

Abstract
Summary: Viral infections affect wheezing and asthma in children and adults of all ages. In infancy, wheezing illnesses are usually viral in origin, and children with more severe wheezing episodes are more likely to develop recurrent episodes of asthma and to develop asthma later in childhood. Children who develop allergen‐specific immunoglobulin E (allergic sensitization) and those who wheeze with human rhinoviruses (HRV) are at especially high risk for asthma. In older children and adults, HRV infections generally cause relatively mild respiratory illnesses and yet contribute to acute and potentially severe exacerbations in patients with asthma. These findings underline the importance of understanding the synergistic nature of allergic sensitization and infections with HRV in infants relative to the onset of asthma and in children and adults with respect to exacerbations of asthma. This review discusses clinical and experimental evidence of virus–allergen interactions and evaluates theories which relate immunologic responses to respiratory viruses and allergens to the pathogenesis and disease activity of asthma. Greater understanding of the relationship between viral respiratory infections, allergic inflammation, and asthma is likely to suggest new strategies for the prevention and treatment of asthma.
Keywords: rhinovirus, virus, allergy, asthma, inflammation
Introduction
Viral infections affect wheezing and asthma in children and adults of all ages. In infancy, wheezing illnesses are usually viral in origin, and children with more severe wheezing episodes are more likely to develop recurrent episodes of asthma and to develop asthma later in childhood. The etiology of the viral infection affects the long‐term prognosis, and preschool children who wheeze with human rhinoviruses (HRV) are particularly likely to subsequently develop asthma. The other major risk factor for asthma in this age group is the early onset of sensitization to respiratory allergies. In older children and adults, most viral respiratory illnesses are relatively mild and yet contribute to acute and potentially severe exacerbations in patients with asthma. In fact, there is considerable evidence that viral respiratory infections and respiratory allergies are the two most significant risk factors for exacerbations leading to acute care visits or hospitalization. These findings underline the importance of understanding the synergistic nature of allergic sensitization and infections with HRV in infants relative to the onset of asthma and in children and adults with respect to exacerbations of asthma. In this article, we review clinical evidence of virus–allergen interactions and discuss experimental evidence and theories relating immunologic responses to respiratory viruses and allergic airway inflammation to the pathogenesis and disease activity of asthma.
Rhinovirus biology and replication cycle
Classification
HRV were classified into 100 serotypes based on growth in tissue culture and inhibition by specific antisera, and these canonical strains were classified into groups ‘A’ and ‘B’ based on similarity of partial genetic sequences and responses to certain antiviral medications. Data from studies using molecular diagnostics in the US (1, 2, 3, 4), Australia (5, 6), Asia (7, 8), and Europe (9) demonstrate that there are at least 60 more HRV strains than had been previously appreciated (10). Analysis of full genome sequences of these newly identified HRV indicates that the majority belong to a unique species now designated HRV‐C (11). Like several other newly discovered respiratory viruses, HRV‐C do not grow in standard tissue culture, and this characteristic explains why their discovery required the development of molecular diagnostics.
Rhinovirus replication
HRVs are small (30 nM diameter) non‐enveloped viruses of the Picornaviridae family, containing single stranded RNA that can be translated directly into protein (positive strand). The viral capsid is composed of four proteins (VP1, VP2, VP3, VP4) that assemble into protomers, and each capsid contains 60 protomers. Variations in the structure of these proteins (particularly VP1 and VP2) accounts for the large number of viral serotypes and strains. The capsid binds to specific proteins on the surface of airway cells. Of the 99 HRV serotypes, 84 bind to intercellular adhesion molecule‐1 (ICAM‐1) and the others bind to members of the low‐density lipoprotein receptor. Analysis of a limited number of genomes suggested that HRV‐C probably binds to unique cellular receptors (11). Recently, HRV‐C have been grown in vitro by culturing sinus mucosal biopsy specimens in organ culture, and this system was used to demonstrate that HRV‐C viruses bind to receptors that are distinct from those used by other HRV (12).
Once bound to the cell, HRVs are internalized into endosomes, and acidification of this compartment leads to dissociation of the VP4 protein from the rest of the capsid and initiates release of HRV RNA into the cell. In permissive airway epithelial cells, the viral RNA is translated to a single polyprotein, and HRV proteases (2A and 3C) direct the processing of the polyprotein into mature non‐structural proteins and structural proteins. The HRV polymerase directs RNA replication, and both single‐ and double‐stranded viral RNA are present in the cell during this process. Once sufficient quantities of positive strand RNA and capsid proteins are synthesized, progeny virions are assembled, and are released upon lysis of the cell.
In addition to helping to direct replication, viral non‐structural proteins also inhibit host cell processes to favor viral replication. For example, the 2A protease cleaves eukaryotic translation initiation factor 4 gamma (eIF4G) in the ribosomal protein complex; this prevents the translation of capped RNA (the majority of cellular mRNA). In contrast, the HRV genome has an RNA structure known as the internal ribosome entry site (IRES) which binds to the ribosomal complex independently of eIF4G, and so translation of HRV RNA is unimpeded. The 2A protease also cleaves nuclear pore proteins which impedes communication between the nucleus and cytoplasm. One consequence of these activities is that cellular RNA levels may not reflect protein synthesis. HRV infection also inhibits transcription of cellular RNA but not viral RNA. Like many other viruses, there is evidence that HRV has developed mechanisms to inhibit antiviral responses by cleaving proteins such as RIG‐I and IPS‐1 that are involved in virus recognition pathways (13, 14), and transcription factors related to the antiviral and chemokine responses (e.g. OCT‐1) (15).
Clinical course
HRV infections can be transmitted by either aerosol droplets or contact with infected secretions. Transmission of HRV infections between adults generally requires prolonged contact (16, 17); infants and young children are the most effective vectors and readily transmit HRV infections within families (18). Respiratory symptoms typically develop after 1–2 days in inoculation studies. Early signs of a cold include sore or scratchy throat and malaise, followed by a thin mucoid discharge, sneezing, congestion, and thicker nasal secretions with increased mucus during the peak of the cold. Uncomplicated HRV infections usually peak 2–4 days after inoculation and are resolving by 1 week after inoculation. Infections that last 2 weeks or longer or those that seem to be improving and then worsen again represent either serial infections with different cold viruses (19) or else a secondary bacterial infection following an initial viral infection. HRV infections are often detected in asymptomatic infants and less commonly in adults. Viral detection without symptoms can represent the beginning or end of an infection or true asymptomatic infection. In immune competent individuals, there is no long‐term ‘carrier state’ for HRV.
The discovery of HRV‐C raises new questions as to whether infections with this species of viruses cause distinct patterns of illness. HRV‐C viruses have been closely associated with wheezing illnesses in infants and pneumonia in young children in a number of clinical studies (1, 5, 8, 20, 21). In addition, HRV‐C viruses are frequently detected during exacerbations of asthma in children, and there is some evidence that HRV‐C can cause more severe wheezing episodes compared to HRV‐A and ‐B (1, 22). HRV epidemiology is complex: up to 20 strains of HRV circulate through a community during a single season, and the prevailing HRV strains differ according to season and location, and shift almost completely from year to year (23). The complexity of HRV epidemiology and the large number of HRV strains indicate that long‐term population‐based studies will be required to determine whether HRV‐C are indeed more virulent than other HRV strains.
Pathogenesis of respiratory symptoms
Once infection is established, respiratory symptoms are the result of two processes: destruction of normal airway tissue due to direct effects of the virus, and pro‐inflammatory immune responses to the infection. Virus‐induced damage to the epithelium can disturb airway physiology through a number of different pathways. For example, epithelial edema and shedding together with mucus production can cause airway obstruction and wheezing. Furthermore, HRV infections can inhibit processes associated with epithelial repair (24). In addition, viral replication initiates innate immune responses within the epithelial cell including induction of interferons (IFNs), antiviral effectors, and chemokines that recruit inflammatory cells into the airway (25, 26, 27, 28, 29). The recent development of rodent models of infection with HRV and related picornaviruses will provide systems to test theories about immunopathogenesis in vivo (30, 31, 32). Identification of antiviral responses that determine susceptibility to HRV are now under study in a number of laboratories.
For viruses such as HRV that infect relatively few cells in the airway (33, 34), inflammatory responses to the infection are important contributors to respiratory symptoms. The majority of the cells recruited to the airway during acute colds are neutrophils, with smaller numbers of mononuclear cells and in some studies, eosinophils. Products of neutrophil activation are likely involved in obstructing the airways and causing lower airway symptoms. For example, the release of the potent secretagogue elastase from activated neutrophils can upregulate goblet cell secretion of mucus (35). In addition, changes in neutrophils in nasal secretions have been related to respiratory symptoms and virus‐induced increases in airway hyperresponsiveness (enhanced bronchoconstriction in response to irritants), a key feature of asthma (36, 37). These findings suggest that one strategy to reduce the severity of viral illnesses might be to moderate neutrophilic inflammatory responses. Moreover, mononuclear cells are recruited into the upper and lower airway during the early stages of a viral respiratory infection, and serve to limit the extent of infection and to clear virus‐infected epithelial cells. This is consistent with reports of severe viral lower respiratory infections in immunocompromised patients (38).
Severity of colds
A number of risk factors for more severe HRV illnesses have been identified in clinical studies, and in studies in which volunteers are inoculated with safety‐tested viruses under controlled conditions ( Table 1 ). These factors include characteristics of the host; the very young and the elderly, and individuals with chronic lung diseases tend to have worse illnesses with HRV infection. Interestingly, boys are more likely to develop moderate to severe HRV illnesses during early childhood (39). There has been considerable interest in defining the relationship between patterns of antiviral responses and illness outcomes, especially IFNs and factors related to atopy and asthma. These relationships are discussed in detail later in the review. Finally, environmental and lifestyle factors associated with more severe common colds include pollutants, smoking, characteristics of the immune responses, stress, and diet. Notably, there are data to suggest that supplementation with either probiotics (40, 41) or vitamin D (42, 43) could reduce the frequency and severity of common colds.
Table 1.
Factors linked to more severe HRV or common cold infections*
| Host characteristics | Immunologic | Environment and lifestyle |
|---|---|---|
| Chronic lung diseases Asthma COPD Cystic fibrosis | Low interferon responses IFN‐α IFN‐β IFN‐γ IFN‐λ | Tobacco smoke Pollutants (NO2) |
| Age Preschool children Elderly | Allergy Eosinophilia | Diet Vitamin D Probiotics |
| Genetics Gender (young boys) | Epithelial integrity Immune compromise | Stress Day care attendance |
*Adapted with permission from reference (185).
HRV, human rhinoviruses; COPD, chronic obstructive pulmonary disease.
Clinical evidence of virus–allergen interactions
Wheezing in early childhood and the development of asthma
Wheezing illnesses are closely associated with viral respiratory infections in all age groups ( Fig. 1 ). Approximately half of infants experience wheezing, and studies using sensitive PCR‐based viral diagnostics indicate that wheezing illnesses can be caused by a plethora of respiratory viruses ( Table 2 ). The more severe end of the illness spectrum is known as bronchiolitis and includes many clinical features of acute asthma, including wheezing, rapid breathing, and sometimes hypoxia and hypoventilation. Respiratory syncytial virus (RSV) is particularly likely to cause lower respiratory tract infection, and accounts for about half of the cases of bronchiolitis in the winter months. Interestingly, although some respiratory viruses are more or less likely to cause severe illnesses, the clinical manifestations of infection (rhinitis, cough, and in some cases wheeze) are pretty similar. This is remarkable given the major differences in the structure of the viruses, their replication cycles, and to some extent the virus‐induced immune responses.
Figure 1.
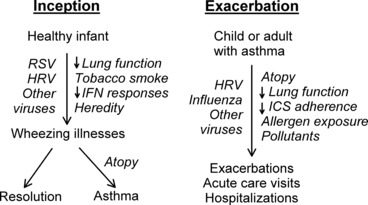
Role of viral infections in the inception and exacerbation of asthma.
Table 2.
Respiratory viruses that cause wheezing illnesses in infants and young children*
| Viruses | Family | Genome |
|---|---|---|
| RSV | Paramyxovirus | ssRNA (−) |
| PIV | Paramyxovirus | ssRNA (−) |
| Influenza | Orthomyxovirus | ssRNA (−) |
| MPV | Pneumovirus | ssRNA (−) |
| HRV A, HRV B, HRV C | Picornavirus | ssRNA (+) |
| Enteroviruses | Picornavirus | ssRNA (+) |
| OC43, NL63, HKU1, SARS | Coronavirus | ssRNA (+) |
| Bocavirus | Parvovirus | ssDNA (−) |
| WU, KIP | Polyomavirus | dsDNA |
| Adenoviruses | Arenovirus | dsDNA |
*Adapted from reference (185).
ss, single stranded; ds, double stranded; (+) and (−) refer to the polarity of the genome.
Recent studies using molecular diagnostics have demonstrated that HRV are common causes of lower respiratory tract infections in infants and young children. This recognition of HRV as a lower airway pathogen is a departure from its original characterization as a virus that exclusively causes the common cold. Soon after its discovery in 1956, HRV was found to replicate best at relatively cool temperatures (33–35 °C), and so it was assumed that infections were limited to the upper airway. Although lung parenchyma is at core temperature (37 °C), airways are considerably cooler, and temperatures in large and medium sized airways are ideal for HRV replication (44). In fact, HRV has been detected in lower airway fluids and cells and after experimental infection of the upper airway (45, 46, 47). Furthermore, there is considerable clinical evidence linking HRV infections to lower respiratory infections in children, including those who are hospitalized for pneumonia (48, 49, 50, 51). A recent year‐long population‐based study of children <5 years of age found that HRV was detected in 26% of children hospitalized for respiratory symptoms or fever (49). HRV‐related hospitalization rates were especially high for infants, and children with asthma. These findings suggest the possibility that infections with HRV, like RSV and other respiratory viruses, can directly injure airway tissues during the acute infection. One feature that distinguishes HRV from other respiratory viruses is the large number of distinct strains, including 100 traditional serotypes, and an estimated 60 HRV‐C species viruses that are grouped by genotype (10). As a result, HRV infections occur frequently in young children, and children prone to wheezing with respiratory viruses can experience recurrent episodes of wheezing with distinct HRV strains.
