

Summary
Impaired kidney function and subsequent skeletal responses play a critical role in disrupting phosphate balance in chronic kidney disease (CKD) patients with mineral and bone disorder (CKD-MBD). In patients with CKD-MBD, the inability of the kidney to maintain normal mineral ion balance affects bone remodeling to induce skeletal fracture and extraskeletal vascular calcification. In physiological conditions, bone-derived fibroblast growth factor 23 (FGF23) acts on the kidney to reduce serum phosphate and 1,25-dihydroxyvitamin D levels. In humans, increased bioactivity of FGF23 leads to increased urinary phosphate excretion, which induces hypophosphatemic diseases (e.g., rickets/osteomalacia). However, reduced FGF23 activity is associated with hyperphosphatemic diseases (e.g., tumoral calcinosis). In patients with CKD, high serum levels of FGF23 fail to reduce serum phosphate levels and lead to numerous complications, including vascular calcification, one of the important determinants of mortality of CKD-MBD patients. Of particular significance, molecular, biochemical and morphological changes in patients with CKD-MBD are mostly due to osteo-renal dysregulation of mineral ion metabolism. Furthermore, hyperphosphatemia can partly contribute to the development of secondary hyperparathyroidism in patients with CKD-MBD. Relatively new pharmacological agents including sevelamer hydrochloride, calcitriol analogs and cinacalcet hydrochloride are used either alone, or in combination, to minimize hyperphosphatemia and hyperparathyroidism associated complications to improve morbidity and mortality of CKD-MBD patients. This article will briefly summarize how osteo-renal miscommunication can induce phosphate toxicity, resulting in extensive tissue injuries.
Keywords: Klotho, fibroblast growth factor 23, vitamin D, parathyroid hormone, chronic kidney disease
INTRODUCTION
Bone provides the structural framework for the human body, protects internal organs and maintains mineral ion balance. Bone is a target organ for several hormones such as parathyroid hormone (PTH), 1,25-dihydroxyvitamin D [1,25(OH)2D], calcitonin and sex hormones. Recent studies found important roles of bone-derived factors including osteocalcin and fibroblast growth factor 23 (FGF23) in systemic regulation of glucose and phosphate metabolism, respectively (1–5). Optimal phosphate balance is essential for normal skeletal growth, development and maintenance. However, maintaining optimal phosphate levels is a significant clinical challenge in various osteo-renal diseases because phosphate toxicity (excessive retention of phosphate in the body) has a wide range of harmful effects and because its therapeutic control is difficult, particularly in patients with chronic kidney disease (CKD).
Phosphate balance in the body is coordinately regulated by complex cross-organ interactions that mainly involve the kidney, intestine, bone and parathyroid gland; functional impairments in any of these organs can lead to abnormal phosphate levels (Table 1). The body’s daily-required amount of phosphate is maintained through intestinal absorption from consumed foods, whereas the serum level of phosphate is maintained primarily by renal reabsorption/excretion of phosphate. In addition according to need, bone can also contribute to this homeostasis by providing phosphate thorough promoting skeletal resorption. The intestinal absorption and renal reabsorption of phosphate are partly mediated by the sodium-dependent phosphate (NaPi) transporter system that includes NaPi-2a (SLC34A1, kidney), NaPi-2b (SLC34A2, intestine) and NaPi-2c (SLC34A3, kidney). An electrogenic NaPi-2a transports three sodium ions into the proximal tubular epithelial cell for each phosphorus ion, whereas an electroneutral NaPi-2c transports two sodium ions for each phosphorus ion (7). It is believed that the basolateral Na+,K+-ATPase activity creates a low intracellular sodium microenvironment that facilitates phosphorus transport in the proximal tubular epithelial cells by the NaPi system (8–10). As for NaPi-2b, the sodium-phosphate transport cycle involves both voltage-dependent and independent stages that lead to the influx of three sodium ions with each phosphorus ion (11). Hormones that are either produced in the bone or act on the bone can affect the activities of the NaPi system. Our understanding of the FGF23-klotho system has provided new mechanistic insights into homeostatic regulation of phosphate metabolism through osteo-renal communication (12, 13).
Table 1.
| Hypophosphatemia |
| Autosomal dominant hypophosphatemic rickets |
| Autosomal recessive hypophosphatemic rickets |
| Chronic diarrhea |
| Diabetic ketoacidosis |
| Diuretics |
| Hormones (insulin, glucagon, cortisol) |
| Hyperparathyroidism |
| Metabolic acidosis |
| Renal transplantation |
| Renal tubular defects (Fanconi syndrome) |
| Respiratory alkalosis |
| Sepsis |
| Severe dietary deficiency |
| Tumor-induced rickets/osteomalacia |
| Vitamin D resistance/deficiency |
| X-linked hypophosphatemic rickets |
| Hyperphosphatemia |
| Acromegaly |
| Bisphosphonate therapy |
| Bowel infarction |
| Hemolysis |
| Hypoparathyroidism |
| Intravenous/oral phosphate therapy |
| Magnesium deficiency |
| Metabolic acidosis |
| Phosphate enema |
| Renal failure |
| Respiratory acidosis |
| Rhabdomyolysis |
| Tumor calcinosis |
| Tumor lysis syndrome |
| Vitamin D intoxication |
OSTEO-RENAL COMMUNICATION
Osteo-renal communication is vital for physiologic maintenance of both calcium and phosphate levels. Two hormones that act on bone, PTH and 1,25(OH)2D, markedly influence serum calcium and phosphate levels by affecting the rate of intestinal absorption, renal reabsorption and bone formation and resorption. A delicate balance between intracellular calcium-phosphate concentration and skeletal hydroxyapatite formation is also an important determinant of calcium-phosphate balance (14–19). PTH acts on bone to provoke resorption and to induce phosphaturia in the kidney; it also facilitates an increase in circulating levels of 1,25(OH)2D, which in turn enhance intestinal phosphate absorption. It is important to mention that PTH has a bimodal response to bone; in addition to its commonly known catabolic effects, a modest amount of PTH, usually provided by intermittent administration, can exert anabolic effects on bone (20–22). Although phosphate metabolism is partly regulated by both PTH and 1,25(OH)2D, the synthesis and release of these two hormones are mostly controlled by the serum levels of calcium (23). There are a number of review articles summarizing the current understanding of calcium metabolism, and this brief article will emphasize on osteo-renal regulation of phosphate metabolism.
Serum phosphate levels are mostly maintained by fine-tuning the reabsorption rate of renal phosphate. Filtered phosphate from the glomeruli is mainly reabsorbed in the proximal part of the tubules where NaPi-2a and NaPi-2c cotransporters mediate reabsorption (10, 24). The numbers of factors that can regulate NaPi activities are growing, and experimental studies have shown that PTH, FGF23 and klotho can suppress NaPi activities to increase urinary phosphate loss (10, 25–29), whereas 1,25(OH)2D can stimulate NaPi-2b to increase intestinal phosphate uptake (30). Even though both PTH and 1,25(OH)2D are involved in phosphate metabolism, there are human urinary phosphate wasting diseases with relatively normal levels of PTH and 1,25(OH)2D, suggesting the presence of novel phosphaturic factors. The search for such factors inducing phosphaturia in these osteo-renal diseases resulted in the identification of the FGF23-klotho system (12, 13).
