

Abstract
This chapter will review the literature on cobalamin- and corrinoid-containing enzymes. These enzymes fall into two broad classes, those using methylcobalamin or related methylcorrinoids as prosthetic groups and catalyzing methyltransfer reactions, and those using adenosylcobalamin as the prosthetic group and catalyzing the generation of substrate radicals that in turn undergo rearrangements and/or eliminations.
Keywords: methyltransferase, methylcobalamin, adenosylcobalamin
1. INTRODUCTION—WHAT IS A CORRINOID?
The structure of cobalamin, or dimethylbenzimidazolylcobamide, is shown in Fig. 1. In cob(III)alamin derivatives like methyl- or adenosylcobalamin the cobalt is in the +3 oxidation state and is typically six coordinate. Four nitrogens from the corrin macrocycle serve as the equatorial ligands, and a substituent of the corrin ring known as the nucleotide loop, which terminates in a dimethylbenzimidazole base, provides the lower axial or α ligand to the cobalt. Cobamides in which the benzimidazole moiety is coordinated to the cobalt are referred to as “base-on” cobamides. The upper axial or β ligand, shown as R in Fig. 1, is a methyl group in methylcobalamin, an adenosyl group in adenosylcobalamin (AdoCbl), or may be occupied by an exchangeable ligand such as water in aquacobalamin.
Figure 1. The structure of cobalamin.

R is a methyl group in methylcobalamin or an adenosyl group in adenosylcobalamin.
In the kingdoms Archaea and Prokarya, cobalamin is only one of many variants grouped under the name corrinoids. Some of these variants involve changes in the structure of the dimethybenzimidazole (DMB) nucleotide base, such as 5′-methoxybenzimidazole cobamide, while other variants involve replacement of the DMB base by compounds such as adenine (pseudovitamin B12) or p-cresol (p-cresolylcobamide). The latter two bases can not be coordinated to the cobalt of the free cobamide, which instead contain a water in the lower axial position and is referred to as a base-off corrinoid.
Corrinoid-dependent methyltransferases are found in all three kingdoms of life, and in all cases, the cofactor is bound to its enzyme in a base-off manner. It is probably for this reason that so much variation in the nucleotide loop is tolerated. In a subset of corrinoid-dependent methyltransferases, the corrinoid is bound with a histidine (His) from the protein as the lower axial ligand, and this mode of binding is referred to as base-off, His-on. The first observation of this form of binding was made by Ragsdale and his colleagues 1, who characterized the corrinoid in the corrinoid iron-sulfur protein from Moorella thermoaceticum by EPR and Mossbauer spectroscopy. Extracts of the bacterium Sporomusa ovata were subsequently shown to contain the base-off corrinoid p-cresolyl-cob(II)amide in the Stupperich laboratory 2. When a protein containing the bound corrinoid was isolated, the corrinoid was found to exhibit the electron paragmagnetic resonance (EPR) properties of a base-on corrinoid in the +2 oxidation state. When the bacterial cells were grown on [15N]-His, the EPR spectrum was altered, indicating that the axial nitrogen ligand of the corrinoid was derived from the imidazole group of His. However after release from the protein, the corrinoid remained in the base-off form.
AdoCbl-dependent enzymes are only found in the kingdoms Eukaryota and Prokaryota, and cobalamin is the only corrinoid to be found in these enzymes. As will be discussed further in the second major section of this chapter, AdoCbl-dependent enzymes may contain the cobalamin bound either in the base-on form with the DMB ligand coordinated to the cobalt, or in the base-off, His-on form.
As the cobalt in corrinoids is reduced, the preferred coordination number decreases. Corrinoids in the +2 oxidation state are preferentially five-coordinate, with only one axial ligand, while corrinoids in the +1 oxidation state are preferentially four-coordinate, with no axial ligands.
2. CORRINOID-DEPENDENT METHYLTRANSFERASES
2.1. Overview of the metabolic roles of corrinoid-dependent methyltransferases
In humans, only one corrinoid-dependent methyl transferase, methionine synthase, is found, and this appears to be the only such corrinoid-dependent methyltransferase to be found in the kingdom Eukarya. However, in the kingdoms Prokarya and Archea, a wide variety of corrinoid-dependent methyltransferases play central roles in metabolism, particularly in organisms that grow under anaerobic conditons. We will begin by briefly enumerating these methyltransferases and their roles in carbon assimilation and energy generation.
Many organisms belonging to these two kingdoms make use of the reactions in the Wood-Ljungdahl pathway (Fig. 2), first elucidated by the studies of Harland Wood and Lars Ljungdahl and their groups (recently reviewed in 3). The enzymes in the Eastern branch of the Wood-Ljungdahl pathway catalyze the reduction of CO2 to form methyl groups that are intially bound to tetrahydrofolate or tetrahydropterin analogues of tetrahydrofolate. The reducing power that is needed is provided by three hydride ion transfers. In organisms that can grow on CO2 and hydrogen, hydrogenases catalyze the reversible conversion of hydrogen gas to hydride and a proton. The Western branch of the Wood-Ljungdahl pathway involves the reduction of CO2 to CO, catalyzed by CO dehydrogenase, and the incorporation of CO into a methyl-nickel bond to form an acetyl-Ni at the Ni-Ni metal center of acylCoA synthase. The acetyl group generated by carbonyation can then be transferred to coenzyme A to form acetylCoA. While none of the reactions of the Eastern or Western branches of the Wood-Ljungdahl pathway involve corrinoids, the corrinoid iron-sulfur protein (highlighted in red in Fig. 2) plays a central role in transferring the methyl group generated in the Eastern branch to the nickel in acylCoA synthase. All of the reactions in the Wood-Ljungdahl pathway are reversible, and some organisms will run portions of the Wood-Ljungdahl pathway in reverse, as we shall see.
Figure 2. The role of corrinoid methyltransferases in the central metabolic pathways of anaerobic eubacteria and methanogens.

The corrinoid methyl transferases are labeled in red. Red arrows indicate pathways, not shown in detail, that also involve corrinoid proteins. The boxed reactions are not part of the Wood-Ljungdahl pathway but are unique to methanogens. R′ is tetrahydrofolate or a tetrahydropterin analogue of tetrahydrofolate. The charge on the nickel that serves as the methyl acceptor on acylCoA synthase/decarbonylase is shown as +1, but whether it is Ni+1 or Ni0 rremains a matter for debate 175, 176.
A subgroup of organisms in the kingdom Archaea are obligate anaerobes that derive their carbon from CO2 when grown in the presence of hydrogen and produce methane as the final product. These organisms are known as methanogens. Methanogens convert a portion of the methyl groups generated in the Eastern branch of the Wood-Ljungdahl pathway into methane. This reaction occurs in two irreversible steps, in which the methyl group is first transferred to coenzyme M (ethanethiolsulfonate) by coenzyme M methyltransferases, and then reduced to methane by coenzyme M reductase. These two steps are energy generating, and are coupled to the creation of an ion gradient across the cell membrane that is used to generate energy for cellular growth. The energy-conserving coenzyme M methyltransferase in the methanogen Methanobacterium thermoautotropicum is a complex containing a corrinoid protein. The complex catalyzes the transfer of a methyl group from methyltetrahydromethanopterin, a methyltetrahydrofolate analogue found in this organism, to the cobalt of the corrinoid protein, and thence to the sulfur of coenzyme M to form methylcoenzyme M.
The basic pattern for corrinoid-dependent methyl transferases is shown in Fig. 3. These enzymes comprise a minimum of three modules, a central module that binds the corrinoid and modules that present the methyl donor to the corrinoid in the cobalt(I) oxidation state, and activate the donor if necessary, and that present the methyl acceptor to the methylcorrinoid and activate it if necessary. These three modules may reside on three separate proteins, or they may be present as modules on a single polypeptide or on several polypeptides. A common feature of these methyltransferases is that they must stabilize both the methylcorrinoid and the corrinoid in the cobalt(I) oxidation state. Furthermore, they must be capable of undergoing conformational changes that allow the corrinoid prosthetic group access to both donor and acceptor modules.
Figure 3.

Basic pattern for corrinoid methyltransferases.
Some methanogens can also grow on acetate by converting it to acetylCoA and then reversing the acylCoA synthase reaction to decarbonylate the acetyl group and produce CO and the methylated form of the corrinoid iron-sulfur protein. The CO is oxidized to CO2 with the generation of a hydride ion. The reversal of the methyl transfers catalyzed by the corrinoid iron-sulfur protein complex will then produce a methyltetrahydropterin, which will be converted to methane. The hydride needed for the conversion of methylcoenzyme M to methane is generated by the oxidation of CO.
Members of the genus Methanosarcina can also use other simple one-carbon compounds as sources of carbon and energy, including methylamines, methylthiols, and methanol. The methyl groups of these compounds are transferred to coenzyme M by specific non-energy conserving corrinoid methyltransferases that follow the basic pattern shown in Fig. 3. Methanogens growing on substrates other than acetate synthesize acetylCoA from methyltetrahydropterins and CO2 by reversal of the corrinoid iron-sulfur complex methyltransfers to generate methyl-Ni on acyl CoA synthase, followed by the acylCoA decarbonylase reaction and oxidation of the resultant CO. From the acetylCoA thus formed, all other carbon-containing cellular components are generated.
Acetogenic bacteria do not generate energy by methanogenesis, but rather generate energy by the anaerobic fermentation of glucose or by anaerobic growth on hydrogen and CO2. Glucose is converted to two molecules of pyruvate, which in turn is decarboxylated to form two molecules of acetyl CoA in a reaction coupled to the generation of ATP from ADP and inorganic phosphate. They use the Eastern branch of the Wood-Ljungdahl pathway to reduce CO2 to a methylgroup and the Western branch of the pathway to produce CO. These two reagents are then coupled to form an additional molecule of acetylCoA, from which all other carbon-containing compounds are generated.
A variant use of the Wood-Ljungdahl pathway is made by hydrogenogenic bacteria such as Carboxydothermus hydrogenoformans, which can grow on CO as the sole source of energy and carbon under anerobic conditions. Some of the CO is oxidized to CO2 by the action of CO dehydrogenase, with protons serving as the terminal electron acceptors. The Eastern branch of the Wood-Ljungdahl pathway is used for production of methyl groups, using hydride equivalents generated by oxidation of CO to CO2. The remainder of the CO is converted to acetyl CoA by the action of acylCoA synthase and the corrinoid iron-sulfur protein.
Another variant use of the Wood-Ljungdahl pathway is provided by bacteria that use aromatic O-methylethers as the source of both carbon and energy such as Sporomusa ovata. Corrinoid aromatic O-demethylases catalyze transfer of the methyl group to tetrahydrofolate. The methyl group can then be oxidized to formate and/or CO2 by reversal of the Eastern branch of the pathway with the accompanying generation of reducing equivalents, and then converted to acetate using corrinoid iron-sulfur protein and acylCoA synthase.
Finally corrinoid-dependent reductive dehalogenases found in Prokaryota use the corrinoid protein to catalyze the anaerobic dehalogenation of a variety of halogenated alkyl and aryl compounds. Since the products of such dehalogenations will vary with the halogenated substrate, the reductive dehalogenases are not shown on the central metabolic scheme in Fig. 2.
2.2 Cobalamin-dependent methionine synthase
Cobalamin-dependent methionine synthase (MetH) catalyzes the reaction shown in eq. 1. The enzyme is found in many members of the kingdom Prokaryota, including Escherichia coli, but has not been found in the Archaea. It is one of only two B12-dependent enzymes found in humans and other mammals, and is widely distributed among the animal Eukaryota. Due to its overexpression in recombinant form 4, 5 and the resultant ease of purification under aerobic conditions, large amounts of purified E. coli protein have been available for biochemical and structural characterization. It was one of the first corrinoid proteins to be characterized, and has subsequently been extensively studied in the laboratories of Herbert Weissbach, Frank Huennekens, and more recently in my own laboratory.
Eq. 1.
The enzyme consists of four modules, that are arranged linearly with single interdomain linkers to form a single 136 kDa polypeptide. The N-terminal module, the methyl donor module in the parlance used in Fig. 3, binds and activates methyltetrahydrofolate and presents it to the cobalamin prosthetic group for methyl transfer. The next module in the sequence is the methyl acceptor module, this module binds and activates homocysteine and presents it to methylcobalamin to allow methyl transfer to form methionine. The third module is the cobalamin-binding module and also contains a four helix bundle at the N-terminus of the domain responsible for binding the upper face of the cobalamin. The final module binds adenosylmethionine (AdoMet) and is required for reductive activation of the protein. Its raison d’être requires discussion of the reactions catalyzed by the enzyme, which are shown in Fig. 4. MetH is active during aerobic growth in E. coli, and also under in vitro turnover in microaerophilic conditions. In vitro, the cob(I)alamin form of the enzyme is oxidized to the inactive species cob(II)alamin about once in every 2000 turnovers 6. Return of the prosthetic group to the active methylcobalamin form requires a reductive remethylation. In E. coli, the electron is provided by the electron transfer protein flavodoxin, the fldA gene product 7, 8. The reduction potential of the flavodoxin semiquinone/hydroquinone is −440 mV vs. the standard hydrogen electrode 9, and the quinone/semiquinone potential, which is probably the more relevant one for cells grown under microaerophilic conditions, is −260 mV. In contrast, the cob(II)alamin/ cob(I)alamin reduction potential is −490 mV at pH 710. Thus the reduction of cob(II)alamin is a highly endergonic reaction and must be driven to completion by coupling to a highly exergonic methyl transfer. For this purpose, AdoMet is used as the methyl donor for reductive activation. The transfer of the methyl group of AdoMet is associated with a driving force of about 17 kcal/mol, helping to assure that the reductive remethyation proceeds quantitatively. The C-terminal domain of MetH binds AdoMet and catalyzes this alternate methyl donor reaction, and is designated as the reactivation module. Indeed, if this module is removed from the protein by limited proteolysis of the native enzyme, the methylated enzyme turns over until all the cobalamin accumulates as cob(II)alamin, at which point the enzyme can no longer be reactivated 6.
Figure 4. Reactions catalyzed by methionine synthase.