About a third of infants who have an acute wheezing illness go on to develop recurrent wheezing, leading to the speculation that viral respiratory illnesses in early life promote asthma. A causal role for viruses in asthma seems plausible because the lungs and the immune system are growing and developing during infancy, and might be especially vulnerable to pathogens during this period of time (52). Alternatively, it is also possible that the relationship is not causal, and that virus‐induced wheezing episodes instead reveal a preexisting tendency for asthma secondary to impaired lung physiology or antiviral responses. In fact, immunologic and lung‐specific risk factors for more severe viral respiratory illnesses have been identified and are discussed in detail later in this chapter. This theory suggests that some children are either born with or soon acquire abnormal lung physiology, and then infection with a respiratory virus, such as HRV, presents the first noxious stimulus to provoke wheezing. A third possibility, which combines elements of the first two, is a ‘two hit hypothesis’ in which viral infections promote asthma mainly in predisposed children (53, 54). For example, infants who are predisposed to have poor antiviral responses would develop more severe illness during infections with respiratory viruses. These more severe infections could lead to increased damage of the lower airways during a particularly important growth phase of the lungs. According to this hypothesis, recurrent lower respiratory infections could hinder proper lung growth and development, leading to changes in airway structure (e.g. fibrosis) that promote asthma.
This line of reasoning is supported by experimental findings that infections with viruses such as RSV and HRV in vivo and in vitro increase the synthesis of factors that can influence lung growth, development and repair (27, 52, 55, 56, 57, 58). The pathophysiology of asthma is closely related to airway remodeling, a collection of features that reflect structural changes in the airway that promote acute and chronic airway obstruction. These structural changes include hypertrophy of airway smooth muscle, increased fibrosis, reduced barrier function, increased number of mucus‐secreting cells and proliferation of airway vasculature within the epithelium (59). These changes in airway structure are not present in most wheezing infants but appear to develop in childhood (60, 61). In addition, acute infections with HRV or other viruses can induce the synthesis of factors that regulate airway growth, angiogenesis, fibrosis, and repair ( Table 3 ). Furthermore, viral infections can upregulate neurotropins that regulate the development of the airway neural network (62). This effect on neural mechanisms could affect bronchoconstrictive responses to non‐specific irritants, including future viral infections. In fact, increased sensitivity to inhaled irritants (a.k.a. bronchial hyperresponsiveness) is a key feature of asthma. How single or repeated bouts of virus‐induced overexpression of these regulators of lung development and remodeling affects the ultimate lung structure and function is not known, but is of interest regarding the long‐term effects of viral infection on lung function and asthma. These questions may be best addressed in animal models, and models to study effects of HRV infections in the mouse have been published (30, 31), as well as methods for serial passage of mouse epithelial cells for propagation of HRV (63). In addition, mengovirus, another member of the Picornaviridae family, is a natural rodent pathogen that causes systemic infections that resemble poliovirus. This virus has been attenuated to replicate, but not kill, epithelial cells, and when inoculated into the airway, replicates for several days and causes a mild respiratory illness that resembles HRV infection in the human (32).
Table 3.
Virus‐induced cytokines and growth factors
| Airway development | Fibrosis | Angiogenesis |
|---|---|---|
| NO (186) | TGF‐β (187, 188) | Amphiregulin (55) |
| IL‐6 (189) | MMP‐9 (57, 190) | VEGF (55, 191) |
| MMP‐12 (27) | ||
| FGF (192) |
MMP, matrix metalloprotease; VEGF, vascular endothelial growth factor.
A number of other risk factors for developing asthma after recurrent wheezing in the first few years of life have been identified. Long‐term studies have demonstrated that infants hospitalized with bronchiolitis have a 2–3‐fold increase in the risk of developing asthma later in childhood (64, 65). This risk is further increased by a family history of asthma, the development of allergic diseases such as allergic rhinitis or atopic dermatitis, blood eosinophilia, and male gender (54). Several predictive indices that incorporate these risk factors have been developed order to estimate the prognosis of infants with recurrent wheezing (66, 67, 68).
The specific etiology of virus‐induced wheezing episode also influences the risk of subsequent asthma. Several studies suggest that infants who wheeze with HRV may have an especially high risk for subsequent asthma. For example, a case control study conducted in Finland demonstrated that infants hospitalized with HRV‐induced wheezing were found to have a particularly high risk for subsequent asthma, and this relationship persisted at least through adolescence (69, 70). This finding is further supported by the results of two birth cohort studies. The Childhood Origins of Asthma (COAST) study is a high‐risk birth cohort study in which families with at least one parent with allergies or asthma were enrolled prenatally, and both immune development and respiratory illnesses were prospectively evaluated (53). Through the use of PCR‐based diagnostics, viral etiologies were identified in 90% of wheezing illnesses. Notably, moderate to severe HRV infections (with and without wheezing) during infancy were a significant risk factor (odds ratio = 10) for persistent wheezing at age 3 years (71). Moreover, HRV wheezing illnesses in the first 3 years of life were significantly associated with the development of asthma at age 6 years (72). The combination of allergic sensitization and HRV‐induced wheezing by age 3 years was associated with the highest risk of developing asthma. Similarly, in an Australian birth cohort study, Kusel and colleagues (73) enrolled 198 babies and compared viral respiratory illnesses in the first year of life to subsequent respiratory outcomes. Wheezing illnesses with either HRV or RSV in infancy were associated with asthma at age 5 years. Interestingly, these associations were only significant in the children with early onset (by age 2 years) allergic sensitization. Collectively, these results highlight the role of virus‐induced wheezing in infancy, and HRV in particular, in determining the risk for subsequent asthma.
Exacerbations of asthma
For decades, clinicians had suspected that respiratory infections were a major cause of asthma exacerbations, which are characterized by acute onset of obstruction of the small airways, leading to clinical manifestations such as wheezing and shortness of breath. Beginning in the mid‐1990s, studies utilizing polymerase chain reaction (PCR)‐based viral diagnostics found that viral respiratory infections were detected in up to 85% of exacerbations of asthma in children and about half of exacerbations in adults (74, 75). Furthermore, approximately two‐thirds of the infections associated with asthma exacerbations were caused by HRV. Similar findings have been reported for children and adults hospitalized for acute asthma (76). Exacerbations of asthma are seasonal, and in temperate climates there are strong peaks in asthma morbidity in September, shortly after children return to school, and also in the spring (77). These spring and fall peaks in hospitalizations correspond closely to patterns of HRV isolation within the community (78), suggesting a causal relationship. In contrast, influenza and RSV infections are more likely to be associated with acute asthma symptoms in the wintertime.
It is interesting that individuals with asthma do not necessarily have more colds, and neither the severity nor duration of virus‐induced upper respiratory symptoms are enhanced during experimental HRV infections in adults with asthma (79, 80). In contrast to findings in the upper airway, a prospective study of colds in couples consisting of one asthmatic and one normal individual demonstrated that colds cause greater duration and severity of lower respiratory symptoms in subjects with asthma (79). These findings suggest that asthma‐related differences in the expression of respiratory viral infections are specific to the lower airway.
These studies provide evidence of a strong relationship between viral infections, particularly those due to HRV, and acute exacerbations of asthma. In most instances, exacerbations are caused by multiple factors. Accordingly, HRV inoculation of volunteers with stable asthma does not usually provoke acute asthma symptoms (80, 81). Other factors that commonly contribute to exacerbations of asthma are allergy, allergen exposure, indoor pollutants (e.g. tobacco smoke, NO2), outdoor pollutants (e.g. particulates, ozone), stress, and other infections (e.g. sinusitis). In addition, lack of use of an asthma control medication for persistent asthma is associated with increased risk of exacerbation. Some of these factors are also associated with increased severity of colds in the absence of asthma ( Table 1 ), suggesting the possibility that they may enhance viral replication or amplify the effects of virus‐induced inflammation.
Allergy appears to be the strongest risk factor for developing virus‐induced exacerbations of asthma. For example, in cross‐sectional studies performed at a tertiary pediatric emergency department, allergy, and viral infection (most commonly HRV) were both identified as risk factors for wheezing. Children who had HRV infection detected in combination with either eosinophilia or a positive radioallergosorbent test (RAST) to common aeroallergens had the strongest odds of wheezing (82, 83). In addition, a recent study performed routine sampling of nasal secretions in children with asthma during periods of peak HRV prevalence, and approximately two‐thirds of these children also were sensitized to respiratory allergens (23). Allergic sensitization was associated with similar rates of viral detection in nasal secretions, but the children with allergic asthma had illnesses of greater duration and severity, and were more likely to experience loss of asthma control (moderate asthma exacerbations).
Other studies have tested whether allergen exposure increases the risk of wheezing in children and adults with allergic asthma. In a case‐control study of 84 asthmatics admitted for acute exacerbations versus stable asthmatics and patients hospitalized for non‐respiratory diagnoses, a significantly higher proportion of children hospitalized for an acute exacerbation were infected with a respiratory virus (44%), compared with children with stable asthma (18%) and children with non‐respiratory hospital admissions (17%; both P < 0.001) (83, 84). The combination of allergic sensitization and allergen exposure was also found more frequently in asthma hospitalization (76%) compared to the two control groups (48% and 28% respectively, P < 0.001). In the multivariate analysis, the combination of these risk factors dramatically increased the risk of hospital admission (OR 19.4, P < 0.001), demonstrating a synergistic relationship. Similar findings were reported in a case‐control study of adults; the combination of sensitization, high exposure to one or more indoor allergens, and viral detection considerably increased the risk of being admitted to the hospital with asthma (odds ratio 8.4, 95% confidence interval 2.1 to 32.8; P = 0.002) (85). Finally, in the Inner City Asthma Study (ICAS), children who were sensitized and highly exposed to cockroach allergen were more likely to be admitted to hospital, have more unscheduled medical visits, and more time off school than either non‐exposed or non‐sensitized children (86). Virology was not performed in ICAS, but it can be presumed that viral infections were important contributors to asthma morbidity in this population.
There is considerable interest in determining whether respiratory viruses and pathogenic bacteria work together to destabilize chronic lung conditions. In addition to provoking acute exacerbations of asthma, HRV infections can also increase lower airway obstruction in individuals with other chronic airway diseases such as chronic obstructive lung disease (COPD) and cystic fibrosis (87). Whether allergy also plays a role in exacerbations of these disorders has not been determined. Traditionally, it has been observed that viral URI or LRI commonly precede bacterial infections, and mechanisms for viral infections to promote secondary bacterial infections have been described. More recently, the converse mechanism has received a lot of attention: can bacterial infections (or colonization) increase susceptibility to viral respiratory infection and illness? Interestingly, individuals with COPD require very small amounts of virus to induce moderate colds after experimental inoculation (88). Moreover, new genomic techniques have disproved the longstanding assumption that the lower airway is sterile, and two recent studies indicate that there are unique features of lower airway flora in asthma (89, 90). Finally, colonization of the upper airway with bacterial pathogens in the first 3 months of life has been related to virus‐induced wheeze and increased risk for subsequent asthma (91). Collectively, these findings suggest that viruses and bacteria may interact in determining the risk for onset of asthma, and in the causation of acute exacerbations in children and adults with established disease.
HRV‐induced inflammatory mechanisms
Cellular inflammation and cell–cell interactions
Airway epithelia cover the entire surface of the respiratory tract in contact with air, and function as a barrier to potential pathogens and foreign particles (92). The epithelial cell is of primary importance during viral respiratory infections, because it serves as the host cell for viral replication and also initiates innate immune responses. It has been demonstrated that other cells may become infected during viral infections. For example, airway smooth muscle cells can be infected by virus in tissue culture, suggesting a potential mechanism for viral infections to cause bronchospasm and airway hyperresponsiveness (93, 94), although this has not yet been verified in vivo. HRV also replicates readily in fibroblasts in vitro (95), suggesting that infections could involve fibroblasts if the airway epithelium is compromised.
Earlier work on cytokine release following HRV infection focused on epithelial cells; these cells are the principal site of virus replication, and internalization/replication of viral RNA appears necessary for optimal epithelial cell mediator release (96, 97, 98). However, during HRV infection, only a small proportion of epithelial cells become infected (33, 34, 45, 99). As HRV‐induced cytokine levels in vivo are markedly elevated, this finding suggests that other cell types and/or mechanisms (besides direct virus‐induced cytokine release by epithelial cells) are critical for airway responses to HRV. Epithelial cells and fibroblasts are in close contact at the basement membrane (95), and macrophages and dendritic cells (DCs) contact epithelial cells in the lumen and within the epithelial tissue, respectively. Moreover, activation of monocytic cells represents an early step in the natural immunity towards a viral infection (100), and close contact of monocytic cells with both upper and lower airway epithelium suggests that these cells influence immune responses to HRV in vivo, and could be relevant to asthma exacerbations.
Findings of experimental and natural cold studies
A variety of experimental models, including experimental cold studies, ex vivo HRV infection studies, and animal model systems, have been used to define the cells and cytokines that are involved in regulating the inflammatory response to HRV infections. Nasal lavage samples from patients experiencing the first signs of a natural cold (2–7 days postsymptom) revealed an increase in bradykinin and lysylbradykinin, which are potent vasoactive peptides, and whose levels correlate with vascular permeability (101). Furthermore, increased levels of the proinflammatory cytokine interleukin‐1β (IL‐1β) in nasal secretions of volunteers during experimentally induced colds correlate with the concentrations of kinins and the severity of the cold (102). Levandowski and colleagues (103) found a direct correlation between nasopharyngeal symptoms and the percentage of lymphocytes and phagocytes (e.g. monocytes) in nasal secretions in induced colds. Other responses that have been consistently observed in nasal secretions include increased levels of chemokines such as IL‐8 (CXCL8) and IFN gamma‐induced protein 10 kDa (IP‐10, CXCL10) (97, 104). Generally, concentrations of cytokines and mediators correlate reasonably well with both viral replication and symptom scores during the acute cold (101, 102, 103, 105).
