency on the following grounds. (i) Corticothalamic fibers ending in the RTN of rodents (Fig. 1A) are thinner than thalamocortical fibers and do not exhibit multiple diameters. Larger diameter corticothalamic fibers arising from layer V cells do not collateralize in RTN (1, 2). (ii) The mean latency of antidromic action potentials in VP neurons in response to cortical stimuli was 2.25 ± 0.35 msec, indicating that RTN EPSCs with similarly short latencies arose from antidromic stimulation of collaterals of these fibers. (iii) The latency distribution of EPSCs in RTN cells showed two peaks at 2–3 and 6–7 msec (Fig. 1B). In recordings from RTN neurons, therefore, only EPSCs with latencies greater than 6.0 msec were considered to result from corticothalamic fiber stimulation. When RTN EPSCs with latencies less than 6 msec were excluded, mean EPSC latency in RTN neurons was not different statistically from mean EPSC latency in VP neurons, indicating that EPSCs resulted from stimulation of the same axons.
Figure 1.

(A) Thalamocortical slice preparation and an injected layer VI corticothalamic neuron with its axon giving off collaterals in RTN and continuing into VP. (Bars = 50 μm). (B) Latencies of synaptic responses in RTN neurons following stimulation of layer VI. The early peak represents responses to antidromic activation of thalamocortical collaterals, and the later peak represents responses to orthodromic activation of slower conducting corticothalamic fibers.
Minimal Corticothalamic EPSC Amplitudes Are Higher in RTN than in VP Neurons.
When all criteria to include only EPSCs resulting from orthodromic stimulation of corticothalamic fibers were met, and stimulus strength was adjusted to 10–15% above threshold to elicit a stable (≈50%) mixture of failures and successes, mean failure rate in VP neurons was 61 ± 13% (n = 28) and in RTN neurons 44 ± 24% (n = 19). Peak amplitudes of corticothalamic EPSCs fluctuated considerably (Fig. 2 A and C). At −70 mV holding potential, mean peak amplitude of corticothalamic EPSCs (excluding failures) was 8.96 ± 3.30 pA in VP neurons (n = 28) and 21.65 ± 5.58 pA (n = 20) in RTN neurons (Fig. 2B). These amplitudes correspond to conductance changes of 128 ± 47 pS for VP neurons and 309 ± 80 pS for RTN neurons. The differences were highly significant statistically (P < 0.0001) and indicate that corticothalamic input to RTN neurons is 2.42 times stronger than that to VP neurons (Fig. 2 Upper Inset).
Figure 2.

(A) Overlay of 10 voltage-clamp traces from a VP neuron (Left) and an RTN neuron (Right), following minimal stimulation of corticothalamic fibers, showing EPSC successes and failures. Minimal EPSC amplitudes are larger in RTN neurons than in VP neurons. (B Left) Average of EPSC successes recorded from the VP neuron, averaged from responses to 500 stimuli. (B Right) Average of EPSC successes recorded from the RTN neuron in A, averaged from responses to 1,000 stimuli. The average EPSC in VP and RTN neurons exhibits fast rise and decay times. (C) Graph illustrating the trial-to-trial variability in EPSC peak amplitudes recorded in the VP (Left) and RTN (Right) neurons showing the large number of failures and small size of EPSCs. (D) Distribution of EPSC amplitudes recorded in the VP (Left) and RTN (Right) neurons showing comparatively narrow amplitude distribution of EPSC successes in the VP neuron and range of amplitudes of EPSC successes in the RTN neuron. (Upper Inset) Mean conductance of EPSC successes in VP and RTN cells recorded in artificial cerebrospinal fluid and low-Ca2+/high-Mg2+ solutions. Asterisks indicate a statistically significant difference. (Lower Inset) Rise and decay times of EPSC successes in VP and RTN neurons. Slight differences are not significant statistically.