The underlying molecular mechanisms of hypophosphatemia and subsequent skeletal anomalies in patients with X-linked hypophosphatemic rickets/osteomalacia (XLH), autosomal dominant hypophosphatemic rickets (ADHR) and tumor-induced rickets/osteomalacia (TIO) have been extensively studied in the last decade. The idea of the presence of a humoral phosphaturic factor came from the observation that renal phosphate reabsorption defects in patients with XLH was not corrected by renal transplantation from a healthy donor (31). In a similar line of observation, when the kidney of a Hyp mouse (a murine homolog of human XLH) with hypophosphatemia due to impaired renal phosphate reabsorption was transplanted to a wild-type mouse, the phosphate reabsorption defects of Hyp mice kidney no longer persists. This again suggests the presence of a humoral phosphaturic factor (32). The extensive genetic, molecular and biochemical studies on hypophosphatemic diseases such as XLH, ADHR and TIO have lead to the identification of FGF23 as a phosphaturic factor (12, 13).
FGF23-KLOTHO SYSTEM
FGF23 is a bone-derived protein with a FGF receptor (FGFR)-binding domain in the N-terminus and a potential klotho-interacting site at the C-terminus (33). The klotho gene encodes a type 1 membrane protein (8) with an extracellular domain that can be released from the plasma membrane by disintegrin and metalloproteinases (ADAM-10 and ADAM-17) (34). Klotho is present predominantly in the distal convoluted tubules of the kidney, the parathyroid gland and the epithelium of the choroid plexus in the brain (35). The restricted expression of klotho provides the tissue specificity for FGF23 function. The FGF23-FGFR interaction needs klotho as a cofactor to activate signaling phosphoproteins that include but are not limited to ERK and AKT (36, 37). 1,25(OH)2D can induce the expression of FGF23 in osteocytes, which in the presence of klotho can act on the kidney to suppress the expression of the 1-alpha hydroxylase [1α(OH)ase] gene and thereby reduce production of 1,25(OH)2D (38–40). It appears likely that the mutual regulation of FGF23 and 1,25(OH)2D forms a regulatory loop that plays a major role in maintaining mineral homeostasis (Fig. 1). Of relevance, osteocytes are derived from osteoblasts that become embedded in the bone matrix and are believed to be the source of numerous important endocrine factors including FGF23. Osteocytes also act as mechanosensors and can regulate bone formation by secreting sclerostin, which antagonizes canonical Wnt signaling (41, 42). In physiological conditions, osteo-renal communication through 1,25(OH)2D and FGF23 may fine-tune the levels of these essential factors to keep normal homeostatic balance of mineral ions (Fig. 1). Of physiologic significance, when renal function is preserved, an increased serum level of bone-derived FGF23 coincides with increased urinary excretion of phosphate.
Figure 1.
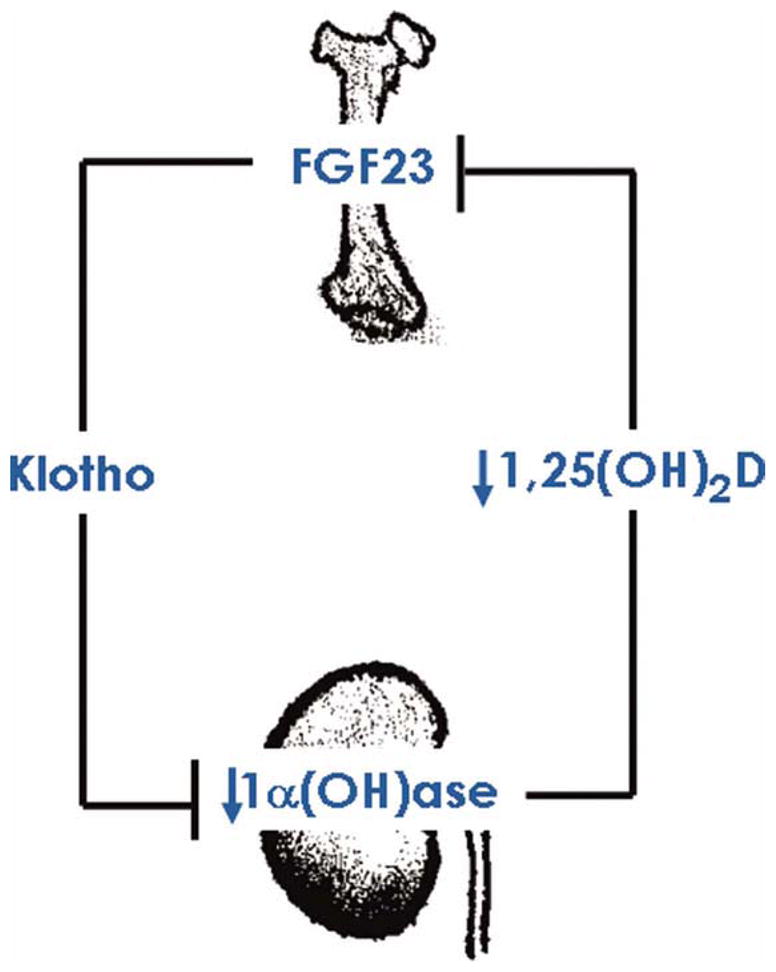
Simplified diagram showing osteo-renal communication of FGF23 and vitamin D. The osteocyte-derived FGF23 in presence of klotho can act on the kidney to suppress the expression of the 1α(OH)ase gene, and it thereby can reduce production of 1,25(OH)2D. Reduced levels of 1,25(OH)2D may have inhibitory effects on FGF23 production in bone. In physiological conditions, osteo-renal communication through 1,25(OH)2D and FGF23 may fine-tune their levels to maintain normal mineral ion balance. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Activation of the FGF23-klotho system promotes phosphate excretion and can induce phosphaturia by reducing NaPi-2a and NaPi-2c cotransporter activity (26, 28). Whether FGF23 can directly suppress NaPi cotransporter activity or FGF23-regulated molecules facilitate such suppression will need further clarification (43). Klotho is dominantly expressed in the distal tubules with week expression in proximal tubules (29). How a protein mostly produced in the distal tubules can influence the functions of proximal tubules is an ongoing area of research. Studies have shown that following FGF23 injection in mice, the activation of signaling phosphoproteins is detected mostly in the distal tubules (44). This observation raised the possibility that FGF23 might initially act on klotho-expressing distal tubules, generating a factor to exert phosphaturic effects on adjacent proximal tubules; some investigators speculated that secreted klotho protein might be one of such generated factors. Secreted klotho can facilitate deglycosylation of N-linked glycans on NaPi-2a protein to make it susceptible to degradation, reducing their numbers and activity in the proximal tubules (29). Hence, klotho might induce phosphaturia not only by acting as a coreceptor for FGF23, but may also do so by modulating the functions of NaPi proteins (29).