Experimental infection has also been used to gain additional insights about differences in HRV‐induced responses in the upper and lower airways of volunteers with allergies and/or stable asthma. One other key question is whether HRV provokes asthma by infecting the lower airway, or if there are other mechanisms that link infection of the upper airway with potentiation of systemic or lung inflammatory responses. In tissue culture, HRV appear to replicate at least as well or perhaps even better in epithelial cells derived from either upper versus lower airway (106, 107). In addition, HRV has been detected in lower airway cells and secretions by RT‐PCR, in situ hybridization, and immunostaining of mucosal biopsies after experimental inoculation (45, 46, 47). Following experimental inoculation, about one‐third of adults have levels of virus detected in sputum that are equal to or exceed virus detected in nasal secretions, whether or not asthma is present. Finally, experimentally‐induced HRV infections can produce lower airway inflammation, including increased neutrophils in bronchial lavage fluid (108), influx of T cells and eosinophils into lower airway epithelium (109), and enhanced epithelial expression of ICAM‐1 (110). These findings provide strong evidence that HRV can replicate in the large lower airways, and suggest that virus‐induced cellular inflammation, mucus, and tissue edema could directly induce airway obstruction and closure.
Mouse models of HRV infection
Only recently has there been the generation of mouse models to study the systemic effects of HRV infection. HRVs differ in their cellular targets and receptor binding, and while minor group HRVs can bind to murine low density lipoprotein receptors (LDLRs) (111), major group HRV serotypes bind to human but not rodent ICAM‐1 (112). Thus, the development of a transgenic mouse possessing a chimeric mouse–human ICAM‐1 was necessary for the study of major group HRV serotypes in mice (30). In agreement with human studies, major group HRV infection of human ICAM‐1 transgenic mice leads to the increased detection of neutrophils and lymphocytes in the lungs (30), and this same response is also observed in the lungs of mice after minor group HRV administration (30, 31). In addition, inoculation of mice with minor group HRV induces the expression/release of mucins (Muc5AC and Muc5B), IFNs (IFN‐α family members and IFN‐β), and a similar range of cytokines and chemokines (30, 31). One limitation of the mouse models is that viral replication occurs for a relatively brief period of time (<24 h as compared to days in humans).
The mouse model has been used to explore mechanisms of virus effects on allergic airway inflammation. After allergic sensitization, HRV infection increases the infiltration of numerous leukocytes (including neutrophils, eosinophils, and macrophages) and increases airway cholinergic hyperresponsiveness (113). When macrophages are depleted from allergen‐challenged mice, HRV infection results in diminished airway responsiveness and reduced eosinophil recruitment to the lungs, supporting a role for macrophages in the cellular inflammation associated with HRV infection (113). HRV treated macrophages from allergen‐challenged mice exhibit increased expression of alternatively activated macrophage markers (i.e. arginase‐1, Ym‐1, Mgl‐2 and IL10), suggesting that this subset of macrophages contributes to enhanced inflammatory responses to virus in the context of allergy (113).
Viral recognition pathways
HRV receptors
Rhinovirus infection occurs mainly through receptor‐mediated endocytosis. The majority of HRV‐A and all the HRV‐B strains bind to ICAM‐1 (CD54) and are referred to as major group HRV (11, 112). Approximately 10% of the HRV‐A strains do not bind to ICAM‐1 but instead bind to members of the LDLR family, specifically LDLR, very LDLR (VLDLR) and/or α2‐macroglobulin receptor/LDLR‐related protein (LRP), and are referred to as minor group rhinoviruses (114, 115). Many labs have utilized have utilized soluble ICAM‐1 or ICAM‐1 antibodies to prevent HRV from binding to cells and initiating cellular signaling. In addition, viral RNA can be damaged by treatment with neutral red dye; this treatment preserves viral binding but severely inhibits viral replication (28). Studies using these controls support a role for ICAM‐1 binding in downstream major group HRV signaling (28, 116). In contrast to HRV signaling events that are dependent on replication (e.g. activation of TLR3), binding of HRV to surface receptors on various cells types can initiate signaling events that lead to the production of cytokines or chemokines (28, 116). Interestingly, the cell surface receptor(s) that recognize HRV‐C strains have yet to be identified, and appear different from those used by HRV‐A and –B (11, 12).
Innate immune receptors
The innate immune response to HRV is likely to be a major determinant of both infectivity and severity of illness. In addition to the importance of ICAM‐1 and LDLR receptors in HRV signaling, there is also evidence for a role of membrane bound and cytoplasmic RNA sensing molecules. TLR3 and TLR7 are localized to endosomal and plasma membranes and bind double‐stranded and single‐stranded RNA respectively. Infection of BEAS‐2B epithelial cells with HRV‐16, a major group HRV‐A serotype, has been shown to increase the expression of TLR3 (117). It has also been reported that HRV‐induced IL‐8 promoter activity is attenuated when TLR3 levels are knocked down in 16HBE14o epithelial cells, and transfection of HEK 293 with TLR3 leads to increased HRV‐induced IL‐8 production (118). These studies support a role for TLR3 in HRV signaling, although many of the studies have modeled signaling events induced by HRV with synthetic dsRNA ligand (PolyI:C), or have utilized epithelial cells lines instead of primary cultures of airway cells (26, 98, 118, 119). Notably, signaling events initiated by PolyI:C and HRV can have significant differences (117).
Several intracellular molecules also serve to detect HRV, including dsRNA/protein kinase receptor (PKR) and the cytoplasmic RNA helicases retinoic acid inducible gene protein‐1 (RIG‐1) and melanoma differentiation‐associated gene (MDA‐5). For example, studies by Chen and colleagues (98) found that PKR activation is essential for HRV‐mediated IFN release from primary epithelial cells. This same group provided evidence that the TLR adapter protein, TIR‐domain‐containing adapter‐inducing IFN‐β (TRIF), and MDA‐5 also contribute to HRV‐mediated cytokine release (26). A recent study found that TLR3 is constitutively expressed on airway epithelial cells, and activation of TLR3 leads to induction of RIG‐1 and MDA‐5. All three of these molecules (TLR3, RIG‐1 and MDA‐5) were found to cooperate in the upregulation of innate IFN responses to HRV infection (120). Other investigators have reported that RIG‐1 is less important than other molecules in for detection of picornavirus RNA (26).
Rhinovirus‐induced cell signaling
HRV signaling initiated by viral replication
Many HRV‐induced events are not detected with cells that have been challenged with virus treated with UV radiation, and thus appear to require viral replication. These responses include the expression and/or release of chemokines (26, 96, 97, 121, 122), cytokines (121, 122), IFNs (26, 116, 123), antiviral pathways (26, 27, 98, 117), ICAM‐1 (124), mucin (119, 125), defensins (126, 127), and matrix metalloprotease‐9 (MMP‐9) (57). These findings suggest that active replication in an important component of HRV‐mediated signaling; however, UV inactivation can disrupt the coat proteins of the virus and affect binding of virus to host cells (authors’ unpublished data). In addition to cell activation initiated by viral RNA, expression of HRV 3C protease, a cysteine protease expressed only in actively infected cells, has also been linked to cell activation and the expression of several cytokines, including IL‐8 and GM‐CSF in response to infection (128).
HRV‐induced signaling mechanisms in epithelial cells
The binding of HRV to surface receptors on various cells types can induce specific signaling events leading to cell activation, with the majority of studies performed in epithelial cells (see Fig. 2A) (28, 129, 130). For example, HRV‐16 binding to ICAM‐1 on epithelial cells induces the activation of the tyrosine kinases Src and Syk and the subsequent activation of the phosphatidylinositol 3‐kinase (PI3K)/Akt signaling cascade (29, 131, 132). The activation of PI3K/Akt by major group HRV infection has been linked to NF‐κB activation and subsequent CXCL8 release from epithelial cells (131). In further support of a role for the PI3K/Akt pathway, mice inoculated with minor group HRV had increased Akt phosphorylation in the lung, which was abrogated by pretreatment with a PI3K inhibitor (31). Furthermore, HRV‐induced Syk activation is critical in replication‐independent p38 mitogen‐activated protein kinase (MAPK) activation and subsequent IL‐8 expression (121, 133). HRV activation of p38 activation also contributes to production of IL‐6, G‐CSF, GM‐CSF, CXCL1 (GROα), CXCL5 (ENA‐78) and vascular endothelial growth factor (VEGF) (55, 121). The expression of the angiogenic factor VEGF after HRV infection is also regulated by extracellular signal‐regulated kinases‐1/2 (ERK1/2) MAPK pathways, which are critical for the induction of MUC5AC and CXCL8 expression (55, 134). Conversely, ERK1/2 kinase inhibition enhances HRV‐dependent IRF1, IRF3, CXCL10 and CCL5 expression (125, 136).
Figure 2.
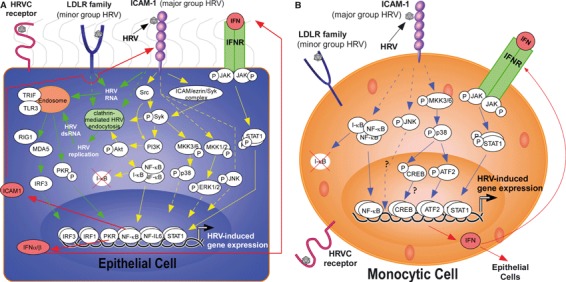
HRV‐induced signaling pathways in epithelial and monocytic cells. Major group HRV binds to ICAM‐1 and minor group HRV binds to LDLR receptors on the surface of cells, such as epithelial cells and leukocytes that would be abundant in the airway, to induce downstream signaling. The receptor and signaling pathways induced by HRVC serotypes has yet to be elucidated. (A) Upon ligation of HRV to surface receptors on epithelial cells, direct uncoating of viral RNA or clathrin‐mediated endocytosis leads to the release/replication of HRV RNA into the cytoplasm. This viral RNA is detected by endosomal receptors, such as TLR3, to propagate downstream signaling and induction of gene expression, including the production of interferons that can exert an autocrine/paracrine effect by binding to interferon receptors and triggering a JAK/STAT signaling cascade. Replication‐independent signaling is also induced upon HRV infection, and includes the activation of Src, Syk, and MAPK signaling cascades. Yellow solid lines: data to support link; Dashed lines: indirectly connected (may have signaling molecules in between); Red lines: signaling induced by product of HRV‐induced gene transcription; Green lines: replication‐dependent signaling. (B) Unlike epithelial cells, HRV‐signaling in monocytic cells is thought to be replication independent, as viral replication in these cells is inefficient. In monocytic cells, such as alveolar macrophages and peripheral blood monocytes, HRV induces the activation of both NF‐κB and the stress‐activated protein kinases (SAPK) cascades. Transcriptional activation after HRV binding leads to the induction of gene expression, including IFNs that bind to IFN receptors on the surface of both monocytic and epithelial cells, to induce downstream JAK/STAT signaling and further gene expression. Blue solid lines: data to support link; Dashed lines: indirectly connected (may have signaling molecules in between). It has yet to be determined whether HRV‐induced JNK and CREB activation are essential for the expression of cytokines.
Numerous transcription factors have been linked to HRV‐mediated events in epithelial cells, including the activation of members of the nuclear factor κB (NF‐κB), signal transducer and activator of transcription (STAT), and IFN regulatory factor (IRF) families.
The transcription factor NF‐κB has been shown by numerous groups to be upstream of epithelial cell mediator release (55, 57). Activation of NF‐κB requires its translocation into the nucleus after liberation from IκB. Infection of epithelial cells with major group HRV increases NF‐κB binding activity and has been implicated in HRV‐mediated IL‐6, ICAM, CXCL8, CXCL10 and MMP‐9 expression (55, 57, 124, 128, 137, 138, 139, 140, 141). Activation of NF‐κB requires its translocation into the nucleus after liberation from IκB. Hewson and colleagues (125) found that only cells infected with HRV had NF‐κB (p65) in their nuclei. PKR has been reported to associate with NF‐κB in the nucleus and regulate HRV‐mediated CCL5, CXCL8 and IL‐6 expression (139). The induction of MUC5AC by HRV was also found to be indirectly dependent on NF‐κB, as it is required for the induction of MMPs (125). The HRV‐mediated activation of NF‐κB in epithelial cells has been postulated to be the result of rapid superoxide production and the activation of the PI3K/Akt and PKR signaling cascades (122, 130, 142).
The Janus kinase (JAK)/STAT signaling cascade has also been implicated in HRV‐induced signaling. Chen and colleagues (98) report that HRV‐16 is able to induce STAT1 phosphorylation, and preincubation with a JAK1 inhibitor attenuated HRV‐induced gene expression from human tracheobronchial epithelial cells. The expression of STAT1 is thought to be dependent on HRV replication and the subsequent release of interferons (98).
A role for the IRF transcription factor family in HRV‐mediated signaling events in epithelial cells has been supported. The induction of CCL5 release by major group HRV is downstream of IRF1, whereas the expression of IRF3 in minor group HRV‐induced gene expression has been implicated in the induction of IFN‐β IFN‐γ, IRF7, RIG1, MDA5, and CXCL10 expression, but not IL‐8 or GM‐CSF (26, 136). Furthermore, HRV induced CXCL10 release has been shown to be dependent on both IRF1 and IRF3 (136).
Infection of epithelial cells with HRV also induces the release of reactive oxygen species, which is proposed to be upstream of the release of a subset of HRV‐induced mediators. Rhinovirus infection leads to the proteolytic activation of xanthine oxidase, depletes intracellular reduced glutathione, and induces a rapid increase of intracellular superoxide anion, which has been proposed to be upstream of NF‐κB activation and ICAM‐1 expression (142, 143). Conversely, another free radical, nitric oxide (NO), has been implicated in the suppression of HRV‐induced events in epithelial cells, such as NF‐κB activation and IRF1, IL‐6, CXCL8 and CXCL10 expression (138, 141, 144).
Autocrine/paracrine signaling after HRV exposure has also been attributed to mediator release. Neutralization of IFN‐β was shown to significantly block the induction of numerous genes that were upregulated by major group HRV, which supports a role for HRV‐induced IFN‐β release in subsequent gene transcription (98). Furthermore, mucin expression in response to HRV infection is proposed to be the result of epidermal growth factor receptor (EGFR) activation by an increase in EGFR ligands (1).
