

Abstract
Galanin is a neuropeptide with multiple inhibitory actions on neurotransmission and memory. In Alzheimer's disease (AD), increased galanin-containing fibers hyperinnervate cholinergic neurons within the basal forebrain in association with a decline in cognition. We generated transgenic mice (GAL-tg) that overexpress galanin under the control of the dopamine β-hydroxylase promoter to study the neurochemical and behavioral sequelae of a mouse model of galanin overexpression in AD. Overexpression of galanin was associated with a reduction in the number of identifiable neurons producing acetylcholine in the horizontal limb of the diagonal band. Behavioral phenotyping indicated that GAL-tgs displayed normal general health and sensory and motor abilities; however, GAL-tg mice showed selective performance deficits on the Morris spatial navigational task and the social transmission of food preference olfactory memory test. These results suggest that elevated expression of galanin contributes to the neurochemical and cognitive impairments characteristic of AD.
Galanin acts as an inhibitory neuromodulator in the neural circuitry mediating learning and memory (1–3). Galanin and its receptors are distributed in brain regions that regulate cognitive processes, including the basal forebrain, hippocampus and cerebral cortex (4–6). Galanin reduces the evoked release of neurotransmitters, including glutamate, acetylcholine, norepinephrine, and dopamine (7–11), inhibits adenylate cyclase activity (12), and lowers the firing rate of serotonergic and noradrenergic neurons in the dorsal raphe and locus coeruleus, respectively (13, 14). In hippocampal slices, galanin reduces long-term potentiation and cholinergically induced excitatory postsynaptic potentials (15, 16). In rodents, the administration of galanin into the lateral ventricles, hippocampus, and medial septum/diagonal band impairs performance during learning and memory tasks, including delayed nonmatching to position, the Morris water maze, T-maze delayed alternation, starburst radial maze, spontaneous alternation, and passive avoidance tests (17–24).
In Alzheimer's disease (AD), most neurotransmitters decline in association with neurodegeneration; however, galanin is a notable exception. The expression of galanin progressively increases in the basal forebrain in AD (25–27), and galanin-containing fibers and terminals form a dense plexus surrounding the remaining cholinergic cell bodies within the nucleus basalis of Meynert, reaching concentrations twice that of age-matched controls (28). In addition, high levels of galanin continue to be expressed in the surviving neurons of the locus coeruleus in AD (29, 30). The overexpression of galanin in AD may contribute to the cognitive deficits characteristic of this disease. To test the hypothesis that galanin plays a role in the cognitive decline associated with AD, we developed a line of transgenic mice that overexpress galanin [GAL-transgenic (tg)] in discrete regions of the nervous system and evaluated the neurochemical and behavioral consequences of this genetic manipulation.
Materials and Methods
All experiments were approved by the National Institute of Mental Health or University of Washington Animal Care and Use Committees and conformed to National Institutes of Health and U.S. Department of Agriculture guidelines.
Generation of Galanin Transgenic Mice.
To overexpress galanin in a manner resembling the hyperinnervation of galanin seen in the AD forebrain, galanin gene expression was targeted to noradrenergic neurons. The mouse galanin gene was coupled to the human dopamine β-hydroxylase (hDBH) promoter, as previously reported (Fig. 1A) (31). For neurochemical and behavioral phenotyping, GAL-tg mice were backcrossed into C57BL/6J for seven generations, to avoid complications of mixed genetic background and strains with unusual alleles relevant to memory tasks (32, 33). All mice in these studies were genotyped by dot hybridization of genomic DNA to confirm the presence of the hDBH transgene, as previously described (Fig. 1B) (34).
Figure 1.

Galanin/human dopamine β-hydroxylase (hDBH) DNA construct (A) and representative dot hybridizations demonstrate hDBH DNA present in the GAL-tg but not in WT (B).
Gene Expression.
In situ hybridization assays were performed as previously reported (35). Analysis of galanin mRNA levels in the locus coeruleus (LC) of GAL-tg and wild-type (WT) mice (male 2–4 months old; n = 7 each) was performed in Experiment (Exp.) 1. Qualitative assessment of galanin mRNA expression in the GAL-tg compared with WT brain (11- to 12-month-old female; n = 5 each) was performed in Exp. 2. Coronal sections (20 μm) were hybridized with 35S-labeled riboprobes complementary to the coding sequences of the rat galanin gene (680 bp cDNA, kindly provided by Maria Vrontakis, University of Manitoba, Winnipeg, Canada) in Exp. 1, and the mouse galanin gene (493 bp cDNA kindly provided by James Hyde, University of Kentucky, Lexington, KY) in Exp. 2. After 16-h hybridizations (0.25 μg/ml⋅kb of probe), slides were dipped in NTB-2 emulsion (Eastman Kodak) and exposed for 7 (Exp. 1) or 23 days (Exp. 2). Slides were counterstained with cresyl violet and viewed under light- and dark-field microscopy for the presence of silver grain clusters. Relative optical density of LC signals was determined by computerized image analysis (Imaging Research, St. Catherine's, ON, Canada) (35). Specificities of antisense probes were determined by hybridization with mouse and rat 35S-labeled sense probes, yielding no detectable signal.