During primary turnover, the enzyme-bound cobalamin cycles between methylcobalamin and cob(I)alamin forms as the prosthetic group is alternately methylated by methyltetrahydrofolate (CH3-H4folate) and demethylated by transfer of the methyl group to homocysteine (Hcy). During turnover under microaerophilic conditions, the cob(I)alamin form of the enzyme is oxidized to cob(II)alamin about once in every 2000 turnovers. This form of the enzyme is inactive, and reactivation requires a reductive methylation in which the reduction of cob(II)alamin to cob(I)alamin is coupled to a highly exergonic methylation using adenosylmethionine (AdoMet) as the methyl donor.
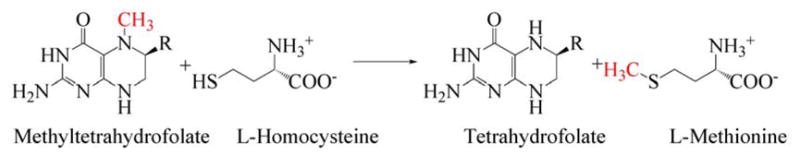
Further insights into the complicated catalytic and reactivation cycles of methionine synthase came as x-ray structures were determined for fragments of methionine synthase in the laboratory of Martha Ludwig. The entire enzyme has never been crystallized, presumably because of the conformational lability of the enzyme. The first fragment to be crystallized was the cobalamin-binding module11. This was the first structure of cobalamin bound to a protein, and it revealed the remarkable displacement of the DMB axial ligand by a histidine residue from the protein--what we now call the base-off His-on state of cobalamin. As shown in Fig. 5, His759 is coordinated to the cobalt of methylcobalamin and also linked by a network of hydrogen bonds to the carboxyl group of Asp757, which in turn is hydrogen-bonded to the hydroxyl of Ser810. The binding of the prosthetic group in the base-off His-on form was shown to be associated with a signature His-x-Asp-xxGly---41/42--- Ser-x-Leu-25/26---Gly-Gly sequence that had originally been identified in glutamate mutase 12.
Figure 5. Coordination of methylcobalamin in the cobalamin-binding module of MetH.
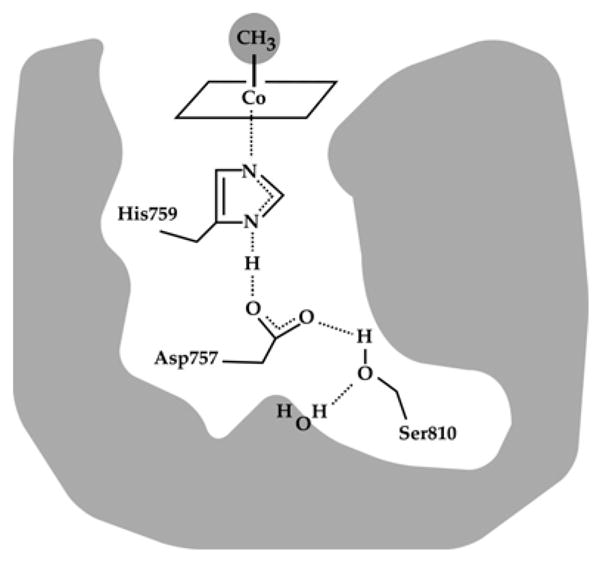
A hydrogen bonded network comprising His 759, Asp757 and Ser810 links His759 to the external solvent; these three residues which are absolutely conserved in all MetH enzymes, are referred to as the ligand triad. (Reprinted with permission, from 177)
The β-face of the cobalamin prosthetic group in the structure is shielded by a four helix bundle that forms the N-terminal portion of the module sequence and is referred to as the “cap”. Thus far, only indirect evidence suggests that this conformation of the intact MetH protein exists in solution 13.
The structure of the C-terminal reactivation module of MetH was determined next 14, and then a structure was obtained of the entire C-terminal half of the protein comprising the cobalamin-binding and reactivation modules 15. This structure was determined with a fragment of His759Gly MetH, and revealed that the fragment had crystallized in a conformation in which the β-face of the cobalamin prosthetic group was now in contact with the reactivation module (Fig. 6, right). Although, His759 is absent in this structure, the distance between Cα of Gly759 and the cobalt of the cob(II)alamin prosthetic group is 2.3 Å greater than in the Cap:Cob conformation assumed by the isolated cobalamin-binding module. This increased distance would be predicted to cause the cobalamin of the wild-type enzyme to assume a base-off His-off conformation, enforced in part by the juxtaposition of residues from the AdoMet-binding module between the cobalamin-binding domain and the corrin ring. Indeed, the methylated form of the wild-type enzyme has been shown to undergo interconversions between His-on and His-off forms that are induced by binding of ligands, shifts in temperature, or the binding of flavodoxin 15, 16. These interconversions are thought to reflect rearrangements of the four modules of methionine synthase, as shown in Fig. 7.
Figure 6. Structures of the N-terminal and C-terminal halves of methionine synthase.
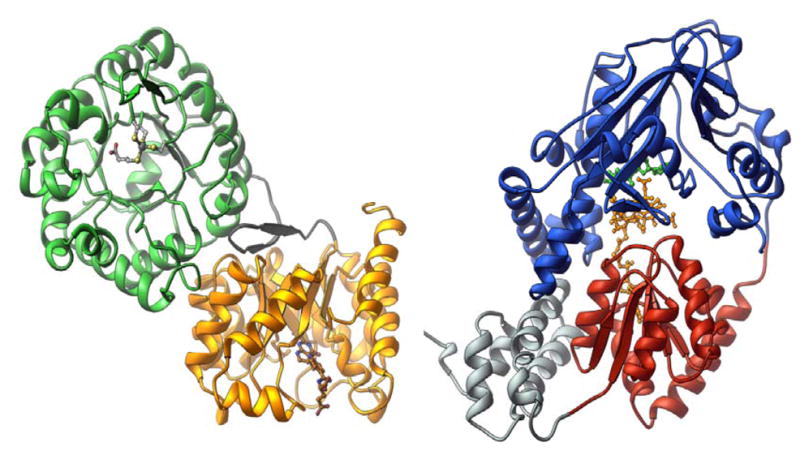
The structure of the homocysteine-binding (green) and folate-binding (gold) modules of MetH was determined with protein from Thermotoga maritima 17, and the structure of the cobalamin-binding (red) and reactivation (blue) modules of MetH was determined with the His759Gly mutant of the enzyme from Escherichia coli 178. The cobalamin-binding module also contains a four helix bundle, referred to as the “cap” and shown in grey.
Figure 7. Conformational states of methionine synthase.
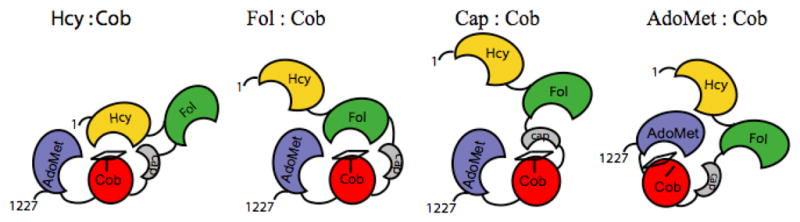
The four modules are shown in gold (Hcy-binding), green (folate-binding), red (cobalamin-binding domain) and gray (cap domain), and blue (AdoMet-binding). The corrin ring of methylcobalamin is indicated by the rectangle on top of the cobalamin-binding domain, and His759 is indicated by the vertical line. In the AdoMet:Cob conformation, the histidine is displaced as indicated in the cartoon, and the corrin ring tilts away from the cobalamin-binding domain and from Cα of His 759. Reprinted from 20.
The right hand side of Fig. 6 shows the structure of the N-terminal substrate-binding modules of methionine synthase from Thermotoga maritima17. The homocysteine- and folate-binding modules of methionine synthase are both α8β8 barrels, with their openings positioned orthogonally with respect to each other. There is a large buried surface area between the two barrels, strongly suggesting that they move as a unit rather than individually. Thus, as cartooned in Fig. 7, large modular rearrangements are required to allow the cobalamin to access the Hcy-binding and Fol-binding modules alternately during catalysis.
Originally, it was proposed that the base-off His-on state of cobalamin in methionine synthase might accelerate the methyl transfer reactions 11. However, mounting evidence suggests that the primary role of the ligand replacement is to facilitate the conformational changes necessary for catalysis. In the initial studies, the wild-type His759 enzyme was compared with mutations of each residue of the ligand triad: His759Gly, Asp757Glu and Asp757Asn, and Ser810Ala. While the His759Gly mutant was inactive in steady-state assays, the Asp757 mutants showed kcat values that were 4–6% of the wild-type enzyme and that for the Ser810Ala mutant was 56% 5. When the approach to steady-state was examined by enzyme-monitored turnover, the Asp757 mutants were 33 to 54% as fast as the wild-type enzyme and the Ser810Ala mutant was 61% as fast, indicating that these mutants were barely compromised in establishing the initial steady-state distribution of methylcobalamin and cob(I)alamin enzyme forms. The rate constant for AdoMet- and reduced flavodoxin-dependent reactivation of enzyme in the cob(II)alamin form, which occurs in state 4 of Fig. 7, was also measured. The His759Gly mutant was methylated 14× faster than wild-type enzyme, as was the Asp757Glu mutant. The Asp757Asn and Ser810Ala mutants were methylated about twice as fast wild-type enzyme. Based on these data, Jarrett 18 proposed that mutations of residues in the ligand triad might alter the distribution of states shown in Fig. 7, and that, as the ligation of the histidine was weakened and then finally abolished, the distribution would increasingly favor the AdoMet:Cob conformation. In agreement with this proposal, it was found that the EPR spectra of wild-type and mutant enzymes in the cob(II)alamin form increasingly favored the His-off conformation in the order: His759Gly (100%)> Asp757Glu (65%)>Asp757Asn (25%)>wild-type (15%)>Ser810Ala (5%).
The next contribution to our understanding was the demonstration that addition of oxidized flavodoxin to methionine synthase in the cob(II)alamin form resulted in the conversion of the His-on state of the prosthetic group to the His-off state, as shown by the loss of superhyperfine coupling in the EPR spectrum of the cobalamin 9. Further studies 19 established that enzyme in the presumably His-off four-coordinate cob(I)alamin form could not interconvert between catalytic conformations (states 1, 2, and 3 in figure 7) and the AdoMet:Cob conformation (state 4 in Figure 7). If MetH in the cob(I)alamin state is produced by reduction of cob(II)alamin, the enzyme thus formed reacts with AdoMet but not with methyltetrahydrofolate, suggesting that the enzyme can assume the AdoMet:Cob conformation but not the Fol:Cob conformation. Conversely, if cob(I)alamin is generated by demethylation of enzyme in the methylcobalamin state in the presence of homocysteine, the cob(I)alamin reacts with methyltetrahydrofolate 30,000-fold more rapidly than it reacts with AdoMet, suggesting that it can readily assume the Fol:Cob conformation but has very limited access to the AdoMet:Cob conformation. Further evidence that the two forms of cob(I)alamin are in different protein conformations came from the observation that limited proteolysis of the native enzyme resulted in different patterns. The pattern seen with cob(I)alamin enzyme generated by reduction was also seen with wild-type cob(II)alamin enzyme with flavodoxin bound (previously shown to be His-off 9) and also with cob(II)alamin bound to the His759Gly mutant. In contrast, the cob(I)alamin enzyme generated by demethylation with homocysteine showed the same cleavage pattern as wild-type enzyme in the methylcobalamin and cob(II)alamin (in the absence of flavodoxin) forms. Thus the results suggested that the first cleavage pattern was characteristic of enzyme in the AdoMet:Cob conformation, while the second cleavage pattern was characteristic of enzyme in one of the catalytic conformations (states 1, 2, and 3 in Fig. 7).
The results described thus far indicated that enzyme in the cob(I)alamin form can not freely interconvert between catalytic and reactivation conformations, while enzyme in the cob(II)alamin form interconverts readily on addition of flavodoxin. Bandarian 15 discovered that enzyme in the methylcobalamin form can also be induced to interconvert between catalytic and reactivation conformations, and that the reactivation conformation is associated with an absorbance spectrum typical of base-off methycobalamin. He showed that the His-off/His-on equilibrium was <5:95 for the full length wild-type enzyme, while it was 12:88 for the Asp757Glu mutant, confirming that this ligand triad mutation weakens the ligation of His759 to the cobalt. Addition of AdoHcy, which is bound at the interface between the AdoMet module and the Cob module in the AdoMet:Cob conformation of the protein, shifts the equilibrium to favor the His-off state, consistent with the argument that the His-off state is asssociated with the AdoMet:Cob conformation. Addition of methyltetrahydrofolate also shifts the equilibrium to favor the His-off state, presumably because the methyl group of methyltetrahydrofolate is in steric conflict with the methyl group of methylcobalamin in the Fol:Cob state, which is therefore disfavored. The two ligands exert their effects on the equilbrium independently, favoring the His-off state with free energy changes of 0.9 and 0.6 kcal/mol respectively. The picture that emerges is of a delicately balanced equilbrium between alternate conformations of methionine synthase, with small free energy changes induced by ligand binding able to shift the distribution of conformers because these ligands have different affinities for the different states.
The next advance in our understanding of the dynamic equilibrium of conformers in MetH came from the studies of Fleischhacker 16. She examined the effect of substitutions in the upper axial (β) ligand of cob(III)alamin on the conformational equilibrium. She showed that methionine synthase in the propylcobalamin form had a His-off/His-on equilibrium of 31:69 in the absence of ligands, while the His-off form was undetectable when the enzyme prosthetic group was aquacobalamin. The His-off/His-on equilibrium was predicted by the ligand trans influence, with the more electron-donating propyl substituent favoring the AdoMet:Cob conformation. These studies provided a rationale for the base-off His-on substitution. The histidine ligand serves as a protein sensor of the oxidation and ligation state of the cobalamin, biasing the equilibrium in accord with the net formal charge on the cobalt and its resultant effect on the histidine ligation.
The cob(II)alamin state of methionine synthase showed properties intermediate between methylcobalamin and propylcobalamin in its ability to enter the AdoMet:Cob conformation. Oxidation of the prosthetic group to the cob(II)alamin state would lead to an enhanced propensity to enter the AdoMet:Cob conformation, which is favored by addition of flavodoxin and/or by addition of AdoMet (there is no steric conflict between cob(II)alamin and AdoMet). Once the prosthetic group is reduced and methylated, the resultant His-off methylcobalamin is converted to the His-on state and thus returns to the catalytic cycle.
A further role for the His759 ligand was discovered when the structure of a C-terminal fragment of “wild-type” methylated enzyme was determined 20. To stabilize the AdoMet:Cob confomation of the enzyme, a disulfide crosslink was introduced between the “cap” and the cobalamin-binding domain by mutation 21, 22 of Ile690 and Gly743 to cysteine residues. The crystal structure of this fragment revealed that it was indeed in the AdoMet:Cob conformation, but that the histidine had now moved about 7Å away from the cobalt and was now involved in intermodular hydrogen bonding with the AdoMet-binding domain (Figure 8). These unanticipated intermodular contacts would be expected to stabilize the His-off forms of the wild-type enzyme in the AdoMet:Cob conformation by as much as 3–5 kcal/mol.
Figure 8. Intermodular contacts between His 759 and residues in the AdoMet-binding reactivation module of MetH.
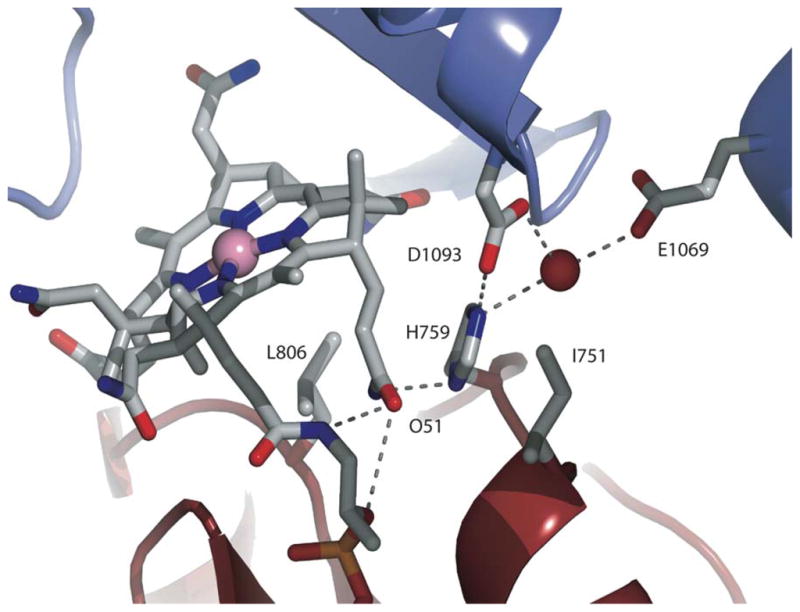
Nε2 of His759 interacts with the AdoMet module directly through a hydrogen bond to Asp1093 and via a water-mediated hydrogen bond to Glu1069. Nδ1 of His759 forms a hydrogen bond with the amide of the propionamide side chain of ring B of the cobalamin (not shown). Adapted from 20.
2.3 Corrinoid-dependent methyltransferases in Methanosarcina spp
Methanogens in the genus Methanosarcina use protein complexes containing corrinoid-binding proteins to catabolize simple one-carbon compounds such as methylamines and methylthiols as well as methanol. These complexes typically consist of a substrate-specific methyltransferase, a cognate corrinoid protein that receives the methyl group, and a second methyltransferase that catalyzes the transfer of the methyl group from the corrinoid protein to coenzyme M (ethane thiol sulfonate). More recently, tetramethylammonium-coenzyme M methyltransferase activity has been identified in Methanococcoides sp. 23. Fig. 9 diagrams the individual complexes that have been studied: methanol-coenzyme M methyltransferase 24, dimethylsulfide-coenzyme M methyltransferase 25, monomethylamine-coenzyme M methyltransferase 26, 27, dimethylamine-coenzyme M methyltransferase 28, trimethylamine-coenzyme M methyltransferase 29, and tetramethylammonium-coenzyme M methyltransferase 30. However, the genome sequence of Methanosarcina acetivorans contains 10 sequences with homologies to substrate-specific methyltransferases, 15 putative corrinoid protein sequences, and 14 sequences with homologies to coenzyme M methyltransferases. The substrates for many of these proteins remain unidentified 31.
Figure 9.

Complexes catalyzing methyl transfer from methyl donors to coenzyme M. The gene product designations are shown above each protein. The first two letters, mt, indicate involvement of the gene product in methyl transfer, the third letter indicates the substrate: a for methanol, s for methylthiols, m for monomethylamine, b for dimethylamine, t for trimethylamine and q for tetramethylammonium. The final letter designates the polypeptide function: where B is the substrate-specific methyltransferase that methylates the corrinoid protein with a methyl group derived from substrate, C is the corrinoid binding polypeptide and A is the CoM-methylating protein, as originally suggested by Krzycki and coworkers 29.
The reactions catalyzed by these cytoplasmic enzymes bear striking similarity to the overall reaction catalyzed by cobalamin-dependent methionine synthase, and indeed the corrinoid-binding proteins in those complexes that have been sequenced show homology with the cobalamin-binding domain of methionine synthase, including the characteristic Asp-X-His-X-X-Gly motif indicative of a corrinoid cofactor with a histidine axial ligand. However, the methylcorrinoid-coenzyme M methyltransferase proteins do not show significant sequence homology with the homocysteine-binding domain of methionine synthase, but instead show sequence similarity with uroporphyrinogen decarboxylase (UroD) 32. The substrate-methylcorrinoid methyl transferases neither resemble methionine synthase domains nor each other. The genes specifying MtmC, MtbC, and MttC all contain an in-frame UAG stop codon in the middle of the open reading frame 29, and this UAG has been shown to specify pyrrolysine 33. This unique residue is located at the active site of MtmB and is thought to be involved in activation of the amine substrate for methyl transfer. Despite the lack of sequence similarity between MtmB and the methyltetrahydrofolate-binding domain of MetH, their overall structures are similar.
Of this group of enzymes, the most mechanistically characterized system is the Mta complex catalyzing methanol-coenzyme M methyl transfer. As mentioned above, MtaC, shows sequence homology with other corrinoid proteins involved in cytoplasmic methyl transfers to coenzyme M, and with the cobalamin-binding module of MetH. The active-site histidine responsible for the base-off His-on ligation of the 5-hydroxybenzimidazolylcobamide corrinoid of MtaC was shown to be His 136, the histidine in the signature Asp-X-His-X-X-Gly sequence 24. MtaC is isolated in a complex with MtaB, and the complex was shown to catalyze methylation of the corrinoid prosthetic group using methanol as the methyl donor 34.
Recently, an x-ray structure of the MtaBC complex has been determined 35. Thus far, this is the only structure of a methyl transferase corrinoid-binding protein in complex with one of its substrate binding domains. MtaC is indeed structurally related to the cobalamin-binding module of MetH, and as in that structure it contains both a four helix bundle (the cap) and the Rossmann domain responsible for cobalamin binding. As in the structure of the C-terminal fragment of His759Gly MetH, the cap is displaced to allow juxtaposition of MtaB with the Rossmann domain. MtaB is composed of an α8β8 barrel with similarities to the substrate-binding domains of MetH, and a unique helical layer that is not seen in MetH.
A zinc atom is located at the C-terminus of the barrel in a deep funnel shaped pocket (Fig. 10); this zinc was previously shown to be required for activity 36. In the MtmBC structure the barrel is positioned over the Rossmann domain so as to position the zinc above the cobalt of the corrinoid prosthetic group and to define the methanol binding site at the interface. The catalytic zinc ion is ligated by Glu164, Cys220, and Cys269, and although it exhibits approximately tetrahedral geometry, is apparently lacking a fourth ligand. The authors assume that methanol binds to the fourth site on the zinc through its hydroxyl group. Additional electron density, which has been modeled as a potassium ion, is located 3.1 Å away from the zinc, and this putative potassium ion is also ligated by Glu164 as well as by other oxygen ligands. The authors suggest that the methanol will actually bridge between the potassium ion and the zinc. His136, the cobalt of the corrinoid, the methyl group and the oxygen of methanol and the catalytic zinc all form a line, favoring an SN2 mechanism in which the Co(I) state of the cofactor nucleophilically attacks the methyl group of methanol with the departing hydroxyl group remaining ligated to the zinc. In agreement with this proposed mechanism, which would result in inversion of configuration of the methyl group, the overall stereochemistry of the methyl group transfer from methanol to coenzyme M, which would require two successive nucleophilic displacements, proceeds with retention of stereochemistry 37.
Figure 10. Proposed catalysis of methyl transfer from methanol to the corrinoid cofactor in the MtmBC complex as deduced from the x-ray structure of the complex.