HRV‐induced signaling mechanisms in monocytic cells
In contrast to the numerous HRV signaling studies in epithelial cells, much less is known about the signaling mechanisms that are induced upon HRV binding to immune cells. The signaling mechanisms that have been elucidated in leukocytes to date have been determined in monocytic cells. Considering that little‐to‐no replication of HRV has been observed in airway macrophages or blood monocytes (129, 145), the signaling pathways are likely to vary considerably from epithelial cell signaling, which is thought to rely heavily on viral replication. Compared to the elaborate signaling mechanisms that have been elucidated in epithelial cells, signaling in monocytic cells has not been extensively explored, and have been exclusively performed with major group HRV (see Fig. 2B). We have observed that CCL2 protein release from primary monocytic cells is linked to the activation of the transcription factor NF‐κB and phosphorylation of the transcription ATF‐2 (146). NF‐κB activation, but not the p38 MAPK, has been reported to be involved in HRV‐mediated TNF‐α release by monocytic cells (145). Furthermore, HRV16 induces a significant increase in IFN‐α release and STAT1 tyrosine phosphorylation in human monocytes, and neutralization of the type I IFN receptor or inhibition of JAK or p38 MAPK activity strongly attenuates HRV16‐stimulated STAT1 phosphorylation and CXCL10 release (28). We have also observed the phosphorylation of JNK and the transcription factor cyclicAMP response binding protein (CREB) after HRV‐16 treatment of monocytic cells, but the consequence of these phosphorylation events have yet to be elucidated (unpublished data).
HRV effects on cytokines and mediators
The epithelial cell serves as the host cell for viral replication, and initiates numerous innate immune responses ( Table 4 ). Epithelial cells generally release IFNs rapidly in response to viral infections as a first line of defense (147). Indeed, infection of epithelial cells with HRV induces the expression and release of multiple IFNs to combat infection (26, 98, 123, 148). In addition to the release of IFNs, epithelial cells produce a large array of cytokines, chemokines, and other inflammatory mediators to induce the influx of leukocytes and cause respiratory inflammation. To facilitate HRV‐mediated signaling, the expression of certain signaling molecules and transcription factors from epithelial cells is induced in response to HRV. Additionally, exposure of epithelial cells to HRV also induces the release of growth factors, which can serve to prolong the life of infiltrating leukocytes, and may contribute to airway remodeling seen in the lungs of asthmatics. It is worth noting that more comprehensive HRV‐induced gene expression profiles are provided in a few studies (27, 98, 149).
Table 4.
Epithelial cell responses to HRV infection*
| HRV serotype | mRNA/gene expression | Protein expression/release | Cell type/source | Reference | |
|---|---|---|---|---|---|
| Interferons | |||||
| IFN‐α2 | 1b, 16 | X | BEC | (123) | |
| IFN‐β | 1b, 39 | X | BEAS‐2B, BEC | (26) | |
| 1b, 16 | X | X (HRV‐16) | BEC | (98) | |
| 16 | X | X | BEC (brushing) | (148) | |
| 1b, 16 | X | X (BEAS‐2B) | BEAS‐2B, BEC | (123) | |
| IFN‐β | 1b | X (gene exp) | BEC | (120) | |
| IFN‐λ1 | 1b, 39 | X | BEAS‐2B, BEC | (26) | |
| 1b, 16 | X | X (BEAS‐2B) | BEAS‐2B, BEC | (123) | |
| IFN‐λ2/3 | 1b, 39 | X | BEAS‐2B, BEC | (26) | |
| 1b, 16 | X | BEC | (123) | ||
| IFN‐λ2/3 | 1b | X (gene exp) | BEC | (120) | |
| Chemokines | |||||
| CCL2 (MCP‐1) | 16 | X | Nasal epithelial scraping | (27) | |
| CCL5 (RANTES) | 16, 49 | X | X | BEC (lung transplant) | (96) |
| 16, 49 | X | X | BEC, A549 | (106) | |
| 16 | X | TEC | (99) | ||
| 16 | X | BEC (brushings) | (155) | ||
| 1b, 16 | X | BEC | (123) | ||
| CCL5 | 1b, 9, 16 | X (Prtn Exp) | BEAS‐2B | (122) | |
| CCL5 | 16 | X (gene exp) | X (Prtn Exp) | BEC | (134) |
| CCL5 | 16 | X (gene exp) | X (Prtn Exp) | BEC | (135) |
| CCL5 | 1b | X (gene exp) | BEC | (120) | |
| CCL8 (MCP‐2) | 16 | X | Nasal epithelial scraping | (27) | |
| CCL20 (LARC) | 16 | X | Nasal epi scraping | (27) | |
| 1a | X | BEC (brushing) | (149) | ||
| CXCL1 (GROα) | 39 | X | X | BEAS‐2B | (121) |
| CXCL2 (MIP‐2α) | 1a | X | BEC (brushing) | (149) | |
| CXCL3 (MIP‐2β) | 1a | X | BEC (brushing) | (149) | |
| ENA‐78 (CXCL5) | 39 | X | 16HBE14o‐, TEC | (140) | |
| 16 | X (BEC) | X | BEC (brushing), BEAS‐2B | (193) | |
| 39 | X | BEAS‐2B | (121) | ||
| CXCL5 | 1b | X (gene exp) | BEC | (120) | |
| CXCL9 (IP‐9) | 16 | X | Nasal epithelial scraping | (27) | |
| CXCL10 (IP‐10) | 16 | X | Nasal epithelial scraping | (27) | |
| 16 | X | X | BEC, BEAS‐2B | (97) | |
| 1b, 39 | X | BEAS‐2B, BEC | (26) | ||
| 16 | X | X | BEC, BEAS‐2B | (194) | |
| 16 | X | BEC (brushings) | (155) | ||
| 1b, 16 | X | BEC | (98) | ||
| CXCL10 (IP10) | 16 | X(gene exp) | X(Prtn exp) | BEC | (135) |
| CXCL10 (IP10) | 1b | X(gene exp) | BEC | (120) | |
| CXCL11 (I‐TAC) | 16 | X | Nasal epithelial scraping | (27) | |
| 1b, 16 | X | BEC | (98) | ||
| CXCL13 (BCA‐1) | 16 | X | Nasal epithelial scraping | (27) | |
| Cytokines | |||||
| IL‐1β | 2, 14 | X | TEC | (195) | |
| 16 | X | A549 | (127) | ||
| IL‐1Ra | 16 | X | A549 | (127) | |
| IL‐6 | 14 | X | BEAS‐2B | (196) | |
| 2, 14 | X | TEC | (195) | ||
| 16 | X | TEC | (99) | ||
| 16 | X | BEC (brushings) | (155) | ||
| 39 | X | BEAS‐2B | (121) | ||
| IL‐6 | 1b, 9, 16 | X(Prtn exp) | BEAS‐2B | (122) | |
| IL‐6 | 1a, 14, 16, 39 | X(Gene exp ‐ HRV‐14‐16) | X(Prtn exp) | BEAS‐2B | (144) |
| IL‐8 | 1b, 39 | X | BEAS‐2B, BEC | (26) | |
| 2, 14 | X | TEC | (196) | ||
| 16 | X (B2B) | BEAS‐2B | (55) | ||
| 16 | X | Nasal lavage | (197) | ||
| 14 | X | BEAS‐2B | (196) | ||
| 39 | X | 16HBE14o‐ | (132) | ||
| 39 | X | 16HBE14o‐, TEC | (140) | ||
| 16 | X (BEC) | X | BEC (brushing), BEAS‐2B | (193) | |
| 39 | X | X | BEAS‐2B | (121) | |
| 16 | X | TEC | (99) | ||
| 16 | X | BEC (brushings) | (155) | ||
| 1a | X | BEC (brushing) | (149) | ||
| IL‐8 (CXCL8) | 16 | X(gene exp) | X(Prtn exp) | BEC | (134) |
| IL‐8 (CXCL8) | 1b, 9, 16 | X(Prtn exp) | BEAS‐2B | (122) | |
| IL‐8 (CXCL8) | 1a, 14, 16, 39 | X(gene exp ‐ HRV‐14‐16) | X(Prtn exp) | BEAS‐2B | (144) |
| IL‐8 (CXCL8) | 1b | X(gene exp) | BEC | (120) | |
| IL1F9 | 1a | X | BEC (brushing) | (149) | |
| ISG15 | 1b, 16 | X | BEC | (98) | |
| TNF‐α | 2, 14 | X | TEC | (195) | |
| 16 | X | TEC | (99) | ||
| 16 | X | BEC (brushings) | (155) | ||
| Signaling and transcription factors | |||||
| STAT1 | 1b, 16 | X | BEC | (98) | |
| IRF1 | 16 | X(gene exp) | X(Prtn exp) | BEAS‐2B | (141) |
| IRF1 | 16 | X(gene exp) | X(Prtn exp) | BEAS‐2B, BEC | (136) |
| IRF7 | 1b, 39 | X | BEAS‐2B, BEC | (26) | |
| 1b, 16 | X | BEC | (98) | ||
| RIG1 | 1b, 39 | X | BEAS‐2B, BEC | (26) | |
| 1b, 16 | X | BEC | (98) | ||
| 16 | X | Nasal epithelial scraping | (27) | ||
| RIG1 | 1b | X(gene exp) | X(Prtn exp) | BEC | (120) |
| MDA5 | 1b, 39 | X | BEAS‐2B, BEC | (26) | |
| 16 | X | Nasal epithelial scraping | (27) | ||
| 1b, 16 | X | BEC | (98) | ||
| MDA5 | 1b | X(gene exp) | X(Prtn exp) | BEC | (120) |
| SOCS3 | 1a | X | BEC (brushing) | (149) | |
| 16 | X | Nasal epithelial scraping | (27) | ||
| PKR | 16 | X | TEC | (99) | |
| 1b, 16 | X | BEC | (98) | ||
| Potential antivirals | |||||
| Vipirin | 16 | X | X | Nasal epithelial scraping | (27) |
| 1b, 16 | BEC | (98) | |||
| MX1 | 1b, 16 | BEC | (98) | ||
| 16 | Nasal epithelial scraping | (27) | |||
| OAS1 | 1b, 16 | X | BEC | (98) | |
| 16 | X | Nasal epithelial scraping | (27) | ||
| Reactive oxygen species | |||||
| NOS‐2 | 16 | X | X | BEC | (197) |
| Superoxide anion | 16 | X | A549, BEC (brushing) | (143) | |
| Anti‐microbial | |||||
| β‐defensin 2 | 1a, 2, 14, 16 | X | X | A549 | (127) |
| β‐defensin 2 | 16 | X(gene exp) | X(Prtn exp) | BEC | (134) |
| Proteases | |||||
| MMP‐9 | 1a, 16 | X | X | BEC (lung tissue), B2B | (57) |
| Growth factors | |||||
| Activin A | 16 | X | BEC, BEAS‐2B | (55) | |
| Amphiregulin | 16 | X | BEC, BEAS‐2B | (55) | |
| CSF2 | 1a | X | BEC (brushing) | (149) | |
| CSF3 | 1a | X | BEC (brushing) | (149) | |
| G‐CSF | 39 | X | X | BEAS‐2B | (121) |
| GMCSF | 1b, 39 | X | BEAS‐2B, BEC | (26) | |
| 16 | X (B2B) | X | BEC, BEAS‐2B | (198) | |
| 14 | X | BEAS‐2B | (196) | ||
| 39 | X | X | BEAS‐2B | (121) | |
| GM‐CSF | 16 | X (gene exp) | X(Prtn exp) | BEAS‐2B | (138) |
| VEGF | 1b, 9, 16 | X (B2B) | X | BEC, BEAS‐2B | (158) |
| 16 | X (B2B) | X | BEC, BEAS‐2B | (55) | |
| Inhibitors of apoptosis | |||||
| TNFAIP3 | 1a | X | BEC (brushing) | (149) | |
| Mucins | |||||
| MUC5AC | 1b, 9, 16 | X(gene exp) | X(Prtn exp) | BEC, NCI‐H292 | (125) |
*For microarray studies, not all of the data from these studies was included in this list.
BEC, primary bronchial epithelial cells (from transplant donors, unless noted as brushings); TEC, primary tracheal epithelial cells; VEGF, vascular endothelial growth factor; MMP, matrix metalloprotease.
Monocytes, macrophages, T lymphocytes, and presumably DCs secrete proinflammatory cytokines such as IL‐1, IL‐8, TNF‐α, IL‐10, IFN‐α, and IFN‐γ in response to HRV infection ( Table 5 ); these cytokines activate other cells in the environment and are potent inducers of adhesion molecules. Furthermore, exposure of leukocytes to HRV also modulates the expression of surface adhesion receptors, which alters leukocyte migration (150, 151, 152, 153). Concentrations of CCL2, a chemotactic factor for monocytes, T lymphocytes, and basophils are markedly elevated in BALF from asthmatic individuals compared to BALF from control individuals (154). Moreover, levels of the cytokine CXCL10, a chemotactic factor for CXCR3+ cells such as NK cells and T lymphocytes, have been associated with HRV‐induced pathology in asthmatics (97, 155). Interestingly, we have observed that monocytic cells (i.e. peripheral blood monocytes and BALF macrophages) are able to robustly release CCL2 and CXCL10 upon HRV‐16 treatment (28, 146), supporting the notion that these phagocytes are a source of these chemokines during a natural cold. In an atopic asthma model, experimental HRV inoculation of BALF macrophages induces the expression of mediators (i.e. Arginase‐1, Ym‐1, Mgl2) that are indicative of a less inflammatory, more fibrotic, macrophage population, supporting a more Th‐2 dominant environment (113).
Table 5.
