

Abstract
The pathogenesis of RA, a disabling autoimmune disease, is incompletely understood. Early in the development of RA there appears to be loss of immune homeostasis and regulation, and premature immunosenescence. While identification of risk factors and understanding of the phases of RA pathogenesis are advancing, means of accurately predicting an individual’s risk of developing RA are currently lacking. Telomere length has been proposed as a potential new biomarker for the development of RA that could enhance prediction of this serious disease. Studies examining telomere length in relation to RA have found that telomere erosion appears to proceed more rapidly in subjects with RA than in healthy controls, and that telomere lengths are shorter in those with the RA-risk HLA-shared epitope genes. These studies have been small, however, with retrospective or cross-sectional designs. The potential role of telomere shortening as an independent biomarker for future RA risk, perhaps strongly genetically determined by HLA-SE genes, after controlling for known risk factors such as smoking, body mass index and immunosuppressant medication use, as well as systemic inflammation, is an unanswered question.
Keywords: immunosenescence, rheumatoid arthritis, telomere, biomarker, risk factor, aging, autoimmune
1.1 Pathogenesis of Rheumatoid Arthritis
Rheumatoid arthritis (RA) is a chronic inflammatory polyarthritis that progressively destroys synovial joints and causes systemic complications and early mortality(1). While RA affects approximately 1% of the world’s population(2), at present there is no known cure. RA therapies have improved in the past decade, but new immunosuppressive agents are potentially toxic; long-term prognosis remains poor and average life expectancy is reduced by 3 to 18 years(3). The direct costs of treatment, and the indirect costs of disability and lost productivity are high(4).
Active RA is characterized by extensive systemic inflammation, increased levels of circulating inflammatory cytokines, and synovial hyperplasia with infiltration by lymphocytes, monocytes, macrophages, and fibroblasts. Aberrant T cell activation occurs early, with CD4+ T cells stimulating monocytes and macrophages to produce inflammatory cytokines, as well as proteolytic enzymes that destroy synovium, cartilage, and underlying bone(5, 6). T cells infiltrating rheumatoid synovium are oligoclonal, implicating an antigen-driven process(7, 8), but the inciting antigen or antigens are unidentified. Activated T cells signal B cells to increase production of immunoglobulins, including rheumatoid factor (RF). Active RA is characterized by production and release of pro-inflammatory cytokines such as tumor necrosis factor(TNF)-α and IL-6 (9). TNFα is a critical cytokine in RA pathogenesis(10) and TNF-antagonists are remarkably effective treatments for RA(11, 12). IL-6 produced by T cells, monocytes, macrophages and synovial fibroblasts(13) also causes joint destruction and systemic symptoms, and correlates with RA severity(14). IL-6 levels are elevated in subjects with early untreated(15) and preclinical RA(16, 17)
1.2 Oxidative stress in RA
Increased oxidative stress has also been documented in patients with RA compared to controls and is likely due to polymorphonuclear leukocyte and lymphocyte production of reactive oxygen intermediates(18, 19). Levels of lymphocyte 8-oxo-7-hydrodeoxyguanosine (8-oxodG), a promutagenic DNA lesion induced by reactive oxygen intermediates, were significantly higher in 98 RA patients than in 68 healthy controls(18). Lymphocytes from RA patients, but not those with scleroderma, also showed cellular hypersensitivity to the toxic effects of hydrogen peroxide. Increased DNA damage and increased susceptibility to cytotoxic killing by hydrogen peroxide in RA lymphocytes from patients have been explained by defective repair of DNA damage and increased production of reactive oxygen intermediates in inflammation(18).
1.3 “Normal” Immunosenescence
Normal aging of the immune system, or immunosenescence, is characterized by changes in T cell subsets, cellular and molecular alterations and thymic atrophy. It results in a decline of T and B cell function and loss of ability to recognize “self” and “foreign” antigens(20). A specific skewing of the T cell repertoire has been demonstrated with aging, with an increase in memory T cells. A large proportion of these memory T cells have lost normal expression of the surface CD28 receptor, which is involved in dendritic cell-T cell interactions central to the recognition of self vs. foreign antigens, and have acquired expression of other surface markers (including killer immunoglobulin-like receptors, NKG2D receptors and lymphocyte function-associated antigen)(21, 22). These CD28− T cells have a lowered threshold for antigen-specific activation, and more cytotoxic granules, while aging dendritic cells in response secrete higher levels of inflammatory cytokines and can induce T-cell proliferation even in response to self-peptides. Altered apoptosis, increased cytokine secretion, and the altered T cell repertoire is thought to give rise to a chronic inflammatory state, producing an “autoimmune-risk phenotype”(20).