RIA.
An RIA for total galanin content in WT (n = 3) and GAL-tg (n = 3) forebrain was conducted as previously described (36), with minor modifications. Galanin antiserum (diluted 1:80,000) was directed against rat galanin and showed a 35 and 15% crossreactivity with porcine and human galanin, respectively, and no crossreactivity with any other neuropeptides examined. Limits of detection were 1.2 fmol/tube. Intraassay variability was <10%.
Stereological and Histological Analysis.
For stereological cell counting, age-matched (6-month-old) male WT (n = 5) and GAL-tg (n = 5) mice were perfused transcardially with 4% paraformaldehyde in phosphate buffer (PB), cryoprotected in 30% PB, sucrose and cut at 40 μm on a freezing microtome. Immunohistochemistry was performed with human polyclonal choline acetyltransferase (ChAT) antisera (1:1,000; a gift of L. Hirsch, University of Kentucky, Lexington, KY) and polyclonal galanin antisera (1:10,000; Peninsula Laboratories) (37). Total numbers of ChAT-immunoreactive neurons within cholinergic basal forebrain (CBF) subfields were determined with the optical fractionator method (38). Briefly, the CBF was outlined at low magnification (×4 objective), and at least 30% of the outlined region was measured with dissector frames (4420 μm2) and a ×100 planar oil immersion lens (1.4 numerical aperture). A systematic random design was used. Although the average section thickness was 30 μm, neurons were counted only within a 20-μm tissue height, with top and bottom guard heights of 5 μm. Antibody penetration was uniform throughout the entire section thickness (38).
Behavioral Phenotyping.
Mice (n = 11 male GAL-tg, 10 female GAL-tg, 16 male WT, 9 female WT, age-matched littermates) were evaluated on a multitiered behavioral phenotyping strategy, as previously described (39–41). All mice were evaluated on standard quantitative measures of general health, neurological reflexes, and sensory and motor abilities, to avoid false positives caused by either potential health problems or specific deficits in sensory or motor functions. Learning and memory were evaluated in the cohort of GAL-tg and WT littermate control mice at 8, 16, and 24 months of age in the Morris water task, with the use of standard methods and equipment, as previously reported (41) (published as supplemental data on the PNAS web site, www.pnas.org). The same mice were tested at age 26 months with the social transmission of food preference memory task, with methods as previously described (42, 43) and as detailed in supplemental data.
Statistical Analyses.
Behavioral data were analyzed by repeated-measures ANOVA. Newman–Keuls post hoc analysis compared probe trial scores for trained vs. nontrained quadrants. Student's unpaired t test was used to assess galanin mRNA and peptide concentrations, numbers of ChAT-containing cells, and measures of general health, reflexes, acoustic startle, and rotarod performance. Results of statistical tests were considered significant at P < 0.05.
Results
Overexpression of Galanin mRNA and Galanin Peptide in the GAL-tg.
Quantitative analysis of galanin expression revealed an approximately 5-fold increase in galanin mRNA signal levels in the locus coeruleus of GAL-tg mice compared with WTs (P < 0.0001; Fig. 2), and in other brainstem nuclei known to express DBH (44). Some ectopic expression of galanin mRNA was noted in the piriform and entorhinal cortices and in the subiculum, which may reflect the modest promiscuity of this hDBH promoter, as previously discussed (45).
Figure 2.

Photomicrographs illustrating typical galanin mRNA expression in the locus coeruleus of WT (n = 7) (A) and GAL-tg (n = 7) (B). Galanin mRNA levels were approximately 5-fold higher in GAL-tg. (Bar = 250 μm.)
RIA confirmed a corresponding elevation in galanin peptide content, which was approximately 2-fold higher in the forebrain of GAL-tgs compared with their WT controls (7.3 ± 0.1 vs. 3.8 ± 0.3 pmol/g wet tissue weight, respectively; P < 0.01). Immunocytochemistry for galanin-like immunoreactivity revealed increased intensity of staining for cell bodies in the locus coeruleus, medial septum, and entorhinal and piriform cortices, and increased fiber density in the hippocampus and septum (Figs. 3 and 4).
Figure 3.

Low-power photomicrographs illustrating examples of galanin-like immunoreactivity in the hippocampus of a WT (A) and GAL-tg (B), corresponding to atlas plate 56 of the Franklin and Paxinos atlas (66), and showing increased galaninergic fiber density in the dentate gyrus and lacunosum moleculare layers in the GAL-tg compared with WT. (Bar = 1,000 μm.)
Figure 4.

Photomicrographs of the septal region of a WT (A–C) and GAL-tg (D–F). (A) High-power image of the medial septal region showing scattered thin galanin-containing fibers in a WT. Note the lightly labeled galanin-containing cell bodies (arrowheads). (B) Low-power image of septal region in a WT. Boxes indicate the regions from which high-power photomicrographs were taken. (C) High-power image of the lateral septum in a WT. (D) High-power photomicrograph of thickened galanin-containing fibers in the medial septal region of a GAL-tg. Note the darkly labeled galanin-containing neuron (arrowhead). (E) Low-power image of septal region in a GAL-tg. Boxes indicate the regions from which high-power photomicrographs were taken. (F) High-power image showing thickened and twisted galanin-containing fibers in the lateral septum of a GAL-tg. These are similar to those seen in patients with AD. CC, corpus callosum; CD, caudate nucleus; LS, lateral septum; LV, lateral ventricle; MS, medial septum. (Bars: A, C, D, F = 50 μm; B and E = 200 μm.)
