

Abstract
Phosphorus is an essential nutrient required for critical biological reactions that maintain the normal homoeostatic control of the cell. This element is an important component of different cellular structures, including nucleic acids and cell membranes. Adequate phosphorus balance is vital for maintaining basic cellular functions, ranging from energy metabolism to cell signalling. In addition, many intracellular pathways utilize phosphate ions for important cellular reactions; therefore, homoeostatic control of phosphate is one of the most delicate biological regulations. Impaired phosphorus balance can affect the functionality of almost every human system, including musculoskeletal and cardiovascular systems, ultimately leading to an increase in morbidity and mortality of the affected patients. Human and experimental studies have found that delicate balance among circulating factors, like vitamin D, PTH (parathyroid hormone) and FGF23 (fibroblast growth factor 23), are essential for regulation of physiological phosphate balance. Dysregulation of these factors, either alone or in combination, can induce phosphorus imbalance. Recent studies have shown that suppression of the FGF23–klotho system can lead to hyperphosphataemia with extensive tissue damage caused by phosphate toxicity. The cause and consequences of phosphate toxicity will be briefly summarized in the present review.
Keywords: calcium, fibroblast growth factor 23 (FGF23), klotho, parathyroid hormone (PTH), sodium-dependent phosphate transporter, vitamin D
INTRODUCTION
Phosphorus is an essential nutrient for the body and is routinely consumed through food. After consumption, phosphorus is usually bound with oxygen and exists as phosphate in the body. Both organic and inorganic forms of phosphate are present in regularly consumed foods such as meats, fish, eggs, milk/dairy products and vegetables. The amount of total phosphate ingestion can be significantly influenced by processed food and/or beverage intake, as phosphate metabolites are used as additives in these items. Following a meal, inorganic phosphate can be rapidly absorbed across the small intestine and enter the blood stream causing an elevation in blood phosphate levels. The net efficiency of intestinal phosphate absorption is more than twice that of calcium absorption. An increase in serum levels of inorganic phosphate usually reduce serum levels of ionic calcium by forming a calcium–phosphate complex; such reduced ionic calcium concentration in turn stimulates release of PTH (parathyroid hormone) in an attempt to restore the serum calcium balance. In contrast, dietary phosphate deficiency, mostly due to malnutrition, not only induces hypophosphataemia, but can also impair the bone mineralization process and eventually lead to the development of rickets [1,2]. Optimal phosphorus and calcium balance is important for skeletal growth, development and maintenance [3,4]. Despite the essential role of phosphate in living cells, the molecular regulation of intra- and extra-cellular phosphorus metabolism is poorly understood and is an active area of research.
PHOSPHATE METABOLISM
Phosphorus is widely distributed in the body. More than 80 % of total phosphate is present in the bone and teeth in the form of apatite. The remaining phosphate is mostly present in the viscera and skeletal muscle, with a very small amount in the extracellular fluids (<0.1 %) [5–8]. Intracellular phosphate ions are essential for oxidative phosphorylation and approx. 20 % of cellular phosphate is present in the mitochondria. Approx. 30 % of total cellular phosphate is stored in the ER (endoplasmic reticulum) and is used in phosphorylation of various proteins. The remaining cellular phosphate is present in the nucleus, Golgi complex and lysosomes. Transporting phosphate in and out of the cell according to the need of the body is a complex process and the exact molecular mechanisms of such delicate transport are not yet clear. It is necessary to mention that cells also use phosphate to transport cellular energy through the formation of ATP by oxidative phosphorylation. In addition, glucose and triacylglycerol (triglyceide) synthesis utilize phosphate to form glucose 6-phosphate and glycerol 3-phosphate respectively.
Phosphorylation is an essential cellular reaction, where addition of a phosphate group to a molecule/protein (including enzymes or receptors), can determine the biological activity of that particular molecule. Phosphorylation is catalysed by specific protein kinases, whereas dephosphorylation is catalysed by phosphatases. More than 2000 chemical reactions in living cells use phosphate [9] and optimal intra- and extra-cellular phosphate balance is essential for efficacy of such biochemical reactions. Phosphate balance is tightly controlled by complex cross-organ communication among the kidney, intestine, bone and parathyroid gland (Figure 1) [6,10–14]. Our understanding of intestinal phosphate transport is mostly based on experimental studies that have shown that the bulk of phosphate absorption takes place in the duodenum and jejunum in rats, and in the ileum in mice [15]. NaPi (sodium-dependent phosphate) transport, involving NaPi-2b, is believed to actively regulate the intestinal phosphate absorption. Both 1,25-dihydroxyvitamin D and dietary phosphate can influence intestinal NaPi-2b activities [16]. For instance, low-phosphate diet, through inducing the renal expression of 1α(OH)ase can increase circulating 1,25-dihydroxyvitamin D levels, which in turn can enhance intestinal NaPi-2b protein expression to increase intestinal phosphate absorption. In a similar line of observation, the absorption ability of NaPi-2b knockout mice was 50 % less than that of wild-type animals following acute administration of phosphate [17]. However, the human relevance of the experimental studies need additional studies, as inactivating mutations of the human NaPi-2b gene did not show any considerable reduction in serum phosphate levels [18]. In general, the body’s daily need of phosphate is covered by intestinal absorption from consumed food, and the serum level of phosphate is delicately maintained by renal excretion of phosphate.