Differences in amplitude of corticothalamic EPSCs in RTN and VP neurons were not determined by differences in series, and input resistance for these resistances were not significantly different in RTN and VP neurons (data not shown). Rise and decay times of the EPSCs also did not differ significantly (Fig. 2 Lower Inset). The mean 20–80% rise time was 0.612 ± 0.14 msec (range = 0.429–0.984 msec) in VP neurons (n = 28) and 0.664 ± 0.26 msec in RTN neurons (range = 0.335–1.11 msec). The decay time course of EPSCs could be best fit to a single exponential function in VP and RTN neurons. The mean single exponential decay time constant was 2.59 ± 1.17 msec in VP neurons (range = 1.16–6.25 msec; n = 27) and 2.73 ± 1.52 msec in RTN neurons (range = 1.01–7.46 msec; n = 19).
Paired whole-cell recordings from 50-layer VI–VP neuron pairs resulted in three pairs (6%) in which single action potentials in the layer VI neuron elicited EPSPs in the VP neuron (Fig. 3). Only a small proportion of the action potentials in the layer VI neuron elicited an EPSP in the VP neuron (Fig. 3C). Failure rate was 68 ± 7%. Mean peak amplitude of EPSPs when failures were included was 85 ± 8 μV. Mean peak amplitude when failures were excluded was 254 ± 30 μV. The latency of the EPSPs was 8.67 ± 2.02 msec, reflecting the slow conduction velocity of the thin corticothalamic axons (Fig. 1B) and justifying the 6-msec latency cutoff.
Figure 3.

(A) Intrinsic firing patterns of a regular-spiking layer VI neuron (Upper) and a synaptically coupled VP neuron (Lower) revealed by injection of current. (B Upper) Averaged trace of single action potentials induced by injection of current into the layer VI neuron. (B Lower) Average of unitary EPSP successes recorded in the VP neuron and induced by single action potentials in the synaptically coupled layer VI neuron. (The neurons were coupled in the corticothalamic direction only). (C Upper) Trial-to-trial variability in unitary EPSP amplitude showing large number of trials in which presynaptic action potential did not induce an EPSP. (C Lower) Distribution of unitary EPSP amplitudes (excluding failures).
Unitary Corticothalamic EPSPs Elicited in VP Neurons Show Paired-Pulse Facilitation.
In paired recordings (n = 3) in which single action potentials in a layer VI neuron elicited EPSPs in a synaptically coupled VP neuron, pairs of action potentials separated by 50 or 100 msec in the layer VI neuron elicited dramatic paired-pulse facilitation in the VP neuron (Fig. 4A). The amplitude of the second EPSP was 340% greater than that of the first EPSP at a 50-msec interspike interval and 224% greater at a 100-msec interspike interval. The paired-pulse facilitation was accompanied by a comparable decrease in the failure rate (Fig. 4B). The failure rate of the first EPSP was 333% greater than the failure rate of the second EPSP at 50-msec interspike interval and 185% greater at 100-msec interspike interval (data not shown). These results suggest that paired-pulse facilitation results from an increase in probability of transmitter release following the second action potential.
Figure 4.

(A Left) Averaged trace of pairs of action potentials separated by 50 msec and induced in a layer VI neuron. (A Right) Averaged trace of pairs of EPSPs recorded in a synaptically coupled VP neuron induced by the pairs of action potentials, showing paired-pulse facilitation. Capacitative-coupling artifacts have been blanked. (B) Trial-to-trial variability in EPSP amplitude following the first (Right) and second (Left) action potentials illustrated in A. (C) Distribution of unitary EPSP amplitudes after first (Right) and second (Left) action potentials showing large decrease in failures following second action potential.
Differences in Quantal Size Underlie Differences in Minimal EPSC Amplitudes in RTN and VP Neurons.
