

Abstract
Risk management is the systematic application of management policies, procedures, and practices to the tasks of analyzing, evaluating, controlling and monitoring risk (the effect of uncertainty on objectives). Clinical laboratories conduct a number of activities that could be considered risk management including verification of performance of new tests, troubleshooting instrument problems and responding to physician complaints. Development of a quality control plan for a laboratory test requires a process map of the testing process with consideration for weak steps in the preanalytic, analytic and postanalytic phases of testing where there is an increased probability of errors. Control processes that either prevent or improve the detection of errors can be implemented at these weak points in the testing process to enhance the overall quality of the test result. This manuscript is based on a presentation at the 2nd International Symposium on Point of Care Testing held at King Faisal Specialist Hospital in Riyadh, Saudi Arabia on October 12-13, 2010. Risk management principles will be reviewed and progress towards adopting a new Clinical and Laboratory Standards Institute Guideline for developing laboratory quality control plans based on risk management will be discussed.
Introduction to Risk and Risk Management
Risk is defined in ISO31000 as the effect of uncertainty on objectives, whether positive or negative.1 For healthcare, risk is generally understood to mean the chance of suffering or encountering harm or loss.2 So, risk is essentially the potential for harm to occur to a patient or the possibility of an error that can lead to patient harm. Risk can be estimated through a combination of the probability of harm and the severity of that harm.3 There are two methods to reduce the risk of harm to a patient:
prevent the error from occurring which averts harm to the patient, or
detect the error before it can harm the patient.
Risk management is the identification, assessment, and prioritization of risks followed by coordinated and economical application of resources to minimize, monitor, and control the probability and/or impact of unfortunate events or to maximize the realization of opportunities.4,5 Risk management is essentially the systematic application of management policies, procedures, and practices to the tasks of analyzing, evaluating, controlling and monitoring risk.6
The terms risk and risk management may seem unfamiliar in the clinical laboratory, but technical staff and laboratory directors conduct a number of activities that could be considered risk management in the day-to-day operations of a laboratory. The performance of new tests is evaluated before use in patient care, control sample failures are investigated for instrument and reagent problems, and management responds to physician complaints. When incorrect results are reported, the staff must determine and correct the cause, and report the correct results. If patients were treated based on incorrect results, management must estimate the harm that occurred to the patient and take steps to prevent similar incidents in the future. So, risk management is not a new concept, just a formal description of activities that laboratories are already doing as part of their quality assurance program to prevent errors and reduce harm to a patient.
Sources of Laboratory Error
Understanding weaknesses in the testing process is a first step to developing a quality control plan based on risk management. Laboratories should create a process map that outlines all the steps of the testing process from physician order to reporting the result. A process map basically follows the path of the sample from the patient through transportation, receipt and analysis in the lab to reporting of result. This process map should include preanalytic, analytic, and postanalytic processes required to generate a test result that can be acted on by a clinician.
Weak steps in the testing processes are those that have a higher probability of generating an error. These can be identified through prior experience with similar instrumentation or from information collected from the manufacturer and other users about the test and method, how the test will be utilized in diagnosing or managing the patient, the laboratory environment and staff who will perform the test, and local regulatory and accreditation requirements that mandate control over specific aspects of the testing process (Figure 1). This information will be utilized to develop a quality control plan specific to the device, the laboratory, and the health-care setting that reduces the risk of harm to the patient, and meets regulatory requirements for quality of testing by the laboratory. To identify weaknesses in the testing process that could lead to error, laboratories need to acknowledge that all medical devices can fail when subjected to the right conditions (environment, operator or device sources of error). Realizing those conditions that may cause device failure and taking steps to protect a testing device from exposure to those conditions is the foundation of a quality control plan.
Figure 1.