Behavior.
General health, body weight, and home cage behaviors were normal in the GAL-tg (Table 1 of supplemental data). Neurological reflexes, including eye blink, ear twitch, and righting reflex, were normal. Performance during sensory and motor tasks, including olfaction, acoustic startle, hot plate, Digiscan (Accuscan, Columbus, OH) open field and accelerating rotarod, was not significantly different between GAL-tg and WT (Fig. 5). These apparently normal baseline functions indicated that the GAL-tg were physically able to perform the procedures required for the cognitive tasks.
Figure 5.

Motor and sensory abilities were normal in GAL-tg mice. No differences between genotypes were detected in A, either general exploratory locomotion or habituation to a novel open field over a 2-h session; (B) vertical rearing in the open field; (C) motor coordination and balance on an accelerating rotarod over a 5- min session; (D) acoustic startle flinch amplitude; (E) pain sensitivity on the hot plate test; (F) olfactory ability on a 10-min scent preference test. *, P = 0.004, familiar vs. unfamiliar scent.
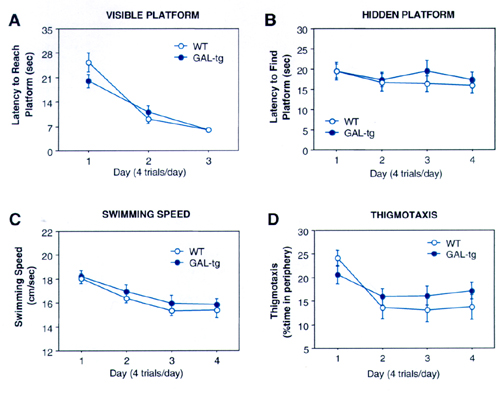
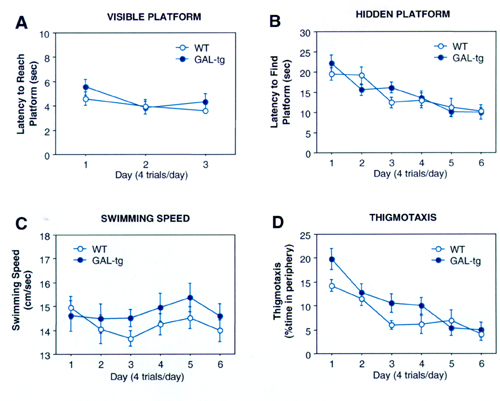
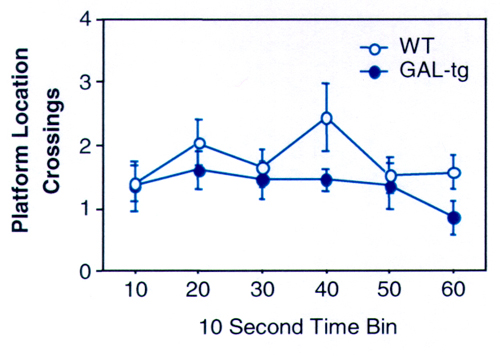
GAL-tg mice failed to display selective search during the Morris probe trial, in the absence of the escape platform, at 8, 16, and 24 months of age (Fig. 6 E–G). WT controls displayed normal selective search by preferentially crossing the location where the platform had been during the training trials (F3,69 = 5.06, P < 0.005 at 8 months; F3,72 = 15.36, P < 0.001 at 16 months; F3,57 = 8.09, P < 0.001 at 24 months). WT displayed a higher percent preference for the trained quadrant at 8 (F1,43 = 8.63; P < 0.005) and 16 months of age (F1,43 = 4.62; P < 0.05). Preference was not significantly different between genotypes at 24 months of age (F1,33 = 1.76; P = 0.19), perhaps reflecting either the general cognitive decline in both genotypes with aging or the reduced “n ” and statistical power, with natural attrition. No differences were detected between genotypes on the visible and hidden platform training trials. Swim speed, swim pattern, and thigmotaxis were not significantly different between genotypes at any age, on the visible platform, hidden platform, or probe trials. Analysis of swim pattern by 10-sec time bins during the probe trial revealed a similar temporal pattern of approaching or swimming away from the previous location of the platform (supplemental data, www.pnas.org), although total crossings over the previously trained location of the hidden platform differed between genotypes. This profile indicates that the GAL-tg were competent on all procedural components of the Morris water task but were unable to remember the environmental room cues necessary to solve the spatial navigation task.
Figure 6.