In both animal and human studies, chronic or pathological activation of the FGF23-klotho system consistently leads to hypophosphatemia. For instance, hypophosphatemia in patients with autosomal recessive hypophosphatemic rickets (ARHR) is accompanied by high serum levels of FGF23 (45). Similarly, FGF23 transgenic mice develop hypophosphatemia due to persistent phosphaturia, whereas Fgf23 knockout mice develop hyperphosphatemia due to renal retention of phosphate. Of relevance, the biochemical and morphological features of Fgf23 knockout mice mimic the clinical symptoms of patients with familial tumoral calcinosis (FTC), in which an inactivating mutation of the FGF23 gene leads to the development of hyperphosphatemia (46). The identification of the FGF23-klotho system has markedly enhanced our understanding of phosphate metabolism via the bone-kidney endocrine axis. In various disease conditions, the miscommunication of the bone-kidney axis leads to abnormal phosphate metabolism and associated complications.
OSTEO-RENAL MISCOMMUNICATION
A miscommunication between bone-derived FGF23 and kidney-derived klotho in patients with CKD can induce excessive phosphate accumulation in the body; in patients with CKD, advanced structural impairment of the kidney and reduced expression of klotho are responsible for the failure of FGF23 to reduce phosphate levels (47). Such excessive retention of phosphate in the body can cause a wide range of cellular and tissue injuries (Fig. 2). For example, vascular calcification in patients with CKD is mainly due to hyperphosphatemia (50–52). Similar vascular pathology in the form of cardiovascular calcification is also observed in various animal models including hyperphosphatemic klotho knockout mice (27, 53). Notably, reducing the phosphate burden from klotho knockout mice is sufficient to markedly reduce vascular calcification, despite the presence of considerably higher serum calcium and 1,25-dihydroxyvitamin D levels (27, 52, 53). Of particular importance, hyperphosphatemia is identified as independent risk factors for high mortality in CKD patients (54).
Figure 2.

Simplified diagram showing how osteo-renal miscommunication can impair serum phosphate balance to induce organ pathology. Modified from earlier publications (48, 49). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Elevated serum calcium and phosphate concentrations are usually considered main contributors to arterial calcification, and the estimation of calcium-phosphorus (CaxPi) product has long been used to predict vascular pathology in patients with CKD (55, 56). However, in some studies, instead of CaxPi product, a correlation was found only with serum phosphate levels in patients undergoing hemodialysis treatment with vascular calcification (57). In fact, the clinical utility of CaxPi product has been questioned in recent studies (58). It would be more reasonable to take into account different variables (namely calcium, phosphate and PTH) instead of calculating CaxPi products to determine the eventual outcome of CKD patients (59). Of clinical importance, in contrast to hyperphosphatemia, associations between serum calcium levels and cardiovascular events or disease outcomes of CKD patients are not yet clearly established. Although, a few studies have shown that hypercalcemia may have predictive value in determining the negative outcome of CKD patients (60, 61).
It is important to note that klotho is essential in FGF23-mediated regulation of phosphate. Functionally bioactive FGF23 protein failed to reduce serum phosphate levels in mice lacking klotho activity (either klotho knockout mice or Fgf23/klotho double knockout mice) (48, 62). Furthermore, genetically eliminating klotho function from hypophosphatemic phex mutant mice resulted in the generation of hyperphosphatemic phex/klotho double mutant mice, despite markedly increased serum FGF23 levels in double mutant mice (52, 49). In a similar line of observation, inactivation of klotho from hypophosphatemic FGF23 transgenic mice led to the development of hyperphosphatemia (63). In a human study with tumoral calcinosis, a loss-of-function homozygous missense mutation in the Klotho gene showed severe hyperphosphatemia despite high serum FGF23 levels in the affected patient (64). It is clear from the above-cited human and experimental studies that klotho is indispensable for FGF23-mediated homeostatic control of phosphate metabolism (3, 5, 48, 62, 65–67) and that osteo-renal miscommunication in the form of FGF23-klotho dysregulation can lead to phosphate imbalance (5, 43, 68, 69). It is necessary to mention that the effects of FGF23 on extra-renal tissues are not yet clearly defined. For example, FGF23 suppresses PTH secretion, probably through interacting with klotho (70, 71) (Fig. 3), whereas in response to hypocalcemia, klotho facilitates PTH secretion (8). Further studies are needed to understand these complex and often opposing effects. Recently, Hofman-Bang et al. showed increased expression of klotho, FGFR1 (IIIC) and Na+/K+-ATPase in the parathyroid glands of rats with CKD, although the investigator did find variability in the parathyroid expression of klotho and FGFR1, depending on the duration and stage of CKD (72). One of the likely scenarios is that increased levels of klotho may influence Na+/K+-ATPase to induce PTH production in response to hypocalcemia (72); whereas the FGF23-klotho-FGFR1 complex in the parathyroid gland may fine-tune such production by reducing PTH secretion in an effort to keep the homeostatic balance of mineral ions. These complex molecular interactions in the advanced stages of CKD may not be able to suppress PTH production, perhaps due to reduced expression of both klotho and FGFR expression, leading to secondary hyperparathyroidism (73, 74). The inability to influence PTH secretion following in vivo suppression of the parathyroid gland Na+/K+-ATPase activity, in response to acute hypocalcemia adds additional complexity in PTH metabolism (75).
Figure 3.
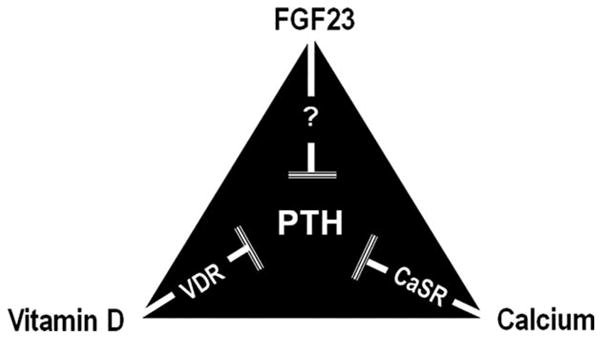
Simplified diagram showing factors that can exert inhibitory effects on PTH production, depending on the homeostatic balance of mineral ions. Please note that other factors can also directly influence PTH production but for simplicity, only relevant factors that are discussed in this article are included in this figure. Both vitamin D and calcium can exert inhibitory effects on PTH production by interacting with vitamin D receptor (VDR) and calcium-sensing receptors (CaSR), respectively. FGF23, possibly through interacting with klotho, may also exert inhibitory effects on PTH secretion.