Reproduced, with permission, from 35. The electron density indicated by X has been modeled as a potassium ion.
MtaA catalyzes methyl transfer from MtaC to coenzyme M and also from exogenous methylcobalamin to coenzyme M 38. Like the other proteins that catalyze alkyl transfer to thiols 39, MtaA contains a catalytically essential zinc ion that serves as the binding site for coenzyme M 40. EXAFS analysis indicated that the substrate-free MtbA zinc was ligated by one sulfur ligand and three oxygen or nitrogen ligands, and that binding of coenzyme M was associated with replacement of one of the oxygen/nitrogen ligands by the sulfur of coenzyme M41.
The MtaA isozyme MtbA also contains a catalytically essential zinc ion that serves as the binding site for coenzyme M 42. EXAFS analysis indicated that the substrate-free MtbA zinc was ligated by two sulfur ligands and two oxygen or nitrogen ligands, and that binding of coenzyme M was associated with replacement of one of the oxygen/nitrogen ligands by the sulfur of coenzyme M 42. These two isozymes share only 40% sequence identity, and presumably one of the cysteine ligands of MtaA is not conserved in MtaB.
2.4 Membrane-associated energy-conserving corrinoid methyltransferase
N5-Methyltetrahydromethanopterin:coenzyme M methyltransferase is an integral membrane protein that catalyzes an energy-conserving step in methane formation from CO2 and/or acetate in Methanobacterium thermoautotrophicum. The enzyme catalyzes methyl transfer from methyltetrahydromethanopterin, a methyl donor similar in structure to methyltetrahydrofolate, to the thiol of coenzyme M. This reaction is exergonic (ΔG′° = −30 kJ/mol) and is coupled to extrusion of a sodium ion across the cell membrane 43. The enzyme was purified to homogeneity 44 and shown to comprise eight different subunits 45. The corrinoid-containing subunit MtrA was intially identified as part of the complex by immunoprecipitation 46, and subsequently was shown to contain one equivalent of 5′-hydroxybenz-imidazolyl-cobalamide 47 bound with a histidine ligand to the cobalt 48. The histidine ligand was shown to be His84 by mutagenesis, but interestingly there is no sequence homology between MtrA and the cobalamin-binding region of methionine synthase or the corrinoid proteins of the non-energy conserving cytoplasmic methyltransferases discussed in the previous section 49.
Mechanistic studies on this very large membrane-bound protein complex are challenging indeed. The Thauer laboratory succeeded in demonstrating that enzyme in the Co(II) form was inactive, and could be activated by incubation with titanium citrate and methyltetrahydromethanopterin, suggesting that the Co(I) form of the cofactor was required for reaction. Enzyme demethylation in the presence of coenzyme M did not require the presence of titanium citrate. The authors concluded that the basic mechanism was similar to that of MetH, in which the prosthetic group cycles in catalysis between cob(I)alamin and methylcobalamin forms 50. As is the case with MetH and with the non-energy conserving cytoplasmic corrinoid methyltransferase, the complex can catalyze the methylation and demethylation of exogenous cobalamin as well as of the endogenous MtrA corrinoid. Only the half reaction involving transfer of the methyl group from the methylcobalamin to coenzyme M was sodium-ion dependent 51.
2.5 The corrinoid iron-sulfur protein
Acetogenic bacteria like members of the genus Clostridium produce acetate as the sole fermentation product during growth on either glucose or H2 and CO2. These organisms employ the Wood-Ljungdahl pathway (Fig. 2), in which CO2 derived from acetate is converted to formate and then sequentially reduced to a methyl group bound to tetrahydrofolate in the “eastern branch” of the pathway, and CO2 is converted to CO in the reaction catalyzed by CO dehydrogenase in the “western branch” of the pathway. The corrinoid iron-sulfur protein accepts the methyl group of methyltetrahydofolate and transfers it to a nickel center on acyl CoA synthase, which then catalyzes the carbonylation of the methyl-nickel bond and the subsequent transfer of the acetyl group to the thiol of coenzyme A 3. The corrinoid iron-sulfur protein consists of two subunits. The large subunit, AcsC, contains a 4Fe-4S cluster, while both subunits are necessary for tight binding of the corrinoid prosthetic group. A separate methyltransferase, AcsE, catalyzes the methylation of the corrinoid using methyltetrahydrofolate as the methyl donor. Hydrogenogenic bacteria, which can utilize CO as a sole source of carbon and energy under anaerobic growth conditions, employ the Wood-Ljungdahl pathway for assimilation of carbon. They also contain a corrinoid iron-sulfur protein consisting of two subunits and a separate methyltransferase 52.
Methanogens also synthesize acetyl-CoA from methyltetrahydromethanopterin or methyltetrahydrosarcinopterin and CO2, but during methanogenesis they run the reaction in reverse, transferring the methyl group of acetylCoA to analogues of tetrahydrolate and generating carbon monoxide. The anabolic and catabolic acylCoA synthase/decarbonylase complexes appear to be similar, and a corrinoid has been shown to be involved in catabolism by the anabolic complex by inhibition with propyl iodide and relief of inhibition by photolysis 53, 54. Methanogens express a corrinoid iron-sulfur protein as part of this acetyl CoA decarbonylase/synthase multienzyme complex. As the name suggests, this complex catalyzes both acetyl CoA synthesis and cleavage. Members of the genus Methanosarcina can grow on acetate by metabolizing it to CO2 and methane, with acetyl CoA as an intermediate. The overall decomposition of acetyl CoA is shown by eq. 2, where H4SPT is tetrahydrosarcinapterin, an analogue of tetrahydrofolate, and H:− represents hydride. The reducing equivalents produced in this reaction are used to reduce the methyl group of methyltetrahydrosarcinapterin to methane following transfer of the methyl group to coenzyme M 55.
Eq. 2.

Corrinoid iron-sulfur proteins have been isolated and characterized from Clostridium thermoaceticum (renamed Moorella thermoaceticum) 1, 56, from the methanogenic archaeon Methanosarcina thermophila 57 and from the hydrogenogenic bacterium Carboxydothermus hydrogenoformans 52. The acetyl CoA decarbonylase/synthase complex from Methanosarcina thermophila has been purified to homogeneity 58–60 and shown to contain the two subunits of the corrinoid iron-sulfur protein CdhE (the γ subunit, 63 kDa) and CdhD (the δ subunit, 53 kDa). These subunits show sequence homology to the two subunits, AcsC and AcsD, of the corrinoid iron-sulfur protein in acetogens 61 and to the CfsA and CfsB subunits from C. hydrogenoformans 62, although the smaller δ subunit is considerably larger than the small subunits in acetogens and hydrogenogenic bacteria. The γδ complex exhibits methyltransferase activity. In acetogens and hydrogenogenic bacteria, transfer of the methyl group from methyltetrahydrofolate to the corrinoid iron-sulfur protein requires a separate protein, but in methanogens this activity appears to be integrated into CdhD and/or CdhE.
The spectroscopic properties of the corrinoid iron-sulfur protein from M. thermoaceticum have been particularly extensively characterized. Tight binding of the corrinoid, which is 5-hydroxybenzimidazolyl-cobamide in M. thermoaceticum, requires the presence of both large and small subunits. The sequences of the genes specifying the two subunits have no homology with that specifying the cobalamin binding module of MetH and lack the DxHxxG motif associated with binding of cobalamin to MetH in the base-off His-on form 56. The corrinoid is bound to the protein in the base-off form and its spectral properties indicate the absence of a nitrogen ligand in the lower axial position in both the methylcob(III)amide and cob(II)amide redox states of the corrinoid. In marked contrast, Maupin-Furlow and Ferry noted that the purified CdhD subunit of the M. thermophila protein could be reconstituted with hydroxocobalamin in the base-on configuration, while reconstitution of purified CdhE protein (the large subunit) led to base-off cobalamin binding 57.
The spectrum of the methylated protein was determined in the presence of sodium mersalyl, an organic mercurial reagent which disrupts the iron-sulfur center and allows unobstructed study of the cobalt chromophore. The spectrum exhibits a peak at ~440 nm, which is consistent with a base-off methylcorrinoid, and release of the cofactor from the protein leads to an absorbance spectrum with a maximum of 537 nm, typical of a base-on methylcorrinoid 1.
The enzyme is isolated in the cob(II)amide form, and its EPR spectrum exhibits eight singlet peaks centered around g=2 1, 56. This spectrum is diagnostic of base-off cob(II)amides, and is due to the hyperfine splitting imposed by the spin 7/2 of the cobalt nucleus. The absence of superhyperfine splitting, which would lead to an octet of triplets rather than singlets, demonstrates that nitrogen is not coordinated to 5-hydroxybenzimidazolyl-cobamide in the protein. Similar results have been observed for the corrinoid/iron-sulfur component of the acylCoA synthase/decarbonylase complex from Methanosarcina thermophila 63.
In a collaboration between the laboratories of Thomas Brunold and Stephen Ragsdale, the axial ligation of the corrinoid cofactor has been determined using a combination of magnetic circular dichroism, EPR, resonance Raman and computational chemistry 64. This analysis demonstrated that both the methylcob(III)amide and cob(II)inamide states of the prosthetic group are present in the base-off form, with an axial water ligand. These spectrocopies can not distinguish between β and α axial ligation of the water in the cob(II)inamide state, but presumably the water is in the α position when the prosthetic group is methylated.
The role of the iron-sulfur center of the corrinoid iron-sulfur protein was also examined in the M. thermoaceticum enzyme. The cobalt(I) form of the enzyme undergoes oxidation to form an inactive cobalt(II) species about once in every 100 turnovers 65. The iron-sulfur center of the corrinoid iron-sulfur protein has been shown to be required for reductive reactivation of the inactivated cob(II)amide prosthetic group, but is not required for the methyl transfers catalyzed by the active enzyme 66. This cluster has a redox potential of −523 mV vs. the standard hydrogen electrode 61, which is nearly isopotential with the cob(II)amide/cob(I)amide couple of the enzyme-bound corrinoid. For this reason, efficient reduction does not require coupling to ATP hydrolysis or methyl transfer from AdoMet. The oxidized iron-sulfur cluster can then be reduced by carbon monoxide/carbon monoxide dehydrogenase, by hydrogen/hydrogenase, or by a low-potential reduced ferredoxin 66.
The structure of the corrinoid iron-sulfur protein from C. hydrogeno-formans was recently determined 62. This structure revealed that the large subunit has three domains: an N-terminal domain that binds the 4Fe-4S cluster, a central α8β8 barrel, and a C-terminal domain that binds the cobalamin in the expected “base-off” conformation. The small subunit was also an α8β8 barrel. The small subunit packs against the uppper face of the cobalamin prosthetic group. This binding mode for the cobalamin is in agreement with earlier observations that the M. thermoaceticum corrinoid is bound most tightly when both subunits are present 56. No protein ligand to the cobalamin was identified, consistent with its binding in a base-off conformation, but a water ligand was seen in the upper axial (β) position. The structure could not distinguish between cobalt in the +2 or +3 oxidation state of the cofactor. The prosthetic group ligation seen in this structure is in excellent agreement with the spectroscopy reported for the M. thermoaceticum enzyme 64, and with the previously puzzling observations of Maupin-Furlow and Ferry on the M. thermophila protein, who observed base-on binding of hydroxocobalamin to the large subunit and base-on binding to the small subunit 57.
In a DALI search (a program that looks for structural similarity in proteins) to identify structures with high similarity to the subunits of the corrinoid iron-sulfur protein, the highest similarity was found to be with the folate-binding domain of MetH, which is similar to both of the α8β8 barrels. Structural similarity was also seen between these two barrels and the methyltransferase AcsE from M. thermoaceticum 67. The C-terminal domain of the large subunit was also found to exhibit structural similarity to the cobalamin-binding domain of MetH.
Catalysis of the methyl transfers from methyltetrahydrofolate or its analogues to the nickel site of acylCoA synthase requires that the corrinoid iron-sulfur protein interact first with the methyltransferase to receive the methyl group of methyltetrahydrofolate and then with the metal site on acylCoA synthase to form methyl-Ni. Reactivation of the inactive cobalt(II) fom of the enzyme presumably requires a third conformation in which the iron-sulfur center is juxtaposed with the β-face of the corrinoid prosthetic group. Thus the corrinoid iron-sulfur protein must undergo a series of conformational changes that may resemble those seen for MetH.
What advantages does the base-off state of the prosthetic group confer to the corrinoid iron-sulfur protein? Base-off cob(II)alamin, formed at low pH, is more facilely reduced than base-on cob(II)alamin 68 and the enzyme-bound 5-methoxybenzimidazolylcob(II)amide is also more readily reduced than the free base-on cofactor 61, as is the enzyme-bound cob(II)alamin found in the M. thermophila 63 and M. barkeri 59 proteins. In the case of methionine synthase, the cob(II)alamin form of the enzyme can be readily interconverted between base-on and base-off forms, while the methylcobalamin form of the enzyme is present predominantly as the base-on form. However, the methylcobinamide form of the corrinoid iron-sulfur protein is completely base-off.
The acetogenic acylCoA synthase shares another property with methionine synthase and other corrinoid-dependent methyl transferases, namely the ability to react with exogenous corrinoids as well as with their physiological corrinoid partner proteins. The Ragsdale group has shown that acylCoA synthase from M. thermoaceticum reacts 2000-times faster with methylcobinamide, which lacks a lower axial base, than with methylcobalamin 69. These results may suggest a late transition state for this methyl transfer, with substantial bond breaking leading to cob(I)amide character.
2.6 Aromatic O-demethylases
Many acetogenic bacteria derive both carbon and energy by demethylating aromatic methyl ethers. The pathway involves transfer of the methyl group from the ether to methyltetrahydrofolate, which then can enter the Western branch of the Wood-Ljungdahl pathway and be converted into the methyl group of acetate. Methyl groups bound to methyltetrahydrofolate can also be oxidized to formate and/or CO2 by the reverse of the Eastern branch of the Wood-Ljungdahl pathway. The reversal of the Eastern branch to CO2 generates six hydride equivalents per methyl group oxidized, which in turn can be used to reduce 3 moles of CO2 to CO to form 3 mol of acetyl CoA in the Western branch. The overall stoichiometry is shown in eq. 3.
Eq. 3.

The O-demethylases from Acetobacterium dehalogenans 21, 70 and Moorella thermoacetica71 conform to the basic pattern illustrated in Fig. 3. The four components of the vanillate O-demethylase from A. dehalogenans comprise two methyl transferases, a corrinoid protein and an activating enzyme 21, 70. The first methyltransferase (odmB) catalyzes methyl transfer from vanillate (O-methylhydroxybenzoate) to the corrinoid protein (odmA) and the second methyltransferase catalyzes methyl transfer from the methylated corrinoid protein to tetrahydrofolate. The activating protein uses hydrogenase as the source of reducing equivalents and requires ATP. The amino acid sequence derived from the odmA gene shows about 60% similarity with the cobalamin-binding domain of methionine synthase, including the Asp-x-His-x-x-Gly sequence characteristic of binding cobalamin in the base-off His-on mode.
2.7 Reductive dehalogenases
Free cob(I)alamin is known to react rapidly with alkyl halides like methyl iodide to produce alkyl cobalamins, and in the presence of reducing agents abiotic dehalogenation is catalyzed at significant rates 72. The mechanism of abiotic reductive dechlorination of perchloroethylene has recently been examined 73. The authors concluded that the most likely mechanism was that shown in Fig. 11. This mechanism involves an initial electron transfer from cob(I)alamin to the perchloroethylene with release of chloride ion and formation of a trichlorovinyl radical that would immediately combine with cob(II)alamin to produce a trichlorovinylcobalamin. A further electron transfer would then generate a trichlorovinyl anion and regenerate cob(II)alamin.
Figure 11. Proposed abiotic mechanism for dechlorination of perchloroethylene.