The GAL-tg mice failed either to learn or remember the environmental landmarks necessary to solve the Morris water maze memory task. GAL-tg mice (n = 11 males, 10 females) and WT controls (n = 16 males, 9 females) were tested for cognitive abilities on a spatial navigational task at 8, 16, and 24 months of age. WT mice showed selective search on the probe trial, whereas the GAL-tg did not. GAL-tg were not significantly different from WT controls on acquisition of the visible platform or hidden platform tasks. (A) Visible platform acquisition; (B) hidden platform acquisition; (C) swim speed; (D) thigmotaxis, swimming within 8 cm of the circumference of the pool, A–D at 16 months of age; (E) transfer test at 8 months of age; (F) transfer test at 16 months of age; (G) transfer test at 24 months of age. This highly specific deficit on the probe trials reflects an inability to learn and/or recall the association between the environmental room cues and the spatial location of the hidden platform. Data are expressed as the mean ± SEM; *, P < 0.05 compared with the trained quadrant.
In the olfactory memory task (Fig. 7), WT observer mice showed a significant preference for the cued food odor (F1,17 = 11.28, P = 0.01). Although an independent test confirmed that GAL-tg could smell a novel odor (Fig. 5F), the GAL-tg showed no preference for the previously cued food odor 24 h after the end of the exposure to the demonstrator mouse (F1,21 = 2.12, not significant).
Figure 7.

Social transmission of food preference over a 24 h delay was normal in WT control mice (n = 9), which consumed more chow mixed with the previously cued scent than with the novel uncued scent. Social transmission of food preference was significantly impaired in the GAL-tg (n = 11), which did not show a significant preference for chow mixed with the scent previously cued by the demonstrator mouse. Data are expressed as the mean ± SEM; *, P = 0.010, cued vs. noncued.
Number of Cholinergic Neurons in the Basal Forebrain.
To investigate the cellular mechanisms underlying the observed cognitive deficit in GAL-tg, we examined the effect of galanin overexpression on the number of identifiable cholinergic neurons within the medial septum (MS), vertical (VDB), and horizontal (HDB) limbs of the diagonal band, and nucleus basalis of Meynert (NB). These neurons provide the major cholinergic innervation to the hippocampus and entire cortical mantle. The numbers of ChAT-containing cells were dramatically reduced in the HDB of GAL-tg, compared with WT controls (P < 0.0002, Fig. 8 Inset), whereas no differences were detected in numbers of ChAT-containing cells between genotypes in MS, VDB, or NB.
Figure 8.

Photomicrographs and histogram (Inset) illustrating the total number of identifiable neurons containing ChAT within the horizontal limb of the diagonal band between young adult WT (n = 5) and GAL-tg (n = 5). (Bar = 50 μm.) Data are expressed as the mean ± SEM; *, P < 0.0002.
Discussion
Targeted overexpression of the mouse galanin gene coupled to the hDBH promoter produced transgenic mice with anatomically discrete overexpression of galanin mRNA and galanin peptide. Increased density of galanin-containing fibers appeared in the hippocampus and septum of GAL-tg mice, consistent with the known projections of the locus coeruleus to these regions (6, 44). The distribution and magnitude of galanin overexpression and hyperinnervation simulate the patterns of increased galanin in the locus coeruleus and basal forebrain in AD (26, 27, 29, 30).
The genetically targeted overexpression of galanin in the GAL-tg was associated with a significant reduction in the number of identifiable ChAT-containing neurons in the HDB of these animals. Although the present experiments cannot distinguish between the death of cholinergic neurons and diminished expression of ChAT (to below the limits of detection), the disappearance of detectable cholinergic neurons is reminiscent of the loss observed in the AD basal forebrain (27, 29). The anatomically selective effects of galanin overexpression on subpopulations of cholinergic neurons in the basal forebrain in GAL-tg may reflect the differential distribution of galaninergic fibers that innervate these neurons. In the rat, the central portions of the MS/VDB contain numerous cholinergic cells that are devoid of galanin-containing fibers. The lateral border of the MS/VDB is heavily innervated with galanin-containing fibers but contains very few cholinergic neurons (46). The HDB presents a different profile. High concentrations of galanin-containing terminals overlap with the cholinergic neurons of the HDB, and in the mouse, expression of galanin mRNA is higher in the HDB than in other cholinergic areas (J.G.H., unpublished observations). Moreover, a differential distribution of galanin receptor subtypes has been reported among the basal forebrain nuclei in the rat. One report indicates that only the GalR1 receptor subtype is expressed in the HDB, whereas both the GalR1 and GalR2 subtypes are expressed in the VDB (5). GalR1, acting through inhibitory Gi/o-coupled mechanisms, is implicated in both neuronal development and plasticity, whereas GalR2, acting through an excitatory mechanism, is postulated to promote survival of cholinergic neurons (4). The anatomically discrete localization of galanin receptor subtypes coupled with their reportedly different signal transduction mechanisms and the differential expression of galanin among the basal forebrain nuclei may contribute to the selective reduction in ChAT-containing cholinergic neurons within the HDB of GAL-tg mice.