1.4 Telomeres and Telomere Length
Telomeres are composed of proteins complexed to a hexameric nucleotide (TTAGGG)n repeat sequence at the distal ends of eukaryotic chromosomes that are critical in maintaining the structural integrity of the genome. Telomeres prevent fusion of chromosomal ends, nucleolytic decay, and atypical recombination(23). With each mitotic cell division in normal somatic tissue, telomeric repeats shorten by 30 to 200 bp (24). The progressive loss has been likened to a “molecular clock” reflecting the number of divisions a cell has undergone(25, 26). The rate of telomere attrition is not constant, but a function of age(27), oxidative stress, and antioxidative defenses, as well as cell turnover(28). When telomeres reach a critically truncated length, the cell will either undergo replication-mediated senescence or apoptosis(29). Hence, telomere length is considered a biomarker of biological rather than chronological age(30).
Telomerase, a ribonucleoprotein enzyme, maintains telomere length in germline and stem cells. The protein catalytic subunit of telomerase, telomerase reverse transcriptase (TERT), adds telomere repeats to the ends of chromosomes by reverse transcribing an RNA template (TERC) to DNA. Telomerase activity is generally low in replicative somatic tissues, but high in germ cells, in which chromosomal integrity is critical for fecundity(31–33). Telomere erosion associated with advancing age is determined in part by genetics, and in part by the balance of cell turnover, telomere damage and telomerase repair activity(34). Telomeres and telomerase are central to the biology of cancer, stem cells and aging(35).
1.5 Epidemiologic Associations between Telomere Length, Exposures and Disease Risk
Cigarette smoking potentially contributes to accelerated telomere shortening in humans by enhancing oxidative stress(36–38). In a study by Valdes and colleagues, age-adjusted telomere length was approximately 5 bp shorter for every pack-year smoked: 40 pack-years of smoking corresponded to 7.4 years of age-related shortening in telomere length(36). Morla and colleagues also observed a dose-response relationship between cumulative lifetime exposure to tobacco smoking and telomere length(37). In addition, obesity, likely acting via increased systemic inflammation, and oxidative stress has been associated with decreased telomere length in several studies(36, 39). Telomeres have been shown to be significantly longer in women compared to men, after adjusting for age and smoking(38, 40), but the reasons for this are not known.
Telomere length in peripheral blood leukocytes (PBLs) has emerged as a biomarker of aging and risk of age-related diseases such as cancer. In a study by Wu and colleagues, shorter telomere length was significantly correlated with baseline DNA damage as measured by the Comet assay and with mutagen sensitivity in lymphocytes after gamma-irradiation or exposure to benzo[a]pyrene diol epoxide, suggesting that telomere length is a marker of DNA damage and of susceptibility to such damage(41). Relationships between PBL telomere length and risk of several different age-related diseases, including bladder, skin and esophageal cancers, Parkinson’s disease and cognitive decline have been investigated(38, 42–47). Interestingly, telomere shortening was significantly associated with the risk of some, but not all, of these disease states, pointing to specific pathways of cellular senescence and disease pathogenesis. These findings suggest that truncated telomeres in PBLs are specific risk markers for certain diseases, rather than a generic marker of cellular aging.