Redrawn, with permission from 73.
With this mechanism as a guide, we can now examine the studies on corrinoid-dependent enzymes that catalyze dehalogenation reactions in anaerobic bacteria. Many of the organisms capable of aryl halide conversion belong to the sulfidogenic bacteria, which can reductively dechlorinate polychlorinated phenols and benzoates, tetrachloroethylene and trichlorethylene (reviewed in 74). The perchloroethylene reductive dehalogenases have been particularly well characterized. These enzymes are typically membrane anchored, and contain two Fe4S4 or Fe3S4 clusters in addition to a corrinoid, which is present in the base-off form 75, 76. The redox potentials of the iron sulfur centers were lower than that of the Co(II)/Co(I) couple of the corrinoid, which would favor reduction of the corrinoid 75, 76. An ortho-chlorophenol reductive dehalogenase was purified from Desulfitobacterium dehalogenans and shown to have similar properties 77, including the presence of base-off corrinoid as isolated in the Co+2 oxidation state. This protein contained one Fe4S4 and one Fe3S4 cluster, with the Fe4S4 cluster having the lower potential. More recently a meta- and para-chlorophenol reductive dehalogenase was purified, cloned and sequenced from Desulfitobacterium frappieri78. The CprA protein sequence contains two iron-sulfur binding motifs, and a Glu-Tyr-His-Tyr-Asn-Gly motif (EYHYNG) that is related to the Asp-x-His-x-x-Gly motif found in base-off His-on cobalamin-binding proteins. It will be of great interest to know whether this histidine is indeed a ligand to the cobalt under some conditions. Given the reaction mechanism proposed in Fig. 11, binding of cobalamin in the base-off mode should greatly facilitate the reduction of both cob(II)alamin and trichlorovinyl-cobalamin.
The enzymes responsible for reductive dehalogenation of chloromethane have also been studied. These enzymes catalyze methyl transfer from chloromethane to tetrahydrofolate. In Methylobacterium sp., the reductive dehalogenase comprises two components, CmuA and CmuB. CmuA is a two domain methyltransferase/corrinoid-binding protein and contains cob(II)alamin as isolated 79. The N-terminal domain shows sequence similarity to methycobamide:coenzyme M methyltransferases, while the C-terminal domain shows sequence similarity to the cobalamin-binding domain of methionine synthase. However the Asp-x-His-x-x-Gly sequence characteristic for base-on His-off cobalamin binding is replaced by Asn-Thr-Gln-x-x-Gly in the sequence alignment, suggesting a base-off mode of cobalamin binding 80. CmuB shows methylcobalamin:tetrahydrofolate methyltransferase activity and shares sequence similarity with MtrH of the methyltetrahydromethanopterin:coenzyme M methyl transferase complex and with the methyltetrahydrofolate-binding domain of MetH 81. There is no evidence for iron-sulfur cluster motifs in the sequences of CmuA or CmuB and the spectra of the enzymes as isolated, or after reduction, do not reveal the presence of iron-sulfur clusters.
2.8 Modes of activation of corrinoid-dependent methyltransferases
A majority of the corrinoid-dependent methyltransferases requires a reductive activation, even those proteins native to strictly anaerobic organisms. In MetH, reductive activation is necessary to retrieve the inactive cob(II)alamin prosthetic group and return it to the catalytic cycle, but it is less certain whether such oxidative inactivation actually occurs under strictly anaerobic growth conditions. Recent studies on the incorporation of the cobalamin cofactor into the apoenzyme suggest an alternative function for these activation proteins or modules, namely a role as a chaperone to assist the enzyme in binding the prosthetic group 82–84.
In the methyltransferases that bind the cobalamin prosthetic group in the base-off or base-off His-on state, incorporation of the prosthetic group requires that the dimethylbenzimidazole first be dissociated from the cobalamin. The free energy required for replacement of dimethylbenzimidazole by water has been measured for a series of cobalamins 85, 86. For aquocob(III)alamin, the base-off form is disfavored by 10.4 kcal/mol, while for cob(II)alamin, the base-off form is only disfavored by 3.3 kcal/mol. Thus on thermodynamic grounds alone, we would expect the initial formation of base-off cobalamin to occur at the cob(II)alamin oxidation state and to be an endergonic process.
Table 1 summarizes what is known about the activation systems in corrinoid-dependent methyltransferases. I will again begin by discussing what is known about the activation of methionine synthase, and then proceed to describe studies on the ATP-dependent activation systems found in other corrinoid-dependent methyl transferases. The activation of the corrinoid iron/sulfur protein, which does not require either ATP or AdoMet, has already been discussed in section 2.5.
TABLE I.
Requirements for reductive activation of corrinoid-dependent methyltransferases
| Enzyme complex (corrinoid cofactor) | Organism | Activating protein(s) or domains | Cofactors/reductants | Reference |
|---|---|---|---|---|
| Methionine synthase (cobalamin) |
Escherichia coli Homo sapiens |
AdoMet domain AdoMet domain |
AdoMet/flavodoxin AdoMet/methionine synthase reductase |
[85] [88, 90] |
| Methanol:coenzyme M methyltransferase (5-OH-benzimidazolylcobamide) | Methanosarcina barkeri | MAP | ATP/H2/hydrogenase | [81, 91] |
| Methylamine:coenzyme M methyltransferases (5-OH-benzimidazolylcobamide) |
Methanosarcina barkeri Methanosarcina acetovorans |
RamA (MA0150) | ATP/ | [26] |
| Energy-conserving methyltetrahydromethanopterin: coenzyme M methyltransferase (5-OH-benzimidazolylcobamide) | Methanobacterium thermoautotrophicum | not identified | ATP/reduced ferredoxin | [92] |
| Veratrol:H4folate and vanillate:H4folate O-methyltransferases (cobalamin) | Acetobacterium dehalogenans | not identified | ATP H2/hydrogenase ferredoxin | [93, 94] |
| Corrinoid Fe/S protein (5-methoxybenzimidazolyl | Moorella thermoaceticum | requires Fe4S4 center on large subunit | CO/CO dehydrogenase ferredoxin | [61] |
In MetH from E. coli, activation of enzyme in the cob(II)alamin form requires AdoMet 87 and a reducing system. While early studies of the enzyme employed exogenous reductants, isolation of the components responsible for reduction in crude bacterial extracts in the laboratory of Frank Huennekens led to the identification of flavodoxin, flavodoxin (ferredoxin) reductase, and NADPH as the components responsible for MetH reduction in these extracts 88. Incubation of MetH, isolated in the cob(II)alamin form with AdoMet bound, with flavodoxin, flavodoxin reductase and excess NADPH led to formation of enzyme in the methylcobalamin form 89. As mentioned previously, AdoMet is bound to the C-terminal module of methionine synthase 6, 14, which is required for reductive activation. This C-terminal module also contains determinants for the binding of flavodoxin 90.
In 1997, Hoover reported that incubation of MetH in the cob(II)alamin form with oxidized flavodoxin led to the formation of base-off cob(II)alamin 9. This shift required stoichiometric concentrations of methionine synthase and flavodoxin. When the structure of the C-terminal half of His759Gly methionine synthase in the AdoMet:Cob conformation was determined, it was apparent that assumption of this conformation required that the cobalamin assume a base-off conformation. Thus oxidized flavodoxin, which is incapable of electron transfer, is assuming a role as a chaperone to facilitate the formation of base-off cobalamin in the AdoMet:Cob conformation when the enzyme is in the cob(II)alamin form. This is of course the conformation needed for reductive remethylation of the cofactor. If the enzyme is in the aquocob(III)alamin form, flavodoxin binds tightly, but the shift to the base-off conformation does not occur.
Mammals lack flavodoxin and flavodoxin (ferredoxin) reductase. For the activation of methionine synthase they instead employ a fusion protein with an N-terminal domain homologous to flavodoxin and a C-terminal domain homologous to flavodoxin reductase 91. This protein, methionine synthase reductase, is required for the in vivo activation of methionine synthase, and patients with severe deficiencies of this enzyme present with symptoms resembling those of patients with severe deficiencies of methionine synthase itself 92. Methionine synthase reductase is not only required for the reductive reactivation of human methionine synthase. The oxidized reductase substantially stabilizes the apoenzyme, which rapidly undergoes irreversible denaturation on incubation at 37 °C 83. Furthermore, methionine synthase reductase greatly increases the yield of holoenzyme formed on incubation of apo-methionine synthase with aquacobalamin and dithiothreitol 83. These findings suggest a chaperone-like function for methionine synthase reductase. Thus far, insufficient amounts of human methionine synthase have been available to permit experiments to determine whether methionine synthase reductase also stabilizes holoenzyme in a base-off AdoMet:Cob conformation.
Thus far, AdoMet-dependent reductive activation appears to be unique to methionine synthase. Where activation of other methyltransferases has been shown to occur, reductive activation appears to be coupled to ATP rather than AdoMet. For a listing of methyltransferase activating proteins see Table 1. The best-studied reactivation protein is the methyltransferase-activating protein (MAP) which is involved in the activation of the cytoplasmic methylamine:coenzyme M methyltransferases and methanol:coenzyme M methyltransferase in Methanosarcina barkeri. Sustained activity of these coenzyme M methyltransferases requires ATP, hydrogen, hydrogenase and MAP 84. Incubation of ATP with MAP at 1:1 concentration ratios led to the phosphorylation of MAP. Phosphorylated MAP substituted for ATP in the stoichiometric activation of MT1, the corrinoid-containing methanol:5-hydroxybenzimidazolyl cobamide component of the cytoplasmic methanol:coenzyme M methyltransferase complex. If ATP were present in excess, MAP could catalyze multiple rounds of activation, indicating that ATP hydrolysis occurred during the activation process.
If MT1 in the aquacob(III)inamide form was incubated with hydrogen and hydrogenase, base-on cob(II)amide was formed. Addition of MAP and ATP resulted in formation of a mixture of base-on (60%) and base-off (40%) cob(II)inamide, and if methanol was then added, the prosthetic group was quantitatively converted to the methylcobinamide form 94. Thus, phosphorylated MAP acts as a chaperone, inducing a conformation change in MT1 that favors the formation of base-off cob(II)inamide, similar to the effect of oxidized flavodoxin on MetH in the cob(II)alamin form. The difference is that methanol, the substrate, also serves as the methyl donor in reductive reactivation. A protein thought to be similar or identical to MAP can also serve to activate the corrinoid-proteins in the methylamine methyltransferase complexes 97. However, annotation of the Methanosarcina acetivorans genome sequence 31, 98 indicates that a gene specifying an iron-sulfur protein designated RamM is responsible for activation of methylamine methyltransferases. A number of homologues of ramM have been identified in the M. acetivorans genome, although their functions have not yet been determined.
2.9 Methyl transfer in fosfomycin biosynthesis
The biosynthesis of fosfomycin was initially proposed to involve a unique function for methylcobalamin, namely the transfer of the methyl group as a methyl anion (Fig. 12). However, all characterized methylcobalamin-dependent methyl transferases transfer the methyl group as a methyl cation. The evidence for the involvement of methyl cobalamin as the methyl donor comes from the studies of Seto and Kuzuyama and their colleagues 99, 100. They showed that a mutant strain of Streptomyces with a block in the B12 biosynthetic pathway could not produce fosfomycin, and that feeding of 14C-labeled methylcobalamin to this blocked mutant resulted in 14C-labeled fosfomycin.
Figure 12. Originally proposed mechanism for generation of the methyl group of fosfomycin.

Adapted with permission from 101.
Recently, van der Donk and his colleagues 101 identified the entire biosynthetic cluster for fosfomycin and showed that it induced fosfomycin production in a Streptomyces strain lacking this capability. Analysis of the gene sequence of one of the components of the cluster, fom3, showed that it contained a region with similarities to a B12-binding domain and another region with homology to the family of proteins that utilize AdoMet as a radical generator. The fom3 gene was shown to be essential for the biosynthesis of fosfomycin. Furthermore the authors provided a strong inference that the actual substrate for methylation was hydroxyethylphosphonate rather than phosphonoacetaldehyde, leading to the proposal of a mechanism much more in keeping with known B12 chemistry (Fig. 13).
Figure 13. Mechanism for generation of the methyl group of fosfomycin proposed by van der Donk and colleagues.

Adapted with permission from 101
We must await the purification and characterization of Fom3. But if the proposed mechanism is indeed correct, we may have a fascinating clue to the origin of AdoCbl-dependent enzymes. As pointed out by Sauer and Thauer in their recent review 55, thus far all corrinoid protein characterized from methanogenic archaea have been methyltransferases containing methylcobalamin as a prosthetic group and no AdoCbl-dependent enzymes have been found. Furthermore the cobO gene required for synthesis of AdoCbl appears to be lacking. Fom3, and its analogues in methylation reactions required for the biosynthesis of other antibiotics produced in Streptomyces, may represent a step in the direction of development of the AdoCbl family of enzymes. In the mechanism proposed for Fom3, AdoMet is cleaved to generate an adenosyl radical which abstracts a hydrogen from hydroxyethylphosponate. The substrate radical is then methylated by methylcobalamin leaving cob(II)alamin as the product. Regeneration of methylcobalamin might then occur by way of a process similar to the reactivation of methionine synthase, using an external reductant and AdoMet as the methyl donor. It should be noted that this proposed mechanism would require two molecules of AdoMet per methylation reaction: one to generate the initial radical and a second to serve as a methyl donor.
3. ADENOSYLCOBALAMIN-DEPENDENT REARRANGEMENTS AND ELIMINATIONS
Although the AdoCbl-dependent enzymes initially attracted the greatest attention from organic and inorganic chemists because of their fascinating chemistry, we now know that they represent but a small branch of the corrinoid-dependent enzymes. They are found most frequently in the eubacteria (Kingdom Prokaryota) and just one AdoCbl-dependent enzyme, methylmalonyl CoA mutase, is found in mammals. No AdoCbl-dependent enzymes have yet been identified in the Archaea.
The basic mechanism of AdoCbl-dependent rearrangements is shown in Fig. 14. This mechanism was simultaneously elucidated in Abeles’ laboratory at Brandeis University and Arigoni’s laboratory in Zürich. In classic papers, Frey and Abeles 102 showed that AdoCbl bound to propanediol dehydrase is tritiated as the enzyme reacts with [1-3H]1,2-propanediol and that tritium could subsequently be transferred from the isolated tritiated coenzyme to unlabeled propanediol, and Rétey and Arigoni 103 then showed that AdoCbl that had been labeled with tritium when catalyzing the propanediol dehydrase reaction could also transfer tritium to methylmalonylCoA. Frey, Essenberg and Abeles 104 showed that tritium is transferred from [1-3H]propanediol to the C-5′ position of the AdoCbl of dioldehydrase, and from [5′-3H]AdoCbl to C2 of the product propionaldehyde. They also showed that the hydrogen abstracted from C1 of the substrate becomes equivalent with one or both of the hydrogens of the C5′ position of the cofactor following the initial hydride transfer. Cleavage of the Co-C5′ bond of AdoCbl to form cob(II)alamin and 5′-deoxyadenosine was first observed in the suicide inactivation of dioldehydrase by glycolaldehyde, and in the same paper the substrate propanediol was also shown to induce transient formation of cob(II)alamin 105. At the same time, Rétey, Umani-Ronchi, Seibl and Arigoni 106, used 18O-enriched propanediols to demonstrate the migration of 18O from C2 of propanediol to C1 of propionaldehyde, thus demonstrating the migration of the substituent at C1 (X in Fig. 14) in another classic paper.
Figure 14.
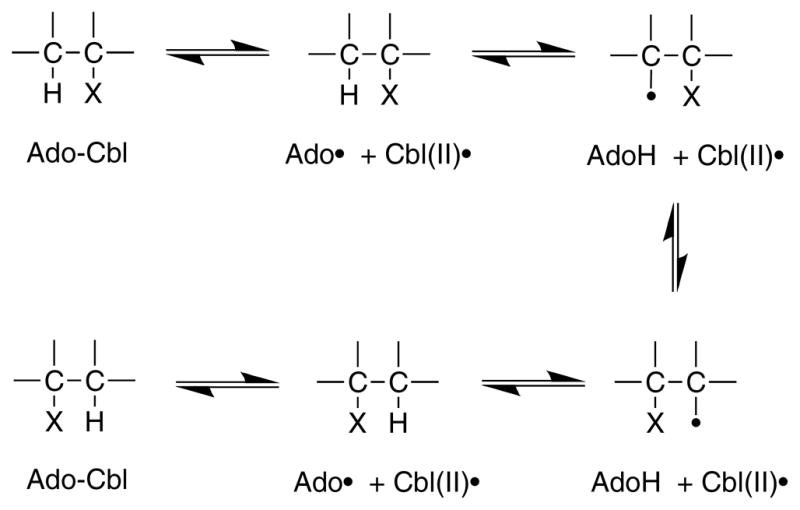
Basic mechanism of AdoCbl-dependent rearrangements
In the sections that follow, I will review the literature on each of the AdoCbl-dependent enzymes in turn. First, however, I wish to emphasize several challenges to understanding the mechanisms of each of these enzymes:
Activation of the C-Co bond of AdoCbl for cleavage. Th: carbon-cobalt bond of AdoCbl is estimated to have a bond dissociation energy of 30 kcal/mol 107. Thus Finke and Hay have estimated that dioldehydrase must lower the barrier for Co-C bond homolysis by at least 14.7 kcal/mol for a rate acceleration of 1010! How this is achieved in any AdoCbl-dependent enzyme remains controversial.
Transfer of H• from substrate to deoxyAdo and from deoxyAdo to product: For some of the AdoCbl-dependent enzymes, the H• must traverse long distances (6–10 Å) during the reaction. We are just now beginning to understand how H• is transferred by the various enzymes.
Catalysis of the migration of X: The issue of whether cob(II)alamin is a participant in catalysis or a bystander is an argument that has persisted over decades. However, recent studies have greatly illuminated the mechanisms of migration, especially in the enzymes that catalyze carbon skeleton rearrangements. It has become clear that the distance between cob(II)alamin and the substrate radical following homolytic cleavage of AdoClb in diol dehydrase and ethanolamine ammonia lyase is too great for the cobalamin to participate in the subsequent rearrangment. However, in the mutases, the distance between cob(II)alamin and substrate radical would be consistent with participation of the cobalamin in the rearrangement 108, and in fact density functional theory calculations support this role for cobalamin 109.
In reviewing the voluminous literature on these enzymes, I have tried to emphasize recent developments in the field, particularly emphasizing what we have learned as x-ray structures of these proteins have been determined. But I have not done justice to the earlier stereochemical experiments that elucidated the details of the overall reactions, nor the extensive characterization of the substrate and product radicals. Rather, I have attempted to focus on the role of the B12 cofactor.
3.1. Enzymes that catalyze carbon skeleton rearrangements
This class of enzymes catalyzes rearrangements that require cleavage of a carbon-carbon bond to allow migration of X. These enzymes are glutamate mutase, methylmalonyl CoA mutase, and isobutyryl CoA mutase. In all three of these enzymes, the AdoCbl cofactor is ligated by a histidine residue from the protein. Indeed, following the cloning of glutamate mutase, Marsh and Holloway first recognized the Asp-X-His-X-X-Gly motif that characterizes His-on ligation in methionine synthase and the enzymes that catalyze carbon skeleton rearrangements 12. While all three enzymes have similar cobalamin-binding domains or subunits, they differ considerably in the structures/sequences of the substrate-binding regions of the proteins.
3.1.1 Glutamate mutase
Glutamate mutase catalyzes the reaction shown in Eq. 4, the conversion of (S)-glutamate to (2S,3S)-3-methylaspartate. In this reaction, a hydrogen on Cβ is exchanged with the glycyl group on Cα. The enzyme is an α2β2 oligomer; the small subunit GlmS (σ) is 14.7 kDa while the large subunit GlmE (ε) is 50 kDa 110. As noted above, the small subunit contains the Asp-X-His-X-X-Gly motif associated with binding of B12 in a DMB-off His-on state. However, neither the large nor the small subunit binds AdoCbl tightly, and the binding site is created at the interface between the two subunits. For some studies, a fusion protein comprising both large and small subunits has been used to avoid the complications of subunit dissociation and loss of activity and AdoCbl 111.
Eq. 4.
The structure of glutamate synthase with AdoCbl substituted by methyl- or cyanocobalamin reveals that the architecture of GlmS is highly similar to that of the B12-binding domain of methionine synthase from E. coli 112. The DMB nucleotide is deeply buried in a hydrophobic pocket in this subunit, and His16, which is carried on the loop between the first strand and the first helix of the barrel, coordinates the α position of the cobalt in B12. As suggested by the conserved motif, His 16 is also hydrogen bonded to Asp14; however the third amino acid in the “ligand triad” is missing and instead Asp14 also forms hydrogen bonds to main-chain amide groups and to a water molecule.
A structure of GlmS apoenzyme has been determined by NMR and provides insights into how the holoenzyme may be formed 113. In this structure, which otherwise resembles the architecture of the holoenzyme small subunit, residues 13–27 form a disordered and highly mobile loop. The region corresponding to the first alpha helix in the holoenzyme, residues 18–27, rapidly interconverts between unstructured forms and an α–helical conformation. The unfolding of the first helix exposes to solvent the cavity where the DMB will reside in the holoenzyme, so that the apoenzyme is preorganized for incorporation of the B12 cofactor. I have long argued that only cobalamins with relatively high propensities to form base-off cobalamin (e.g. methyl- and adenosyl-cobalamin, cob(II)alamin and cob(I)alamin) will lend themselves to incorporation into proteins that bind the cofactor in the base-off His-on state, but this NMR structure adds another element to our understanding of the process by which holoenzyme formation might occur.
The GlmE subunit is an α8β8 barrel, with the open end packed against the β face of the B12 in the holoenzyme structure 112. The crystallization medium contained tartrate, an analogue of methylaspartate, which bound in the barrel in close proximity to the B12 cofactor. In a subsequent paper 114, the structure of active glutamate mutase with AdoCbl and glutamate bound was determined. In this structure, the electron density of the adenine is clearly modeled, but fitting the electron density with the ribose moiety of adenosine requires modeling in a mixture of C2′-endo and C3′-endo conformations (Fig. 15). The C3′-endo conformation places C5′ of the ribose within bonding distance of the cobalt of cobalamin, while the C2′-endo conformation leads to a 4.2 Å distance between C5′ and Co, but places C5′ within 3.3 Å of Cγ of the glutamate substrate. Thus the authors propose that a simple pseudorotation of the ribose between two low-energy conformers leads to cleavage of the carbon-cobalt bond and abstraction of hydrogen from Cγ of glutamate.
Figure 15. Alternate conformations of the ribose of AdoCbl at the active site of glutamate mutase.