EPSC amplitudes rarely displayed clear peaks from which quantal conductance could be identified by conventional means. We therefore determined quantal conductance by decreasing the probability of release to a point at which, according to a Poisson model of transmitter release, the large majority of responses would be due to release of a single quantum. The Poisson model is applicable when quanta are released independently, and the probability of release at each release site is small, conditions that were met when we reduced the [Ca2+]/[Mg2+] ratio (15). When [Ca2+] was decreased from 2 to 1 mM and [Mg2+] was increased from 1.3 to 3 mM, the failure rate of corticothalamic EPSCs increased from 61 ± 13% to 82 ± 6% in VP neurons and from 44 ± 24% to 69 ± 7% in RTN neurons, consistent with a decrease in release probability. According to the Poisson model, the probability (P) of observing a postsynaptic current made of x unit currents in which there are N trials and N0 failures is:
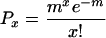 |
where:
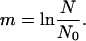 |
Therefore, the probability of observing EPSCs made up of two quanta in low-Ca2+/high-Mg2+ solutions was 1.6% in VP neurons and 4.7% in RTN neurons, suggesting that the large majority of EPSC successes recorded under these conditions were uniquantal events and dependent on release of a single vesicle at a single release site.
The mean amplitude of corticothalamic EPSC successes permitted a reliable estimate of the quantal size at these synapses. Mean amplitude was 7.24 ± 1.78 pA in VP neurons (n = 5) and 18.59 ± 3.37 pA in RTN neurons (n = 4) at −70 mV. These values correspond to conductance changes of 103 ± 25 pS in VP neurons and 266 ± 48 pS in RTN neurons. The difference was highly significant statistically (P < 0.005; Fig. 2 Inset). Hence, differences in quantal size of EPSCs may underlie differences in minimal corticothalamic EPSCs in RTN and VP neurons. It remains unknown, however, if there are differences in number of release sites at the two synapses.
Differences in Number of GluR4 Subunits at Corticothalamic Synapses Correlate with Differences in Quantal Conductance.
Corticothalamic terminals in RTN and VP were identified morphologically by using established criteria (16). Although called ST synapses in RTN and RS synapses in VP, they have identical characteristics: small size (0.2–0.5 μm), spherical synaptic vesicles, few mitochondria, and asymmetrical membrane contacts. They are distinguished readily from terminals of thalamocortical collaterals (in RTN; refs. 17 and 18) and lemniscal fibers (in VP; refs. 16 and 19). In VP and the somatosensory part of the RTN, the number of similar terminals originating from brainstem fibers is extremely small, and they use transmitters other than glutamate (20, 21).
Immunogold particles that labeled GluR2/3 or GluR4 subunits were associated exclusively with asymmetrical synapses including those made by corticothalamic terminals and were localized predominantly at PSDs (Fig. 5). In serial thin-section analysis, the mean number of GluR2/3 particles was 12.0 ± 5.4 at RS synapses in VP and 12.0 ± 3.5 at ST synapses in RTN, indicating that corticothalamic synapses in RTN and VP have approximately equal numbers of GluR2/3 receptor subunits (Fig. 5G). The mean number of GluR4 particles was 9.3 ± 3.5 at RS synapses in VP and 34.5 ± 13.0 at ST synapses in RTN (Figs. 5G and 6), indicating that there are 3.7 times as many GluR4 subunits at corticothalamic synapses in RTN than in VP. This difference was highly significant statistically (P < 0.0001; Fig. 5G). Because the synaptic contacts are equal in size, density of GluR4 receptor subunits differs in the same proportions.
Figure 5.

(A, B, and C) Serial electron micrographs from a single corticothalamic synapse in VP showing small number of GluR4 immunogold-labeled particles at the PSD. (D, E, and F) Serial electron micrographs from a single corticothalamic synapse in RTN showing larger number of GluR4 immunogold particles at the PSD. (Bar = 125 nM.) (G Upper) Mean number of GluR4 subunit-specific particles at serially sectioned corticothalamic synapses in RTN is much higher than at synapses in VP (* P < 0.0001). Mean number of GluR2/3 particles is approximately equal. (G Lower) Tracings of the PSDs and associated immunogold particles from two serially sectioned corticothalamic synapses in VP and two in RTN. Number of particles associated with each section through the PSD is in parentheses. GluR4 particles are associated with all sections through the PSD of the RTN synapse, but some sections through the PSD of the VP synapse typically lack GluR4 particles. GluR2/3 particles have identical distributions in VP and RTN.
Figure 6.