Leukocyte responses to HRV infection
| HRV serotype | mRNA/gene expression | Protein expression/release | Cell type/source | Reference | |
|---|---|---|---|---|---|
| Interferons | |||||
| IFN‐α | 16 | X | X | PBMCs | (123) |
| 16 | X | THP‐1 mac | (145) | ||
| IFN‐β | 16 | X | THP‐1 mac | (145) | |
| 16 | X | X | PBMCs | (123) | |
| IFN‐γ | 16 | X | PBMCs | (150) | |
| 16 | X | CD3+ T cells | (150) | ||
| 16 | X | NK (CD56+) cells | (150) | ||
| 16 | X | Th (CD4+) cells | (165) | ||
| 16, 49 | X | CD4+ T cells | (156) | ||
| 16 | X | PBMCs | (116) | ||
| 1a, 15 | X | Primary tonsilar T cells | (199) | ||
| IFN‐γ | 1b | X(gene exp) | OVA‐challenged murine BAL mac | (113) | |
| IFN‐λ | 16 | X | X | PBMCs | (123) |
| Cytokines | |||||
| IL‐1β | 14 | X | Blood monocytes | (151) | |
| IL‐2 | 1a, 15 | X | Primary tonsilar T cells | (199) | |
| IL‐4 | 1b | X | OVA‐challenged murine BAL mac | (113) | |
| 16, 49 | X | Blood CD4+ T cells | (156) | ||
| 16 | X | PBMCs (asthmatics only) | (116) | ||
| IL‐5 | 16, 49 | X | Blood CD4+ T cells | (156) | |
| IL‐8 | 2, 9, 14 | X | X | PBMCs | (200) |
| IL‐10 | 14 | X | Blood monocytes | (151) | |
| 16 | X | Blood CD4+ T cells | (165) | ||
| 16 | X | PBMCs | (116) | ||
| 1b | X | OVA‐challenged murine BAL mac | (113) | ||
| IL‐12 | 16 | X | PBMCs | (116) | |
| IL‐13 | 16 | X | PBMCs | (116) | |
| 1b | X | OVA‐challenged murine BAL mac | (113) | ||
| TNF‐α | 1b, 9, 16 | X | THP‐1 mac | (145) | |
| 1b | X | Murine BAL mac | (113) | ||
| Chemokines | |||||
| CCL2 | 16 | X | X | THP‐1, blood monocytes, BAL mac | (146) |
| CCL11 (eotaxin) | 1b | X | OVA‐challenged murine BAL mac | (113) | |
| CXCL10 | 16 | X | Blood monocytes, BAL mac | (28) | |
| Growth factors | |||||
| GM‐CSF | 16, 49 | X | CD4+ T cells | (156) | |
| Alternatively activated macrophage markers | |||||
| Arginase‐1 | 1b | X | OVA‐challenged murine BAL mac | (113) | |
| Ym‐1 | 1b | X | OVA‐challenged murine BAL mac | (113) | |
| Mgl2 | 1b | X | OVA‐challenged murine BAL mac | (113) | |
| Receptors | |||||
| CD14* | 16 | X | PMBCs | (152) | |
| CTLA‐4 | 16 | X | Blood Th cells (asthmatics) | (152) | |
| CD80 | 16 | X | Blood monocytes | (152) | |
| CD86 | 16 | X | Blood B cells | (152) | |
| CD25 | 16 | X | PBMCs | (152) | |
| MHC II* | 14 | X | Blood monocytes | (151) | |
| CD69 | 16 | X | Blood CD3+, CD56+, CD19+ cells | (150) | |
| CD54* | 16 | X | Blood monocytes | (150) | |
| B7‐H1 | 14 | Blood monocyte‐derived DC | (153) | ||
*Receptors that are downregulated by HRV treatment.
PBMCs, peripheral blood mononuclear cells; mac, macrophage; NK, natural killer; DC, dendritic cells; Fb, fibroblast.
Lymphocytes are recruited into the upper and lower airway during the early stages of a viral respiratory infection, and it is presumed that innate and adaptive immune responses serve to limit the extent of infection, and to clear virus‐infected epithelial cells. This is consistent with reports of severe viral lower respiratory infections in immunocompromised patients (38). T‐cell responses to HRV are directed both at epitopes shared among multiple serotypes and strain‐specific epitopes (156). HRV infection of peripheral blood mononuclear cells has been shown to inhibit lymphocyte proliferative responses by inhibiting the production of costimulatory molecules and through the induction of IL‐10 (151, 153, 157).
Epithelial cell – mononuclear cell crosstalk
HRV replicates in only a small percentage of epithelial cells at any given time and generally causes little or no cytopathology in primary cells (96). Considering that HRV‐mediated events are thought to be replication‐dependent, these findings suggest that the pathology exhibited upon HRV‐induced respiratory infections may be a consequence of virus‐induced cytokine elaboration and the recruitment of inflammatory cells. The close proximity of epithelial cells and macrophages in vivo suggests that intercellular interactions between these two cell types would likely enhance antiviral responses to HRV infection. In support of this hypothesis, HRV‐mediated VEGF induction from bronchial epithelial cells is enhanced in the presence of HRV‐treated mononuclear cell supernatants (158). Furthermore, co‐culture of human monocytic and bronchial epithelial cells promoted a synergistic augmentation of CXCL10 and CCL2 protein release following HRV16 challenge (159). Transfer of conditioned media from HRV16‐treated monocytic cells to epithelial cultures induced robust CXCL10 release by the epithelial cells, which was attenuated by type IFN receptor blocking antibodies and could be recapitulated by IFNα addition. These data indicate that epithelial CXCL10 release during HRV infection is augmented by a monocytic cell‐dependent mechanism involving type I IFN(s) and provide evidence for crosstalk between immune cells and the epithelium during HRV infection.
Mechanisms of virus–allergen interactions
Several mechanisms have been proposed to explain virus–allergen interactions ( Fig. 3 ). First, underlying defects in immune regulation or epithelial barrier function could promote both viral infections and allergy/asthma (99, 160, 161). The possibility that abnormalities in antiviral activity in asthma exist has been evaluated by a number of investigators by measuring virus‐induced cytokine responses of peripheral blood mononuclear cells. For example, HRV‐induced IFN‐γ responses in allergic individuals were found to be inversely related to viral shedding (162). Interestingly, there is evidence that mononuclear cell production of IFN‐α and IFN‐γ may be impaired in allergic asthma (116), and HRV‐induced IFN‐γ responses in subjects with asthma are positively associated with measures of pulmonary function (163). Furthermore, in airway sputum cells, a more dominant Th1 response (ratio of IFN‐γ/IL‐5 mRNA) was associated with milder colds and a more rapid clearance of the virus (105).
Figure 3.
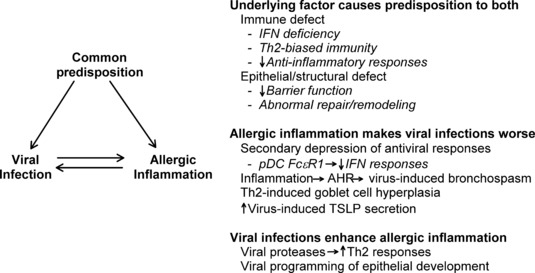
Mechanisms of virus–allergen interactions.
In addition to these differences in mononuclear cell and sputum cell IFN responses, there is evidence that epithelial cell IFN responses may also be diminished in asthma. In the first series of experiments, Wark et al. (148) used bronchoscopy to obtain bronchial epithelial cells (BEC) for culture from normal and asthmatic subjects. The use of BEC is highly relevant as these cells are the primary site for a HRV infection in the lower airway. Using this model, Wark et al. (148) found HRV‐16 replication was increased and IFN‐β responses were diminished in BEC from asthmatic patients. The addition of IFN‐β to the HRV BEC cultures from asthmatic subjects reduced virus replication. Paralleling these observations with IFN‐β, Contoli et al. (164) found decreased IFN‐λ responses in bronchoalveolar airway cells from the same asthmatic subjects. In addition, after experimental inoculation with HRV‐16, the fall in lung function, i.e. forced expiratory volume in one‐second (FEV1), was inversely proportional to IFN‐λ generation previously noted. These data suggest that airway cells in some asthma patients have defective antiviral responses as reflected by a reduced generation of IFNs to HRV‐16.
Not all investigations support this conclusion, and two recent studies were unable to confirm that epithelial cell IFN responses were deficient in asthma (107, 149). Moreover, to date studies of experimentally inoculated volunteers have found no significant differences in HRV shedding related to asthma (80, 81, 165). Many factors can contribute to these differences in observations, particularly the severity of asthma under study. Consequently, additional studies of patients with asthma and naturally acquired colds will be needed to resolve these differences.
One feature of respiratory allergies and asthma is increases in allergic effector cells, such as eosinophils and mast cells, in the airways. Interactions between HRV and cells involved in allergic responses have been studied in vitro. HRV binds to eosinophils, but there is little evidence of cell activation as measured by the release of granular proteins. However, eosinophils are able to stimulate the proliferation of HRV‐specific T‐cell clones (166), and there is evidence that HRV infections can enhance the eosinophilic response to allergen challenge in vivo (167, 168). Common cold infections are generally not associated with increased histamine in nasal secretions (169), however, HRV can infect mast cell lines and enhance allergen‐induced responses in vitro (170).
Both viral infections and allergic inflammation can damage airway epithelium, and this could lead to synergistic effects. On one hand, viral infections compromise the barrier function of the airway epithelium and could lead to enhanced absorption of allergens and/or irritants across the airway wall and increased allergic inflammation. Alternatively, HRV replication in vitro is greater in damaged epithelium (99, 161), and this implies that allergen‐induced damages to airway epithelium could promote greater viral replication and more severe clinical illness. This concept could also apply to pollutants, and explain why exposure to toxic agents (e.g. tobacco smoke, NO2) also increases the risk of viral wheeze (171, 172). Notably, there is evidence that epithelial repair mechanisms are be abnormal in asthma (59), suggesting that noxious environmental stimuli could compromise epithelial barrier function, and thus disproportionately increase susceptibility to HRV replication in asthmatics. In addition, airway epithelium in asthma has distinct features, including increased numbers of mucus‐secreting goblet cells; interestingly, there is laboratory evidence that HRV replication is enhanced in these cells (173).
Second, there is experimental evidence that allergic inflammation could promote more severe viral illnesses. Recent mechanistic studies have provided evidence of antagonistic relationships between allergic inflammation and antiviral immunity (174, 175). For example, Gill and colleagues (175) demonstrated that cross linking of IgE receptors on plasmacytoid DCs, a major contributor to antiviral IFN responses, leads to inhibition of virus‐induced IFN‐α secretion. DCs can also play a key role in allergic sensitization, by directing the development of naive T cells into Th1 versus Th2 cells; the latter secrete cytokines that lead to IgE production (i.e. IL‐4, IL‐13) and eosinophilic inflammation (e.g., IL‐5). Thymic stromal lymphopoeitin (TSLP) is an epithelial‐derived cytokine that acts on DCs to promote Th2 differentiation (176), and this cytokine can be induced by HRV infection through a TLR3‐dependent mechanism (177). Furthermore, TLR‐3‐induced secretion of TSLP is enhanced by IL‐4, suggesting that virus infection in allergic airways could intensify Th2 inflammatory responses (177). Interestingly, TSLP and IFN‐β responses in epithelial cells may be reciprocally regulated (178, 179), suggesting that high levels of TSLP in the airways could potentially impair antiviral responses. Finally, allergic inflammation and viral infections synergistically enhance mucus secretion in mice (180); excess secretion of mucus can lead to plugging and closure of the airways in asthma.
Third, viral infections could enhance allergic inflammation. HRV has two proteases, and the HRV 2A protease enhanced allergen‐induced Th2 inflammation in a mouse model of allergic inflammation (181). Animal models also suggest that viral respiratory infections in early life may establish a pattern of overproduction of key cytokines such as IL‐13, leading to suboptimal antiviral responses, increased risk for respiratory allergies, and changes in airway structure to promote asthma (182). Similarly, in clinical studies of acute exacerbations of asthma, circulating PBMC had increased expression of IFN‐response genes, DC FcεR1 expression and Th2 chemokines, suggesting that innate antiviral responses are linked to enhanced activation of allergic inflammation (183, 184).
Conclusions
Viral infections and allergic airway inflammation have synergistic effects on the risk of developing asthma, and in the pathogenesis of acute exacerbations of asthma. Understanding the interactions between innate antiviral responses and Th2 inflammation would be of great utility in identifying mechanisms of asthma onset and acute airway obstruction. Regardless of the directionality of virus‐allergen interactions, these findings strongly suggest that the optimal strategy to prevent virus‐induced wheeze would be twofold: (i) improve antiviral responses and (ii) reduce allergic sensitization or inflammation.
Acknowledgements
Supported by NIH Grants P01 AI50500 and P01HL070831. Dr. Gern is a consultant for Boehringer Ingelheim, Centocor, GlaxoSmithKline, Synairgen, 3V Biosciences (with stock options), and Eragen Biosciences (with stock options). Each of these companies is interested in antiviral medications or viral diagnostics, but there are no conflicts of interest related to commercial products.