2.1 Immunosenescence and Telomere Length in RA
Among patients with RA, loss of normal immune regulation and “premature immunosenescence” have been described(20, 21, 48, 49). T cell repertoires in RA patients are age-inappropriately skewed compared to age-matched controls, with increased proportions of CD28− T cells, possibly reflecting accelerated thymic output (50). Weyand and colleagues have found that multiple cell lines, including lymphocytes and neutrophils, from RA patients have telomere sequences that are significantly shortened compared with those from age-matched controls(51, 52). They studied T cell repertoire diversity and telomere length comparing 51 patients with established RA meeting American College of Rheumatology (ACR) classification criteria, to 47 healthy controls of the same age, and reported that increased self-replication of T cells in RA patients was indicated by age-inappropriate erosion of telomeres in circulating T cells with almost complete attrition of telomeric reserves in patients 20 to 30 years of age. In this small study, the degree of telomere loss among RA patients was not related to disease duration (53). In a related study, they compared telomere maintenance in circulating hematopoietic progenitor cells in RA, comparing 63 patients with established RA to 48 healthy matched controls(52). Telomeres in hematopoietic progenitor cells from RA patients were markedly shortened (Figure. 1). RA patients who were 20–30 years old had progenitor cell telomeres shortened to approximately 9,000 bp, a length equivalent to that observed in the 50–60-year-old controls. RA-derived progenitor cells lost 45 bp of their telomere ends per year of life, emphasizing an abnormal proliferative turnover of the surviving progenitor cells. Using the same sample of RA patients and controls, the Weyand research group has reported that upon stimulation, RA naïve CD4 T cells are defective in up-regulating telomerase activity due to insufficient induction of TERT(54). These defects were present in untreated patients and were independent from disease activity. Thewissen and colleagues in Germany have reported that, among early RA patients with a disease duration of less than one year, TERT mRNA levels were reduced compared to healthy controls; however chronic RA patients, with a disease duration of more than one year, did not show these reduced TERT mRNA levels(55). Thus, it remains unclear at what point (s) in the development of RA telomerase activity may be insufficient, and whether these defects may be seen even prior to disease onset.
Fig. 1.
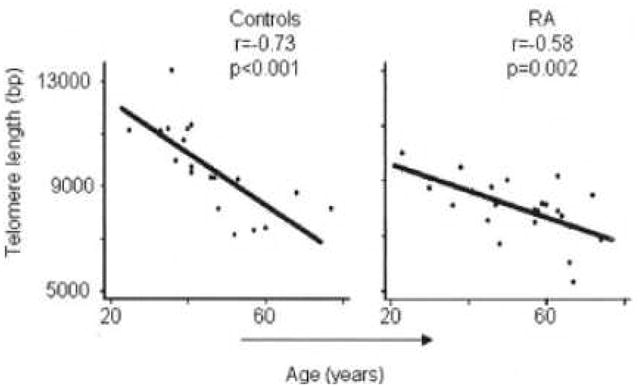
Association between age and telomere length in RA subjects and controls, from Colmegna, 2008 21
2.2. HLA-shared epitope and Telomere Length
Using blood, venous cord blood and semen samples from healthy volunteers, and blood samples from patients with well-established RA, the Weyand group tested whether HLA-shared epitope (SE) alleles were associated with T cell telomere erosion(51). In healthy individuals, HLA-SE alleles were associated with excessive loss of telomeres in CD4+ T cells. Accelerated telomeric erosion occurred before age 20 and reduced homeostatic T cell proliferation was seen in HLA-SE positive adults(51).
2.3 Telomere length in other connective tissue diseases
The handful of studies of telomere length and telomerase activity in other connective tissue diseases has recently been reviewed(26). In a case-control studies of patients with existing autoimmune diseases, including systemic lupus erythematosus(56–58), scleroderma(59), Wegener’s granulomatosis(60) and sarcoidosis(61), compared to healthy matched controls, premature telomere shortening has been found in peripheral blood mononuclear cells and has been associated with disease activity and/or duration..
2.4 Limitations of past telomere length-RA association studies and need for larger epidemiologic studies
Telomere erosion has thus been posited to reflect premature immunosenescence in RA and has been ascribed to excessive proliferative pressure or inadequate telomeric maintenance (49, 52, 53). The Weyand group hypothesizes that telomerase insufficiency in RA results in excessive T cell loss, leading to the observed “aged” T cell repertoire in RA. The past relatively small studies from this group have not observed correlations between RA disease duration or RA disease activity and telomere shortening. Analysis of a small number of patients with RA who had not been treated with any immunosuppressive medications showed their telomeres were eroded to the same degree as those from patients who had received immunosuppressive treatments. As telomere shortening was observed in these RA patients apparently independently of the duration, severity, and activity of the disease and the treatment, RA itself may be associated with intrinsic telomere shortening and bone marrow-derived hematopoietic progenitor cell dysfunction.
The potential for telomere shortening to be a relatively specific biomarker of RA is exciting. Past studies examining telomere shortening in RA have been small, however, with cross-sectional designs. These intriguing studies did not take patient cigarette smoking, reproductive status, or body mass index into account, and included a paucity of untreated patients. Treatments such as corticosteroids, methotrexate, and other immunosuppressant medications undoubtedly have measurable effects upon telomere length. These factors could be important confounders in the relationship between telomere length and RA, but to date no studies of telomere length prior to the onset of RA symptoms have been performed. As inflammation and autoantibodies are present years prior to RA onset, systemic inflammation likely leads to telomere shortening prior to RA. Prior studies have detected abnormalities in subjects affected with RA compared to controls. The retrospective study design used to date cannot address whether abnormalities predate RA onset or whether telomere shortening could be used as a predictive biomarker for RA. The potential role of telomere shortening as an independent biomarker for future RA risk, perhaps strongly genetically determined by HLA-SE genes, after controlling for known risk factors and systemic inflammation, is an unanswered question.