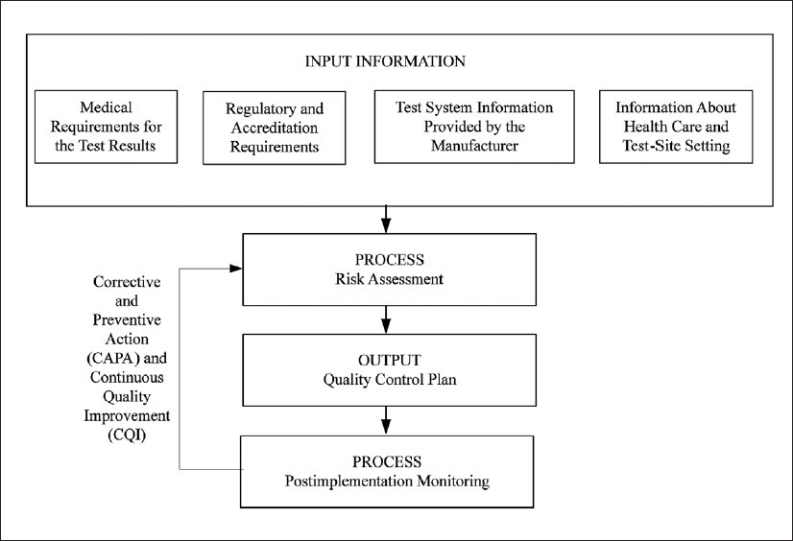
Process to develop and continually improve a quality control plan. (Obtained with permission from CLSI. Laboratory Quality Control Based on Risk Management; Proposed Guideline. CLSI document EP23-P. Wayne, PA: Clinical and Laboratory Standards Institute; 2010.)
Most errors occur in the preanalytic or postanalytic phases of testing, outside of the laboratory and beyond the supervision of laboratory staff.7 Preanalytic concerns should include the physician order (is there an order and was it transcribed correctly), was the patient prepared for the test (if fasting or withholding medication is required), was the appropriate preservative used to collect the specimen, was the specimen collected at the expected time, and was the specimen promptly transported to the laboratory, (protected from freezing or heating)? Reagents, controls, calibrators and other testing supplies must be shipped to the laboratory where they may be exposed to conditions (heating and freezing) that could compromise test results. Postanalytical processes should consider how the test was reported and communicated to the ordering physician since manual transcription is prone to more errors than automated reporting systems using computer interfaces. But computers are also not foolproof, and instrument interfaces have been known to occasionally report incorrect results to the wrong patients or associate results with the wrong test due to glitches in the interface communication. Verbal communication also has the potential to be misunderstood and communication of critical, life-threatening values can be mixed-up unless the results are written down and confirmed by read-back to the caller. The prevalence of errors in the preanlytic and postanalytic phases does not mean that the laboratory and analytic phase is free of errors. Failure to verify instrument performance prior to patient testing, incorrect maintenance, wrong calibrator setpoints, use of expired reagents, aliquotting errors, flawed calculations and dilution factors can all be sources of analytic error that occur within the laboratory.
There are, thus, many sources of error to consider throughout the testing process. Of primary consideration is the impact of the environment, the operator, and the analysis on the quality of test results. Temperature can freeze or overheat sensitive reagents and compromise results, but so can humidity, light and even altitude. Operators can inadvertently misidentify a patient and associate the labeling of the sample with the wrong patient. So the optimum laboratory control processes will be worthless if the specimen is mislabeled with another patient's identification. Testing by clinical personnel at the point of care is more prone to errors than analyses conducted by experienced laboratory professionals with training in error recognition and prevention. Analyzers can fail despite proper operation if incorrect calibrator factors are programmed or samples are incorrectly applied, aliquoted or diluted. So, considerations for common sources of environmental, operator and analyzer error should be considered when developing a quality control plan.
Control Samples
Weak steps in the testing process are sites for control processes to either prevent errors before they happen or detect errors before they can harm the patient. Historical quality control arose from the industrial setting where factories analyzed samples of a product to ensure that the product met specifications and the production line was operating as expected. In the laboratory, a control sample of known analyte concentration, sometimes called a “quality control” sample or “QC” sample, is analyzed like a patient sample. If the instrument produces a result within an acceptable tolerance of the target concentration, then the measurement system is assumed to be stable and operating as expected. The final result from measurement system is the sum of all factors affecting the result including the instrument, reagent, operator and environment.
Control samples have a number of advantages and disadvantages. A control sample is a sample with known concentration that consists of a matrix similar to a patient sample, like plasma, serum or urine. Unfortunately, not all analytes are stable in a biological matrix, like glucose in whole blood or blood gases. So, preservatives and other stabilizers must be added to control samples to ensure the analyte recovery and test results are stable over time. The additives that stabilize control samples can change the manner in which some instrumentation interacts with the specimen, such that control samples behave differently than patient samples in the same test. This non-commutability of control samples prevents their use as accuracy based materials in determining bias between instruments of different makes and manufacturers, unless the sample is certified commutable and accuracy-based. However, stabilized control samples do offer a target range for analytes specific to make and model of measurment system that allow their use in determining ongoing stability of laboratory instrumentation. Control samples can be analyzed each day of testing (or more frequently for high volume testing) and if the test recovers the expected target results, then the laboratory staff know that the system is stable and patient results are acceptable.