The deficits in learning and memory observed in GAL-tg mice are consistent with previous reports of memory loss induced by the central administration of galanin in rats and mice (17–24). Selective deficits on the probe trial of the Morris water task are consistent with our earlier findings in Sprague–Dawley rats, in which intraventricularly administered galanin induced probe trial search deficits, following a normal acquisition curve for the visible and hidden platform training trials (24). Similar dissociations between training and probe trial performance have been reported in other experimental models, including rats with entorhinal cortex lesions and mice with mutations in the genes for adenylyl cyclase, Lis1, and the serotonin 5-HT2C receptor (47–50). This behavioral phenotype may reflect alternative strategies to solve the Morris water task when the platform is present during training, as evidenced by the impairment of spatial memory using environmental cues when the platform is removed. Common alternative strategies include learning to swim faster and to stay within the annulus of the circular pool that contains the platform. However, swim speed was not significantly different between GAL-tg and WT, and swimming in the outer annulus of the pool (thigmotaxis) was not significantly different between genotypes. Another possibility is that GAL-tg mice recognize more quickly than WT controls that the platform location has changed and begin to search other quadrants for its new location. However, our time bin analysis showed that search patterns in both GAL-tg and WT did not change over the course of the probe trial. Normal performance of the GAL-tg on neurological, sensory, and motor tests argues against an interpretation that either motor or sensory deficits contribute to the performance deficits of the GAL-tg on the Morris water task, supporting the inference of these animals having selective cognitive deficits. Dissociation of performance on the acquisition vs. the probe trial components of this task is consistent with current theories of the role of the cortex vs. the hippocampus in cued-place recognition habit learning versus flexible-place navigational declarative learning (47, 51–54). The type of learning and memory impairment exhibited by the GAL-tg mice appears to be selective for the more difficult portion of the Morris task, particularly the ability to store, retrieve, or attend to the more subtle environmental cues necessary to generate a cognitive map and solve the transfer task. Notably, the GAL-tg mice showed similar memory deficits at 8, 16, and 24 months of age. The overexpression of galanin in the GAL-tg and its associated cognitive deficits at these ages is analogous to the moderate-to-severe levels of galanin overexpression (26–30) and memory loss characteristic of the later stages of AD.
Corroboration of cognitive deficits in GAL-tg mice was seen in their poor performance on the social transmission of food preference test. This memory task demands different sensory and motor skills than the Morris task and measures olfactory as opposed to spatial learning and memory (42, 43). Although olfaction was apparently normal in the GAL-tg mice, their ability to remember a familiar scent was absent, providing further support for the conclusion that the overexpression of galanin impairs memory processes. Intriguingly, the HDB (where in these animals severe reductions in ChAT-producing cells were seen) provides the major cholinergic innervation to the olfactory bulb (55).
The molecular and cellular mechanisms underlying the learning and memory deficits seen in GAL-tg mice remain to be fully elucidated; however, recent studies of GAL-tg and GAL null mutant mice suggest that genetically induced modifications of galanin expression alter the excitability of hippocampal circuitry (31). Evoked glutamate release from hippocampal slices is significantly lower in GAL-tg compared with WT controls. Long-term potentiation generated by stimulation of the perforant path is enhanced in galanin knockouts and reduced in GAL-tg compared with WT controls. We infer that efferent projections of noradrenergic neurons in the brainstem that overexpress galanin are the source of the hyperinnervation of galaninergic fibers in the hippocampus and contribute to memory deficits in the GAL-tg. However, it is conceivable that the ectopic expression of galanin in the entorhinal cortex of GAL-tg also contributes to the hyperinnervation and behavioral phenomena observed in these animals, because there are strong projections from the entorhinal cortex to the hippocampus through the perforant pathway (56).
Galanin is not among the genes identified in linkage analyses with AD, and no correlations have been reported between the presence of either amyloid neuritic plaques or neurofibrillary tangles and galanin overexpression in the Alzheimer's brain. However, galanin and β-amyloid peptide synergize to inhibit acetylcholine release from rat cortical synaptosomes (57), and our observation that the number of acetylcholine-producing neurons is reduced in the basal forebrain of the GAL-tg suggests that the induction of galanin and the loss of cholinergic function may be causally linked in AD. The neuroanatomical source of the increased galanin in AD is presently under investigation.
The physiological trigger for galanin overexpression in AD is a fascinating and enigmatic question. Galaninergic systems appear to be highly activated as AD progresses, producing the aberrant thickened and twisted galaninergic fibers, unlike the normal straight fiber profile (25–27). We propose that up-regulation of galanin occurs as a consequence of the primary underlying pathology of the disease. Up-regulation of galanin mRNA is consistently seen after neuronal damage, including axotomy, inflammation, tetrodotoxin, reserpine, and colchicine treatments (58–61). Compensatory and possibly neuroprotective actions of galanin are implied by reports that galanin pretreatment attenuates the physiological consequences of ischemia, concussion, and seizures (7, 62, 63). Also, in a recent description of mice that have a targeted deletion of the galanin gene, O'Meara and coworkers concluded that endogenous galanin plays an important role as a neuroprotective agent (64). Galanin overexpression in AD may reflect an attempt to compensate for neurodegeneration in AD, perhaps early in the development of the disease process. Recently, it has been hypothesized that chronic demands for compensatory adjustment may lead to pathological alterations in cellular function (1, 65). The causative agents in AD may create a milieu wherein galanin-containing neurons must work harder to meet the reparatory demands of neuroprotection (59). However, our findings suggest that the putative redemptory benefits of excess galanin may be offset by the liability of an accompanying loss of cognitive function, possibly attributable to the reduced cholinergic activity in discrete areas of the basal forebrain.