3.1 What if we could identify imminent or impending RA?
Early diagnosis and treatment strategies are critical to minimize disability from joint destruction as treatment with biologic therapies such as anti-tumor necrosis factor(TNF)-α inhibitors slow disease progression(62). Anti-citrullinated peptide antibodies and cytokines are elevated prior to clinical onset(63, 64), but means for accurately predicting RA development in those at risk and therefore intervening before the onset of suffering and irreversible damage are currently lacking. The identification of individuals at high risk for disease could lead to prevention during the pre-clinical period when patients are asymptomatic(63–65).
Telomere shortening, associated with genetic factors, aging and systemic inflammation, is present in RA subjects and could be a potential biomarker of immunosenescence associated with subsequent RA risk. If telomere shortening and telomerase activity are affected years prior to the onset of clinical RA, might there be a window one day for intervention with a telomerase-restoring therapy? Understanding whether telomere abnormalities precede RA onset, and how early prior to RA they occur, is crucial in distinguishing inciting from secondary events in RA pathogenesis. Relationships between genetic risk factors for RA, cytokines and biomarkers of systemic inflammation and oxidative stress, telomere shortening and ultimately RA susceptibility are complex. The potential role of telomere shortening in the development of RA remains an unanswered question.
Take home messages.
Premature aging of the immune system, known as immunosenescence, has been convincingly demonstrated in patients with RA.
Telomeres, the distal ends of eukaryotic chromosomes that are critical in maintaining the structural integrity of the genome, function as a “molecular clock”. Premature telomere shortening has been observed in lymphocytes and hematopoietic stem cells from patients with RA compared to those from healthy controls and is likely related to immunosenescence.
Prospective studies with bloods collected prior to diagnosis of RA are necessary to decipher whether these changes are due to the immune dysregulation of RA itself, or arise prior to the disease, pointing to their involvement in the pathogenesis of disease.
Acknowledgments
Grant Support: NIAMS R01 AR059073 (Costenbader)
Footnotes
Disclosure Statement
None of the authors of this paper has any disclosures or conflicts of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wolfe F, Mitchell DM, Sibley JT, Fries JF, Bloch DA, Williams CA, Spitz PW, Haga M, Kleinheksel SM, Cathey MA. The mortality of rheumatoid arthritis. Arthritis Rheum. 1994;37(4):481–94. doi: 10.1002/art.1780370408. [DOI] [PubMed] [Google Scholar]
- 2.Gabriel SE, Crowson CS, O’Fallon WM. The epidemiology of rheumatoid arthritis in Rochester, Minnesota, 1955–1985. Arthritis Rheum. 1999;42(3):415–420. doi: 10.1002/1529-0131(199904)42:3<415::AID-ANR4>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 3.Pincus T, Callahan LF. Taking mortality in rheumatoid arthritis seriously--predictive markers, socioeconomic status and comorbidity. J Rheumatol. 1986;13(5):841–5. [PubMed] [Google Scholar]
- 4.Yelin E. The costs of rheumatoid arthritis: absolute, incremental, and marginal estimates. J Rheumatol Suppl. 1996;44:47–51. [PubMed] [Google Scholar]
- 5.Miossec P, van den Berg W. Th1/Th2 cytokine balance in arthritis. Arthritis Rheum JID - 0370605. 1997;40(12):2105–2115. doi: 10.1002/art.1780401203. [DOI] [PubMed] [Google Scholar]
- 6.Choy EH, Panayi GS. Cytokine pathways and joint inflammation in rheumatoid arthritis. N Engl J Med. 2001;344(12):907–16. doi: 10.1056/NEJM200103223441207. [DOI] [PubMed] [Google Scholar]