Counts of GluR4 and GluR2/3 particles at randomly sectioned PSDs in RTN and VP. Overlying curves show theoretical Poisson-distributed data with means corresponding to the observed means. Similarity in observed and theoretical distributions implies that quantification of receptor-subunit numbers was based on random exposure of epitopes to the immunoreagents.
In 436 single sections through PSDs in random thin sections, the relative numbers of GluR4 or GluR2/3 particles per PSD were the same proportionally as in the serial section analysis (0.95:1 for GluR2/3 synapses in RTN and VP; 3.2:1 for GluR4 synapses in RTN and VP). The mean particle counts were used to calculate the Poisson probabilities expected for GluR2/3 and GluR4 synapses. These probabilities were quite similar, confirming that the number of immunoreactive particles observed was based on random exposure of the epitopes to the immunoreagents (ref. 22; Fig. 6).
Discussion
The results demonstrate that the efficacy of corticothalamic input to RTN neurons is greater than that to VP neurons. The disynaptic inhibitory postsynaptic potential in VP neurons resulting from corticothalamic excitation of RTN cells quickly overwhelms the small direct corticothalamic EPSP in vitro (14), and in vivo electrical stimulation of the cortex primarily elicits disynaptic inhibitory postsynaptic potentials in relay neurons (13), although this can be due to antidromic invasion of thalamocortical axons, the collaterals of which can induce large EPSPs in RTN neurons (23). When we excluded all EPSCs with latencies less than 6 msec, presumed to result from activation of thalamocortical fibers, latencies of EPSCs evoked by minimal stimulation of cortex were not statistically different in RTN and VP cells, in keeping with activation of RTN and VP neurons by the same corticothalamic axons.
Rise times and decay-time constants of corticothalamic EPSCs in RTN and VP cells were relatively fast, suggesting that EPSCs originated from electrotonically close synaptic terminals on the dendrites. Although corticothalamic synapses are concentrated on distal dendrites of relay neurons (24, 25), the neurons are electrically compact (26, 27) so corticothalamic EPSCs are filtered minimally by dendritic electrotonic length. The electrotonic structure of RTN neurons has not been defined, but because EPSC kinetics were similar in VP and RTN neurons, corticothalamic terminals on both cells are likely to be at similar electrotonic distances from the soma.
Because peaks were unclear in histograms of corticothalamic EPSC amplitudes and because deconvolution of peaks in amplitude distributions may be unreliable for determination of quantal parameters (28, 29), we estimated quantal size by lowering release probability to a point at which Poisson statistics predicted that 90% of successes in VP neurons and 82% of successes in RTN neurons would be uniquantal events. Quantal conductance then was estimated by determining mean amplitude of successes. This mean amplitude was 103 ± 25 pS at corticothalamic synapses on VP neurons and 266 ± 48 pS at synapses on RTN neurons. The values are similar at excitatory synapses on visual cortex interneurons (100 pS; ref. 30), at mossy fiber-CA3 synapses (121–553 pS; ref. 31), and at excitatory synapses on cerebellar granule cells (139 pS; ref. 32).
Because of voltage-clamp limitations, the actual conductance change at the corticothalamic synapses may be greater than estimated. Jonas et al. (31), who recorded EPSCs in mossy fiber-CA3 synapses with rise and decay times similar to ours, estimated that the EPSCs may have been attenuated to 0.32–0.87 of the “real” value. If the same degree of attenuation occurred in our study, then the conductance change at corticothalamic synapses may be as high as 118–322 pS in VP neurons and 306–831 pS in RTN neurons.
Dual whole-cell recordings from synaptically coupled pairs of layer VI and VP neurons demonstrated that corticothalamic transmission is unreliable: 68 ± 7% of action potentials in layer VI neurons failed to elicit an EPSP in VP neurons. The high failure rate may arise from low probability of transmitter release. Even if transmitter release occurs, it may not generate a detectable EPSP because some corticothalamic synapses, although possessing functional N-methyl-D-aspartate receptors, lack AMPA receptors (8).