On the left the ribose assumes a C3′-endo conformation, placing the 5′-carbon within bonding distance of the cobalt of cobalamin and distant from Cγ of the glutamate substrate. On the right the ribose assumes a C2′-endo conformation, leading to breakage of the carbon-cobalt bond and placing C5′ within van der Waal’s distance of Cγ of the substrate.
Fig. 16 shows the mechanism proposed for glutamate mutase. The initial research supporting this mechanism was performed in Horace Barker’s laboratory in Berkeley and has been recently reviewed 110. These studies established the stereochemistry of the reaction, and showed that there was no exchange of the hydrogens of substrate or product with solvent, and no exchange of potential intermediates such as glycine or acrylate. The reaction is unique among the enzymes catalyzing rearrangement of carbon skeletons in that the migrating carbon is sp3 hybridized, as shown in Eq 4. Initial evidence for the fragmentation to form a glycyl radical and acrylate came from the observation that glycine and acrylate inhibit the enzyme synergistically, and induce the formation of an EPR spectrum similar to those observed for the substrate or product radical (reviewed in 110). More recently, direct evidence for formation of glycine and acrylate was obtained by rapid-quench analysis of the reaction, and the rate of formation of these intermediates was shown to be faster than the overall rate of reaction 115.
Figure 16. Proposed mechanism for glutamate mutase.

The adenosyl radical formed by cleavage of the C-Co bond of AdoCbl abstracts a hydrogen from glutamate to yield the glutamyl radical. This leads to the elimination of acrylate and formation of glycyl radical, which in turn condenses with acrylate to form the β-methylaspartate radical. In the final step, the product radical abstracts hydrogen from 5′-deoxyadenosine to regenerate the adenosyl radical, which can now recombine with cob(II)alamin to reform AdoCbl.
3.1.2 Methyleneglutarate mutase
Methyleneglutarate mutase catalyzes a reaction that is very similar to that catalyzed by glutamate mutase, as shown in Eq 5. One key difference however, in the reaction is that the migrating carbon is sp2 hybridized, a property that is shared with all the other enzymes that catalyze carbon skeletal rearrangement except glutamate mutase. This property permits the rearrangement to proceed through a cyclopropylcarbinyl intermediate rather than requiring a fragmentation-recombination as shown in Fig. 17 110. Rearrangements of cyclopropinyl radicals are well precedented in model chemistry and proceed extremely rapidly 116.
Eq. 5.

Figure 17. Mechanism proposed for radical rearrangment in methyleneglutarate mutase.

Because the migrating carbon is sp2, a cyclopropylcarbinyl radical can form to mediate the transfer of the radical between Cα and Cβ116.
The protein is isolated from Clostridium barkeri as a homotetramer of 60 kDa subunits. The deduced amino acid sequence of the protein shows significant sequence homology in its C-terminal region with the cobalamin-binding regions of methylmalonyl CoA mutase, glutamate mutase and MetH, including the conserved Asp-X-His-X-X-Gly sequence that is the hallmark of DMB-off His-on binding of the cobalamin cofactor 117. Mutation of the corresponding residues, His 485 and Asp483, decreases the rate of substrate turnover by >4000-fold and by 2000-fold respectively 118.
3.1.3 Methylmalonyl CoA mutase
Methylmalonyl CoA mutase catalyzes the reaction shown in Eq. 6. The migrating carbon is sp2, allowing radical rearrangement to proceed by way of a cyclopropylcarbinyl radical, as in methyleneglutarate mutase. The bacterial enzymes are αβ heterodimers, with considerable homology between the α and β subunits, while the mammalian enzyme is an α2 homodimer. Only one molecule of AdoCbl is bound per bacterial heterodimer, however. The x-ray structure of the enzyme from Propionobacterium shermanii was the first structure to be determined of a complete cobalamin-binding protein 119. The α and β chains exhibit similar folds, but only the α chain contains bound cofactor. Each chain consists of an N-terminal α8β8 barrel and a C-terminal domain that exhibits a fold similar to the cobalamin-binding domain of methionine synthase. Nε2 of HisA610 coordinates the lower axial position of the B12 cofactor, which appears to be cob(II)alamin in this structure. Nδ1 of HisA610 is hydrogen bonded to the carboxyl oxygen of AspA608, and the other carboxyl oxygen of the Asp608 sidechain is hydrogen bonded to LysA604. As in glutamate mutase and MetH, the DMB substitutent of the corrin ring is deeply buried in a hydrophobic pocket in the cobalamin-binding domain.
Eq. 6.

The N-terminal barrel of the α chain of methylmalonyl CoA mutase is juxtaposed against the β-face of the B12, similar to its position in glutamate mutase 119. The protein was crystallized in the presence of desulphoCoA, a substrate analogue that lacks the terminal thiol and the succinyl group of succinyl CoA. One equivalent of this analogue was bound in a narrow tunnel along the axis of the barrel of the α chain, completely buried in the interior of the barrel 119. Structures of methylmalonyl CoA mutase were subsequently obtained for the substrate-free enzyme and for enzyme in a non-productive complex with CoA 120. These structures, which were similar, revealed that in the absence of productively bound CoA, the α8β8 barrel is split apart and the CoA binding site is accessible to solvent. When CoA binds, the barrel closes up and encapsulates the substrate. The adenosyl group of AdoCbl could be seen in the substrate-free complex, but when the active site closes it is no longer visible, and the TyrA89 sidechain now occupies a position that overlaps with the adenosyl binding region in the substrate-free enzyme. The authors propose that the closing of the active site cavity forces the carbon-cobalt bond cleavage. Support for the role of TyrA89 as a “molecular wedge” comes from studies of TyrA89Phe and TyrA89Ala mutant enzymes that demonstrate 1000-fold reductions in catalytic activity, and the disappearance of cob(II)alamin from the spectrum of the enzyme taken under steady-state conditions 121. In the wild-type enzyme the ratio of AdoCbl:cob(II)alamin is about 4:1 when the enzyme is catalyzing the conversion of methylmalonyl- to succinyl-CoA. These studies enforce the view that the enzyme uses conformational changes driven by the binding of the CoA substrate to break the carbon-cobalt bond of the cofactor, consistent with the earlier observation that substrate binding accelerates carbon-cobalt bond cleavage by a factor of 1012 122.
Banerjee and her coworkers have measured the Co-C homolysis rate of AdoCbl bound to methylmalonyl-CoA mutase 122. When the rates of homolysis are compared in the presence of [CH3]methylmalonyl-CoA and [CD3]methylmalonyl-CoA, the rate in the presence of the deuterated substrate is at least 20-fold slower. One would not expect the deuteration of the substrate to affect the rate of cleavage of AdoCbl unless the situation shown in Fig. 18 were to prevail. The large isotope effect on formation of the substrate radical slows the overall rate of cleavage of AdoCbl. In a subsequent study 123 Chowdhury and Banerjee measured the temperature dependence of the activation parameters for reaction with [CH3]methylmalonyl-CoA, providing a better estimate of magnitude of the kinetic isotope effect at 49.9. Subsequently computational analysis of the reaction confirmed that the cleavage of AdoCbl was indeed a stepwise process, rather than being concerted with hydrogen atom transfer from substrate to the deoxyadenosyl radical formed on cleavage of AdoCbl 124. Surprisingly, these computations predicted that the rate constants k1 and k−1 in Fig. 18 are actually much faster than the rate constant for transfer of the hydrogen atom from the substrate to deoxyadenosine. The equilibrium governing the first step is unfavorable as shown in Fig. 18.
Figure 18. Coupling between cleavage of AdoCbl and formation of the substrate radical in methylmalonyl-CoA mutase.

A rapid but unfavorable equilibrium between AdoCbl and the homolytically cleaved dAdo radical and cob(II)alamin precedes a rate-limiting hydrogen atom abstraction from the substrate 124.
Electron paramagnetic resonance has been used to estimate the distance between cob(II)alamin and a succinyl-CoA radical at the active site and their relative orientations 125. Line broadening induced by heavy atom substitutions in succinyl CoA indicated that the radical was centered on the carbon α to the free carboxyl. The interspin distance was about 6 Å between the two radical centers, and the radical could be modeled in a position very similar to that occupied by succinyl-CoA in a product complex determined by x-ray crystallography.
The interspin distance is large for a system that exhibits such large deuterium kinetic isotope effects, which are well above the classical limit and suggest a significant contribution due to hydrogen atom tunneling. In a recent paper, the contribution of hydrogen tunneling to the radical transfer catalyzed by methylmalonyl-CoA mutase has been rigorously analyzed 126. The authors conclude that the large kinetic isotope effect can only be explained if corner-cutting tunneling decreases the distance over which the system tunnels.
Human methylmalonyl CoA mutase is a mitochondrial enzyme and the only AdoCbl-dependent enzyme in humans, and mitochondrial B12 processing involves involves reduction of cob(II)alamin to cob(I)alamin, conversion of cob(I)alamin to AdoCbl and then transfer to the methylmalonyl CoA mutase apoenzyme. Human adenosyltransferase catalyzes the conversion of cob(I)alamin to AdoCbl using ATP as the source of the adenosyl group 127. However cob(II)alamin can not be used as the substrate, indicating that the adenosyltransferase does not itself catalyze the reduction of cob(II)alamin to cob(I)alamin. If adenosyltransferase is incubated with cob(II)alamin in the presence of methionine synthase reductase, ATP and NADPH, AdoCbl is formed 128. Addition of a cob(I)alamin scavenging agent, iodoacetamide, has no effect on this conversion, indicating that the cob(I)alamin is sequestered. The reasonable assumption is therefore that reduction occurs while cob(II)alamin is bound to adenosyltransferase. However, we note that methionine synthase reductase is probably not the physiological reducing agent for adenosyltransferase: this reaction occurs in mitochondria, while methionine synthase reductase is cytoplasmic, and patients lacking methionine synthase reductase do not show abnormalities in AdoCbl synthesis. The physiological reducing agent for adenosyltransferase thus remains to be identified.
Banerjee and Brunold and their colleagues have shown that adenosyltransferase binds cob(II)alamin is the base-off His-off form 129, which leads to a more favorable potential for reduction to cob(I)alamin. Furthermore, the cob(II)alamin becomes four coordinate, lacking water as a ligand, when ATP is present. It is proposed that adenosyltransferase also functions as a chaperone, transferring the base-off AdoCbl to methylmalonyl CoA mutase 130, which has a higher affinity for the cofactor than adenosyltransferase. While it remains to identify the protein responsible for the reduction of cob(II)alamin to cob(I)alamin in mitochondria, and it remains to determine whether cob(II)alamin is indeed bound to human adenosyltransferase during reduction, the idea of chaperoning this rare and reactive cofactor is highly compelling. The situation should be compared to that in cytoplasmic MetH, where reduction of cob(II)alamin to cob(I)alamin takes place when bound to MetH itself, using electrons from a partner electron transfer protein, and methylation requires AdoMet bound to its own module in methionine synthase.
In addition to the reducing agent and adenosyltransferase required for the activity of methylmalonyl-CoA in human mitochondria, a third component, MMAA, is also strongly stimulatory. (MMAA is the gene designation for this protein.)This protein is a homologue of MeaB, a bacterial protein that is frequently found in operons also containing methylmalonyl-CoA mutase. Mutant strains lacking MeaB are unable to convert methylmalonyl-CoA to succinyl-CoA, although they retain the ability to synthesize AdoCbl 131. Human patients lacking MMAA belong to the cblA complementation group of patients who present with methylmalonic aciduria 132. Several studies have been carried out on MeaB from Methylobacterium extorquens, the organism in which MeaB was first studied. MeaB shows homology with GTPases, a family that includes many enzymes involved in assembly of metal cofactors 131. It forms complexes with holomethylmalonyl-CoA mutase that are enhanced when GTP is bound, and methylmalonyl-CoA mutase stimulates the GTPase activity of MeaB 133. Furthermore the GTP-bound form of MeaB slows the rate of oxidative inactivation of methylmalonyl-CoA mutase (to form aquacob(III)alamin) by about 15-fold 134. However, the physiological role of MeaB and its human homologue MMAA in maintaining methylmalonyl-CoA mutase activity remains to be elucidated.
3.1.4 Isobutyryl CoA mutase
Isobutyryl CoA mutase catalyzes the reaction shown in Eq. 7. This reaction is very similar to the reaction catalyzed by methylmalonyl-CoA mutase, with the carboxyl group in methylmalonyl-CoA being replaced by a methyl group in isobutyryl-CoA. Inactivation of the icmA gene in Streptomyces cinnamonensis leads to a strain that is unable to use valine or isobutyryl-CoA as carbon sources 135. The genes specifying the large and small subunits of isobutyryl-CoA mutase in Streptomyces cinnamonensis have been cloned and sequenced and expressed in E. coli. The icmA gene specifies a 62 kDa large subunit with ~40% sequence identity to the large subunits of bacterial methylmalonyl-CoA mutases 136. However, homologies to the C-terminal cobalamin-binding regions of the latter proteins are lacking. Instead, homologies to the cobalamin-binding regions of methylmalonyl-CoA mutases are found in the icmB gene specifying the 14 kDa small subunit 137. These homologies include the Asp-X-His-X-X-Gly motif associated with DMB-off His-on binding of the cofactor. The purified protein is an α2β2 heterodimer. Given the extensive homologies with methylmalonyl-CoA mutase, it is likely that the catalytic mechanisms of these two proteins will be highly similar. Early studies showed that the enzyme catalyzes an intramolecular rearrangement in which the carbonyl thioester of isobutyryl-CoA undergoes a 1,2-migration to the pro-(S) methyl and is replaced by a hydrogen atom at C(3) of n-butyryl-CoA with overall retention 138.
Eq. 7.

3.1.5 MeaA—A mutase of unknown function
A second protein with extensive homology to methylmalonyl-CoA mutase, MeaA, has been identified in Streptomyces collinus and Methylobacterium extorquens. The meaA gene from S. collinus has been cloned and sequenced 139. It specifies a putative 74 kDa protein with 40% homology to methylmalonyl-CoA mutase and significant homology to the large and small subunits of isobutyryl-CoA mutase. Growth studies suggest that MeaA is crucial for the production of methylmalonyl-CoA in Streptomyces cinnamonensis, although the actual reaction catalyzed remains to be elucidated.
3.2. Aminomutases and Diol Dehydrases—Isomerization and Elimination
Aminomutases catalyze the 1,2 exchange of an amino group with a hydrogen atom, while diol dehydrases catalyze a 1,2 exchange between a hydroxyl group with a hydrogen atom. In a subset of the aminomutase reactions and all the diol dehydrase reactions, the 1,2 exchange is followed by elimination of water. Two enzymes of this type that have been particularly well characterized are diol dehydrase and ethanolamine ammonia lyase, and these will be discussed first. We will then turn to the aminomutases that do not catalyze a subsequent elimination, lysine 2,3-aminomutase and ornithine aminomutase.
3.2.1. Diol dehydrase
Diol dehydrase catalyzes the conversion of (S)-1,2 propanediol (eq. 8) and of (R)-1,2 propanediol (eq. 9) to propionaldehyde and water. While the lack of stereoselection between (S) and (R)-1,2-propanediols is unusual, the reader will note that the stereochemistry of the two reactions is different 106, 140. As discussed at the beginning of the section on AdoCbl-dependent mutases, the role of the AdoCbl cofactor as a radical generator was first elucidated for diol dehydrase by observing the exchange of tritium between the 5′-position of AdoCbl and the substrate.
Eq. 8.

Eq. 9.