- 7.Goronzy JJ, Bartz-Bazzanella P, Hu W, Jendro MC, Walser-Kuntz DR, Weyand CM. Dominant clonotypes in the repertoire of peripheral CD4+ T cells in rheumatoid arthritis. J Clin Invest. 1994;94(5):2068–76. doi: 10.1172/JCI117561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Waase I, Kayser C, Carlson PJ, Goronzy JJ, Weyand CM. Oligoclonal T cell proliferation in patients with rheumatoid arthritis and their unaffected siblings. Arthritis Rheum. 1996;39(6):904–13. doi: 10.1002/art.1780390606. [DOI] [PubMed] [Google Scholar]
- 9.Morinobu A, Gadina M, Strober W, Visconti R, Fornace A, Montagna C, Feldman GM, Nishikomori R, O’Shea JJ. STAT4 serine phosphorylation is critical for IL-12-induced IFN-gamma production but not for cell proliferation. Proc Natl Acad Sci U S A. 2002;99(19):12281–6. doi: 10.1073/pnas.182618999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Firestein GS. Evolving concepts of rheumatoid arthritis. Nature. 2003;423(6937):356–61. doi: 10.1038/nature01661. [DOI] [PubMed] [Google Scholar]
- 11.Brennan FM, Maini RN, Feldmann M. TNF alpha--a pivotal role in rheumatoid arthritis? Br J Rheumatol. 1992;31(5):293–8. doi: 10.1093/rheumatology/31.5.293. [DOI] [PubMed] [Google Scholar]
- 12.Feldmann M, Brennan FM, Foxwell BM, Taylor PC, Williams RO, Maini RN. Anti-TNF therapy: where have we got to in 2005? J Autoimmun. 2005;25 (Suppl):26–8. doi: 10.1016/j.jaut.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 13.Van Snick J. Interleukin-6: an overview. Annu Rev Immunol. 1990;8:253–78. doi: 10.1146/annurev.iy.08.040190.001345. [DOI] [PubMed] [Google Scholar]
- 14.Robak T, Gladalska A, Stepien H, Robak E. Serum levels of interleukin-6 type cytokines and soluble interleukin-6 receptor in patients with rheumatoid arthritis. Mediators of Inflammation. 1998;7(5):347–353. doi: 10.1080/09629359890875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vazquez-Del Mercado M, Delgado-Rizo V, Munoz-Valle JF, Orozco-Alcala J, Volk HD, Armendariz-Borunda J. Expression of interleukin-1 beta, tumor necrosis factor alpha, interleukins-6, -10 and -4, and metalloproteases by freshly isolated mononuclear cells from early never-treated and non-acute treated rheumatoid arthritis patients. Clin Exp Rheumatol. 1999;17(5):575–83. [PubMed] [Google Scholar]
- 16.Karlson EW, Chibnik LB, Tworoger SS, Lee IM, Buring JE, Shadick NA, Manson JE, Costenbader KH. Biomarkers of inflammation and development of rheumatoid arthritis in women from two prospective cohort studies. Arthritis Rheum. 2009;60(3):641–52. doi: 10.1002/art.24350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jorgensen KT, Wiik A, Pedersen M, Hedegaard CJ, Vestergaard BF, Gislefoss RE, Kvien TK, Wohlfahrt J, Bendtzen K, Frisch M. Cytokines, autoantibodies and viral antibodies in premorbid and postdiagnostic sera from patients with rheumatoid arthritis: case-control study nested in a cohort of Norwegian blood donors. Ann Rheum Dis. 2008;67(6):860–6. doi: 10.1136/ard.2007.073825. [DOI] [PubMed] [Google Scholar]
- 18.Bashir S, Harris G, Denman MA, Blake DR, Winyard PG. Oxidative DNA damage and cellular sensitivity to oxidative stress in human autoimmune diseases. Ann Rheum Dis. 1993;52(9):659–66. doi: 10.1136/ard.52.9.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Agostini M, Di Marco B, Nocentini G, Delfino DV. Oxidative stress and apoptosis in immune diseases. Int J Immunopathol Pharmacol. 2002;15(3):157–164. doi: 10.1177/039463200201500301. [DOI] [PubMed] [Google Scholar]