Unfortunately, when control samples fail to recover expected results, something has failed in the testing process and the laboratory must troubleshoot the source of failure and correct it before patient testing can resume. Troubleshooting will work if testing occurs in batches of specimens where results can be held until control results (analyzed with each batch of specimens) are examined and compared to expected target concentrations. If the control results are acceptable, then the patient results can be released. This type of batch analysis can work for low volume patient testing where test results may not be needed on an immediate basis by the physician. With high volume automation and stat testing, patient results are continuously released using autoverification rules based on periodic analysis of control samples interspersed with patient specimens. Should a control sample fail to recover expected results, the laboratory must stop patient testing, correct the problem, then reanalyze patient specimens back to when the system was reporting acceptable results. Repeat testing can be expensive for both labor and reagent costs. Once the problem is found and corrected, some results may need to be corrected, and this can lead to physicians questioning the quality of the laboratory.
Control samples, however, are a good means of detecting systematic errors, but perform poorly in detecting random errors. Systematic errors are those that affect every test in a constant and predictable manner. Many instruments utilize bottles of liquid reagent to perform hundreds of tests. These reagents may stay on an instrument for a number of hours to days, so analysis of control samples periodically or with each day of testing confirms that the reagent is still viable. Analysis of control samples does a good job at detecting errors that affect control samples the same as patient samples, like reagent deterioration, errors in reagent preparation, improper storage, incorrect operator technique, and wrong pipette or calibration settings. However, if a single patient sample should have a clot or drug that interferes with the reagent, the analysis of control samples will not be impacted and cannot detect the error. Such random errors affect individual samples in an unpredictable fashion, like clots, bubbles and interfering drugs and substances. Analysis of control samples does a poor job at detecting random errors.
Optimally, the laboratory needs control processes that function more than periodically. The laboratory needs to get to fully automated analyzers that prevent errors upfront and provide assured quality with every specimen.
Other Control Processes
Newer instrumentation has a variety of control processes built-in the device. There are analyzers with electronic controls and system checks performed automatically to detect electronic operation as well as reagent function. Blood gas analyzers, like the Instrumentation Laboratories GEM and Radiometer's ABL80, detect baseline sensor signals before and after each specimen. If each sensor does not display a characteristic signal, the flow cells may be blocked by a clot or bubble, and the system can initiate corrective action flushing, back-flushing the flow cell until the sensors return to expected operation. If not, the analyzer can shut down individual sensors or the entire sensor array until manual staff intervention. These processes occur with each sample to detect certain types of errors, specimen clots and bubbles that may block specific sensors specimen flow path. Other types of control processes may be engineered by the manufacturer into each test card or strip of unit-use testing devices, like the positive/negative control area on stool guaiac cards, and the control line on pregnancy, rapid strep and drug tests. These built-in controls test the viability of the reagents on the test (storage and expiration), adequate sample application, absence of interfering substances (clots, viscous urines, and drug adulterants), as well as timing and appropriate visual interpretation of test results by the operator with each test. Still other types of control processes may include barcoding of reagents to prevent use past the expiration date, device lockouts that prevent operation if control samples have not been analyzed or fail to recover expected target concentration, security codes to prevent inadvertent changes to instrument settings, and disposable pipette tips to prevent sample carryover. There are thus a variety of control processes available on different models of laboratory instrumentation, and each process is intended to reduce the risk of specific types of errors.
No single control process can therefore cover all devices and types of errors. Laboratory devices differ in design, technology, function and intended use. Some devices have internal checks which are performed automatically with every specimen, while the possibility of other errors is reduced through engineering by the manufacturer into the device. For example, the barcoding of expiration date and lot number on each bottle of reagent prevents use of reagents past their stamped expiration date and the requirement of entering lot number into the instrument reduces the possibility of using a lot number whose performance has not been previously verified. Barcoding of expiration date and lot number, however, does not verify the stability of reagents once they are opened on the instrument. Periodic analysis of control samples may better determine open bottle stability. So, the historical analysis of control samples has provided labs with some degree of assurance and analysis of control samples over the past several decades and will continue to plan an important role in future quality assurance in combination with the built-in controls and on-board chemical and biological control processes found in newer devices.