When failure rate of EPSPs is reduced during paired-pulse facilitation, the corticothalamic synapse can adjust EPSP amplitude. Other studies have shown frequency-dependent facilitation at corticothalamic synapses in response to repetitive stimulation (10, 33, 34). Changes in failure rate during facilitation at cortical and spinal cord synapses are thought to result from increase in probability of release at synapses with initially low release probabilities (35, 36). Changes in release probability during short-term facilitation probably result from residual calcium remaining in the terminal following the first action potential (37).
With immuno-electron microscopic procedures of the type used, the number of immunogold particles bears a close relationship to the number of receptor subunits present (38, 39). There was no difference in the relative numbers of GluR2/3 particles at corticothalamic synapses in VP and RTN, but there were 3.7 times as many GluR4 particles at corticothalamic synapses in RTN than in VP. This reflects the higher expression of GluR4 mRNA and protein in the RTN (40–44). GluR2/3 immunoreactivity probably is due to the presence of GluR3 in both RTN and VP, because GluR2 along with GluR1 is expressed negligibly in both nuclei. Therefore, AMPA receptors at corticothalamic synapses in RTN and VP are likely to be heteromers of GluR3 and GluR4 subunits. The larger number of GluR4 subunits undoubtedly determines that quantal conductance at the RTN corticothalamic synapse is greater than at the VP synapse, because single-channel conductance of recombinant GluR4 receptors is substantially higher than that of receptors formed by homo- or heteromeric combinations of other AMPA subunits (45). The relative increase in GluR4-containing receptors should provide the predicted greater strength of the corticothalamic synapse in the RTN, helping to promote large scale synchrony of thalamic and cortical neurons (46, 47).
At hippocampal mossy fiber synapses, it is hypothesized that each immunogold particle corresponds to 2.3 AMPA receptors (31, 38). Using this ratio, our findings suggest approximately 21 GluR4-containing AMPA receptors at VP corticothalamic synapses and approximately 79 at RTN corticothalamic synapses. Jonas et al. (31), by using fast application of glutamate to somatic patches of hippocampal granule cells in combination with noise fluctuation analysis, estimated an 8.5 ± 2.1 pS AMPA receptor conductance, a maximal open probability of 0.71 ± 0.06, and receptor occupancy of 85%. If single-channel properties of AMPA receptors at corticothalamic synapses on VP and RTN neurons have similar characteristics, and if each immunogold particle corresponds to 2.3 receptors, then the estimated quantal conductances would be 110 pS at VP corticothalamic synapses and 407 pS at RTN corticothalamic synapses, which are similar to the measured conductances.
Corticothalamic synapses made by branches of the same axons on RTN and VP cells acquire different weights by postsynaptic mechanisms based on AMPA-receptor subunit composition. The synaptic heterogeneity thus produced provides a basis for the ability of the cortex to generate large-scale synchrony in the thalamocortical network and for the preeminence of the cortex in regulating the thalamus.
Acknowledgments
We thank Drs. F. Schweizer, C. Stricker, K. McAllister, and S. Bolea for advice, Phong Nguyen and Nick Singh for technical support, and Dr. Y. He and Mr. W. G. M. Janssen for advice on the cryo-infiltration method. This work was supported by Grant NS39094 from the National Institutes of Health, U.S. Public Health Service. Peyman Golshani is an M.D./Ph.D. student.
Abbreviations
- RTN
reticular nucleus
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- EPSP
excitatory postsynaptic potential
- VP
ventral posterior thalamic nucleus
- EPSC
excitatory postsynaptic conductance
- PSD
postsynaptic density
- ST
corticothalamic synapse in RT
- RS
corticothalamic synapse in VP
Footnotes
See commentary on page 3625.