In contrast to the AdoCbl-dependent enzymes that catalyze carbon skeleton rearrangements, diol dehydrase and ethanolamine ammonia lyase bind AdoCbl in the DMB-on form. This was initially demonstrated by labeling the DMB moiety of AdoCbl with 15N and then determining the EPR spectrum of the enzyme after homolytic cleavage of the carbon-cobalt bond was induced with the suicide substrate 2-methyl-1,2-propanediol 141. The x-ray structure of diol dehydrase confirmed this conclusion 142. Diol dehydrase is an α2β2γ2 dimer, and the cobalamin is bound at the interface of the α and β subunits of each monomer. The α subunit is an α8β8 barrel, with the cofactor bound at the C-terminal ends of the central β strands and the substrate 1,2-propanediol bound more deeply in the barrel. The lower face of the cofactor, with its DMB nucleotide coordinated to the cobalt, interacts primarily with the β subunit. Monovalent cations were reported to be essential cofactors for diol dehydrase 143, and the structure revealed a potassium ion coordinated to the two hydroxyls of the substrate. The distance between the cobalt of the cofactor and C1 and C2 of propanediol were 8.4 Å and 9.0 Å respectively, in good agreement with data estimating the distance between the radical intermediate observed during steady-state turnover and cob(II)alamin as about 9 Å 144. However, this places the 5′-deoxyadenosyl radical that would be generated by homolytic cleavage of AdoCbl too far from the substrate to permit substrate radical formation. Toraya and his colleagues proposed that a simple rotation of the 5′-deoxyadenosyl radical around the glycosidic bond would bring C5′ within 2 Å of C1 of the substrate 145, as shown in Fig. 19.
Figure 19. Mechanism for hydroxyl migration in diol dehydrase.
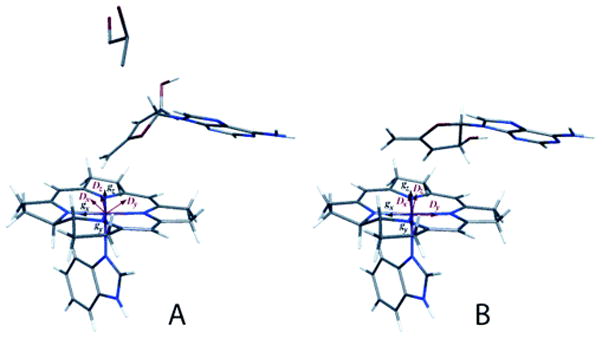
Reprinted, with permission from 149. Electron paramagnetic analysis of diol dehydrase reconstituted with 3′,4′-anhydroAdoCbl and subjected to homolytic cleavage to form the anhydroadenosyl radical and cob(II)alamin in the presence (A) and absence (B) of (R,S)-1,2-propanediol was used to model the structures. In the absence of substrate, the anhydroribosyl moity rotates about 60° relative to its position in the presence of substrate, bringing the radical into a position that would be appropiate for hydrogen atom abstraction from substrate and that would not permit formation of a carbon-cobalt bond.
Subsequent studies clarified the nature of the radical intermediate observed during steady-state turnover of diol dehydrase with 1,2-propanediol. EPR spectroscopy of radicals derived from 13C- and deuterium-labeled substrates established that the radical center resided on C1 146. Thus the intermediate is a substrate-derived radical generated by hydrogen atom abstraction from C1. Further insight into the structure of the radical came from EPR studies of the effects of incorporation of solvent deuterium on the radical signal. These studies indicated that the unpaired electron on the radical center couples with the solvent exchangeable proton on the hydroxyl group at C1 147. The latter studies have important implications for the mechanism of hydroxyl migration from C1 to C2, as will be discussed below.
The deuterium kinetic isotope effect on kcat observed with [1-2H]-1,2-propanediol is 12, indicating that hydrogen abstraction from C1 of the substrate is rate-limiting in catalysis. However, accumulation of a 5′-deoxyadenosyl radical is not seen in either steady-state or stopped-flow analyses. This radical would be expected to accumulate, if hydrogen abstraction from the substrate were rate limiting. A satisfactory resolution to this dilemma is cartooned in eq. 10. The unfavorable equilibrium between AdoCbl and its homolytic cleavage products prevents the accumulation of observable 5′-deoxyadenosyl radical, and leads to the observed kinetic isotope effect on the accumulation of the substrate-derived radical intermediate in the rate-limiting step of catalysis. Also consistent with this proposal is the observation that detectable formation of cob(II)alamin requires the presence of substrate 148. Support for this postulate also comes from studies with diol dehydrase reconstituted with 3′-4′ anhydroAdoCbl (Fig. 20). The introduction of a double bond adjacent to the radical center generated by homolytic cleavage of this cofactor analogue greatly stabilizes the resulting radical, and results in its accumulation during reaction with 1,2-propanediol 149. In fact, homolytic cleavage of 3′-4′-anhydroAdoCbl bound to diol dehydrase is also observed in the absence of substrate.
Eq. 10.

Figure 20.

Structure of 3′,4′-anhydroadenosylcobalamin.
Still to be resolved is the mechanism of hydroxyl migration from C1 to C2 following hydrogen atom abstraction from C1 of 1,2-propanediol. A variety of mechanisms have been proposed (Fig. 21) involving (a) general base catalysis, (b) general acid catalysis, (c) partial protonation (hydrogen bonding) (d) electrophilic catalysis by the activating potassium ion, or (e) combined general acid/base (push-pull) catalysis (discussed in 147). The demonstration that the proton remains on the C1 hydroxyl group of the substrate-derived radical argues against the base-catalyzed mechanism for rearrangement 147. Activation of the enzyme by thallous ion instead of K+, which introduces a spin ½ metal in close proximity to the substrate radical, fails to lead to spin coupling with the substrate-derived radical, also disfavoring a role for K+ in catalyzing the hydroxyl migration 147. His143 is in hydrogen bonding distance to the hydroxyl on C2 of the substrate and Glu170 is in hydrogen bonding distance to the hydroxyl on C1. Mutation of these residues to alanine reduces kcat by 77-fold and 38,000-fold respectively 150, making the push-pull mechanism shown in (e) of Fig. 21 highly attractive.
Figure 21. Proposed mechanisms for hydroxyl migration in diol dehydrase.

Reprinted with permission from Schwartz et al. 147. The mechanisms proposed involve (a) general base catalysis, (b) general acid catalysis, (c) partial protonation (hydrogen bonding) (d) electrophilic catalysis by the activating potassium ion, or (e) combined general acid/base (push-pull) catalysis.
What then is the role of the essential monocation activator? Recent studies have shown that K+ activates the spontaneous cleavage of AdoCbl in the absence of substrate 151. The reaction is observed as the formation of aquacob(III)alamin and requires both O2 and K+. These studies suggest that the binding energy of K+ is used to enforce a conformational change in the enzyme that strains the carbon-cobalt bond of AdoCbl, accelerating cleavage. This conclusion is in agreement with conclusions drawn from crystallographic studies of diol dehydrase with the coenzyme analogue adeninylpentylcobalamin bound 152. This analogue has an increased distance between the adenosine and the cobalamin. These studies suggested that in diol dehydrase with K+ bound, the AdoCbl would be bound in a strained conformation even in the absence of substrate.
Spontaneous cleavage of AdoCbl bound to diol dehydrase would result in inactivation in the cellular milieu, which contains both oxygen and potassium ions, because the aquacobalamin is extremely tightly bound and this form of the enzyme is inactive. Mori and Toraya 153 have identified a reactivating factor, a chaperone-like protein with ATPase activity. In the presence of ADP, the chaperone mediates the release of aquacobalamin from the inactive enzyme and binds tightly to the aponzyme, while in the presence of ATP, the apoenzyme is released and can be reconstituted with AdoCbl. This chaperone comprises two subunits, gene products of the ddrA and ddrB genes in Klebsiella oxytoca.
3.2.2. Ethanolamine ammonia lyase
Ethanolamine ammonia lyase catalyzes the conversion of ethanolamine to acetaldehyde and ammonia. The reaction proceeds as shown in eq. 11. The enzyme is anα6β6 oligomer, and recent studies suggest that a mol of AdoCbl is bound per αβ protomer 154. The predicted masses of the α and β subunits from E. coli are 50 and 32 kDa, respectively. Although the subunits show little homology with those of other B12-dependent enzymes, the larger subunit does show limited homology with the subunits of methylmalonyl CoA mutase from P. shermanii 154, and recent modeling of the large subunit of the enzyme from Salmonella typhimurium suggests that this subunit is indeed a β8α8 barrel 155. By analogy with diol dehydrase, we might expect the AdoCbl to be bound at the interface between the small and large subunits. As in diol dehydrase, the AdoCbl is bound in the “DMB-on” form 156.
Eq. 11.
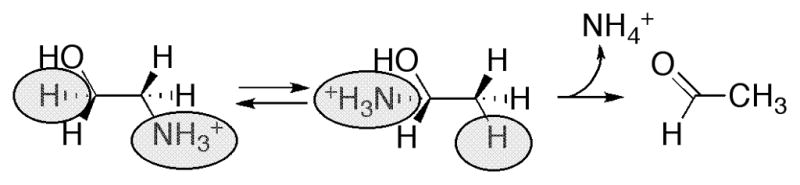
The x-ray structure of ethanolamine ammonia lyase has not been determined, but sophisticated spectroscopic studies have revealed its mechanistic similarity with diol dehydrase and also provided some unique insights into the catalytic mechanism. One of the first issues was whether hydrogen transfer actually occurred directly between the AdoCbl radical and the substrate, or whether an intermediary such as a protein radical might be involved. Following cleavage of AdoCbl in the presence of substrate, the substrate-derived radical was found to be more than 10 Å from the unpaired electron of cob(II)alamin 157. In a landmark study, electron nuclear double resonance and specific 2H and 13C-labeling of the substrate was used to show that the methyl group of 5′-dAdo was positioned 3.4 Å from C1′ of the substrate radical, in a perfect position to mediate direct hydrogen transfer between cofactor and substrate 158. Similar conclusions were reached using pulsed EPR 159 Thus the 5′-carbon of AdoCbl migrates ~7 Å from its position when the cofactor is intact, to its position when the substrate radical is formed.
One major difference between diol dehydrase and ethanolamine ammonia lyase is that, while the stereochemistry of the diol dehydrase reaction requires that the hydroxyl group migrates from C1 to C2 prior to elimination of water, there is no such compelling evidence that the amino group of ethanolamine migrates rather than simply undergoing elimination from C2 after hydrogen atom abstraction at C1. However, arguing by analogy with all the other AdoCbl-dependent isomerases, we might expect that such migration does actually occur. A second difference is the apparent absence of a requirement for a cation (potassium ion or ammonia) for catalysis. In the modeling of the active site of the Salmonella enzyme, Sun and Warncke suggest that the guanidinium sidechain of Arg160 occupies the same position as the potassium ion in the active site of diol dehydrase 155.
3.2.3. Lysine 5,6-aminomutase
Lysine 5,6-amino mutase was initially characterized in the laboratory of Theresa Stadtman, and was shown to be an AdoCbl-dependent enzyme that catalyzed a 1,2-migration of the e-amino group of D-lysine with concomitant reverse migration of a hydrogen atom, as shown in eq. 12160. The stereochemistry of the reaction has not yet been determined, so we do not yet know which of the two hydrogens on the δ-carbon of D-lysine migrates. More recently, the enzyme was cloned, sequenced, expressed and purified from Clostridum sticklandii161. In agreement with earlier studies, in which the enzyme was isolated as a 170 kDa complex of 55 and 30 kDa subunits, the enzyme is an α2β2 heterodimer composed of 57 kDa α subunits and 29 kDa β subunits. The small subunit contains the Asp-x-His-x-x-Gly sequence characteristic of His-on binding of the AdoCbl cofactor.
Eq. 12.

The substrate D-lysine forms an external aldimine linkage (Fig. 22) with the pyridoxal phosphate cofactor that is required for enzyme activity 162. The pyridoxal phosphate is proposed to stabilize radical intermediates formed following hydrogen atom abstraction from the substrate as shown in Fig. 22 163.
Figure 22.

Proposed role for pyridoxal phosphate in the reaction catalyzed by lysine 5,6-aminomutase.
The x-ray structure of the enzyme 164 revealed that the large subunit is an α8β8 barrel and positions the pyridoxal phosphate cofactor at the C-terminal end of the barrel strands. The crystal structure was obtained in the absence of D-lysine, and revealed that the pyridoxal phosphate was linked as an internal aldimine with Lys 144 of the small subunit. The AdoCbl is sandwiched between the large and small subunits, in the predicted DMB-off His-on conformation. However, the AdoCbl is far from the pyridoxal phosphate binding site in this “resting” conformation of the enzyme. The covalent bond between pyridoxal phosphate and Lys144β locks the two subunits together in a conformation that leaves the active site accessible to solvent and substrate and prevents cleavage of AdoCbl. The authors propose that substrate binding will lead to a rearrangement that will sequester the active site and position AdoCbl appropriately for catalysis.
3.2.4. Ornithine 4,5-aminomutase
This enzyme catalyzes the reaction shown in eq. 13. Until very recently, the enzymatic reaction had only been studied in crude extracts, where the product was shown to be (2R,4S)-diaminopentanoate. However the stereochemistry of the migrating hydrogen atom in ornithine has not been determined. In 2001, the oraE and oraS genes specifying the large and small subunits of D-ornithine amino mutase were cloned and sequenced from Clostridium sticklandii 165, and in 2004, the enzyme was successfully expressed in E. coli and purified to homogeneity 166. Despite the lack of sequence homology with other AdoCbl-dependent enzymes, the properties of the purified ornithine aminomutase are very similar to those of lysine 5,6-aminomutase. The enzyme is an α2β2 heterodimer, and requires pyridoxal phosphate for activity in addition to AdoCbl.
Eq. 13.
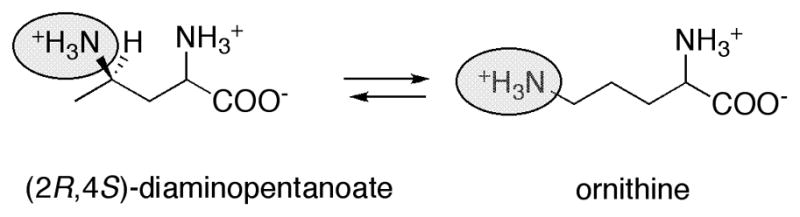
3.3.1 Adenosylcobalamin-dependent ribonucleotide triphosphate reductase
Ribonucleotide triphosphate reductase catalyzes the reaction shown in Eq. 14, which results in oxidation of two active site thiols to form a disulfide. Regeneration of the enzyme by reduction of the disulfide bond is required for sustained turnover, and is accomplished by a series of electron transfers from NADPH to thioredoxin reductase, to thioredoxin, and then to ribonucleotide triphosphate reductase as shown in eq. 15.
Eq. 14.

Eq. 15.

Enzyme catalysis requires AdoCbl, which serves as a radical generator as it does in other AdoCbl-dependent enzymes. However, a unique feature of ribonucleotide triphosphate reductase is that hydrogen atom abstraction from the substrate is not catalyzed by the 5′-deoxyadenosyl radical, but rather by a thiyl radical generated by hydrogen atom abstraction from a cysteine residue on the protein. The enzyme from Lactobacillus leichmanii has been most extensively studied.
In contrast to the intramolecular tritium transfer seen in other AdoCbl-dependent enzymes, if ribonucleotide triphosphate reductase labeled with tritium in the 5′ position of AdoCbl is incubated with the allosteric activator dGTP and reductant, the label is quantitatively transferred to solvent 167. Stubbe and her colleagues then showed using single turnover experiments that ribonucleotide triphosphate reductase catalyzes the cleavage of the 3′-carbon hydrogen bond of UTP with quantitative release of label to solvent when the 3′-position is tritiated 168. The authors proposed a working hypothesis for the enzyme that involved the generation of a protein radical that in turn generated the substrate radical by direct hydrogen atom abstraction of the 3′-hydrogen of UTP. Cysteine 408 was subsequently shown to be the site of formation of the protein radical 169, and rapid freeze quench electron paramagnetic resonance of an intermediate in the exchange of the 5′-hydrogens of AdoCbl with solvent catalyzed by ribonucleotide triphosphate reductase containing specifically deuterated cysteines demonstrated a cysteine-centered radical that was spin-coupled to cob(II)alamin. These studies also demonstrated that the formation and decay of the cysteine-based radical signal proceeded at rates consistent with its involvement in catalysis. The detailed mechanism proposed for the catalytic cycle 170 is shown in Fig. 23.
Figure 23.