Developing a quality plan surrounding a laboratory device requires a partnership between the manufacturer and the laboratory.8 Information about the function of instrument control processes is needed from the manufacturer to increase the user's understanding of overall device quality assurance requirements and so informed decisions can be made regarding which control processes are suitable for certain errors in the laboratory setting.8 Some sources of error may be detected automatically by the device and prevented, while others may require the laboratory to do something, like analyze control samples periodically and on receipt of reagent shipments, or perform specific maintenance. Clear communication of potential sources of error and delineation of laboratory and manufacturer roles for how to detect and prevent errors is needed in order to develop a quality control plan.
Developing a Quality Control Plan
The Clinical and Laboratory Standards Institute (CLSI) is developing a guideline; Laboratory Quality Control Based on Risk Management. This guideline, EP23, describes good laboratory practice for developing a quality control plan based on manufacturer's risk mitigation information, applicable regulatory and accreditation requirements, and the individual healthcare and laboratory setting. Information collected about the instrument from the manufacturer, peer literature and other users of the product is combined with information about the individual healthcare and laboratory setting, and the unique regulatory and accreditation requirements and processed through a risk assessment to develop a laboratory specific quality control plan (Figure 1). This plan is an optimized balance of control sample analysis combined with manufacturer engineered control processes in the instrument and laboratory implemented control processes to minimize risk of error and harm to a patient when using the instrument for laboratory testing. Once implemented, the QC plan is monitored for continued errors and physician complaints. When trends are apparent, the source of errors is investigated and this new information is processed through a new risk assessment to determine if changes to the QC plan are needed to maintain risk to a clinically acceptable level. This is the corrective action and continual improvement cycle (Figure 1).
A risk assessment starts with identification of a potential risk or error (called hazard identification). Once identified, the probability and severity of harm are estimated. Take, for example, the risk of an untrained operator using a point-of-care testing device. The hazard is “operation by an untrained operator”. The probability of harm can be estimated as frequent=once per week, probable=once per month, or remote=once per year or greater. The severity of harm if the device is run by an untrained operator is unknown, but could be serious=injury or impairment requiring medical intervention, rather than negligible=inconvenience/discomfort or minor=temporary injury or impairment not requiring medical treatment. The risk can be estimated by combining probability of harm with severity of harm in a simple 3×3 matrix to evaluate the clinical acceptability of the risk (Table 1). More detailed 4×4, 5×5, or even 10×10 matrices can be developed to estimate risk, but these necessarily require more granular determinations of the exact probability and severity of harm to the patient. In risk management literature, the ability to detect an error, detectability, is also factored into the estimate, but for simplicity, detectability can be assumed to be zero or worse case scenario. Thus, risk in our simple example will depend only on prevention of the error or severity of harm if an error occurs.
Table 1.
Risk Acceptability Matrix
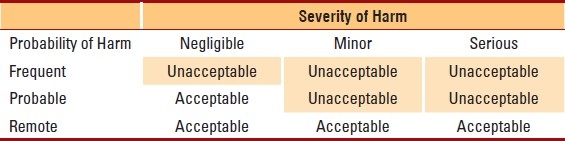
In our example, the risk of an untrained operator using the device will depend on the setting. In a central laboratory where the test is performed by medical technologists, all who are well supervised and experienced, the probability of this hazard is remote. However, in a point-of-care setting where operation is by a variety of clinical staff, possibly in a shared department location where there is little supervision, anyone can walk up to the device and attempt to run a test. In this setting the risk of an untrained operator performing testing is much greater (probable to frequent probability of harm). In both settings, if an error occurs from operation by an untrained operator, the harm could be serious to the patient (depending on the test and how the test is used in medical management). Combining the probability and severity of harm, we can estimate the risk in a laboratory setting to be clinically acceptable, but in the point-of-care setting risk is unacceptable and additional control measures will be needed to reduce the risk (Table 1).
Now, consider an instrument that has operator lockout features where the operator must enter their identification before the instrument will unlock and allow testing. If only operator identifications on a list of trained operators can unlock the instrument, the probability of harm may be reduced to probable or even remote, and thus the risk of using such a device with operator lockout features is now clinically acceptable. This example demonstrates how a manufacturer engineered process, like operator lockout, can reduce the risk of certain errors to improve the quality of test results.