From the perspective of quality of life for the Alzheimer's patient, it will be important to understand the consequences of galanin overexpression, in its context as both a neuroprotectant and a potential adversary of cognition. The transgenic mouse model of galanin overexpression provides an opportunity to explore the neurobiological actions of endogenous galanin and to investigate the therapeutic potential of galanin receptor antagonists in the treatment of AD.
Supplementary Material
Acknowledgments
We thank J. Syed, T. Teal, D. Teklemichael, and K. Gobeske for their technical assistance, and R. Morris, H. Eichenbaum, G. Wenk, and A. Markowska for their insights into interpretation of behavioral tests. We thank M. Ghatei and S. Bloom for performing the RIA for galanin. We thank R. Palmiter for providing the DBH promoter, and R. Palmiter and B. Hille for critical commentary. This work was supported by the Pediatric Epilepsy Research Center and the Alzheimer's Research Center at the University of Washington (R.A.S.), National Science Foundation Grant IBN97201 (R.A.S.), National Institutes of Health Grants HD27142 (R.A.S.), 3U54-HD12629 (R.A.S.), AG05136 (R.A.S.), AG10668 (E.J.M.), AG09466 (E.J.M.), and the National Institute of Mental Health Intramural Research Program (A.H., C.C.W., M.L., M.G., and J.N.C.).
Abbreviations
- AD
Alzheimer's disease
- GAL-tg
galanin transgenic
- WT
wild type
- hDBH
human dopamine β-hydroxylase
- ChAT
choline acetyltransferase
- MS
medial septum
- VDB
vertical limb diagonal band
- HDB
horizontal limb diagonal band
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Hökfelt T, Broberger C, Xu Z Q, Sergeyev V, Ubink R, Diez M. Neuropharmacology. 2000;39:1337–1356. doi: 10.1016/s0028-3908(00)00010-1. [DOI] [PubMed] [Google Scholar]
- 2.Hökfelt T, Bartfai T, Crawley J. Galanin: Basic Research Discoveries and Therapeutic Implications. New York: NY Acad. Sci.; 1998. [PubMed] [Google Scholar]
- 3.Kask K, Langel Ü, Bartfai T. Cell Mol Neurobiol. 1995;15:653–673. doi: 10.1007/BF02071130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Branchek T A, Smith K E, Gerald C, Walker M W. Trends Pharmacol Sci. 2000;21:109–117. doi: 10.1016/s0165-6147(00)01446-2. [DOI] [PubMed] [Google Scholar]
- 5.O'Donnell D, Ahmad S, Wahlestedt C, Walker P. J Comp Neurol. 1999;409:469–481. [PubMed] [Google Scholar]
- 6.Xu Z Q, Shi T J, Hökfelt T. J Comp Neurol. 1998;392:227–251. doi: 10.1002/(sici)1096-9861(19980309)392:2<227::aid-cne6>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 7.Mazarati A M, Liu H, Soomets U, Sankar R, Shin D, Katsumori H, Langel Ü, Wasterlain C G. J Neurosci. 1998;18:10070–10077. doi: 10.1523/JNEUROSCI.18-23-10070.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kinney G A, Emmerson P J, Miller R J. J Neurosci. 1998;18:3489–3500. doi: 10.1523/JNEUROSCI.18-10-03489.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fisone G, Wu C F, Consolo S, Nordström Ö, Brynne N, Bartfai T, Melander T, Hökfelt T. Proc Natl Acad Sci USA. 1987;84:7339–7343. doi: 10.1073/pnas.84.20.7339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robinson J K, Zocchi A, Pert A, Crawley J N. Brain Res. 1996;709:81–87. doi: 10.1016/0006-8993(95)01307-5. [DOI] [PubMed] [Google Scholar]
- 11.Gopalan C, Tian Y, Moore K E, Lookingland K J. Neuroendocrinology. 1993;58:287–293. doi: 10.1159/000126552. [DOI] [PubMed] [Google Scholar]
- 12.Karelson E, Langel Ü. Neuropeptides. 1998;32:197–210. doi: 10.1016/s0143-4179(98)90038-5. [DOI] [PubMed] [Google Scholar]
- 13.Xu Z Q, Zhang X, Pieribone V A, Grillner S, Hökfelt T. Neuroscience. 1998;87:79–94. doi: 10.1016/s0306-4522(98)00151-1. [DOI] [PubMed] [Google Scholar]
- 14.Pieribone V A, Xu Z Q, Zhang X, Grillner S, Bartfai T, Hökfelt T. Neuroscience. 1995;64:861–874. doi: 10.1016/0306-4522(94)00450-j. [DOI] [PubMed] [Google Scholar]