Proposed mechanism for AdoCbl-dependent ribonucleotide reductase.
Following the cloning and sequencing of ribonucleotide triphosphate reductase from L. leichmanii,171 a crystal structure of the enzyme was obtained with the AdoCbl analogue adeninylpentylcobalamin bound 172. The monomeric enzyme folds as a ten stranded α/β barrel with Cys408 positioned at the tip of a hairpin loop at the bottom of the barrel. The AdoCbl analogue is bound with DMB in the “base-on” conformation, as previously predicted 173.
One issue that has recently been clarified concerns the mechanism of formation of the thiyl radical, which could be generated in a stepwise fashion by cleavage of AdoCbl to form 5′-deoxyadenosyl radical and cob(II)alamin, or in a concerted fashion in which a 5′-deoxyadenosyl radical is not an intermediate. When ribonucleotide reductase was incubated with stereoselectively deuterated (5′R)-[5′-2H1]AdoCbl and the allosteric activator dGTP, the 5′-deuterium was stereochemically scrambled 174. This scrambling occurred in both the wild-type enzyme and the Cys408Ala mutant, even though exchange of deuterium with solvent did not occur in the mutant enzyme. The implication is that transient cleavage of the Co-C5′ bond of AdoCbl occurs in the presence of the allosteric activator even in the absence of Cys408.
The second mechanistic issue concerns the sequence by which the active-site disulfide is reduced at the end of each turnover. As previously mentioned, the electrons ultimately come from NADPH, and are transferred to the redox active disulfide of thioredoxin by thioredoxin reductase. Reduction of ribonucleotide triphosphate reductase by thioredoxin requires two cysteines at the C-terminus of the protein, Cys731 and 736 169, and these two cysteines are thought to shuttle reducing equivalents between reduced thioredoxin and the active site disulfide. This C-terminal extension is disordered in the crystal structure of the enzyme 172.
4. CONCLUDING REMARKS
The author hopes that this review highlights the explosion of information recently obtained about the growing family of characterized corrinoid-dependent enzymes, and particularly the mounting insights available from structural analyses of these proteins. We now appreciate the vital role that corrinoid-dependent methyltransferases play in central metabolic pathways in prokaryotes and Archaea. A challenge for the future will be to reach a detailed understanding of how the membrane-associated energy conserving corrinoid methyl transferase couples sodium ion transport with methyl transfer from methylcobalamin to the thiol of coenzyme M. The elegant spectroscopies that have been applied to the AdoCbl-dependent enzymes have greatly clarified the nature of the radical intermediates and structural analyses have revealed the type of molecular motions required to allow hydrogen atom transfer from substrate to the deoxyadenosyl radical and back to product. These complicated enzymes have posed a challenge to enzymologists and chemists for the past 70 years, and it has been thrilling to review the elegant experiments that step-by-step revealed the details of catalysis.
Acknowledgments
Research in the author’s laboratory is funded by National Institutes of Health Grant GM24908.
Abbreviations and definitions
- AdoCbl
adenosylcobalamin
- AdoMet
S-adenosylmethionine
- DMB
dimethylbenzimidazole
- EPR
electron paramagnetic resonance
- MetH
cobalamin-dependent methionine synthase from E. coli
- NMR
nuclear magnetic resonance
References
- 1.Ragsdale SW, Lindahl PA, Munck E. J Biol Chem. 1987;262:14289–14297. [PubMed] [Google Scholar]
- 2.Stupperich E, Eisenger HJ, Albracht SPJ. Eur J Biochem. 1990;193:105–109. doi: 10.1111/j.1432-1033.1990.tb19310.x. [DOI] [PubMed] [Google Scholar]
- 3.Ragsdale SW. In: Chemistry and biochemistry of B12. Banerjee R, editor. John Wiley; New York: 1999. pp. 633–653. [Google Scholar]
- 4.Banerjee RV, Johnston NL, Sobeski JK, Datta P, Matthews RG. J Biol Chem. 1989;264:13888–13895. [PubMed] [Google Scholar]
- 5.Amaratunga M, Fluhr K, Jarrett JT, Drennan CL, Ludwig ML, Matthews RG, Scholten JD. Biochemistry. 1996;35:2453–2463. doi: 10.1021/bi952388u. [DOI] [PubMed] [Google Scholar]
- 6.Drummond JT, Huang S, Blumenthal RM, Matthews RG. Biochemistry. 1993;32:9290–9295. doi: 10.1021/bi00087a005. [DOI] [PubMed] [Google Scholar]
- 7.Fujii K, Galivan JH, Huennekens FM. Arch Biochem Biophys. 1977;178:662–670. doi: 10.1016/0003-9861(77)90238-7. [DOI] [PubMed] [Google Scholar]
- 8.Osborne C, Chen L-M, Matthews RG. J Bacteriol. 1991;173:1729–1737. doi: 10.1128/jb.173.5.1729-1737.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoover DM, Jarrett JT, Sands RH, Dunham WR, Ludwig ML, Matthews RG. Biochemistry. 1997;36:127–138. doi: 10.1021/bi961693s. [DOI] [PubMed] [Google Scholar]
- 10.Jarrett JT, Choi CY, Matthews RG. Biochemistry. 1997;36:15739–15748. doi: 10.1021/bi971987t. [DOI] [PubMed] [Google Scholar]
- 11.Drennan CL, Huang S, Drummond JT, Matthews RG, Ludwig ML. Science. 1994;266:1669–1674. doi: 10.1126/science.7992050. [DOI] [PubMed] [Google Scholar]
- 12.Marsh ENG, Holloway DE. FEBS Letters. 1992;310:167–170. doi: 10.1016/0014-5793(92)81321-c. [DOI] [PubMed] [Google Scholar]
- 13.Jarrett JT, Drennan CL, Amaratunga M, Scholten JD, Ludwig ML, Matthews RG. J Bioorgan Med Chem. 1996;4:1237–1246. doi: 10.1016/0968-0896(96)00119-8. [DOI] [PubMed] [Google Scholar]
- 14.Dixon MM, Huang S, Matthews RG, Ludwig M. Structure. 1996;4:1263–1275. doi: 10.1016/s0969-2126(96)00135-9. [DOI] [PubMed] [Google Scholar]
- 15.Bandarian V, Ludwig ML, Matthews RG. Proc Natl Acad Sci, U S A. 2003;100:8156–8163. doi: 10.1073/pnas.1133218100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fleischhacker AS, Matthews RG. Biochemistry. 2007;46:12382–12392. doi: 10.1021/bi701367c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Evans JC, Huddler DP, Hilgers MT, Romanchuk G, Matthews RG, Ludwig ML. Proc Natl Acad Sci, U S A. 2004;101:3729–3736. doi: 10.1073/pnas.0308082100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jarrett JT, Amaratunga M, Drennan CL, Scholten JD, Sands RH, Ludwig ML, Matthews RG. Biochemistry. 1996;35:2464–2475. doi: 10.1021/bi952389m. [DOI] [PubMed] [Google Scholar]
- 19.Jarrett JT, Huang S, Matthews RG. Biochemistry. 1998;37:5372–5382. doi: 10.1021/bi9730893. [DOI] [PubMed] [Google Scholar]
- 20.Datta S, Koutmos M, Pattridge KA, Ludwig ML, Matthews RG. Proc Natl Acad Sci U S A. 2008;105:4115–4120. doi: 10.1073/pnas.0800329105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaufmann F, Wohlfarth G, Diekert G. Eur J Biochem. 1998;257:5125–5521. doi: 10.1046/j.1432-1327.1998.2570515.x. [DOI] [PubMed] [Google Scholar]
- 22.Kaufmann F, Wohlfarth G, Diekert G. Eur J Biochem. 1998;253:706–711. doi: 10.1046/j.1432-1327.1998.2530706.x. [DOI] [PubMed] [Google Scholar]
- 23.Tanaka K. J Ferment Bioeng. 1994;78:386–388. [Google Scholar]
- 24.Sauer K, Thauer TK. Eur J Biochem. 1998;253:698–705. doi: 10.1046/j.1432-1327.1998.2530698.x. [DOI] [PubMed] [Google Scholar]
- 25.Tallant TC, Paul L, Krzycki JA. J Biol Chem. 2001;276:4485–4493. doi: 10.1074/jbc.M007514200. [DOI] [PubMed] [Google Scholar]
- 26.Burke SA, Krzycki JA. J Biol Chem. 1997;272:16570–16577. doi: 10.1074/jbc.272.26.16570. [DOI] [PubMed] [Google Scholar]
- 27.Burke SA, Lo SL, Krzycki JA. J Bacteriol. 1998;180:3432–3440. doi: 10.1128/jb.180.13.3432-3440.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferguson DJ, Jr, Gorlatova N, Grahame DA, Krzycki JA. J Biol Chem. 2000;275:29053–29060. doi: 10.1074/jbc.M910218199. [DOI] [PubMed] [Google Scholar]
- 29.Paul L, Ferguson DJ, Jr, Kryzycki JA. J Bacteriol. 2000;182:2520–2529. doi: 10.1128/jb.182.9.2520-2529.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Galagan JE, Nusbaum C, Roy A, Endrizzi MG, Macdonald P, FitzHugh W, Calvo S, Engels R, Smirnov S, Atnoor D, Brown A, Allen N, Naylor J, Stange-Thomann N, DeArellano K, Johnson R, Linton L, McEwan P, McKernan K, Talamas J, Tirrell A, Ye W, Zimmer A, Barber RD, Cann I, Graham DE, Grahame DA, Guss AM, Hedderich R, Ingram-Smith C, Kuettner HC, Krzycki JA, Leigh JA, Li W, Liu J, Mukhopadhyay B, Reeve JN, Smith K, Springer TA, Umayam LA, White O, White RH, Conway de Macario E, Ferry JG, Jarrell KF, Jing H, Macario AJ, Paulsen I, Pritchett M, Sowers KR, Swanson RV, Zinder SH, Lander E, Metcalf WW, Birren B. Genome Res. 2002;12:532–542. doi: 10.1101/gr.223902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Galagan JE, et al. Genome Research. 2002;12:532–542. doi: 10.1101/gr.223902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harms U, Thauer RK. Eur J Biochem. 1996;325:653–659. doi: 10.1111/j.1432-1033.1996.00653.x. [DOI] [PubMed] [Google Scholar]
- 33.Hao B, Gong W, Ferguson TK, James CM, Krzycki JA, Chan MK. Science. 2002;296:1462–1466. doi: 10.1126/science.1069556. [DOI] [PubMed] [Google Scholar]
- 34.Sauer K, Harms U, Thauer RK. Eur J Biochem. 1997;243:670–677. doi: 10.1111/j.1432-1033.1997.t01-1-00670.x. [DOI] [PubMed] [Google Scholar]
- 35.Hagemeier CH, Krer M, Thauer RK, Warkentin B, Ermler U. Proc Natl Acad Sci, U S A. 2006;103:18917–18922. doi: 10.1073/pnas.0603650103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sauer K, Thauer RK. Eur J Biochem. 1997;249:280–285. doi: 10.1111/j.1432-1033.1997.t01-1-00280.x. [DOI] [PubMed] [Google Scholar]
- 37.Zydowsky LD, Zydowsky TM, Haas ES, Brown JW, Reeve JN, Floss HG. J Am Chem Soc. 1987;109:7922–7923. [Google Scholar]
- 38.Grahame DA. J Biol Chem. 1989;264:12890–12894. [PubMed] [Google Scholar]
- 39.Goulding CW, Matthews RG. Current Opin Chem Biol. 1997;1:332–339. doi: 10.1016/s1367-5931(97)80070-1. [DOI] [PubMed] [Google Scholar]
- 40.Sauer K, Thauer RK. Eur J Biochem. 2000;267:2498–2504. doi: 10.1046/j.1432-1327.2000.01245.x. [DOI] [PubMed] [Google Scholar]
- 41.Krüer M, Haumann M, Meyer-Klaucke W, Thauer RK, Dau H. Eur J Biochem. 2002;269:2117–2123. doi: 10.1046/j.1432-1033.2002.02860.x. [DOI] [PubMed] [Google Scholar]
- 42.Gencic S, LeClerc GM, Gorlatova N, Peariso K, Penner-Hahn JE, Grahame DA. Biochemistry. 2001;50:13068–13078. doi: 10.1021/bi0112917. [DOI] [PubMed] [Google Scholar]
- 43.Blaut M, Müller V, Gottschalk G. J Bioenerg Biomembr. 1992;24:529–546. doi: 10.1007/BF00762346. [DOI] [PubMed] [Google Scholar]
- 44.Gärtner P, Ecker A, Fischer R, Linder D, Fuchs G, Thauer R. Eur J Biochem. 1993;213:537–545. doi: 10.1111/j.1432-1033.1993.tb17792.x. [DOI] [PubMed] [Google Scholar]
- 45.Harms U, Weiss DS, Gartner P, Linder D, Thauer RK. Eur J Biochem. 1995;228:640–648. doi: 10.1111/j.1432-1033.1995.0640m.x. [DOI] [PubMed] [Google Scholar]
- 46.Stupperich E, Juza A, Hoppert M, Mayer F. Eur J Biochem. 1993;217:115–121. doi: 10.1111/j.1432-1033.1993.tb18225.x. [DOI] [PubMed] [Google Scholar]
- 47.Kräutler B, Moll J, Thauer RK. Eur J Biochem. 1987;162:275–278. doi: 10.1111/j.1432-1033.1987.tb10596.x. [DOI] [PubMed] [Google Scholar]
- 48.Harms U, Thauer RK. Eur J Biochem. 1996;241:149–154. doi: 10.1111/j.1432-1033.1996.0149t.x. [DOI] [PubMed] [Google Scholar]
- 49.Harms U, Thauer RK. Eur J Biochem. 1997;250:783–788. doi: 10.1111/j.1432-1033.1997.00783.x. [DOI] [PubMed] [Google Scholar]
- 50.Gärtner P, Weiss DS, Harms U, Thauer RK. Eur J Biochem. 1994;226:465–472. doi: 10.1111/j.1432-1033.1994.tb20071.x. [DOI] [PubMed] [Google Scholar]
- 51.Weiss DS, Gärtner P, Thauer RK. Eur J Biochem. 1994;226:799–809. doi: 10.1111/j.1432-1033.1994.00799.x. [DOI] [PubMed] [Google Scholar]
- 52.Svetlitchnyi V, Dobbek H, Meyer-Klaucke W, Meins T, Thiele B, Romer P, Huber R, Meyer O. Proc Natl Acad Sci, U S A. 2004;101:446–451. doi: 10.1073/pnas.0304262101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Holder U, Schmidt D-E, Stupperich E, Fuchs G. Arch Microbiol. 1985:141. [Google Scholar]
- 54.Eikmanns B, Fuchs G, Thauer RK. Eur J Biochem. 1985;146:149–154. doi: 10.1111/j.1432-1033.1985.tb08631.x. [DOI] [PubMed] [Google Scholar]
- 55.Sauer K, Thauer RK. In: Chemistry and biochemistry of B12. Banerjee R, editor. John Wiley & Sons, Inc; New York: 1999. pp. 655–679. [Google Scholar]
- 56.Lu W-P, Schiau I, Cunningham JR, Ragsdale SW. J Biol Chem. 1993;268:5605–5614. [PubMed] [Google Scholar]
- 57.Maupin-Furlow J, Ferry JG. J Bacteriol. 1996;178:340–346. doi: 10.1128/jb.178.2.340-346.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grahame DA. J Biol Chem. 1991;266:22227–22233. [PubMed] [Google Scholar]
- 59.Grahame DA. Biochemistry. 1993;32:10786–10793. doi: 10.1021/bi00091a033. [DOI] [PubMed] [Google Scholar]
- 60.Terlesky KC, Nelson MJK, Ferry JG. J Bacteriol. 1986;168:1053–1058. doi: 10.1128/jb.168.3.1053-1058.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Harder SR, Lu W-P, Feinberg BA, Ragsdale SW. Biochemistry. 1989;28:9080–9087. doi: 10.1021/bi00449a019. [DOI] [PubMed] [Google Scholar]
- 62.Svetlitchnaia T, Svetlitchnyi V, Meyer O, Dobbek H. Proc Natl Acad Sci, U S A. 2006;103:14331–14336. doi: 10.1073/pnas.0601420103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jablonski PE, Lu W-P, Ragsdale SW, Ferry JG. J Biol Chem. 1993;268:325–329. [PubMed] [Google Scholar]
- 64.Stich TA, Seravalli J, Venkateshrao S, Spiro TG, Ragsdale SW, Brunold TC. J Am Chem Soc. 2006;128:5010–5020. doi: 10.1021/ja054690o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Menon S, Ragsdale SW. J Biol Chem. 1999;274:11513–11518. doi: 10.1074/jbc.274.17.11513. [DOI] [PubMed] [Google Scholar]
- 66.Menon S, Ragsdale SW. Biochemistry. 1998;37:5689–5698. doi: 10.1021/bi9727996. [DOI] [PubMed] [Google Scholar]
- 67.Doukov T, Seravelli J, Stezowski JJ, Ragsdale SW. Structure. 2000;8:817–830. doi: 10.1016/s0969-2126(00)00172-6. [DOI] [PubMed] [Google Scholar]
- 68.Lexa D, Saveant J-M. Acc Chem Res. 1983;16:235–243. [Google Scholar]
- 69.Seravalli J, Brown KL, Ragsdale SW. J Am Chem Soc. 2001;128:1786–1787. doi: 10.1021/ja005709k. [DOI] [PubMed] [Google Scholar]
- 70.Kaufmann F, Wohlfarth G, Diekert G. Eur J Biochem. 2008;253:706–711. doi: 10.1046/j.1432-1327.1998.2530706.x. [DOI] [PubMed] [Google Scholar]
- 71.Naidu D, Ragsdale SW. J Bacteriol. 2001;183:3272–3281. doi: 10.1128/JB.183.11.3276-3281.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Krone UE, Thauer RK, Hogenkamp HPC. Biochemistry. 1989;28:4908–4914. doi: 10.1021/bi00452a027. [DOI] [PubMed] [Google Scholar]
- 73.McCauley KM, Pratt DA, Wilson SR, Shey J, Burkey TJ, van der Donk WA. J Am Chem Soc. 2005;127:1126–1136. doi: 10.1021/ja048573p. [DOI] [PubMed] [Google Scholar]
- 74.Wohlfarth G, Diekert G. In: Chemistry and Biochemistry of B12. Banerjee R, editor. Wiley; New York: 1999. pp. 871–893. [Google Scholar]
- 75.Schumacher W, Holliger C, Zehnder AJB, Hagen WR. FEBS Lett. 1997;409:421–425. doi: 10.1016/s0014-5793(97)00520-6. [DOI] [PubMed] [Google Scholar]