- 20.Prelog M. Aging of the immune system: a risk factor for autoimmunity? Autoimmun Rev. 2006;5(2):136–9. doi: 10.1016/j.autrev.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 21.Lindstrom TM, Robinson WH. Rheumatoid arthritis: a role for immunosenescence? J Am Geriatr Soc. 2010;58(8):1565–75. doi: 10.1111/j.1532-5415.2010.02965.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weng NP, Akbar AN, Goronzy J. CD28(−) T cells: their role in the age-associated decline of immune function. Trends Immunol. 2009;30(7):306–12. doi: 10.1016/j.it.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stewart SA, Weinberg RA. Telomeres: cancer to human aging. Annu Rev Cell Dev Biol. 2006;22:531–57. doi: 10.1146/annurev.cellbio.22.010305.104518. [DOI] [PubMed] [Google Scholar]
- 24.Shay JW, Wright WE. Telomerase activity in human cancer. Curr Opin Oncol. 1996;8(1):66–71. doi: 10.1097/00001622-199601000-00012. [DOI] [PubMed] [Google Scholar]
- 25.Harley CB. Human ageing and telomeres. Ciba Found Symp. 1997;211:129–39. doi: 10.1002/9780470515433.ch9. discussion 139–44. [DOI] [PubMed] [Google Scholar]
- 26.Georgin-Lavialle S, Aouba A, Mouthon L, Londono-Vallejo JA, Lepelletier Y, Gabet AS, Hermine O. The telomere/telomerase system in autoimmune and systemic immune-mediated diseases. Autoimmun Rev. 2010;9(10):646–51. doi: 10.1016/j.autrev.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 27.Iwama H, Ohyashiki K, Ohyashiki JH, Hayashi S, Yahata N, Ando K, Toyama K, Hoshika A, Takasaki M, Mori M, Shay JW. Telomeric length and telomerase activity vary with age in peripheral blood cells obtained from normal individuals. Hum Genet. 1998;102(4):397–402. doi: 10.1007/s004390050711. [DOI] [PubMed] [Google Scholar]
- 28.von Zglinicki T. Oxidative stress shortens telomeres. Trends Biochem Sci. 2002;27(7):339–44. doi: 10.1016/s0968-0004(02)02110-2. [DOI] [PubMed] [Google Scholar]
- 29.d’Adda di Fagagna F. Living on a break: cellular senescence as a DNA-damage response. Nat Rev Cancer. 2008;8(7):512–22. doi: 10.1038/nrc2440. [DOI] [PubMed] [Google Scholar]
- 30.Blasco MA. Telomeres and human disease: ageing, cancer and beyond. Nat Rev Genet. 2005;6(8):611–22. doi: 10.1038/nrg1656. [DOI] [PubMed] [Google Scholar]
- 31.Harley CB. Telomerases. Pathol Biol (Paris) 1994;42(4):342–5. [PubMed] [Google Scholar]
- 32.Newbold RF. Genetic control of telomerase and replicative senescence in human and rodent cells. Ciba Found Symp. 1997;211:177–89. doi: 10.1002/9780470515433.ch12. discussion 189–97. [DOI] [PubMed] [Google Scholar]
- 33.Wright WE, Piatyszek MA, Rainey WE, Byrd W, Shay JW. Telomerase activity in human germline and embryonic tissues and cells. Dev Genet. 1996;18(2):173–9. doi: 10.1002/(SICI)1520-6408(1996)18:2<173::AID-DVG10>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 34.Gilson E, Londono-Vallejo A. Telomere length profiles in humans: all ends are not equal. Cell Cycle. 2007;6(20):2486–94. doi: 10.4161/cc.6.20.4798. [DOI] [PubMed] [Google Scholar]
- 35.Artandi SE. Telomeres, telomerase, and human disease. N Engl J Med. 2006;355(12):1195–7. doi: 10.1056/NEJMp068187. [DOI] [PubMed] [Google Scholar]
- 36.Valdes AM, Andrew T, Gardner JP, Kimura M, Oelsner E, Cherkas LF, Aviv A, Spector TD. Obesity, cigarette smoking, and telomere length in women. Lancet. 2005;366(9486):662–4. doi: 10.1016/S0140-6736(05)66630-5. [DOI] [PubMed] [Google Scholar]
- 37.Morla M, Busquets X, Pons J, Sauleda J, MacNee W, Agusti AG. Telomere shortening in smokers with and without COPD. Eur Respir J. 2006;27(3):525–8. doi: 10.1183/09031936.06.00087005. [DOI] [PubMed] [Google Scholar]
- 38.McGrath M, Wong JY, Michaud D, Hunter DJ, De Vivo I. Telomere length, cigarette smoking, and bladder cancer risk in men and women. Cancer Epidemiol Biomarkers Prev. 2007;16(4):815–9. doi: 10.1158/1055-9965.EPI-06-0961. [DOI] [PubMed] [Google Scholar]