References
- 1.Bourassa J, Pinault D, Deschênes M. Eur J Neurosci. 1995;7:19–30. doi: 10.1111/j.1460-9568.1995.tb01016.x. [DOI] [PubMed] [Google Scholar]
- 2.Hoogland P V, Welker E, Van Der Loos H. Exp Brain Res. 1987;68:73–87. doi: 10.1007/BF00255235. [DOI] [PubMed] [Google Scholar]
- 3.Steriade M. Adv Neurol. 1998;77:105–134. [PubMed] [Google Scholar]
- 4.Deschênes M, Hu B. Eur J Neurosci. 1990;2:140–152. doi: 10.1111/j.1460-9568.1990.tb00406.x. [DOI] [PubMed] [Google Scholar]
- 5.McCormick D A, Von Krosigk M. Proc Natl Acad Sci USA. 1992;89:2774–2778. doi: 10.1073/pnas.89.7.2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eaton S A, Salt T E. Neuroscience. 1996;73:1–5. doi: 10.1016/0306-4522(96)00123-6. [DOI] [PubMed] [Google Scholar]
- 7.Kao C-Q, Coulter D A. J Neurophysiol. 1997;77:2661–2676. doi: 10.1152/jn.1997.77.5.2661. [DOI] [PubMed] [Google Scholar]
- 8.Golshani P, Warren R A, Jones E G. J Neurophysiol. 1998;80:143–154. doi: 10.1152/jn.1998.80.1.143. [DOI] [PubMed] [Google Scholar]
- 9.Von Krosigk M, Bal T, McCormick D A. Science. 1993;261:361–364. doi: 10.1126/science.8392750. [DOI] [PubMed] [Google Scholar]
- 10.Von Krosigk M, Monckton J E, Reiner P B, McCormick D A. Neuroscience. 1999;91:7–20. doi: 10.1016/s0306-4522(98)00557-0. [DOI] [PubMed] [Google Scholar]
- 11.Warren R A, Agmon A, Jones E G. J Neurophysiol. 1994;72:1993–2003. doi: 10.1152/jn.1994.72.4.1993. [DOI] [PubMed] [Google Scholar]
- 12.Contreras D, Destexhe A, Sejnowski T J, Steriade M. Science. 1996;274:771–774. doi: 10.1126/science.274.5288.771. [DOI] [PubMed] [Google Scholar]
- 13.Destexhe A, Contreras D, Steriade M. J Neurophysiol. 1998;79:999–1016. doi: 10.1152/jn.1998.79.2.999. [DOI] [PubMed] [Google Scholar]
- 14.Warren R A, Jones E G. J Neurosci. 1997;17:277–295. doi: 10.1523/JNEUROSCI.17-01-00277.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Del Castillo J, Katz B. J Physiol (London) 1954;192:407–436. [Google Scholar]
- 16.Liu X-B. J Comp Neurol. 1997;388:587–602. doi: 10.1002/(sici)1096-9861(19971201)388:4<587::aid-cne7>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 17.Harris R M. J Comp Neurol. 1987;258:397–406. doi: 10.1002/cne.902580308. [DOI] [PubMed] [Google Scholar]
- 18.Liu X-B, Jones E G. J Comp Neurol. 1999;414:67–79. [PubMed] [Google Scholar]
- 19.Peschanski M, Roudier F, Ralston H J, Besson J M. Somatosens Res. 1985;3:75–87. doi: 10.3109/07367228509144578. [DOI] [PubMed] [Google Scholar]
- 20.Liu X-B, Jones E G. Exp Brain Res. 1991;85:507–518. doi: 10.1007/BF00231734. [DOI] [PubMed] [Google Scholar]
- 21.Usunoff K G, Kharazia V N, Valtschanoff J G, Schmidt H H, Weinberg R J. Anat Embryol. 1999;200:265–281. doi: 10.1007/s004290050278. [DOI] [PubMed] [Google Scholar]
- 22.Kharazia V N, Weinberg R J. J Comp Neurol. 1999;412:292–302. doi: 10.1002/(sici)1096-9861(19990920)412:2<292::aid-cne8>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 23.Kim U, McCormick D A. J Neurosci. 1998;18:9500–9516. doi: 10.1523/JNEUROSCI.18-22-09500.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jones E G, Powell T P S. Proc R Soc London Ser B. 1969;172:173–185. doi: 10.1098/rspb.1969.0018. [DOI] [PubMed] [Google Scholar]
- 25.Liu X-B, Honda C N, Jones E G. J Comp Neurol. 1995;352:69–91. doi: 10.1002/cne.903520106. [DOI] [PubMed] [Google Scholar]