References
- 1. Miller EK, et al. A novel group of rhinoviruses is associated with asthma hospitalizations. J Allergy Clin Immunol 2009;123:98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kistler AL, et al. Genome‐wide diversity and selective pressure in the human rhinovirus. Virol J 2007;4:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lamson D, et al. MassTag polymerase‐chain‐reaction detection of respiratory pathogens, including a new rhinovirus genotype, that caused influenza‐like illness in New York State during 2004–2005. J Infect Dis 2006;194:1398–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kistler A, et al. Pan‐viral screening of respiratory tract infections in adults with and without asthma reveals unexpected human coronavirus and human rhinovirus diversity. J Infect Dis 2007;196:817–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McErlean P, Shackelton LA, Lambert SB, Nissen MD, Sloots TP, Mackay IM. Characterisation of a newly identified human rhinovirus, HRV‐QPM, discovered in infants with bronchiolitis. J Clin Virol 2007;39:67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McErlean P, et al. Distinguishing molecular features and clinical characteristics of a putative new rhinovirus species, human rhinovirus C (HRV C). PLoS ONE 2008;3:e1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cheuk DK, Tang IW, Chan KH, Woo PC, Peiris MJ, Chiu SS. Rhinovirus infection in hospitalized children in Hong Kong: a prospective study. Pediatr Infect Dis J 2007;26:995–1000. [DOI] [PubMed] [Google Scholar]
- 8. Jin Y, et al. Prevalence and Clinical Characterization of a Newly Identified Human Rhinovirus C Species in Children with Acute Respiratory Tract Infection. J Clin Microbiol 2009;47:2895–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Renwick N, et al. A recently identified rhinovirus genotype is associated with severe respiratory‐tract infection in children in Germany. J Infect Dis 2007;196:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Simmonds P, McIntyre C, Savolainen‐Kopra C, Tapparel C, Mackay IM, Hovi T. Proposals for the classification of human rhinovirus species C into genotypically assigned types. J Gen Virol 2010;91:2409–2419. [DOI] [PubMed] [Google Scholar]
- 11. Palmenberg AC, et al. Sequencing and analyses of all known human rhinovirus genomes reveals structure and evolution. Science 2009;324:55–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bochkov YA, et al. Molecular modeling, organ culture and reverse genetics for the emerging respiratory pathogen human rhinovirus. C Nat Med 2011; Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Drahos J, Racaniello VR. Cleavage of IPS‐1 in cells infected with human rhinovirus. J Virol 2009;83:11581–11587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Barral PM, Sarkar D, Fisher PB, Racaniello VR. RIG‐I is cleaved during picornavirus infection. Virology 2009;391:171–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Amineva SP, Aminev AG, Palmenberg AC, Gern JE. Rhinovirus 3C protease precursors 3CD and 3CD’ localize to the nuclei of infected cells. J Gen Virol 2004;85:2969–2979. [DOI] [PubMed] [Google Scholar]
- 16. D’Alessio DJ, Peterson JA, Dick CR, Dick EC. Transmission of experimental rhinovirus colds in volunteer married couples. J Infect Dis 1976;133:28–36. [DOI] [PubMed] [Google Scholar]
- 17. Meschievitz CK, Schultz SB, Dick EC. A model for obtaining predictable natural transmission of rhinoviruses in human volunteers. J Infect Dis 1984;150:195–201. [DOI] [PubMed] [Google Scholar]
- 18. Peltola V, Waris M, Osterback R, Susi P, Ruuskanen O, Hyypia T. Rhinovirus transmission within families with children: incidence of symptomatic and asymptomatic infections. J Infect Dis 2008;197:382–389. [DOI] [PubMed] [Google Scholar]
- 19. Jartti T, Lee WM, Pappas T, Evans M, Lemanske RF Jr, Gern JE. Serial viral infections in infants with recurrent respiratory illnesses. Eur Respir J 2008;32:314–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lau SK, et al. Clinical features and complete genome characterization of a distinct human rhinovirus (HRV) genetic cluster, probably representing a previously undetected HRV species, HRV‐C, associated with acute respiratory illness in children. J Clin Microbiol 2007;45:3655–3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee WM, et al. A diverse group of previously unrecognized human rhinoviruses are common causes of respiratory illnesses in infants. PLoS ONE 2007;2:e966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bizzintino J, et al. Association between human rhinovirus C and severity of acute asthma in children. Eur Respir J 2011;37:1037–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Olenec JP, et al. Weekly monitoring of children with asthma for infections and illness during common cold seasons. J Allergy Clin Immunol 2010;125:1001–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bossios A, et al. Rhinovirus infection induces cytotoxicity and delays wound healing in bronchial epithelial cells. Respir Res 2005;6:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gern JE, French DA, Grindle KA, Brockman‐Schneider RA, Konno S, Busse WW. Double‐stranded RNA induces the synthesis of specific chemokines by bronchial epithelial cells. Am J Respir Cell Mol Biol 2003;28:731–737. [DOI] [PubMed] [Google Scholar]
- 26. Wang Q, et al. Role of double‐stranded RNA pattern recognition receptors in rhinovirus‐induced airway epithelial cell responses. J Immunol 2009;183:6989–6997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Proud D, et al. Gene expression profiles during in vivo human rhinovirus infection: insights into the host response. Am J Respir Crit Care Med 2008;178:962–968. [DOI] [PubMed] [Google Scholar]
- 28. Korpi‐Steiner NL, Bates ME, Lee WM, Hall DJ, Bertics PJ. Human rhinovirus induces robust IP‐10 release by monocytic cells, which is independent of viral replication but linked to type I interferon receptor ligation and STAT1 activation. J Leukoc Biol 2006;80:1364–1374. [DOI] [PubMed] [Google Scholar]
- 29. Lau C, et al. Syk associates with clathrin and mediates phosphatidylinositol 3‐kinase activation during human rhinovirus internalization. J Immunol 2008;180:870–880. [DOI] [PubMed] [Google Scholar]
- 30. Bartlett NW, et al. Mouse models of rhinovirus‐induced disease and exacerbation of allergic airway inflammation. Nat Med 2008;14:199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Newcomb DC, et al. Human rhinovirus 1B exposure induces phosphatidylinositol 3‐kinase‐dependent airway inflammation in mice. Am J Respir Crit Care Med 2008;177:1111–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rosenthal LA, Amineva SP, Szakaly RJ, Lemanske RF Jr, Gern JE, Sorkness RL. A rat model of picornavirus‐induced airway infection and inflammation. Virol J 2009;6:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Arruda E, Boyle TR, Winther B, Pevear DC, Gwaltney JM, Hayden FG. Localization of human rhinovirus replication in the upper respiratory tract by in situ hybridization. J Infect Dis 1995;171:1329–1333. [DOI] [PubMed] [Google Scholar]
- 34. Mosser AG, et al. Similar frequency of rhinovirus‐infectable cells in upper and lower airway epithelium. J Infect Dis 2002;185:734–743. [DOI] [PubMed] [Google Scholar]
- 35. Cardell LO, Agusti C, Takeyama K, Stjarne P, Nadel JA. LTB(4)‐induced nasal gland serous cell secretion mediated by neutrophil elastase. Am J Respir Crit Care Med 1999;160:411–414. [DOI] [PubMed] [Google Scholar]
- 36. Grünberg K, et al. Effect of experimental rhinovirus 16 colds on airway hyperresponsiveness to histamine and interleukin‐8 in nasal lavage in asthmatic subjects in vivo. Clin Exp Allergy 1997;27:36–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gern JE, et al. Relationships among specific viral pathogens, virus‐induced interleukin‐ 8, and respiratory symptoms in infancy. Pediatr Allergy Immunol 2002;13:386–393. [DOI] [PubMed] [Google Scholar]
- 38. Malcolm E, Arruda E, Hayden FG, Kaiser L. Clinical features of patients with acute respiratory illness and rhinovirus in their bronchoalveolar lavages. J Clin Virol 2001;21:9–16. [DOI] [PubMed] [Google Scholar]
- 39. Uekert SJ, et al. Sex‐related differences in immune development and the expression of atopy in early childhood. J Allergy Clin Immunol 2006;118:1375–1381. [DOI] [PubMed] [Google Scholar]
- 40. Rautava S, Salminen S, Isolauri E. Specific probiotics in reducing the risk of acute infections in infancy–a randomised, double‐blind, placebo‐controlled study. Br J Nutr 2009;101:1722–1726. [DOI] [PubMed] [Google Scholar]
- 41. Leyer GJ, Li S, Mubasher ME, Reifer C, Ouwehand AC. Probiotic effects on cold and influenza‐like symptom incidence and duration in children. Pediatr 2009;124:e172–e179. [DOI] [PubMed] [Google Scholar]
- 42. Ginde AA, Mansbach JM, Camargo CA Jr. Association between serum 25‐hydroxyvitamin D level and upper respiratory tract infection in the Third National Health and Nutrition Examination Survey. Arch Intern Med 2009;169:384–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sabetta JR, DePetrillo P, Cipriani RJ, Smardin J, Burns LA, Landry ML. Serum 25‐hydroxyvitamin d and the incidence of acute viral respiratory tract infections in healthy adults. PLoS ONE 2010;5:e11088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McFadden ER Jr, et al. Thermal mapping of the airways in humans. J Appl Physiol 1985;58:564–570. [DOI] [PubMed] [Google Scholar]
- 45. Mosser AG, et al. Quantitative and qualitative analysis of rhinovirus infection in bronchial tissues. Am J Respir Crit Care Med 2005;171:645–651. [DOI] [PubMed] [Google Scholar]
- 46. Gern JE, Galagan DM, Jarjour NN, Dick EC, Busse WW. Detection of rhinovirus RNA in lower airway cells during experimentally‐induced infection. Am J Respir Crit Care Med 1997;155:1159–1161. [DOI] [PubMed] [Google Scholar]
- 47. Papadopoulos NG, et al. Rhinoviruses infect the lower airways. J Infect Dis 2000;181:1875–1884. [DOI] [PubMed] [Google Scholar]
- 48. Tsolia MN, et al. Etiology of community‐acquired pneumonia in hospitalized school‐age children: evidence for high prevalence of viral infections. Clin Infect Dis 2004;39:681–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Miller EK, et al. Rhinovirus‐associated hospitalizations in young children. J Infect Dis 2007;195:773–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Regamey N, Kaiser L. Rhinovirus infections in infants: is respiratory syncytial virus ready for the challenge? Eur Respir J 2008;32:249–251. [DOI] [PubMed] [Google Scholar]
- 51. Kusel MM, de Klerk NH, Holt PG, Kebadze T, Johnston SL, Sly PD. Role of respiratory viruses in acute upper and lower respiratory tract illness in the first year of life: a birth cohort study. Pediatr Infect Dis J 2006;25:680–686. [DOI] [PubMed] [Google Scholar]
- 52. Gern JE, Rosenthal LA, Sorkness RL, Lemanske RF Jr. Effects of viral respiratory infections on lung development and childhood asthma. J Allergy Clin Immunol 2005;115:668–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lemanske RF Jr. The childhood origins of asthma (COAST) study. Pediatr Allergy Immunol 2002;13(Suppl):38–43. [DOI] [PubMed] [Google Scholar]
- 54. Sly PD, et al. Early identification of atopy in the prediction of persistent asthma in children. Lancet 2008;372:1100–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Leigh R, et al. Human rhinovirus infection enhances airway epithelial cell production of growth factors involved in airway remodeling. J Allergy Clin Immunol 2008;121:1238–1245. [DOI] [PubMed] [Google Scholar]
- 56. Kuo C, et al. Rhinovirus infection induces expression of airway remodelling factors in vitro and in vivo. Respirology 2011;16:367–377. [DOI] [PubMed] [Google Scholar]
- 57. Tacon CE, Wiehler S, Holden NS, Newton R, Proud D, Leigh R. Human rhinovirus infection up‐regulates MMP‐9 production in airway epithelial cells via NF‐{kappa}B. Am J Respir Cell Mol Biol 2010;43:201–209. [DOI] [PubMed] [Google Scholar]
- 58. Jang YJ, Wang JH, Kim JS, Kwon HJ, Yeo NK, Lee BJ. Levocetirizine inhibits rhinovirus‐induced ICAM‐1 and cytokine expression and viral replication in airway epithelial cells. Antiviral Res 2009;81:226–233. [DOI] [PubMed] [Google Scholar]
- 59. Holgate ST, Roberts G, Arshad HS, Howarth PH, Davies DE. The role of the airway epithelium and its interaction with environmental factors in asthma pathogenesis. Proc Am Thorac Soc 2009;6:655–659. [DOI] [PubMed] [Google Scholar]
- 60. Saglani S, et al. Airway remodeling and inflammation in symptomatic infants with reversible airflow obstruction. Am J Respir Crit Care Med 2005;171:722–727. [DOI] [PubMed] [Google Scholar]
- 61. Fedorov IA, Wilson SJ, Davies DE, Holgate ST. Epithelial stress and structural remodelling in childhood asthma. Thorax 2005;60:389–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tortorolo L, et al. Neurotrophin overexpression in lower airways of infants with respiratory syncytial virus infection. Am J Respir Crit Care Med 2005;172:233–237. [DOI] [PubMed] [Google Scholar]
- 63. Brockman‐Schneider RA, Amineva SP, Bulat MV, Gern JE. Serial culture of murine primary airway epithelial cells and ex vivo replication of human rhinoviruses. J Immunol Methods 2008;339:264–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Peebles RS Jr. Viral infections, atopy, and asthma: is there a causal relationship?. J Allergy Clin Immunol 2004;113:S15–S18. [DOI] [PubMed] [Google Scholar]
- 65. Singh AM, Moore PE, Gern JE, Lemanske RF Jr, Hartert TV. Bronchiolitis to asthma: a review and call for studies of gene‐virus interactions in asthma causation. Am J Respir Crit Care Med 2007;175:108–119. [DOI] [PubMed] [Google Scholar]
- 66. Castro‐Rodriguez JA, Holberg CJ, Wright AL, Martinez FD. A clinical index to define risk of asthma in young children with recurrent wheezing. Am J Respir Crit Care Med 2000;162:1403–1406. [DOI] [PubMed] [Google Scholar]
- 67. Kurukulaaratchy RJ, Matthews S, Holgate ST, Arshad SH. Predicting persistent disease among children who wheeze during early life. Eur Respir J 2003;22:767–771. [DOI] [PubMed] [Google Scholar]
- 68. Caudri D, et al. Predicting the long‐term prognosis of children with symptoms suggestive of asthma at preschool age. J Allergy Clin Immunol 2009;124:903–910. [DOI] [PubMed] [Google Scholar]