- 39.Zannolli R, Mohn A, Buoni S, Pietrobelli A, Messina M, Chiarelli F, Miracco C. Telomere length and obesity. Acta Paediatr. 2008;97(7):952–4. doi: 10.1111/j.1651-2227.2008.00783.x. [DOI] [PubMed] [Google Scholar]
- 40.Mayer S, Bruderlein S, Perner S, Waibel I, Holdenried A, Ciloglu N, Hasel C, Mattfeldt T, Nielsen KV, Moller P. Sex-specific telomere length profiles and age-dependent erosion dynamics of individual chromosome arms in humans. Cytogenet Genome Res. 2006;112(3–4):194–201. doi: 10.1159/000089870. [DOI] [PubMed] [Google Scholar]
- 41.Wu X, Amos CI, Zhu Y, Zhao H, Grossman BH, Shay JW, Luo S, Hong WK, Spitz MR. Telomere dysfunction: a potential cancer predisposition factor. J Natl Cancer Inst. 2003;95(16):1211–8. doi: 10.1093/jnci/djg011. [DOI] [PubMed] [Google Scholar]
- 42.Shammas MA, Qazi A, Batchu RB, Bertheau RC, Wong JY, Rao MY, Prasad M, Chanda D, Ponnazhagan S, Anderson KC, Steffes CP, Munshi NC, De Vivo I, Beer DG, Gryaznov S, Weaver DW, Goyal RK. Telomere maintenance in laser capture microdissection-purified Barrett’s adenocarcinoma cells and effect of telomerase inhibition in vivo. Clin Cancer Res. 2008;14(15):4971–80. doi: 10.1158/1078-0432.CCR-08-0473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Han J, Qureshi AA, Prescott J, Guo Q, Ye L, Hunter DJ, De Vivo I. A prospective study of telomere length and the risk of skin cancer. J Invest Dermatol. 2009;129(2):415–21. doi: 10.1038/jid.2008.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grodstein F, van Oijen M, Irizarry MC, Rosas HD, Hyman BT, Growdon JH, De Vivo I. Shorter telomeres may mark early risk of dementia: preliminary analysis of 62 participants from the nurses’ health study. PLoS ONE. 2008;3(2):e1590. doi: 10.1371/journal.pone.0001590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang H, Chen H, Gao X, McGrath M, Deer D, De Vivo I, Schwarzschild MA, Ascherio A. Telomere length and risk of Parkinson’s disease. Mov Disord. 2008;23(2):302–5. doi: 10.1002/mds.21867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.De Vivo I, Prescott J, Wong JY, Kraft P, Hankinson SE, Hunter DJ. A prospective study of relative telomere length and postmenopausal breast cancer risk. Cancer Epidemiol Biomarkers Prev. 2009;18(4):1152–6. doi: 10.1158/1055-9965.EPI-08-0998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Prescott J, McGrath M, Lee IM, Buring JE, De Vivo I. Telomere length and genetic analyses in population-based studies of endometrial cancer risk. Cancer. 116(18):4275–82. doi: 10.1002/cncr.25328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thewissen M, Stinissen P. New concepts on the pathogenesis of autoimmune diseases: a role for immune homeostasis, immunoregulation, and immunosenescence. Crit Rev Immunol. 2008;28(5):363–76. doi: 10.1615/critrevimmunol.v28.i5.10. [DOI] [PubMed] [Google Scholar]
- 49.Weyand CM, Goronzy JJ. Premature immunosenescence in rheumatoid arthritis. J Rheumatol. 2002;29(6):1141–6. [PubMed] [Google Scholar]
- 50.Thewissen M, Somers V, Venken K, Linsen L, van Paassen P, Geusens P, Damoiseaux J, Stinissen P. Analyses of immunosenescent markers in patients with autoimmune disease. Clin Immunol. 2007;123(2):209–18. doi: 10.1016/j.clim.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 51.Schonland SO, Lopez C, Widmann T, Zimmer J, Bryl E, Goronzy JJ, Weyand CM. Premature telomeric loss in rheumatoid arthritis is genetically determined and involves both myeloid and lymphoid cell lineages. Proc Natl Acad Sci U S A. 2003;100(23):13471–6. doi: 10.1073/pnas.2233561100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Colmegna I, Diaz-Borjon A, Fujii H, Schaefer L, Goronzy JJ, Weyand CM. Defective proliferative capacity and accelerated telomeric loss of hematopoietic progenitor cells in rheumatoid arthritis. Arthritis Rheum. 