One other issue that requires further study is the determination of whether PTH can directly induce FGF23 production through PTH receptors present in bone cells. In patients with Jansen’s metaphyseal chondrodysplasia, the constitutively active PTH/PTHrP receptor leads to asymptomatic hypercalcemia and hypophosphatemia despite low or undetectable serum levels of PTH and PTHrP (76). Interestingly, the serum FGF23 levels in these patients are increased, and whether constitutively active PTH/PTHrP receptors can increase production of such FGF23 is an area that needs exploration. Similarly, in FGF23 overexpressing mice, high serum levels of PTH are detected (77), while in the mouse primary hyperparathyroidism model, PTH has been shown to increase FGF23 levels (78). Of relevance, secondary hyperparathyroidism in patients with CKD is traditionally viewed as a consequence of reduced vitamin D activity and/or hyperphosphatemia; the discovery of the FGF23-klotho system has not only added additional insights (as briefly mentioned above) but has also helped us to understand how osteo-renal dysregulation can induce phosphate toxicity.
PHOSPHATE TOXICITY
Phosphate toxicity from excessive serum accumulation of phosphate has recently been shown to accelerate the mammalian aging process by inflicting wide-spread tissue injuries, reducing overall survival (79). Klotho-knockout mice develop phosphate toxicity as early as 3 weeks of age resulting in reduced body weight, kyphosis, hypogonadism and infertility, generalized tissue atrophy and reduced life span (27, 53, 62, 48, 79–82). Increased renal activity of NaPi-2a and NaPi-2c in klotho knockout mice leads to an increased renal retention of phosphate, which induces phosphate toxicity. Indeed, the widespread aging phenotypes in short-lived hyperphosphatemic klotho-knockout mice can be reduced by lowering phosphate burden in NaPi2a/klotho double knockout mice to prolong their survival (79). The hyperphosphatemic klotho-knockout mice are infertile, and reducing serum phosphate levels by eliminating NaPi-2a activity (i.e., NaPi2a/klotho double knockout mice) restored their fertility. More importantly, the NaPi2a/klotho double knockout mice lost fertility when fed a high phosphate diet, clearly suggesting that phosphate toxicity can affect fertility and thereby influence the aging process. Again, compared to the normal phosphate diet (0.6%) fed NaPi2a/klotho double knockout mice, high phosphate diet (1.2%) fed NaPi2a/klotho mutant mice develop phosphate toxicity, with premature aging-like features including generalized tissue atrophy and death by 15 weeks of age (79). These in vivo experimental observations clearly imply that phosphate toxicity can contribute to the progression of the mammalian aging process. In addition to affecting the aging process, increased dietary phosphate intake has recently been shown to stimulate lung tumor growth and size (83).
How excessive phosphate can induce toxicity resulting in tissue injury is unclear. One likely scenario is that it can exert cytotoxic effects to compromise the functional ability of various organ systems. Phosphate toxicity can induce apoptotic cell death in various tissues, and such cell death can be suppressed by reducing phosphate burden in mice (79). It is possible that cell death induced by phosphate toxicity can subsequently trigger inflammatory responses to enhance organ damage. As phosphate is essential for cell signaling activities, its imbalance may impair the homeostatic control of signaling activities that may eventually lead to cell and tissue damage. In a similar line of observation, excessive dietary phosphate has been found to activate the AKT-mediated signaling network that promotes lung tumorigenesis (83).
Failure to maintain optimal phosphate balance has a significant clinical consequence in CKD patients. Phosphate-restricted diet along with dialysis treatment is usually not enough to control serum phosphate levels and are recommended with phosphorus binders to further reduce serum phosphate levels. With time, new formulations of phosphorus binders have been developed to minimize side effects. For instance, once widely used aluminum-based phosphorus binders can induce anemia, myopathies, dementia and bone anomalies (84). Calcium-based binders, though somewhat effective, are also linked to vascular calcification in patients undergoing hemodialysis treatment (55). Recently, sevelamer hydrochloride, a noncalcium and non-aluminum-based binder have been shown to reduce vascular injury better than calcium-based binders (85, 86). Similarly, dialysis patients receiving calcium-based binders showed a significantly higher mortality than did sevelamer users (87). Further studies to determine the long-term safety of clinically used drugs, as well as investigational drugs including sevelamer carbonate (88) and lanthanum carbonate (89), will aid in designing the optimal therapeutic choice to help reduce serum phosphate levels to within the desirable range and to minimize associated complications.
CONCLUSION
Osteo-renal communication through bone-derived FGF23 and kidney-derived klotho is essential for physiologic regulation of phosphate balance and vitamin D homeostasis. In contrast, osteo-renal miscommunication through dysregulation of FGF23-klotho system provokes phosphate imbalance and induces pathological changes in a wide range of organs/tissues including blood vessels, bone and kidney. Of clinical importance, phosphate toxicity induced by excessive exogenous phosphate administration in humans can be fatal (90–93). Acute phosphate toxicity can provoke hypocalcemia and associated complications, whereas chronic phosphate toxicity can induce ectopic calcification. Taking into account human and experimental observations, maintaining phosphate balance appears to be important for a healthy life and for longevity, as phosphate imbalance due to osteo-renal miscommunication can induce serious debilitating complications.
Acknowledgments
Several original research works that formed the basis of this article are performed by Razzaque lab members (Dr. Teruyo Nakatani PhD, Dr. Mutsuko Ohnishi MD, PhD, Dr. Shigeko Kato, PhD, Dr. Kazuyoshi Uchihashi, MD, PhD, Dr. Junko Akiyoshi, MD, and Dr. Khadijah Turkistani, BDS) at the Harvard School of Dental Medicine, Boston, MA, and supported by a grant (R01-DK077276 to M.S.R.) from the National Institute of Diabetes and Digestive and Kidney Diseases. A part of this review article is based on earlier publications (5, 49). The author thanks Dr. William Horne, PhD, for critically reading the manuscript, and providing useful suggestions.