- 69. Kotaniemi‐Syrjanen A, Vainionpaa R, Reijonen TM, Waris M, Korhonen K, Korppi M. Rhinovirus‐induced wheezing in infancy–the first sign of childhood asthma? J Allergy Clin Immunol 2003;111:66–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hyvarinen MK, Kotaniemi‐Syrjanen A, Reijonen TM, Korhonen K, Korppi MO. Teenage asthma after severe early childhood wheezing: an 11‐year prospective follow‐up. Pediatr Pulmonol 2005;40:316–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lemanske RF Jr, et al. Rhinovirus illnesses during infancy predict subsequent childhood wheezing. J Allergy Clin Immunol 2005;116:571–577. [DOI] [PubMed] [Google Scholar]
- 72. Jackson DJ, et al. Wheezing rhinovirus illnesses in early life predict asthma development in high‐risk children. Am J Respir Crit Care Med 2008;178:667–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kusel MM, et al. Early‐life respiratory viral infections, atopic sensitization, and risk of subsequent development of persistent asthma. J Allergy Clin Immunol 2007;119:1105–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Johnston SL, et al. Community study of role of viral infections in exacerbations of asthma in 9–11 year old children. BMJ 1995;310:1225–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Nicholson KG, Kent J, Ireland DC. Respiratory viruses and exacerbations of asthma in adults. BMJ 1993;307:982–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Heymann PW, et al. Viral infections in relation to age, atopy, and season of admission among children hospitalized for wheezing. J Allergy Clin Immunol 2004;114:239–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Johnston NW, Johnston SL, Norman GR, Dai J, Sears MR. The September epidemic of asthma hospitalization: school children as disease vectors. J Allergy Clin Immunol 2006;117:557–562. [DOI] [PubMed] [Google Scholar]
- 78. Johnston SL, et al. The relationship between upper respiratory infections and hospital admissions for asthma: a time trend analysis. Am J Respir Crit Care Med 1996;154:654–660. [DOI] [PubMed] [Google Scholar]
- 79. Corne JM, et al. Frequency, severity, and duration of rhinovirus infections in asthmatic and non‐asthmatic individuals: a longitudinal cohort study. Lancet 2002;359:831–834. [DOI] [PubMed] [Google Scholar]
- 80. DeMore JP, et al. Similar colds in subjects with allergic asthma and nonatopic subjects after inoculation with rhinovirus‐16. J Allergy Clin Immunol 2009;124:245–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Fleming HE, et al. Rhinovirus‐16 colds in healthy and in asthmatic subjects: similar changes in upper and lower airways. Am J Respir Crit Care Med 1999;160:100–108. [DOI] [PubMed] [Google Scholar]
- 82. Duff AL, et al. Risk factors for acute wheezing in infants and children: viruses, passive smoke, and IgE antibodies to inhalant allergens. Pediatrics 1993;92:535–540. [PubMed] [Google Scholar]
- 83. Rakes GP, et al. Rhinovirus and respiratory syncytial virus in wheezing children requiring emergency care. IgE and eosinophil analyses. Am J Respir Crit Care Med 1999;159:785–790. [DOI] [PubMed] [Google Scholar]
- 84. Murray CS, et al. Study of modifiable risk factors for asthma exacerbations: virus infection and allergen exposure increase the risk of asthma hospital admissions in children. Thorax 2006;61:376–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Green RM, Custovic A, Sanderson G, Hunter J, Johnston SL, Woodcock A. Synergism between allergens and viruses and risk of hospital admission with asthma: case‐control study. BMJ 2002;324:763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Rosenstreich DL, et al. The role of cockroach allergy and exposure to cockroach allergen in causing morbidity among inner‐city children with asthma. N Engl J Med 1997;336:1356–1363. [DOI] [PubMed] [Google Scholar]
- 87. Smyth AR, Smyth RL, Tong CYW, Hart CA, Heaf DP. Effect of respiratory virus infections including rhinovirus on clinical status in cystic fibrosis. Arch Dis Child 1995;73:117–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Mallia P, Message SD, Kebadze T, Parker H, Kon OM, Johnston SL. An experimental model of rhinovirus induced chronic obstructive pulmonary disease exacerbations: a pilot study. Respir Res 2006;7:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Hilty M, et al. Disordered microbial communities in asthmatic airways. PLoS ONE 2010;5:e8578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Huang YJ, et al. Airway microbiota and bronchial hyperresponsiveness in patients with suboptimally controlled asthma. J Allergy Clin Immunol 2011;127:372–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Bisgaard H, et al. Association of bacteria and viruses with wheezy episodes in young children: prospective birth cohort study. BMJ 2010;341:c4978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Vareille M, Kieninger E, Edwards MR, Regamey N. The airway epithelium: soldier in the fight against respiratory viruses. Clin Microbiol Rev 2011;24:210–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Hakonarson H, Carter C, Maskeri N, Hodinka R, Grunstein MM. Rhinovirus‐mediated changes in airway smooth muscle responsiveness: induced autocrine role of interleukin‐1β. Am J Physiol Lung Cell Mol Physiol 1999;277:L13–L21. [DOI] [PubMed] [Google Scholar]
- 94. Grunstein MM, Hakonarson H, Whelan R, Yu Z, Grunstein JS, Chuang S. Rhinovirus elicits proasthmatic changes in airway responsiveness independently of viral infection. J Allergy Clin Immunol 2001;108:997–1004. [DOI] [PubMed] [Google Scholar]
- 95. Ghildyal R, et al. Rhinovirus infects primary human airway fibroblasts and induces a neutrophil chemokine and a permeability factor. J Med Virol 2005;75:608–615. [DOI] [PubMed] [Google Scholar]
- 96. Schroth MK, et al. Rhinovirus replication causes RANTES production in primary bronchial epithelial cells. Am J Respir Cell Mol Biol 1999;20:1220–1228. [DOI] [PubMed] [Google Scholar]
- 97. Spurrell JC, Wiehler S, Zaheer RS, Sanders SP, Proud D. Human airway epithelial cells produce IP‐10 (CXCL10) in vitro and in vivo upon rhinovirus infection. Am J Physiol Lung Cell Mol Physiol 2005;289:L85–L95. [DOI] [PubMed] [Google Scholar]
- 98. Chen Y, et al. Rhinovirus induces airway epithelial gene expression through double‐stranded RNA and IFN‐dependent pathways. Am J Respir Cell Mol Biol 2006;34:192–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Lopez‐Souza N, et al. Resistance of differentiated human airway epithelium to infection by rhinovirus. Am J Physiol Lung Cell Mol Physiol 2004;286:L373–L381. [DOI] [PubMed] [Google Scholar]
- 100. Kirchberger S, Majdic O, Stockl J. Modulation of the immune system by human rhinoviruses. Int Arch Allergy Immunol 2007;142:1–10. [DOI] [PubMed] [Google Scholar]
- 101. Proud D, Naclerio RM, Gwaltney JM, Hendley JO. Kinins are generated in nasal secretions during natural rhinovirus colds. J Infect Dis 1990;161:120–123. [DOI] [PubMed] [Google Scholar]
- 102. Proud D, Gwaltney JM Jr, Hendley JO, Dinarello CA, Gillis S, Schleimer RP. Increased levels of interleukin‐1 are detected in nasal secretions of volunteers during experimental rhinovirus colds. J Infect Dis 1994;169:1007–1013. [DOI] [PubMed] [Google Scholar]
- 103. Levandowski RA, Weaver CW, Jackson GG. Nasal secretion leukocyte populations determined by flow cytometry during acute rhinovirus infection. J Med Virol 1988;25:423–432. [DOI] [PubMed] [Google Scholar]
- 104. Turner RB, Weingand KW, Yeh CH, Leedy DW. Association between interleukin‐8 concentration in nasal secretions and severity of symptoms of experimental rhinovirus colds. Clin Infect Dis 1998;26:840–846. [DOI] [PubMed] [Google Scholar]
- 105. Gern JE, Vrtis R, Grindle KA, Swenson C, Busse WW. Relationship of upper and lower airway cytokines to outcome of experimental rhinovirus infection. Am J Respir Crit Care Med 2000;162:2226–2231. [DOI] [PubMed] [Google Scholar]
- 106. Konno S, et al. Interferon‐gamma enhances rhinovirus‐induced RANTES secretion by airway epithelial cells. Am J Respir Cell Mol Biol 2002;26:594–601. [DOI] [PubMed] [Google Scholar]
- 107. Lopez‐Souza N, et al. In vitro susceptibility to rhinovirus infection is greater for bronchial than for nasal airway epithelial cells in human subjects. J Allergy Clin Immunol 2009;123:1384–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Jarjour NN, Gern JE, Kelly EAB, Swenson CA, Dick CR, Busse WW. The effect of an experimental rhinovirus 16 infection on bronchial lavage neutrophils. J Allergy Clin Immunol 2000;105:1169–1177. [DOI] [PubMed] [Google Scholar]
- 109. Fraenkel DJ, Bardin PG, Sanderson G, Lampe F, Johnston SL, Holgate ST. Lower airway inflammation during rhinovirus colds in normal and in asthmatic subjects. Am J Respir Crit Care Med 1995;151:879–886. [DOI] [PubMed] [Google Scholar]
- 110. Grünberg K, et al. Experimental rhinovirus 16 infection increases intercellular adhesion molecule‐1 expression in bronchial epithelium of asthmatics regardless of inhaled steroid treatment. Clin Exp Allergy 2000;30:1015–1023. [DOI] [PubMed] [Google Scholar]
- 111. Tuthill TJ, et al. Mouse respiratory epithelial cells support efficient replication of human rhinovirus. J Gen Virol 2003;84:2829–2836. [DOI] [PubMed] [Google Scholar]
- 112. Greve JM, et al. The major human rhinovirus receptor is ICAM‐1. Cell 1989;56:839–847. [DOI] [PubMed] [Google Scholar]
- 113. Nagarkar DR, et al. Rhinovirus infection of allergen‐sensitized and ‐challenged mice induces eotaxin release from functionally polarized macrophages. J Immunol 2010;185:2525–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Hofer F, et al. Members of the low density lipoprotein receptor family mediate cell entry of a minor‐group common cold virus. Proc Natl Acad Sci USA 1994;91:1839–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Marlovits TC, Abrahamsberg C, Blaas D. Very‐low‐density lipoprotein receptor fragment shed from HeLa cells inhibits human rhinovirus infection. J Virol 1998;72:10246–10250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Papadopoulos NG, Stanciu LA, Papi A, Holgate ST, Johnston SL. A defective type 1 response to rhinovirus in atopic asthma. Thorax 2002;57:328–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Hewson CA, Jardine A, Edwards MR, Laza‐Stanca V, Johnston SL. Toll‐like receptor 3 is induced by and mediates antiviral activity against rhinovirus infection of human bronchial epithelial cells. J Virol 2005;79:12273–12279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Sajjan US, et al. H. influenzae potentiates airway epithelial cell responses to rhinovirus by increasing ICAM‐1 and TLR3 expression. FASEB J 2006;20:2121–2123. [DOI] [PubMed] [Google Scholar]
- 119. Zhu L, Lee PK, Lee WM, Zhao Y, Yu D, Chen Y. Rhinovirus‐induced major airway mucin production involves a novel TLR3‐EGFR‐dependent pathway. Am J Respir Cell Mol Biol 2009;40:610–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Slater L, et al. Co‐ordinated role of TLR3, RIG‐I and MDA5 in the innate response to rhinovirus in bronchial epithelium. PLoS Pathog 2010;6:e1001178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Griego SD, Weston CB, Adams JL, Tal‐Singer R, Dillon SB. Role of p38 mitogen‐activated protein kinase in rhinovirus‐induced cytokine production by bronchial epithelial cells. J Immunol 2000;165:5211–5220. [DOI] [PubMed] [Google Scholar]
- 122. Edwards MR, et al. Protein kinase R, IkappaB kinase‐beta and NF‐kappaB are required for human rhinovirus induced pro‐inflammatory cytokine production in bronchial epithelial cells. Mol Immunol 2007;44:1587–1597. [DOI] [PubMed] [Google Scholar]
- 123. Khaitov MR, et al. Respiratory virus induction of alpha‐, beta‐ and lambda‐interferons in bronchial epithelial cells and peripheral blood mononuclear cells. Allergy 2009;64:375–386. [DOI] [PubMed] [Google Scholar]
- 124. Papi A, Johnston SL. Rhinovirus infection induces expression of its own receptor intercellular adhesion molecule 1 (ICAM‐1) via increased NF‐kappaB‐mediated transcription. J Biol Chem 1999;274:9707–9720. [DOI] [PubMed] [Google Scholar]
- 125. Hewson CA, et al. Rhinovirus induces MUC5AC in a human infection model and in vitro via NF‐kappaB and EGFR pathways. Eur Respir J 2010;36:1425–1435. [DOI] [PubMed] [Google Scholar]
- 126. Duits LA, et al. Rhinovirus increases human beta‐defensin‐2 and ‐3 mRNA expression in cultured bronchial epithelial cells. FEMS Immunol Med Microbiol 2003;38:59–64. [DOI] [PubMed] [Google Scholar]
- 127. Proud D, Sanders SP, Wiehler S. Human rhinovirus infection induces airway epithelial cell production of human beta‐defensin 2 both in vitro and in vivo. J Immunol 2004;172:4637–4645. [DOI] [PubMed] [Google Scholar]
- 128. Funkhouser AW, et al. Rhinovirus 16 3C protease induces interleukin‐8 and granulocyte‐macrophage colony‐stimulating factor expression in human bronchial epithelial cells. Pediatr Res 2004;55:13–18. [DOI] [PubMed] [Google Scholar]
- 129. Gern JE, et al. Rhinovirus enters but does not replicate inside monocytes and airway macrophages. J Immunol 1996;156:621–627. [PubMed] [Google Scholar]
- 130. Johnston SL, Papi A, Bates PJ, Mastronarde JG, Monick MM, Hunninghake GW. Low grade rhinovirus infection induces a prolonged release of IL‐8 in pulmonary epithelium. J Immunol 1998;160:6172–6181. [PubMed] [Google Scholar]
- 131. Newcomb DC, et al. Phosphatidylinositol 3‐kinase is required for rhinovirus‐induced airway epithelial cell interleukin‐8 expression. J Biol Chem 2005;280:36952–36961. [DOI] [PubMed] [Google Scholar]
- 132. Bentley JK, Newcomb DC, Goldsmith AM, Jia Y, Sajjan US, Hershenson MB. Rhinovirus activates interleukin‐8 expression via a Src/p110beta phosphatidylinositol 3‐kinase/Akt pathway in human airway epithelial cells. J Virol 2007;81:1186–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Wang X, et al. Syk is downstream of intercellular adhesion molecule‐1 and mediates human rhinovirus activation of p38 MAPK in airway epithelial cells. J Immunol 2006;177:6859–6870. [DOI] [PubMed] [Google Scholar]
- 134. Wiehler S, Proud D. Interleukin‐17A modulates human airway epithelial responses to human rhinovirus infection. Am J Physiol Lung Cell Mol Physiol 2007;293:L505–515. [DOI] [PubMed] [Google Scholar]
- 135. Zaheer RS, Koetzler R, Holden NS, Wiehler S, Proud D. Selective transcriptional down‐regulation of human rhinovirus‐induced production of CXCL10 from airway epithelial cells via the MEK1 pathway. J Immunol 2009;182:4854–4864. [DOI] [PubMed] [Google Scholar]