2008;58(4):990–1000. doi: 10.1002/art.23287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Koetz K, Bryl E, Spickschen K, O’Fallon WM, Goronzy JJ, Weyand CM. T cell homeostasis in patients with rheumatoid arthritis. Proc Natl Acad Sci U S A. 2000;97(16):9203–8. doi: 10.1073/pnas.97.16.9203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fujii H, Shao L, Colmegna I, Goronzy JJ, Weyand CM. Telomerase insufficiency in rheumatoid arthritis. Proc Natl Acad Sci U S A. 2009;106(11):4360–5. doi: 10.1073/pnas.0811332106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thewissen M, Linsen L, Geusens P, Raus J, Stinissen P. Impaired activation-induced telomerase activity in PBMC of early but not chronic rheumatoid arthritis patients. Immunol Lett. 2005;100(2):205–10. doi: 10.1016/j.imlet.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 56.Honda M, Mengesha E, Albano S, Nichols WS, Wallace DJ, Metzger A, Klinenberg JR, Linker-Israeli M. Telomere shortening and decreased replicative potential, contrasted by continued proliferation of telomerase-positive CD8+CD28(lo) T cells in patients with systemic lupus erythematosus. Clin Immunol. 2001;99(2):211–221. doi: 10.1006/clim.2001.5023. [DOI] [PubMed] [Google Scholar]
- 57.Katayama Y, Kohriyama K. Telomerase activity in peripheral blood mononuclear cells of systemic connective tissue diseases. J Rheumatol. 2001;28(2):288–91. [PubMed] [Google Scholar]
- 58.Wu CH, Hsieh SC, Li KJ, Lu MC, Yu CL. Premature telomere shortening in polymorphonuclear neutrophils from patients with systemic lupus erythematosus is related to the lupus disease activity. Lupus. 2007;16(4):265–72. doi: 10.1177/0961203307077155. [DOI] [PubMed] [Google Scholar]
- 59.Artlett CM, Black CM, Briggs DC, Stevens CO, Welsh KI. Telomere reduction in scleroderma patients: a possible cause for chromosomal instability. Br J Rheumatol. 1996;35(8):732–7. doi: 10.1093/rheumatology/35.8.732. [DOI] [PubMed] [Google Scholar]
- 60.Vogt S, Iking-Konert C, Hug F, Andrassy K, Hansch GM. Shortening of telomeres: Evidence for replicative senescence of T cells derived from patients with Wegener’s granulomatosis. Kidney Int. 2003;63(6):2144–51. doi: 10.1046/j.1523-1755.2003.00037.x. [DOI] [PubMed] [Google Scholar]
- 61.Guan JZ, Maeda T, Sugano M, Oyama J, Higuchi Y, Suzuki T, Makino N. An analysis of telomere length in sarcoidosis. J Gerontol A Biol Sci Med Sci. 2007;62(11):1199–203. doi: 10.1093/gerona/62.11.1199. [DOI] [PubMed] [Google Scholar]
- 62.Finckh A, Liang MH, van Herckenrode CM, de Pablo P. Long-term impact of early treatment on radiographic progression in rheumatoid arthritis: A meta-analysis. Arthritis Rheum. 2006;55(6):864–72. doi: 10.1002/art.22353. [DOI] [PubMed] [Google Scholar]
- 63.Rantapaa-Dahlqvist S, de Jong BA, Berglin E, Hallmans G, Wadell G, Stenlund H, Sundin U, van Venrooij WJ. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum. 2003;48(10):2741–9. doi: 10.1002/art.11223. [DOI] [PubMed] [Google Scholar]
- 64.Nielen MM, van Schaardenburg D, Reesink HW, van de Stadt RJ, van der Horst-Bruinsma IE, de Koning MH, Habibuw MR, Vandenbroucke JP, Dijkmans BA. Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum. 2004;50(2):380–6. doi: 10.1002/art.20018. [DOI] [PubMed] [Google Scholar]
- 65.Chibnik LB, Mandl LA, Costenbader KH, Schur PH, Karlson EW. Comparison of threshold cutpoints and continuous measures of anti-cyclic citrullinated peptide antibodies in predicting future rheumatoid arthritis. J Rheumatol. 2009;36(4):706–11. doi: 10.3899/jrheum.080895. [DOI] [PMC free article] [PubMed] [Google Scholar]