Abbreviations
- 1,25(OH)2D
1,25-dihydroxyvitamin D
- ADAM
A disintegrin and metalloproteinases
- ADHR
autosomal dominant hypophosphatemic rickets
- ARHR
autosomal recessive hypophosphatemic rickets
- CaxPi
calcium-phosphorus product
- CKD
chronic kidney disease
- CKD-MBD
chronic kidney disease with mineral and bone disorder
- FGF23
fibroblast growth factor 23
- FGFR
fibroblast growth factor receptor
- FTC
familial tumoral calcinosis
- NaPi
sodium-dependent phosphate transporter
- PTH
parathyroid hormone
- TIO
tumor-induced rickets/osteomalacia
- XLH
X-linked hypophosphatemic rickets/osteomalacia
References
- 1.Rosen CJ, Motyl KJ. No bones about it: insulin modulates skeletal remodeling. Cell. 2010;142:198–200. doi: 10.1016/j.cell.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 2.Razzaque MS. Osteocalcin: a pivotal mediator or an innocent bystander in energy metabolism? Nephrol Dial Transplant. 2011;26:42–45. doi: 10.1093/ndt/gfq721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Razzaque MS. FGF23-mediated regulation of systemic phosphate homeostasis: is Klotho an essential player? Am J Physiol Renal Physiol. 2009;296:F470–476. doi: 10.1152/ajprenal.90538.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fukagawa M, Kazama JJ. FGF23: its role in renal bone disease. Pediatr Nephrol. 2006;21:1802–1806. doi: 10.1007/s00467-006-0230-3. [DOI] [PubMed] [Google Scholar]
- 5.Razzaque MS. The FGF23-Klotho axis: endocrine regulation of phosphate homeostasis. Nat Rev Endocrinol. 2009;5:611–619. doi: 10.1038/nrendo.2009.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ghosh AK, Joshi SR. Disorders of calcium, phosphorus and magnesium metabolism. JAPI. 2008;56:613–621. [PubMed] [Google Scholar]
- 7.Forster IC, Hernando N, Biber J, Murer H. Proximal tubular handling of phosphate: a molecular perspective. Kidney Int. 2006;70:1548–1559. doi: 10.1038/sj.ki.5001813. [DOI] [PubMed] [Google Scholar]
- 8.Nabeshima Y, Imura H. alpha-Klotho: a regulator that integrates calcium homeostasis. Am J Nephrol. 2008;28:455–464. doi: 10.1159/000112824. [DOI] [PubMed] [Google Scholar]
- 9.Razzaque MS. Klotho and Na+,K+-ATPase activity: solving the calcium metabolism dilemma? Nephrol Dial Transplant. 2008;23:459–461. doi: 10.1093/ndt/gfm702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tenenhouse HS. Regulation of phosphorus homeostasis by the type iia na/phosphate cotransporter. Annu Rev Nutr. 2005;25:197–214. doi: 10.1146/annurev.nutr.25.050304.092642. [DOI] [PubMed] [Google Scholar]
- 11.Virkki LV, Biber J, Murer H, Forster IC. Phosphate transporters: a tale of two solute carrier families. Am J Physiol Renal Physiol. 2007;293:F643–654. doi: 10.1152/ajprenal.00228.2007. [DOI] [PubMed] [Google Scholar]
- 12.Yamashita T, Yoshioka M, Itoh N. Identification of a novel fibroblast growth factor, FGF-23, preferentially expressed in the ventrolateral thalamic nucleus of the brain. Biochem Biophys Res Commun. 2000;277:494–498. doi: 10.1006/bbrc.2000.3696. [DOI] [PubMed] [Google Scholar]
- 13.ADHR_Consortium. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. The ADHR Consortium. Nat Genet. 2000;26:345–348. doi: 10.1038/81664. [DOI] [PubMed] [Google Scholar]
- 14.Azari F, Vali H, Guerquin-Kern JL, Wu TD, Croisy A, Sears SK, Tabrizian M, McKee MD. Intracellular precipitation of hydroxyapatite mineral and implications for pathologic calcification. J Struct Biol. 2008;162:468–479. doi: 10.1016/j.jsb.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 15.Wenisch S, Stahl JP, Horas U, Heiss C, Kilian O, Trinkaus K, Hild A, Schnettler R. In vivo mechanisms of hydroxyapatite ceramic degradation by osteoclasts: fine structural microscopy. J Biomed Mater Res A. 2003;67:713–718. doi: 10.1002/jbm.a.10091. [DOI] [PubMed] [Google Scholar]
- 16.Beck GR., Jr Inorganic phosphate as a signaling molecule in osteoblast differentiation. J Cell Biochem. 2003;90:234–243. doi: 10.1002/jcb.10622. [DOI] [PubMed] [Google Scholar]
- 17.Berner YN, Shike M. Consequences of phosphate imbalance. Annu Rev Nutr. 1988;8:121–148. doi: 10.1146/annurev.nu.08.070188.001005. [DOI] [PubMed] [Google Scholar]
- 18.Ooi SW, Smillie AC, Kardos TB, Shepherd MG. Intracellular mineralization of Bacterionema matruchotii. Can J Microbiol. 1981;27:267–270. doi: 10.1139/m81-042. [DOI] [PubMed] [Google Scholar]
- 19.Betts F, Blumenthal NC, Posner AS, Becker GL, Lehninger AL. Atomic structure of intracellular amorphous calcium phosphate deposits. Proc Natl Acad Sci USA. 1975;72:2088–2090. doi: 10.1073/pnas.72.6.2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bauer W, Aub JC, Albright F. Studies of calcium and phosphorus metabolism: v. a study of the bone trabeculae as a readily available reserve supply of calcium. J Exp Med. 1929;49:145–162. doi: 10.1084/jem.49.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lindsay R, Nieves J, Formica C, Henneman E, Woelfert L, Shen V, Dempster D, Cosman F. Randomised controlled study of effect of parathyroid hormone on vertebral-bone mass and fracture incidence among postmenopausal women on oestrogen with osteoporosis. Lancet. 1997;350:550–555. doi: 10.1016/S0140-6736(97)02342-8. [DOI] [PubMed] [Google Scholar]
- 22.Reeve J, Tregear GW, Parsons JA. Priliminary trial of low doses of human parathyroid hormone 1–34 peptide in treatment of osteoporosis. Calcif Tissue Res. 1976;21:469–477. [PubMed] [Google Scholar]
- 23.Taylor JG, Bushinsky DA. Calcium and phosphorus homeostasis. Blood Purif. 2009;27:387–394. doi: 10.1159/000209740. [DOI] [PubMed] [Google Scholar]
- 24.Segawa H, Aranami F, Kaneko I, Tomoe Y, Miyamoto K. The roles of Na/Pi-II transporters in phosphate metabolism. Bone. 2009;45 (Suppl 1):S2–S7. doi: 10.1016/j.bone.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 25.Murer H, Forster I, Hilfiker H, Pfister M, Kaissling B, Lotscher M, Biber J. Cellular/molecular control of renal Na/Pi-co-transport. Kidney Int Suppl. 1998;65:S2–S10. [PubMed] [Google Scholar]