- 136. Zaheer RS, Proud D. Human rhinovirus‐induced epithelial production of CXCL10 is dependent upon IFN regulatory factor‐1. Am J Respir Cell Mol Biol 2010;43:413–421. [DOI] [PubMed] [Google Scholar]
- 137. Zhu Z, Tang W, Gwaltney JMJ, Wu Y, Elias JA. Rhinovirus stimulation of interleukin‐8 in vivo and in vitro: role of NF‐kappaB. Am J Physiol 1997;273:L814–L824. [DOI] [PubMed] [Google Scholar]
- 138. Kim J, Sanders SP, Siekierski ES, Casolaro V, Proud D. Role of NF‐kappa B in cytokine production induced from human airway epithelial cells by rhinovirus infection. J Immunol 2000;165:3384–3392. [DOI] [PubMed] [Google Scholar]
- 139. Edwards MR, et al. Protein kinase R, IkappaB kinase‐beta and NF‐kappaB are required for human rhinovirus induced pro‐inflammatory cytokine production in bronchial epithelial cells. Mol Immunol 2007;44:1587–1597. [DOI] [PubMed] [Google Scholar]
- 140. Newcomb DC, Sajjan US, Nagarkar DR, Goldsmith AM, Bentley JK, Hershenson MB. Cooperative effects of rhinovirus and TNF‐{alpha} on airway epithelial cell chemokine expression. Am J Physiol Lung Cell Mol Physiol 2007;293:L1021–L1028. [DOI] [PubMed] [Google Scholar]
- 141. Koetzler R, Zaheer RS, Wiehler S, Holden NS, Giembycz MA, Proud D. Nitric oxide inhibits human rhinovirus‐induced transcriptional activation of CXCL10 in airway epithelial cells. J Allergy Clin Immunol 2009;123:201–208. [DOI] [PubMed] [Google Scholar]
- 142. Papi A, et al. Reducing agents inhibit rhinovirus‐induced up‐regulation of the rhinovirus receptor intercellular adhesion molecule‐1 (ICAM‐1) in respiratory epithelial cells. FASEB J 2002;16:1934–1936. [DOI] [PubMed] [Google Scholar]
- 143. Papi A, et al. Role of xanthine‐oxidase activation and reduced glutathione depletion in rhinovirus induction of inflammation in respiratory epithelial cells. J Biol Chem 2008;283:28595–28606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Sanders SP, Siekierski ES, Porter JD, Richards SM, Proud D. Nitric oxide inhibits rhinovirus‐induced cytokine production and viral replication in a human respiratory epithelial cell line. J Virol 1998;72:934–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Laza‐Stanca V, Stanciu LA, Message SD, Edwards MR, Gern JE, Johnston SL. Rhinovirus replication in human macrophages induces NF‐kappaB‐dependent tumor necrosis factor alpha production. J Virol 2006;80:8248–8258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Hall DJ, Bates ME, Guar L, Cronan M, Korpi N, Bertics PJ. The role of p38 MAPK in rhinovirus‐induced monocyte chemoattractant protein‐1 production by monocytic‐lineage cells. J Immunol 2005;174:8056–8063. [DOI] [PubMed] [Google Scholar]
- 147. Platanias LC. Mechanisms of type‐I‐ and type‐II‐interferon‐mediated signalling. Nat Rev Immunol 2005;5:375–386. [DOI] [PubMed] [Google Scholar]
- 148. Wark PA, et al. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J Exp Med 2005;201:937–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Bochkov YA, Hanson KM, Keles S, Brockman‐Schneider RA, Jarjour NN, Gern JE. Rhinovirus‐induced modulation of gene expression in bronchial epithelial cells from subjects with asthma. Mucosal Immunol 2010;3:69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Gern JE, Vrtis R, Kelly EAB, Dick EC, Busse WW. Rhinovirus produces nonspecific activation of lymphocytes through a monocyte‐dependent mechanism. J Immunol 1996;157:1605–1612. [PubMed] [Google Scholar]
- 151. Stockl J, Vetr H, Majdic O, Zlabinger G, Kuechler E, Knapp W. Human major group rhinoviruses downmodulate the accessory function of monocytes by inducing IL‐10. J Clin Invest 1999;104:957–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Papadopoulos NG, Stanciu LA, Papi A, Holgate ST, Johnston SL. Rhinovirus‐induced alterations on peripheral blood mononuclear cell phenotype and costimulatory molecule expression in normal and atopic asthmatic subjects. Clin Exp Allergy 2002;32:537–542. [DOI] [PubMed] [Google Scholar]
- 153. Kirchberger S, et al. Human rhinoviruses inhibit the accessory function of dendritic cells by inducing sialoadhesin and B7‐H1 expression. J Immunol 2005;175:1145–1152. [DOI] [PubMed] [Google Scholar]
- 154. Alam R, et al. Increased MCP‐1, RANTES, and MIP‐1alpha in bronchoalveolar lavage fluid of allergic asthmatic patients. Am J Respir Crit Care Med 1996;153:1398–1404. [DOI] [PubMed] [Google Scholar]
- 155. Wark PA, et al. IFN‐gamma‐induced protein 10 is a novel biomarker of rhinovirus‐induced asthma exacerbations. J Allergy Clin Immunol 2007;120:586–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Gern JE, Dick EC, Kelly EAB, Vrtis R, Klein B. Rhinovirus‐specific T cells recognize both shared and serotype‐restricted viral epitopes. J Infect Dis 1997;175:1108–1114. [DOI] [PubMed] [Google Scholar]
- 157. Gern JE, Joseph B, Galagan DM, Dick EC. Rhinovirus inhibits antigen‐specific T cell proliferation through an Intracellular Adhesion Molecule‐1‐dependent mechanism. J Infect Dis 1996;174:1143–1150. [DOI] [PubMed] [Google Scholar]
- 158. Psarras S, et al. Vascular endothelial growth factor‐mediated induction of angiogenesis by human rhinoviruses. J Allergy Clin Immunol 2006;117:291–297. [DOI] [PubMed] [Google Scholar]
- 159. Korpi‐Steiner NL, Valkenaar SM, Bates ME, Evans MD, Gern JE, Bertics PJ. Human monocytic cells direct the robust release of CXCL10 by bronchial epithelial cells during rhinovirus infection. Clin Exp Allergy 2010;40:1203–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Sakamoto M, Ida S, Takishima T. Effect of influenza virus infection on allergic sensitization to aerosolized ovalbumin in mice. J Immunol 1984;132:2614–2617. [PubMed] [Google Scholar]
- 161. Jakiela B, Brockman‐Schneider R, Amineva S, Lee WM, Gern JE. Basal cells of differentiated bronchial epithelium are more susceptible to rhinovirus infection. Am J Respir Cell Mol Biol 2008;38:517–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Parry DE, Busse WW, Sukow KA, Dick CR, Swenson CA, Gern JE. Rhinovirus‐induced peripheral blood mononuclear cell responses and outcome of experimental infection in allergic subjects. J Allergy Clin Immunol 2000;105:692–698. [DOI] [PubMed] [Google Scholar]
- 163. Brooks GD, Buchta KA, Gern JE, Busse WW. Association of rhinovirus‐induced IFN‐γ with increased asthma severity. Am J Respir Crit Care Med 2003;168:1091–1094. [DOI] [PubMed] [Google Scholar]
- 164. Contoli M, et al. Role of deficient type III interferon‐lambda production in asthma exacerbations. Nat Med 2006;12:1023–1056. [DOI] [PubMed] [Google Scholar]
- 165. Message SD, et al. Rhinovirus‐induced lower respiratory illness is increased in asthma and related to virus load and Th1/2 cytokine and IL‐10 production. Proc Natl Acad Sci USA 2008;105:13562–13567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Handzel ZT, et al. Eosinophils bind rhinovirus and activate virus‐specific T cells. J Immunol 1998;160:1279–1284. [PubMed] [Google Scholar]
- 167. Greiff L, et al. Effects of rhinovirus‐induced common colds on granulocyte activity in allergic rhinitis. J Infect 2002;45:227–232. [DOI] [PubMed] [Google Scholar]
- 168. Calhoun WJ, Dick EC, Schwartz LB, Busse WW. A common cold virus, rhinovirus 16, potentiates airway inflammation after segmental antigen bronchoprovocation in allergic subjects. J Clin Invest 1994;94:2200–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Eggleston PA, Hendley JO, Gwaltney JM Jr. Mediators of immediate hypersensitivity in nasal secretions during natural colds and rhinovirus infection. Acta Oto-Laryngologica – Suppl 1984;413:25–35. [DOI] [PubMed] [Google Scholar]
- 170. Hosoda M, et al. Effects of rhinovirus infection on histamine and cytokine production by cell lines from human mast cells and basophils. J Immunol 2002;169:1482–1491. [DOI] [PubMed] [Google Scholar]
- 171. Venarske DL, et al. The relationship of rhinovirus‐associated asthma hospitalizations with inhaled corticosteroids and smoking. J Infect Dis 2006;193:1536–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Chauhan AJ, et al. Personal exposure to nitrogen dioxide (NO2) and the severity of virus‐induced asthma in children. Lancet 2003;361:1939–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Lachowicz‐Scroggins ME, Boushey HA, Finkbeiner WE, Widdicombe JH. Interleukin‐13 induced mucous metaplasia increases susceptibility of human airway epithelium to rhinovirus infection. Am J Respir Cell Mol Biol 2010;43:652–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Tversky JR, Le TV, Bieneman AP, Chichester KL, Hamilton RG, Schroeder JT. Human blood dendritic cells from allergic subjects have impaired capacity to produce interferon‐alpha via Toll‐like receptor 9. Clin Exp Allergy 2008;38:781–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Gill MA, et al. Counterregulation between the FcepsilonRI pathway and antiviral responses in human plasmacytoid dendritic cells. J Immunol 2010;184:5999–6006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Paul WE, Zhu J. How are T(H)2‐type immune responses initiated and amplified? Nat Rev Immunol 2010;10:225–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Kato A, Favoreto S Jr, Avila PC, Schleimer RP. TLR3‐ and Th2 cytokine‐dependent production of thymic stromal lymphopoietin in human airway epithelial cells. J Immunol 2007;179:1080–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Liu YJ. TSLP in epithelial cell and dendritic cell cross talk. Adv Immunol 2009;101:1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Uller L, et al. Double‐stranded RNA induces disproportionate expression of thymic stromal lymphopoietin versus interferon‐beta in bronchial epithelial cells from donors with asthma. Thorax 2010;65:626–632. [DOI] [PubMed] [Google Scholar]
- 180. Hashimoto K, et al. Respiratory syncytial virus in allergic lung inflammation increases Muc5ac and gob‐5. Am J Respir Crit Care Med 2004;170:306–312. [DOI] [PubMed] [Google Scholar]
- 181. Singh M, et al. Human rhinovirus proteinase 2A induces TH1 and TH2 immunity in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol 2010;125:1369–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Benoit LA, Holtzman MJ. New immune pathways from chronic post‐viral lung disease. Ann N Y Acad Sci 2010;1183:195–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Holt PG, Strickland DH. Interactions between innate and adaptive immunity in asthma pathogenesis: new perspectives from studies on acute exacerbations. J Allergy Clin Immunol 2010;125:963–972. [DOI] [PubMed] [Google Scholar]
- 184. Subrata LS, et al. Interactions between innate antiviral and atopic immunoinflammatory pathways precipitate and sustain asthma exacerbations in children. J Immunol 2009;183:2793–2800. [DOI] [PubMed] [Google Scholar]
- 185. Gern JE. Rhinovirus and the initiation of asthma. Curr Opin Allergy Clin Immunol 2009;9:73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Sanders SP. Asthma, viruses, and nitric oxide. Proc Soc Exp Biol Med 1999;220:123–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Uhl EW, Castleman WL, Sorkness RL, Lemanske RF, McAllister PK. Parainfluenza virus‐induced persistence of airway inflammation, fibrosis, and dysfunction associated with TGF‐β1 expression in Brown Norway rats. Am J Respir Crit Care Med 1996;154:1834–1842. [DOI] [PubMed] [Google Scholar]
- 188. Dosanjh A. Transforming growth factor‐beta expression induced by rhinovirus infection in respiratory epithelial cells. Acta Biochim Biophys Sin (Shanghai) 2006;38:911–914. [DOI] [PubMed] [Google Scholar]
- 189. Nogueira‐Silva C, Santos M, Baptista MJ, Moura RS, Correia‐Pinto J. IL‐6 is constitutively expressed during lung morphogenesis and enhances fetal lung explant branching. Pediatr Res 2006;60:530–536. [DOI] [PubMed] [Google Scholar]
- 190. Wang JH, Kwon HJ, Jang YJ. Rhinovirus upregulates matrix metalloproteinase‐2, matrix metalloproteinase‐9, and vascular endothelial growth factor expression in nasal polyp fibroblasts. Laryngoscope 2009;119:1834–1838. [DOI] [PubMed] [Google Scholar]
- 191. Lee CG, et al. Respiratory syncytial virus stimulation of vascular endothelial cell growth Factor/Vascular permeability factor. Am J Resp Cell Mol Biol 2000;23:662–669. [DOI] [PubMed] [Google Scholar]
- 192. Dosanjh A, Rednam S, Martin M. Respiratory syncytial virus augments production of fibroblast growth factor basic in vitro: implications for a possible mechanism of prolonged wheezing after infection. Ped Allergy Immunol 2003;14:437–440. [DOI] [PubMed] [Google Scholar]
- 193. Donninger H, et al. Rhinovirus induction of the CXC chemokine epithelial‐neutrophil activating peptide‐78 in bronchial epithelium. J Infect Dis 2003;187:1809–1817. [DOI] [PubMed] [Google Scholar]
- 194. Koetzler R, Zaheer RS, Wiehler S, Holden NS, Giembycz MA, Proud D. Nitric oxide inhibits human rhinovirus‐induced transcriptional activation of CXCL10 in airway epithelial cells. J Allergy Clin Immunol 2009;123:201–208. [DOI] [PubMed] [Google Scholar]
- 195. Terajima M, et al. Rhinovirus infection of primary cultures of human tracheal epithelium: role of ICAM‐1 and IL‐1beta. Am J Physiol 1997;273:L749–L759. [DOI] [PubMed] [Google Scholar]
- 196. Subauste MC, Jacoby DB, Richards SM, Proud D. Infection of a human respiratory epithelial cell line with rhinovirus – induction of cytokine release and modulation of susceptibility to infection by cytokine exposure. J Clin Invest 1995;96:549–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Sanders SP, Siekierski ES, Richards SM, Porter JD, Imani F, Proud D. Rhinovirus infection induces expression of type 2 nitric oxide synthase in human respiratory epithelial cells in vitro and in vivo. J Allergy Clin Immunol 2001;107:235–243. [DOI] [PubMed] [Google Scholar]
- 198. Sanders SP, Kim J, Connolly KR, Porter JD, Siekierski ES, Proud D. Nitric oxide inhibits rhinovirus‐induced granulocyte macrophage colony‐stimulating factor production in bronchial epithelial cells. Am J Respir Cell Mol Biol 2001;24:317–325. [DOI] [PubMed] [Google Scholar]
- 199. Wimalasundera SS, Katz DR, Chain BM. Characterization of the T cell response to human rhinovirus in children: implications for understanding the immunopathology of the common cold. J Infect Dis 1997;176:755–759. [DOI] [PubMed] [Google Scholar]
- 200. Johnston SL, Papi A, Monick MM, Hunninghake GW. Rhinoviruses induce interleukin‐8 mRNA and protein production in human monocytes. J Infect Dis 1997;175:323–329. [DOI] [PubMed] [Google Scholar]