- 15.Sakurai E, Maeda T, Kaneko S, Akaike A, Satoh M. Neurosci Lett. 1996;212:21–24. doi: 10.1016/0304-3940(96)12772-5. [DOI] [PubMed] [Google Scholar]
- 16.Dutar P, Lamour Y, Nicoll R A. Eur J Pharmacol. 1989;164:355–360. doi: 10.1016/0014-2999(89)90477-9. [DOI] [PubMed] [Google Scholar]
- 17.Mastropaolo J, Nadi N S, Ostrowski N L, Crawley J N. Proc Natl Acad Sci USA. 1988;85:9841–9845. doi: 10.1073/pnas.85.24.9841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sundström E, Archer T, Melander T, Hökfelt T. Neurosci Lett. 1988;88:331–335. doi: 10.1016/0304-3940(88)90233-9. [DOI] [PubMed] [Google Scholar]
- 19.Givens B S, Olton D S, Crawley J N. Brain Res. 1992;582:71–77. doi: 10.1016/0006-8993(92)90318-4. [DOI] [PubMed] [Google Scholar]
- 20.Robinson J K, Crawley J N. Behav Neurosci. 1993;107:458–467. doi: 10.1037//0735-7044.107.3.458. [DOI] [PubMed] [Google Scholar]
- 21.Ögren S O, Kehr J, Schött P A. Neuroscience. 1996;75:1127–1140. doi: 10.1016/0306-4522(96)00215-1. [DOI] [PubMed] [Google Scholar]
- 22.Stefani M R, Gold P E. Brain Res. 1998;813:50–56. doi: 10.1016/s0006-8993(98)00876-2. [DOI] [PubMed] [Google Scholar]
- 23.McDonald M P, Willard L B, Wenk G L, Crawley J N. J Neurosci. 1998;18:5078–5085. doi: 10.1523/JNEUROSCI.18-13-05078.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gleason T C, Dreiling J L, Crawley J N. Neuropeptides. 1999;33:265–270. doi: 10.1054/npep.1999.0044. [DOI] [PubMed] [Google Scholar]
- 25.Bowser R, Kordower J H, Mufson E J. Brain Pathol. 1997;7:723–730. doi: 10.1111/j.1750-3639.1997.tb01058.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chan-Palay V. J Comp Neurol. 1988;273:543–557. doi: 10.1002/cne.902730409. [DOI] [PubMed] [Google Scholar]
- 27.Mufson E J, Cochran E, Benzing W, Kordower J H. Dementia. 1993;4:237–250. doi: 10.1159/000107329. [DOI] [PubMed] [Google Scholar]
- 28.Beal M F, MacGarvey U, Swartz K J. Ann Neurol. 1990;28:157–161. doi: 10.1002/ana.410280207. [DOI] [PubMed] [Google Scholar]
- 29.Chan-Palay V. Prog Brain Res. 1991;88:625–630. doi: 10.1016/s0079-6123(08)63839-x. [DOI] [PubMed] [Google Scholar]
- 30.Miller M A, Kolb P E, Leverenz J B, Peskind E R, Raskind M A. J Neurochem. 1999;73:2028–2036. [PubMed] [Google Scholar]
- 31.Mazarati A M, Hohmann J G, Bacon A, Liu H, Sankar R, Steiner R A, Wynick D, Wasterlain C G. J Neurosci. 2000;20:6276–6281. doi: 10.1523/JNEUROSCI.20-16-06276.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crawley J N, Belknap J K, Collins A, Crabbe J C, Frankel W, Henderson N, Hitzemann R J, Maxson S C, Miner L L, Silva A J, et al. Psychopharmacology. 1997;132:107–124. doi: 10.1007/s002130050327. [DOI] [PubMed] [Google Scholar]
- 33.Wehner J N, Silva A. Ment Retard Dev Disabil Res Rev. 1996;2:243–248. [Google Scholar]
- 34.Cadd G G, Hoyle G W, Quaife C J, Marck B, Matsumoto A M, Brinster R L, Palmiter R D. Mol Endocrinol. 1992;6:1951–1960. doi: 10.1210/mend.6.11.1480181. [DOI] [PubMed] [Google Scholar]
- 35.Chan Y Y, Steiner R A, Clifton D K. Endocrinology. 1996;137:1319–1325. doi: 10.1210/endo.137.4.8625906. [DOI] [PubMed] [Google Scholar]
- 36.O'Halloran D, Jones P M, Steel J H, Gon G, Giaid A, Ghatei M A, Polak J M, Bloom S R. Endocrinology. 1990;127:467–475. doi: 10.1210/endo-127-1-467. [DOI] [PubMed] [Google Scholar]
- 37.de Lacalle S, Kulkarni S, Mufson E J. Exp Neurol. 1997;146:361–366. doi: 10.1006/exnr.1997.6532. [DOI] [PubMed] [Google Scholar]
- 38.Gilmor M L, Erickson J D, Varoqui H, Hersh L B, Bennett D A, Cochran E J, Mufson E J, Levey A I. J Comp Neurol. 1999;411:693–704. [PubMed] [Google Scholar]
- 39.Crawley J N, Paylor R. Horm Behav. 1997;31:197–211. doi: 10.1006/hbeh.1997.1382. [DOI] [PubMed] [Google Scholar]
- 40.Crawley J N. What's Wrong with My Mouse? Behavioral Phenotyping of Transgenic and Knockout Mice. New York: Wiley; 2000. [Google Scholar]
- 41.Paylor R, Nguyen M, Crawley J N, Patrick J, Beaudet A, Orr-Urtreger A. Learn Mem. 1998;5:302–316. [PMC free article] [PubMed] [Google Scholar]