- 26.Gattineni J, Bates C, Twombley K, Dwarakanath V, Robinson ML, Goetz R, Mohammadi M, Baum M. FGF23 decreases renal NaPi-2a and NaPi-2c expression and induces hypophosphatemia in vivo predominantly via FGF receptor 1. Am J Physiol Renal Physiol. 2009;297:F282–291. doi: 10.1152/ajprenal.90742.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ohnishi M, Nakatani T, Lanske B, Razzaque MS. In vivo genetic evidence for suppressing vascular and soft-tissue calcification through the reduction of serum phosphate levels, even in the presence of high serum calcium and 1,25-Dihydroxyvitamin D levels. Circ Cardiovasc Genet. 2009;2:583–590. doi: 10.1161/CIRCGENETICS.108.847814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miyamoto K, Ito M, Kuwahata M, Kato S, Segawa H. Inhibition of intestinal sodium-dependent inorganic phosphate transport by fibroblast growth factor 23. Ther Apher Dial. 2005;9:331–335. doi: 10.1111/j.1744-9987.2005.00292.x. [DOI] [PubMed] [Google Scholar]
- 29.Hu MC, Shi M, Zhang J, Pastor J, Nakatani T, Lanske B, Razzaque MS, Rosenblatt KP, Baum MG, Kuro-o M, Moe OW. Klotho: a novel phosphaturic substance acting as an auto-crine enzyme in the renal proximal tubule. FASEB J. 2010;24:3438–3450. doi: 10.1096/fj.10-154765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sabbagh Y, O’ Brien SP, Song W, Boulanger JH, Stockmann A, Arbeeny C, Schiavi SC. Intestinal Npt2b plays a major role in phosphate absorption and homeostasis. J Am Soc Nephrol. 2009;20:2348–2358. doi: 10.1681/ASN.2009050559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morgan JM, Hawley WL, Chenoweth AI, Retan WJ, Diethelm AG. Renal transplantation in hypophosphatemia with vitamin D-resistant rickets. Arch Intern Med. 1974;134:549–552. [PubMed] [Google Scholar]
- 32.Nesbitt T, Coffman TM, Griffiths R, Drezner MK. Crosstransplantation of kidneys in normal and Hyp mice. Evidence that the Hyp mouse phenotype is unrelated to an intrinsic renal defect. J Clin Invest. 1992;89:1453–1459. doi: 10.1172/JCI115735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamashita T. Structural and biochemical properties of fibroblast growth factor 23. Ther Apher Dial. 2005;9:313–318. doi: 10.1111/j.1744-9987.2005.00288.x. [DOI] [PubMed] [Google Scholar]
- 34.Chen CD, Podvin S, Gillespie E, Leeman SE, Abraham CR. Insulin stimulates the cleavage and release of the extracellular domain of Klotho by ADAM10 and ADAM17. Proc Natl Acad Sci USA. 2007;104:19796–19801. doi: 10.1073/pnas.0709805104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matsumura Y, Aizawa H, Shiraki-Iida T, Nagai R, Kuro-o M, Nabeshima Y. Identification of the human klotho gene and its two transcripts encoding membrane and secreted klotho protein. Biochem Biophys Res Commun. 1998;242:626–630. doi: 10.1006/bbrc.1997.8019. [DOI] [PubMed] [Google Scholar]
- 36.Kuro-o M. Klotho as a regulator of fibroblast growth factor signaling and phosphate/calcium metabolism. Curr Opin Nephrol Hypertens. 2006;15:437–441. doi: 10.1097/01.mnh.0000232885.81142.83. [DOI] [PubMed] [Google Scholar]
- 37.Medici D, Razzaque MS, Deluca S, Rector TL, Hou B, Kang K, Goetz R, Mohammadi M, Kuro OM, Olsen BR, Lanske B. FGF-23-Klotho signaling stimulates proliferation and prevents vitamin D-induced apoptosis. J Cell Biol. 2008;182:459–465. doi: 10.1083/jcb.200803024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kolek OI, Hines ER, Jones MD, LeSueur LK, Lipko MA, Kiela PR, Collins JF, Haussler MR, Ghishan FK. 1alpha,25-Dihydroxyvitamin D3 upregulates FGF23 gene expression in bone: the final link in a renal-gastrointestinal-skeletal axis that controls phosphate transport. Am J Physiol Gastrointest Liver Physiol. 2005;289:G1036–G1042. doi: 10.1152/ajpgi.00243.2005. [DOI] [PubMed] [Google Scholar]
- 39.Ito M, Sakai Y, Furumoto M, Segawa H, Haito S, Yamanaka S, Nakamura R, Kuwahata M, Miyamoto K. Vitamin D and phosphate regulate fibroblast growth factor-23 in K-562 cells. Am J Physiol Endocrinol Metab. 2005;288:E1101–E1109. doi: 10.1152/ajpendo.00502.2004. [DOI] [PubMed] [Google Scholar]
- 40.Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, Fujita T, Nakahara K, Fukumoto S, Yamashita T. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004;19:429–435. doi: 10.1359/JBMR.0301264. [DOI] [PubMed] [Google Scholar]
- 41.Winkler DG, Sutherland MS, Ojala E, Turcott E, Geoghegan JC, Shpektor D, Skonier JE, Yu C, Latham JA. Sclerostin inhibition of Wnt-3a-induced C3H10T1/2 cell differentiation is indirect and mediated by bone morphogenetic proteins. J Biol Chem. 2005;280:2498–2502. doi: 10.1074/jbc.M400524200. [DOI] [PubMed] [Google Scholar]
- 42.Winkler DG, Sutherland MK, Geoghegan JC, Yu C, Hayes T, Skonier JE, Shpektor D, Jonas M, Kovacevich BR, Staehling-Hampton K, Appleby M, Brunkow ME, Latham JA. Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 2003;22:6267–6276. doi: 10.1093/emboj/cdg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cheng C-Y, Kuro-o M, Razzaque MS. Molecular regulation of phosphate metabolism by FGF23-klotho system. Adv Chronic Kidney Dis. 2011 doi: 10.1053/j.ackd.2010.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Farrow EG, Davis SI, Summers LJ, White KE. Initial FGF23-mediated signaling occurs in the distal convoluted tubule. J Am Soc Nephrol. 2009;20:955–960. doi: 10.1681/ASN.2008070783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lorenz-Depiereux B, Bastepe M, Benet-Pages A, Amyere M, Wagenstaller J, Muller-Barth U, Badenhoop K, Kaiser SM, Rittmaster RS, Shlossberg AH, Olivares JL, Loris C, Ramos FJ, Glorieux F, Vikkula M, Juppner H, Strom TM. DMP1 mutations in autosomal recessive hypophosphatemia implicate a bone matrix protein in the regulation of phosphate homeostasis. Nat Genet. 2006;38:1248–1250. doi: 10.1038/ng1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Benet-Pages A, Orlik P, Strom TM, Lorenz-Depiereux B. An FGF23 missense mutation causes familial tumoral calcinosis with hyperphosphatemia. Hum Mol Genet. 2005;14:385–390. doi: 10.1093/hmg/ddi034. [DOI] [PubMed] [Google Scholar]
- 47.Koh N, Fujimori T, Nishiguchi S, Tamori A, Shiomi S, Nakatani T, Sugimura K, Kishimoto T, Kinoshita S, Kuroki T, Nabeshima Y. Severely reduced production of klotho in human chronic renal failure kidney. Biochem Biophys Res Commun. 2001;280:1015–1020. doi: 10.1006/bbrc.2000.4226. [DOI] [PubMed] [Google Scholar]