- 76.Neumann A, Wohlfarth G, Diekert G. J Bacteriol. 1998;180:4140–4145. doi: 10.1128/jb.180.16.4140-4145.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.van de Pas BA, Smidt H, Hagen WR, van der Oost J, Schraa G, Stams AJM, de Vos WM. J Biol Chem. 1999;274:20287–20292. doi: 10.1074/jbc.274.29.20287. [DOI] [PubMed] [Google Scholar]
- 78.Thibodeau J, Gauthier A, Duguay M, Villemur R, Lépine F, Juteau P, Beaudet R. Appl Environ Microbiol. 2004;70:4532–4537. doi: 10.1128/AEM.70.8.4532-4537.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Studer Z, Stupperich E, Vuilleumier S, Leisinger T. Eur J Biochem. 2001;268:2931–2938. doi: 10.1046/j.1432-1327.2001.02182.x. [DOI] [PubMed] [Google Scholar]
- 80.Vannelli T, Messmer M, Studer A, Vuilleumier S, Leisinger T. Proc Natl Acad Sci, USA. 1999;96:4615–4620. doi: 10.1073/pnas.96.8.4615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Studer A, Vuilleumier S, Leisinger T. J Biochem. 1999;264:242–249. doi: 10.1046/j.1432-1327.1999.00629.x. [DOI] [PubMed] [Google Scholar]
- 82.Yamanishi M, Labunska T, Banerjee R. J Am Chem Soc. 2005;127:526–527. doi: 10.1021/ja044365l. [DOI] [PubMed] [Google Scholar]
- 83.Yamada K, Gravel RA, Toraya T, Matthews RG. Proc Natl Acad Sci, U S A. 2006;103:9476–9481. doi: 10.1073/pnas.0603694103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Daas PJH, Wassenaar RW, Willemsen P, Theunissen RJ, Keltjens JT, van der Drift C, Vogels GD. J Biol Chem. 1996;271:22339–22345. doi: 10.1074/jbc.271.37.22339. [DOI] [PubMed] [Google Scholar]
- 85.Brown KL, Peck-Siler S. Inorg Chem. 1988;27:3548–3555. [Google Scholar]
- 86.Brown KL, Zou X. Inorg Chem. 1991;30:4185–4191. [Google Scholar]
- 87.Mangum JH, Scrimgeour KG. Fed Proc. 1962;21:242. [Google Scholar]
- 88.Fujii K, Huennekens FM. J Biol Chem. 1974;249:6745–6753. [PubMed] [Google Scholar]
- 89.Fujii K, Huennekens FM. In: Biochemical aspects of nutrition. Yagi K, editor. Japan Scientific Societies Press; Tokyo: 1979. pp. 173–183. [Google Scholar]
- 90.Hall DE, Jordan-Starck TC, Loo RO, Ludwig ML, Matthews RG. Biochemisty. 2000;39:10711–10719. doi: 10.1021/bi001096c. [DOI] [PubMed] [Google Scholar]
- 91.LeClerc D, Wilson A, Dumas R, Gafuik C, Song D, Watkins D, Heng HHQ, Rommens JM, Scherer SW, Rosenblatt DS, Gravel RA. Proc Natl Acad Sci, U S A. 1998;95:3059–3064. doi: 10.1073/pnas.95.6.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rosenblatt DS. In: The Metabolic Bases of Inherited Disease. Scriver CR, Beaudet AL, Sly WS, Valle D, editors. II. McGraw-Hill; New York: 1995. pp. 3111–3128. [Google Scholar]
- 93.Olteanu H, Banerjee R. J Biol Chem. 2001;276:35558–35563. doi: 10.1074/jbc.M103707200. [DOI] [PubMed] [Google Scholar]
- 94.Daas PJH, Hagen WR, Keltjens JT, Van der Drift C, Vogels GD. J Biol Chem. 1996;271:22346–22351. doi: 10.1074/jbc.271.37.22346. [DOI] [PubMed] [Google Scholar]
- 95.Fischer R, Gärtner P, Yeliseev A, Thauer RK. Arch Microbiol. 1992;158:208–217. doi: 10.1007/BF00290817. [DOI] [PubMed] [Google Scholar]
- 96.Siebert A, Schubert T, Engelmann T, Studenik S, Diekert G. Arch Microbiol. 2005;183:378–384. doi: 10.1007/s00203-005-0001-8. [DOI] [PubMed] [Google Scholar]
- 97.Wassenaar RW, Daas PJH, Geerts WJ, Keltjens JT, van der Drift C. J Bacteriol. 1996;178:6937–6944. doi: 10.1128/jb.178.23.6937-6944.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Srinivasan G, James CM, Krzycki JA. Science. 2002;296:1459–1462. doi: 10.1126/science.1069588. [DOI] [PubMed] [Google Scholar]
- 99.Kuzuyama T, Hidaka T, Kamigiri K, Imaig S, Seto H. J Antibiot. 1992;45:1812–1814. doi: 10.7164/antibiotics.45.1812. [DOI] [PubMed] [Google Scholar]
- 100.Seto H, Hidaka T, Kuzuyama T, Shibahara S, Usui T, Sakanaka O, Imai S. J Antibiot. 1991;44:1286–1288. doi: 10.7164/antibiotics.44.1286. [DOI] [PubMed] [Google Scholar]
- 101.Woodyer RD, Li G, Zhao H, van der Donk WA. Chem Commun. 2007:359–361. doi: 10.1039/b614678c. [DOI] [PubMed] [Google Scholar]
- 102.Frey PA, Abeles RH. J Biol Chem. 1966;241:2732–2733. [PubMed] [Google Scholar]
- 103.Rétey J, Arigoni D. Experientia. 1966;22:783–784. doi: 10.1007/BF01897408. [DOI] [PubMed] [Google Scholar]
- 104.Frey PA, Essenberg MK, Abeles RH. J Biol Chem. 1967;242:5369–5377. [PubMed] [Google Scholar]
- 105.Wagner OW, Lee HA, Jr, Frey PA, Abeles RH. J Biol Chem. 1966;241:1751–1762. [PubMed] [Google Scholar]
- 106.Rétey J, Umani-Rouchi A, Seibl J, Arigoni D. Experientia. 1966;22:502–503. doi: 10.1007/BF01898652. [DOI] [PubMed] [Google Scholar]
- 107.Finke RG, Hay BP. Inorg Chem. 1984;23:3041–3043. [Google Scholar]
- 108.Buckel W, Golding BT, Kratky C. Chem Eur J. 2006;12:352–362. doi: 10.1002/chem.200501074. [DOI] [PubMed] [Google Scholar]
- 109.Kozlowski PM, Kamachi T, Toraya T, Yoshizawa K. Angew Chem Int Ed. 2007;46:980–983. doi: 10.1002/anie.200602977. [DOI] [PubMed] [Google Scholar]
- 110.Buckel W, Bröker G, Bothe H, Pierek A, Golding BT. In: Chemistry and Biochemistry of B12. Banerjee R, editor. Wiley Interscience; New York: 1999. pp. 757–781. [Google Scholar]
- 111.Chen H-P, Marsh EN. Biochemistry. 1997:36. [Google Scholar]
- 112.Reitzer R, Gruber K, Jogi G, Wagner UG, Bothe H, Buckel W, Kratky C. Structure. 1999;7:891–902. doi: 10.1016/s0969-2126(99)80116-6. [DOI] [PubMed] [Google Scholar]
- 113.Tollinger M, Konrat R, Hilbert BH, Marsh EN, Kräutler B. Structure. 1998;6:1021–1033. doi: 10.1016/s0969-2126(98)00103-8. [DOI] [PubMed] [Google Scholar]
- 114.Gruber K, Reitzer R, Kratky C. Angew Chem Int Ed Engl. 2001;40:3377–3380. doi: 10.1002/1521-3773(20010917)40:18<3377::aid-anie3377>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 115.Chih HW, Marsh EN. Biochemistry. 1999;38:13684–13691. doi: 10.1021/bi991064t. [DOI] [PubMed] [Google Scholar]
- 116.Buckel W, Golding BT. Chem Soc Rev. 1996;25:329–338. [Google Scholar]
- 117.Beatrix B, Zelder O, Linder D, Buckel W. Eur J Biochem. 1994;221:101–109. doi: 10.1111/j.1432-1033.1994.tb18718.x. [DOI] [PubMed] [Google Scholar]
- 118.Pierik AJ, Ciceri D, Lopez RF, Kroll F, Broker G, Beatrix B, Buckel W, Golding BT. Biochemistry. 2005;44:10541–10551. doi: 10.1021/bi050049n. [DOI] [PubMed] [Google Scholar]
- 119.Mancia F, Keep NH, Nakagawa A, Leadlay PF, McSweeney S, Rasmussen B, Boseck P, Diat O, Evans PR. Structure. 1996;4:339–350. doi: 10.1016/s0969-2126(96)00037-8. [DOI] [PubMed] [Google Scholar]
- 120.Mancia F, Evans PR. Structure. 1998;6:711–720. doi: 10.1016/s0969-2126(98)00073-2. [DOI] [PubMed] [Google Scholar]
- 121.Vlasie MD, Banerjee R. J Am Chem Soc. 2003;125:5431–5435. doi: 10.1021/ja029420+. [DOI] [PubMed] [Google Scholar]
- 122.Padmakumar R, Banerjee R. Biochemistry. 1997;36:3713–3718. doi: 10.1021/bi962503g. [DOI] [PubMed] [Google Scholar]
- 123.Chowdhury S, Banerjee R. Biochemistry. 2000;39:7998–8006. doi: 10.1021/bi992535e. [DOI] [PubMed] [Google Scholar]
- 124.Kwiecien RA, Khavrutskii IV, Musaev DG, Morokuma K, Banerjee R, Paneth P. J Am Chem Soc. 2006;128:1287–1292. doi: 10.1021/ja056333j. [DOI] [PubMed] [Google Scholar]
- 125.Mansoorabadi SO, Padmakumar R, Fazliddinova N, Vlasie M, Banerjee R, Reed GH. Biochemistry. 2005;44:3153–3158. doi: 10.1021/bi0482102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Dybala-Defratyka A, Paneth P, Banerjee R, Truhlar DG. Proc Natl Acad Sci U S A. 2007;104:10774–10779. doi: 10.1073/pnas.0702188104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Leal NA, Park SD, Kima PE, Bobik TA. J Biol Chem. 2003;278:9227–9234. doi: 10.1074/jbc.M212739200. [DOI] [PubMed] [Google Scholar]
- 128.Leal NA, Olteanu H, Banerjee R, Bobik TA. J Biol Chem. 2004;279:47536–47542. doi: 10.1074/jbc.M405449200. [DOI] [PubMed] [Google Scholar]
- 129.Stich TA, Yamanishi M, Banerjee R, Brunold TC. J Am Chem Soc. 2005;127:7660–7661. doi: 10.1021/ja050546r. [DOI] [PubMed] [Google Scholar]
- 130.Yamanishi M, Vlasie M, Banerjee R. Trends Biochem Sci. 2005;30:304–308. doi: 10.1016/j.tibs.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 131.Korotkova N, Lidstrom ME. J Biol Chem. 2004;279:13652–13658. doi: 10.1074/jbc.M312852200. [DOI] [PubMed] [Google Scholar]
- 132.Dobson CM, Wai T, Leclerc D, Wilson A, Wu X, Dore C, Hudson T, Rosenblatt DS, Gravel RA. Proc Natl Acad Sci U S A. 2002;99:15554–15559. doi: 10.1073/pnas.242614799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Padovani D, Labunska T, Banerjee R. J Biol Chem. 2006;281:17838–17844. doi: 10.1074/jbc.M600047200. [DOI] [PubMed] [Google Scholar]
- 134.Padovani D, Banerjee R. Biochemistry. 2006;45:9300–9306. doi: 10.1021/bi0604532. [DOI] [PubMed] [Google Scholar]
- 135.Vrijbloed JW, Zerbe-Burkhardt K, Ratnatilleke A, Grubelnik-Leiser A, Robinson JA. J Bacteriol. 1999;181:5600–5605. doi: 10.1128/jb.181.18.5600-5605.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zerbe-Burkhardt K, Ratnatilleke A, Philippon N, Birch A, Leiser A, Vrijbloed JW, Hess D, Hunziker P, Robinson JA. J Biol Chem. 1998;273:6508–6517. doi: 10.1074/jbc.273.11.6508. [DOI] [PubMed] [Google Scholar]
- 137.Ratnatilleke A, Vrijbloed JW, Robinson JA. J Biol Chem. 1999;274:31679–31685. doi: 10.1074/jbc.274.44.31679. [DOI] [PubMed] [Google Scholar]
- 138.Gani D, O’Hagan D, Reynolds K, Robinson JA. J Chem Soc Chem Commun. 1985:1002–1004. [Google Scholar]
- 139.Zhang W, Reynolds KA. J Bacteriol. 2001;183:2071–2080. doi: 10.1128/JB.183.6.2071-2080.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zagalak B, Frey PA, Karabatsos GL, Abeles RH. J Biol chem. 1966;241:3028–3035. [PubMed] [Google Scholar]
- 141.Yamanishi M, Yamada S, Muguruma H, Murakami Y, Tobimatsu T, Ishida A, Yamauchi J, Toraya T. Biochemistry. 1998;37:4799–4803. doi: 10.1021/bi972572a. [DOI] [PubMed] [Google Scholar]
- 142.Shibata N, Masuda J, Tobimatsu T, Toraya T, Suto K, Morimoto Y, Yasuoka N. Structure. 1999;7:997–1008. doi: 10.1016/s0969-2126(99)80126-9. [DOI] [PubMed] [Google Scholar]
- 143.Lee HA, Jr, Abeles RH. J Biol Chem. 1963;238:2367–2373. [PubMed] [Google Scholar]
- 144.Schepler KL, Dunham WR, Sands RH, Gee JA, Abeles RH. Biochim Biophys Acta. 1975;397:510–518. doi: 10.1016/0005-2744(75)90141-2. [DOI] [PubMed] [Google Scholar]
- 145.Masuda J, Shibata N, Morimoto Y, Toraya T, Yasuoka N. Structure. 2000;8:775–788. doi: 10.1016/s0969-2126(00)00164-7. [DOI] [PubMed] [Google Scholar]
- 146.Yamanishi M, Ide H, Murakami Y, Toraya T. Biochemistry. 2005;44:2113–2118. doi: 10.1021/bi0481850. [DOI] [PubMed] [Google Scholar]
- 147.Schwartz PA, Lobrutto R, Reed GH, Frey PA. Protein Sci. 2007;16:1157–1164. doi: 10.1110/ps.072768007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wagner OW, Lee HA, Jr, Frey PA, Abeles RH. J Biol Chem. 1966;249:1751–1762. [PubMed] [Google Scholar]
- 149.Mansoorabadi SO, Magnusson OT, Poyner RR, Frey PA, Reed GH. Biochemistry. 2006;45:14362–14370. doi: 10.1021/bi061586q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Toraya T. Chem Rev. 2003;103:2095–2127. doi: 10.1021/cr020428b. [DOI] [PubMed] [Google Scholar]
- 151.Schwartz PA, Frey PA. Biochemistry. 2007;46:7293–7301. doi: 10.1021/bi700078z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Shibata N, Masuda J, Morimoto Y, Yasuoka N, Toraya T. Biochemistry. 2002;41:12607–12617. doi: 10.1021/bi026104z. [DOI] [PubMed] [Google Scholar]
- 153.Mori K, Toraya T. Biochemistry. 1999;38:13170–13178. doi: 10.1021/bi9911738. [DOI] [PubMed] [Google Scholar]
- 154.Bandarian V, Reed GH. In: Chemistry and Biochemistry of B12. Banerjee R, editor. Wiley; New York: 1999. pp. 811–833. [Google Scholar]
- 155.Sun L, Warncke K. Proteins: Structure, Function, and Bioinformatics. 2006;64:308–319. doi: 10.1002/prot.20997. [DOI] [PubMed] [Google Scholar]
- 156.Abend A, Bandarian V, Nitsche R, Stupperich E, Retey J, Reed GH. Arch Biochem Biophys. 1999;370:138–141. doi: 10.1006/abbi.1999.1382. [DOI] [PubMed] [Google Scholar]
- 157.Boas JF, Hicks PR, Pilbrow JR, Smith TD. J Chem Soc, Faraday Trans 2. 1978;74:417–430. [Google Scholar]
- 158.Lobrutto R, Bandarian V, Magnusson OT, Chen X, Schramm VL, Reed GH. Biochemistry. 2001;40:9–14. doi: 10.1021/bi001865s. [DOI] [PubMed] [Google Scholar]
- 159.Warncke K, Utada AS. J Am Chem Soc. 2001;123:8564–8572. doi: 10.1021/ja003658l. [DOI] [PubMed] [Google Scholar]
- 160.Morley CG, Stadtman TC. Biochemistry. 1971;10:2325–2329. doi: 10.1021/bi00788a023. [DOI] [PubMed] [Google Scholar]
- 161.Chang CH, Frey PA. J Biol Chem. 2000;275:106–114. doi: 10.1074/jbc.275.1.106. [DOI] [PubMed] [Google Scholar]
- 162.Morley CD, Stadtman TC. Biochemistry. 1972;11:600–605. doi: 10.1021/bi00754a019. [DOI] [PubMed] [Google Scholar]
- 163.Wetmore SD, Smith DM, Radom L. J Am Chem Soc. 2001;123:8678–8689. doi: 10.1021/ja010211j. [DOI] [PubMed] [Google Scholar]
- 164.Berkovitch F, Behshad E, Tang KH, Enns EA, Frey PA, Drennan CL. Proc Natl Acad Sci U S A. 2004;101:15870–15875. doi: 10.1073/pnas.0407074101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Chen HP, Wu SH, Lin YL, Chen CM, Tsay SS. J Biol Chem. 2001;276:44744–44750. doi: 10.1074/jbc.M108365200. [DOI] [PubMed] [Google Scholar]
- 166.Chen HP, Hsui FC, Lin LY, Ren CT, Wu SH. Eur J Biochem. 2004;271:4293–4297. doi: 10.1111/j.1432-1033.2004.04369.x. [DOI] [PubMed] [Google Scholar]
- 167.Beck WS, Abeles RH, Robinson WG. Biochem Biophys Res Commun. 1966;25:421–425. doi: 10.1016/0006-291x(66)90222-1. [DOI] [PubMed] [Google Scholar]
- 168.Ashley GW, Harris G, Stubbe J. J Biol Chem. 1986;261:3958–3964. [PubMed] [Google Scholar]
- 169.Booker S, Licht S, Broderick J, Stubbe J. Biochemistry. 1994;33:12676–12685. doi: 10.1021/bi00208a019. [DOI] [PubMed] [Google Scholar]
- 170.Licht S, Stubbe J. In: Comprehensive Natural Products Chemstry. Poulter D, editor. Elsevier; Amsterdam: 1999. p. 5. [Google Scholar]
- 171.Booker S, Stubbe J. Proc Natl Acad Sci USA. 1993;90:8352–8356. doi: 10.1073/pnas.90.18.8352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Sintchak MD, Arjara G, Kellogg BA, Stubbe J, Drennan CL. Nat Struct Biol. 2002;9:293–300. doi: 10.1038/nsb774. [DOI] [PubMed] [Google Scholar]
- 173.Lawrence CC, Gerfen GJ, Samano V, Nitsche R, Robins M, Retey J, Stubbe J. J Biol Chem. 1999;274:7039–7042. doi: 10.1074/jbc.274.11.7039. [DOI] [PubMed] [Google Scholar]
- 174.Chen D, Abend A, Stubbe J, Frey PA. Biochemistry. 2003;42:4578–4584. doi: 10.1021/bi030018x. [DOI] [PubMed] [Google Scholar]
- 175.George SJ, Seravalli J, Ragsdale SW. J Am Chem Soc. 2005;127:13500–13501. doi: 10.1021/ja0528329. [DOI] [PubMed] [Google Scholar]
- 176.Lindahl PA. J Biol Inrog Chem. 2004:9. [Google Scholar]
- 177.Matthews RG. Acc Chem Res. 2001;34:681–698. doi: 10.1021/ar0000051. [DOI] [PubMed] [Google Scholar]
- 178.Bandarian V, Pattridge KA, Lennon BW, Huddler DP, Matthews RG, Ludwig ML. Nat Struct Biol. 2002;9:53–56. doi: 10.1038/nsb738. [DOI] [PubMed] [Google Scholar]