This risk assessment process is repeated for each risk or potential for error identified through weak steps in the process map. If the risk of error from manufacturer recommended and engineered control processes is not clinically acceptable, then additional control processes must be implemented by the laboratory to reduce the risk to a clinically acceptable level. The sum of all risks identified and control processes to mitigate those risks (manufacturer provided and laboratory implemented) becomes the laboratory's QC plan specific to this device and laboratory setting. This plan is then checked against regulatory/accreditation requirements to ensure conformance with recommendations and signed by the laboratory director as the QC plan for that instrument. A single QC plan may cover multiple tests on the same instrument or refer to multiple instruments of the same make/model within an institution depending on the clinical application of the specific test results and availability of manufacturer control processes on the device.
Once implemented, the QC plan is monitored for continuous improvement. Physician complaints, instrument failures, or increasing error trends warrant investigation, corrective action and a new risk assessment. The laboratory should ask if the cause of the error is a new risk/hazard not previously considered, or greater probability of error or severity of harm than estimated in the initial risk assessment. This new information can now be factored into the risk analysis to determine if current control processes are adequate to reduce risk to a clinically acceptable level or if additional control processes are required. In this manner, the laboratory's QC plan defines a strategy for continuous improvement and sets benchmarks for monitoring the effectiveness of the QC plan through the frequency of complaints, instrument failures and trends in error rates. The CLSI EP23 document describing how to develop laboratory QC plans based on risk management is currently in the committee voting stages towards publication as an approved guideline.
Summary
Risk management is an industrial process for managing the potential for error, and although the terminology may be unfamiliar, the clinical laboratory performs a variety of risk management activities in day-to-day operation. The CLSI EP23 guideline simply formalizes the process. Newer laboratory instrumentation and point-of-care testing devices incorporate a number of control processes, some that rely on traditional analysis of control samples, and others engineered into the device to check, monitor or otherwise control specific aspects of the instrument operation. Risk management is not a means to reduce or eliminate the frequency of analyzing control samples, since laboratories must minimally meet manufacturer recommendations and accreditation agency regulations. Rather risk management helps laboratories find the optimal balance between traditional quality control (the analysis of control samples) and other control processes such that each risk in operating the instrument is rationalized with a control process to reduce that risk. The sum of all control processes represents the laboratory's QC plan, a plan that is scientifically supported by the risk assessment process. Once implemented the effectiveness of the QC plan is monitored through trends in error rates, and when issues are noted, corrective action is taken, risk is reassessed, and the plan is modified as needed to maintain risk to a clinically acceptable level. In this manner, risk management promotes continuous quality improvement. CLSI EP23 thus translates the industrial principles of risk management for practical use in the clinical laboratory, and will be a useful in support of the laboratory's overall quality management systems.
REFERENCES
- 1.International Organization for Standardization. ISO31000: Risk management – Principles and guidelines. ISO, Geneva, Switzerland. 2009:24. [Google Scholar]
- 2.Webster's Dictionary and Thesaurus. Landoll, Ashland, OH, USA. 1993 [Google Scholar]
- 3.International Organization for Standardization/International Electrotechnical Commission. ISO/IEC Guide 51, Safety aspects - Guidelines for their inclusion in standards. ISO, Geneva, Switzerland. 1999:10. [Google Scholar]
- 4.International Organization for Standardization/International Electrotechnical Commission. ISO/IEC Guide 73, Risk management – Vocabulary. ISO, Geneva, Switzerland. 2009:15. [Google Scholar]
- 5.Hubbard D. The Failure of Risk Management: Why It's Broken and How to Fix It. John Wiley & Sons, Indianapolis, IN, USA. 2009:46. [Google Scholar]
- 6.International Organization for Standardization. ISO14971: Medical devices - Application of risk management to medical devices. ISO, Geneva, Switzerland. 2007:82. [Google Scholar]
- 7.Bonini P, Plebani M, Ceriotti F, Rubboli F. Errors in laboratory medicine. Clin Chem. 2002;48:691–8. [PubMed] [Google Scholar]
- 8.International Organization for Standardization. ISO15198. Clinical laboratory medicine: In vitro diagnostic medical devices. Validation of user quality control procedures by the manufacturer. ISO, Geneva, Switzerland. 2004:10. [Google Scholar]