- 48.Nakatani T, Ohnishi M, Razzaque MS. Inactivation of klotho function induces hyperphosphatemia even in presence of high serum fibroblast growth factor 23 levels in a genetically engineered hypophosphatemic (Hyp) mouse model. FASEB J. 2009;23:3702–3711. doi: 10.1096/fj.08-123992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Razzaque MS. Phosphate toxicity: new insights into an old problem. Clin Sci (Lond) 2011;120:91–97. doi: 10.1042/CS20100377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fukagawa M, Hamada Y, Nakanishi S, Tanaka M. The kidney and bone metabolism: Nephrologists’ point of view. J Bone Miner Metab. 2006;24:434–438. doi: 10.1007/s00774-006-0719-7. [DOI] [PubMed] [Google Scholar]
- 51.Memon F, El-Abbadi M, Nakatani T, Taguchi T, Lanske B, Razzaque MS. Does Fgf23-klotho activity influence vascular and soft tissue calcification through regulating mineral ion metabolism? Kidney Int. 2008;74:566–570. doi: 10.1038/ki.2008.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Razzaque MS. The dualistic role of vitamin D in vascular calcifications. Kidney Int. 2010 doi: 10.1038/ki.2010.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ohnishi M, Nakatani T, Lanske B, Razzaque MS. Reversal of mineral ion homeostasis and soft-tissue calcification of klotho knockout mice by deletion of vitamin D 1alpha-hydroxylase. Kidney Int. 2009;75:1166–1172. doi: 10.1038/ki.2009.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Voormolen N, Noordzij M, Grootendorst DC, Beetz I, Sijpkens YW, van Manen JG, Boeschoten EW, Huisman RM, Krediet RT, Dekker FW. High plasma phosphate as a risk factor for decline in renal function and mortality in pre-dialysis patients. Nephrol Dial Transplant. 2007;22:2909–2916. doi: 10.1093/ndt/gfm286. [DOI] [PubMed] [Google Scholar]
- 55.Frazao JM, Elangovan L, Maung HM, Chesney RW, Acchiardo SR, Bower JD, Kelley BJ, Rodriguez HJ, Norris KC, Robertson JA, Levine BS, Goodman WG, Gentile D, Mazess RB, Kyllo DM, Douglass LL, Bishop CW, Coburn JW. Intermittent doxercalciferol (1alpha-hydroxyvitamin D(2)) therapy for secondary hyperparathyroidism. Am J Kidney Dis. 2000;36:550–561. doi: 10.1053/ajkd.2000.16193. [DOI] [PubMed] [Google Scholar]
- 56.Stompor TP, Pasowicz M, Sulowicz W, Dembinska-Kiec A, Janda K, Wojcik K, Tracz W, Zdzienicka A, Konieczynska M, Klimeczek P, Janusz- Grzybowska E. Trends and dynamics of changes in calcification score over the 1-year observation period in patients on peritoneal dialysis. Am J Kidney Dis. 2004;44:517–528. [PubMed] [Google Scholar]
- 57.Jung HH, Kim SW, Han H. Inflammation, mineral metabolism and progressive coronary artery calcification in patients on haemodialysis. Nephrol Dial Transplant. 2006;21:1915–1920. doi: 10.1093/ndt/gfl118. [DOI] [PubMed] [Google Scholar]
- 58.O’Neill WC. The fallacy of the calcium-phosphorus product. Kidney Int. 2007;72:792–796. doi: 10.1038/sj.ki.5002412. [DOI] [PubMed] [Google Scholar]
- 59.Stevens LA, Djurdjev O, Cardew S, Cameron EC, Levin A. Calcium, phosphate, and parathyroid hormone levels in combination and as a function of dialysis duration predict mortality: evidence for the complexity of the association between mineral metabolism and outcomes. J Am Soc Nephrol. 2004;15:770–779. doi: 10.1097/01.asn.0000113243.24155.2f. [DOI] [PubMed] [Google Scholar]
- 60.Melamed ML, Eustace JA, Plantinga L, Jaar BG, Fink NE, Coresh J, Klag MJ, Powe NR. Changes in serum calcium, phosphate, and PTH and the risk of death in incident dialysis patients: a longitudinal study. Kidney Int. 2006;70:351–357. doi: 10.1038/sj.ki.5001542. [DOI] [PubMed] [Google Scholar]
- 61.Slinin Y, Foley RN, Collins AJ. Calcium, phosphorus, parathyroid hormone, and cardiovascular disease in hemodialysis patients: the USRDS waves 1, 3, and 4 study. J Am Soc Nephrol. 2005;16:1788–1793. doi: 10.1681/ASN.2004040275. [DOI] [PubMed] [Google Scholar]
- 62.Nakatani T, Bara S, Ohnishi M, Densmore MJ, Taguchi T, Goetz R, Mohammadi M, Lanske B, Razzaque MS. In vivo genetic evidence of klotho-dependent functions of FGF23 in regulation of systemic phosphate homeostasis. FASEB J. 2009;23:433–441. doi: 10.1096/fj.08-114397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bai X, Dinghong Q, Miao D, Goltzman D, Karaplis AC. Klotho ablation converts the biochemical and skeletal alterations in FGF23 (R176Q) transgenic mice to a Klotho-deficient phenotype. Am J Physiol Endocrinol Metab. 2009;296:E79–E88. doi: 10.1152/ajpendo.90539.2008. [DOI] [PubMed] [Google Scholar]
- 64.Ichikawa S, Imel EA, Kreiter ML, Yu X, Mackenzie DS, Sorenson AH, Goetz R, Mohammadi M, White KE, Econs MJ. A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J Clin Invest. 2007;117:2684–2691. doi: 10.1172/JCI31330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Goetz R, Nakada Y, Hu MC, Kurosu H, Wang L, Nakatani T, Shi M, Eliseenkova AV, Razzaque MS, Moe OW, Kuro-o M, Mohammadi M. Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-Klotho complex formation. Proc Natl Acad Sci USA. 2010;107:407–412. doi: 10.1073/pnas.0902006107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Razzaque MS. Does FGF23 toxicity influence the outcome of chronic kidney disease? Nephrol Dial Transplant. 2009;24:4–7. doi: 10.1093/ndt/gfn620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Goetz R, Beenken A, Ibrahimi OA, Kalinina J, Olsen SK, Eliseenkova AV, Xu C, Neubert TA, Zhang F, Linhardt RJ, Yu X, White KE, Inagaki T, Kliewer SA, Yamamoto M, Kurosu H, Ogawa Y, Kuro-o M, Lanske B, Razzaque MS, Mohammadi M. Molecular insights into the klotho-dependent, endocrine mode of action of fibroblast growth factor 19 subfamily members. Mol Cell Biol. 2007;27:3417–3428. doi: 10.1128/MCB.02249-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lanske B, Razzaque MS. Mineral metabolism and aging: the fibroblast growth factor 23 enigma. Curr Opin Nephrol Hypertens. 2007;16:311–318. doi: 10.1097/MNH.0b013e3281c55eca. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Razzaque MS, Lanske B. The emerging role of the fibroblast growth factor-23-klotho axis in renal regulation of phosphate homeostasis. J Endocrinol. 2007;194:1–10. doi: 10.1677/JOE-07-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Silver J, Naveh-Many T. FGF23 and the parathyroid glands. Pediatr Nephrol. 2010;25:2241–2245. doi: 10.1007/s00467-010-1565-3. [DOI] [PubMed] [Google Scholar]