- 26.Crunelli V, Leresche N, Parnavelas J G. J Physiol (London) 1987;390:243–245. doi: 10.1113/jphysiol.1987.sp016697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bloomfield S A, Hamos J E, Sherman S M. J Physiol (London) 1987;383:653–692. doi: 10.1113/jphysiol.1987.sp016435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clements J. Nature (London) 1991;353:396. doi: 10.1038/353396a0. [DOI] [PubMed] [Google Scholar]
- 29.Walmsley B. Proc R Soc London Ser B. 1995;261:245–250. doi: 10.1098/rspb.1995.0144. [DOI] [PubMed] [Google Scholar]
- 30.Stern P, Edwards F A, Sakmann B. J Physiol (London) 1992;449:247–278. doi: 10.1113/jphysiol.1992.sp019085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jonas P, Major G, Sakmann B. J Physiol (London) 1993;472:615–663. doi: 10.1113/jphysiol.1993.sp019965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Silver R A, Cull-Candy S G, Takahashi T. J Physiol (London) 1996;494:231–250. doi: 10.1113/jphysiol.1996.sp021487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lindström S, Wrøbel A. Exp Brain Res. 1990;79:313–318. doi: 10.1007/BF00608240. [DOI] [PubMed] [Google Scholar]
- 34.Castro-Alamancos M A, Calcagnotto M E. J Neurosci. 1999;19:9090–9097. doi: 10.1523/JNEUROSCI.19-20-09090.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hirst G D, Redman S J, Wong K. J Physiol (London) 1981;321:97–109. doi: 10.1113/jphysiol.1981.sp013973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thomson A M, Deuchars J, West D C. Neuroscience. 1993;54:347–360. doi: 10.1016/0306-4522(93)90257-g. [DOI] [PubMed] [Google Scholar]
- 37.Kamiyah A, Zucker R. Nature (London) 1994;371:603–606. doi: 10.1038/371603a0. [DOI] [PubMed] [Google Scholar]
- 38.Nusser Z, Cull-Candy S, Farrant M. Neuron. 1997;19:697–709. doi: 10.1016/s0896-6273(00)80382-7. [DOI] [PubMed] [Google Scholar]
- 39.Nusser Z, Lujan R, Laube G, Roberts J D, Molnar E, Somogyi P. Neuron. 1998;21:545–559. doi: 10.1016/s0896-6273(00)80565-6. [DOI] [PubMed] [Google Scholar]
- 40.Petralia R S, Wenthold R J. J Comp Neurol. 1992;318:329–354. doi: 10.1002/cne.903180309. [DOI] [PubMed] [Google Scholar]
- 41.Martin L J, Blackstone C D, Levey A I, Huganir R L, Price D L. Neuroscience. 1993;53:327–358. doi: 10.1016/0306-4522(93)90199-p. [DOI] [PubMed] [Google Scholar]
- 42.Sato K, Kiyama H, Tohyama M. Neuroscience. 1993;52:515–539. doi: 10.1016/0306-4522(93)90403-3. [DOI] [PubMed] [Google Scholar]
- 43.Spreafico R, Frassoni C, Arcelli P, Bataglia G, Wenthold R J, De Biasi S. Dev Brain Res. 1994;82:231–244. doi: 10.1016/0165-3806(94)90166-x. [DOI] [PubMed] [Google Scholar]
- 44.Gold S J, Ambrose-Ingerson J, Horowitz J R, Lynch G, Gall C M. J Comp Neurol. 1997;385:491–502. doi: 10.1002/(sici)1096-9861(19970908)385:4<491::aid-cne1>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 45.Swanson G T, Kamboj S K, Cull-Candy S G. J Neurosci. 1997;17:58–69. doi: 10.1523/JNEUROSCI.17-01-00058.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Buzsáki G, Chrobak J J. Curr Opin Neurobiol. 1995;5:504–510. doi: 10.1016/0959-4388(95)80012-3. [DOI] [PubMed] [Google Scholar]
- 47.Contreras D, Destexhe A, Sejnowski T J, Steriade M. J Neurosci. 1997;17:1179–1196. doi: 10.1523/JNEUROSCI.17-03-01179.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]