- 42.Kogan J H, Frankland P W, Blendy J A, Coblentz J, Marowitz Z, Schutz G, Silva A J. Curr Biol. 1997;7:1–11. doi: 10.1016/s0960-9822(06)00022-4. [DOI] [PubMed] [Google Scholar]
- 43.Valsecchi P, Galef B G. Int J Comp Psychol. 1989;2:245–256. [Google Scholar]
- 44.Swanson L W, Hartman B K. J Comp Neurol. 1975;163:467–505. doi: 10.1002/cne.901630406. [DOI] [PubMed] [Google Scholar]
- 45.Hoyle G W, Mercer E H, Palmiter R D, Brinster R L. J Neurosci. 1994;14:2455–2463. doi: 10.1523/JNEUROSCI.14-05-02455.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Henderson Z, Morris N. J Comp Neurol. 1997;383:82–93. doi: 10.1002/(sici)1096-9861(19970623)383:1<82::aid-cne7>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 47.Schenk F, Morris R G. Exp Brain Res. 1985;58:11–28. doi: 10.1007/BF00238949. [DOI] [PubMed] [Google Scholar]
- 48.Wu Z L, Thomas S A, Villacres E C, Xia Z, Simmons M L, Chavkin C, Palmiter R D, Storm D R. Proc Natl Acad Sci USA. 1995;92:220–224. doi: 10.1073/pnas.92.1.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Paylor R, Hirotsune S, Gambello M J, Yuva-Paylor L, Crawley J N, Wynshaw-Boris A. Learn Mem. 1999;6:521–537. doi: 10.1101/lm.6.5.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tecott L H, Logue S F, Wehner J M, Kauer J A. Proc Natl Acad Sci USA. 1998;95:15026–15031. doi: 10.1073/pnas.95.25.15026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bunsey M, Eichenbaum H. Nature (London) 1996;379:255–257. doi: 10.1038/379255a0. [DOI] [PubMed] [Google Scholar]
- 52.Eichenbaum H, Stewart C, Morris R G. J Neurosci. 1990;10:3531–3542. doi: 10.1523/JNEUROSCI.10-11-03531.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Markowska A L. Neurobiol Aging. 1999;20:177–189. doi: 10.1016/s0197-4580(99)00031-7. [DOI] [PubMed] [Google Scholar]
- 54.Steele R J, Morris R G. Hippocampus. 1999;9:118–136. doi: 10.1002/(SICI)1098-1063(1999)9:2<118::AID-HIPO4>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 55.Sobreviela T, Jaffar S, Mufson E J. Neuroscience. 1998;87:447–461. doi: 10.1016/s0306-4522(98)00153-5. [DOI] [PubMed] [Google Scholar]
- 56.Stanfield B B, Caviness V S, Jr, Cowan W M. J Comp Neurol. 1979;185:461–483. doi: 10.1002/cne.901850304. [DOI] [PubMed] [Google Scholar]
- 57.Wang H, Wild K D, Shank R P, Lee D H. Neuropeptides. 1999;33:197–205. doi: 10.1054/npep.1999.0024. [DOI] [PubMed] [Google Scholar]
- 58.Zhang X, Verge V M, Wiesenfeld-Hallin Z, Piehl F, Hökfelt T. Exp Brain Res. 1993;93:450–461. doi: 10.1007/BF00229360. [DOI] [PubMed] [Google Scholar]
- 59.Hökfelt T, Broberger C, Diez M, Xu Z Q, Shi T, Kopp J, Zhang X, Holmberg K, Landry M, Koistinaho J. Horm Metab Res. 1999;31:330–334. doi: 10.1055/s-2007-978748. [DOI] [PubMed] [Google Scholar]
- 60.Austin M C, Cottingham S L, Paul S M, Crawley J N. Synapse. 1990;6:351–357. doi: 10.1002/syn.890060407. [DOI] [PubMed] [Google Scholar]
- 61.Cortes R, Ceccatelli S, Schalling M, Hökfelt T. Synapse. 1990;6:369–391. doi: 10.1002/syn.890060410. [DOI] [PubMed] [Google Scholar]
- 62.Ben-Ari Y, Lazdunski M. Eur J Pharmacol. 1989;165:331–332. doi: 10.1016/0014-2999(89)90732-2. [DOI] [PubMed] [Google Scholar]
- 63.Liu S, Lyeth B G, Hamm R J. J Neurotrauma. 1994;11:73–82. doi: 10.1089/neu.1994.11.73. [DOI] [PubMed] [Google Scholar]
- 64.O'Meara G, Coumis U, Ma S Y, Kehr J, Mahoney S, Bacon A, Allen S J, Holmes F, Kahl U, Wang F H, et al. Proc Natl Acad Sci USA. 2000;97:11569–11574. doi: 10.1073/pnas.210254597. . (First published October 3, 2000; 10.1073/pnas.210254597) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mesulam M M. Neuron. 1999;24:521–529. doi: 10.1016/s0896-6273(00)81109-5. [DOI] [PubMed] [Google Scholar]
- 66.Franklin K B, Paxinos G. The Mouse Brain in Stereotaxic Coordinates. San Diego: Academic; 1997. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}