- 71.Krajisnik T, Bjorklund P, Marsell R, Ljunggren O, Akerstrom G, Jonsson KB, Westin G, Larsson TE. Fibroblast growth factor-23 regulates parathyroid hormone and 1alpha-hydroxylase expression in cultured bovine parathyroid cells. J Endocrinol. 2007;195:125–131. doi: 10.1677/JOE-07-0267. [DOI] [PubMed] [Google Scholar]
- 72.Hofman-Bang J, Martuseviciene G, Santini MA, Olgaard K, Lewin E. Increased parathyroid expression of klotho in uremic rats. Kidney Int. 2010;78:1119–1127. doi: 10.1038/ki.2010.215. [DOI] [PubMed] [Google Scholar]
- 73.Drueke TB. Klotho, FGF23, and FGF receptors in chronic kidney disease: a yin-yang situation? Kidney Int. 2010;78:1057–1060. doi: 10.1038/ki.2010.339. [DOI] [PubMed] [Google Scholar]
- 74.Canalejo R, Canalejo A, Martinez-Moreno JM, Rodriguez-Ortiz ME, Estepa JC, Mendoza FJ, Munoz-Castaneda JR, Shalhoub V, Almaden Y, Rodriguez M. FGF23 fails to inhibit uremic parathyroid glands. J Am Soc Nephrol. 2010;21:1125–1135. doi: 10.1681/ASN.2009040427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Martuseviciene G, Hofman-Bang J, Clausen T, Olgaard K, Lewin E. The secretory response of parathyroid hormone to acute hypocalcemia in vivo is independent of parathyroid glandular sodium/potassium-ATPase activity. Kidney Int. 2011 doi: 10.1038/ki.2010.501. [DOI] [PubMed] [Google Scholar]
- 76.Brown WW, Juppner H, Langman CB, Price H, Farrow EG, White KE, McCormick KL. Hypophosphatemia with elevations in serum fibroblast growth factor 23 in a child with Jansen’s metaphyseal chondrodysplasia. J Clin Endocrinol Metab. 2009;94:17–20. doi: 10.1210/jc.2008-0220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bai X, Miao D, Li J, Goltzman D, Karaplis AC. Transgenic mice overexpressing human fibroblast growth factor 23 (R176Q) delineate a putative role for parathyroid hormone in renal phosphate wasting disorders. Endocrinology. 2004;145:5269–5279. doi: 10.1210/en.2004-0233. [DOI] [PubMed] [Google Scholar]
- 78.Kawata T, Imanishi Y, Kobayashi K, Miki T, Arnold A, Inaba M, Nishizawa Y. Parathyroid hormone regulates fibroblast growth factor-23 in a mouse model of primary hyperparathyroidism. J Am Soc Nephrol. 2007;18:2683–2688. doi: 10.1681/ASN.2006070783. [DOI] [PubMed] [Google Scholar]
- 79.Ohnishi M, Razzaque MS. Dietary and genetic evidence for phosphate toxicity accelerating mammalian aging. FASEB J. 2010;24:3562–3571. doi: 10.1096/fj.09-152488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nabeshima Y. Klotho: a fundamental regulator of aging. Ageing Res Rev. 2002;1:627–638. doi: 10.1016/s1568-1637(02)00027-2. [DOI] [PubMed] [Google Scholar]
- 81.Kuro-o M. Disease model: human aging. Trends Mol Med. 2001;7:179–181. doi: 10.1016/s1471-4914(01)01921-9. [DOI] [PubMed] [Google Scholar]
- 82.Nabeshima Y. Toward a better understanding of Klotho. Sci Aging Knowledge Environ. 2006:pe11. doi: 10.1126/sageke.2006.8.pe11. [DOI] [PubMed] [Google Scholar]
- 83.Jin H, Xu CX, Lim HT, Park SJ, Shin JY, Chung YS, Park SC, Chang SH, Youn HJ, Lee KH, Lee YS, Ha YC, Chae CH, Beck GR, Jr, Cho MH. High dietary inorganic phosphate increases lung tumorigenesis and alters Akt signaling. Am J Respir Crit Care Med. 2009;179:59–68. doi: 10.1164/rccm.200802-306OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wills MR, Savory J. Aluminium poisoning: dialysis ence-phalopathy, osteomalacia, and anaemia. Lancet. 1983;2:29–34. doi: 10.1016/s0140-6736(83)90014-4. [DOI] [PubMed] [Google Scholar]
- 85.Chertow GM, Burke SK, Raggi P. Sevelamer attenuates the progression of coronary and aortic calcification in hemodialysis patients. Kidney Int. 2002;62:245–252. doi: 10.1046/j.1523-1755.2002.00434.x. [DOI] [PubMed] [Google Scholar]
- 86.Block GA, Spiegel DM, Ehrlich J, Mehta R, Lindbergh J, Dreisbach A, Raggi P. Effects of sevelamer and calcium on coronary artery calcification in patients new to hemodialysis. Kidney Int. 2005;68:1815–1824. doi: 10.1111/j.1523-1755.2005.00600.x. [DOI] [PubMed] [Google Scholar]
- 87.Block GA, Raggi P, Bellasi A, Kooienga L, Spiegel DM. Mortality effect of coronary calcification and phosphate binder choice in incident hemodialysis patients. Kidney Int. 2007;71:438–441. doi: 10.1038/sj.ki.5002059. [DOI] [PubMed] [Google Scholar]
- 88.Duggal A, Hanus M, Zhorov E, Dagher R, Plone MA, Goldberg J, Burke SK. Novel dosage forms and regimens for sevelamer-based phosphate binders. J Ren Nutr. 2006;16:248–252. doi: 10.1053/j.jrn.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 89.Joy MS, Finn WF. Randomized, double-blind, placebo-controlled, dose-titration, phase III study assessing the efficacy and tolerability of lanthanum carbonate: a new phosphate binder for the treatment of hyperphosphatemia. Am J Kidney Dis. 2003;42:96–107. doi: 10.1016/s0272-6386(03)00554-7. [DOI] [PubMed] [Google Scholar]
- 90.Ismail EA, Al-Mutairi G, Al-Anzy H. A fatal small dose of phosphate enema in a young child with no renal or gastrointestinal abnormality. J Pediatr Gastroenterol Nutr. 2000;30:220–221. doi: 10.1097/00005176-200002000-00025. [DOI] [PubMed] [Google Scholar]
- 91.Marraffa JM, Hui A, Stork CM. Severe hyperphosphatemia and hypocalcemia following the rectal administration of a phosphate-containing Fleet pediatric enema. Pediatr Emerg Care. 2004;20:453–456. doi: 10.1097/01.pec.0000132217.65600.52. [DOI] [PubMed] [Google Scholar]
- 92.Perlman JM. Fatal hyperphosphatemia after oral phosphate overdose in a premature infant. Am J Health Syst Pharm. 1997;54:2488–2490. doi: 10.1093/ajhp/54.21.2488. [DOI] [PubMed] [Google Scholar]
- 93.Martin RR, Lisehora GR, Braxton M, Jr, Barcia PJ. Fatal poisoning from sodium phosphate enema. Case report and experimental study. JAMA. 1987;257:2190–2192. [PubMed] [Google Scholar]