Figure 1. Simplified diagram showing muti-organ interactions in regulation of phosphate homoeostasis.
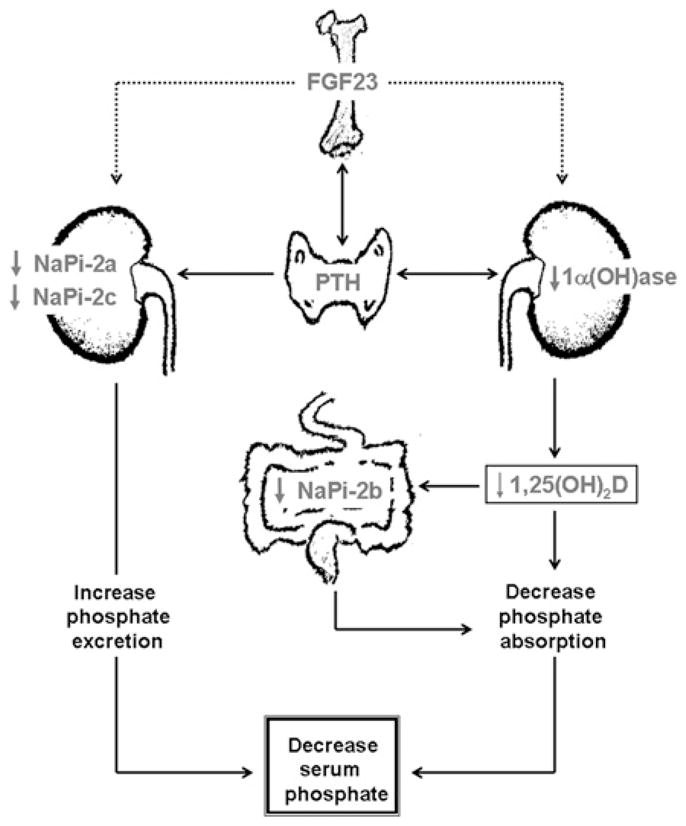
FGF23 produced in the bone cells can suppress renal NaPi-2a and NaPi-2c co-transporter activities to increase the urinary excretion of phosphate. Likewise, FGF23 can also suppress renal expression of 1α(OH)ase to reduce production of 1,25(OH)2D (1,25-dihydroxyvitamin D), which can suppress intestinal NaPi-2b activities to reduce phosphate absorption, resulting in decreased serum phosphate levels [6].
Renal phosphate excretion and reabsorption is partly mediated by the NaPi system – NaPi-2a and NaPi-2c. There is a link between PTH activity and renal proximal tubular epithelial cell membrane integration and retrieval of NaPi-2a that is located in the luminal side of the tubular epithelial cells [19,20]. PTH is a strong inhibitor of NaPi-dependent phosphate reabsorption and thereby facilitates an increase in urinary phosphate excretion [19,20]. The lists of molecules that can regulate NaPi activities are growing and recent studies have shown that FGF23 (fibroblast growth factor 23) and klotho can suppress NaPi activities [21–24]. The amount of dietary phosphate content has a major regulatory effect on renal phosphate reabsorption [25]. Dietary phosphate restriction induces an adaptive increase of intestinal phosphate uptake through a sodium gradient-dependent phosphate transport. Prolonged dietary phosphate restriction also increases NaPi-2a activity and, thereby, increases phosphate reabsorption in the kidney in an attempt to restore the balance [26]. Microtubules (tubulin) are believed to be involved in NaPi-2a activity, possibly by facilitating the rapid translocation of NaPi-2a from intracellular compartments to the cell membrane in response to a low phosphate diet [26]. The molecular events of systemic phosphate metabolism are not yet clearly defined and identification of the FGF23–klotho system as a potent phosphotonin has provided new mechanistic insights into homoeostatic control of phosphate [27,28].
FGF23–KLOTHO SYSTEM
Endocrine regulation of phosphate homoeostasis is a complex process and identification of the FGF23–klotho system has significantly enhanced our understanding of multi-organ interactions during systemic regulation of phosphate homoeostasis. FGF23 is an approx. 30-kDa protein that contains the FGFR (FGF receptor)-binding domain in the N-terminal and a potential klotho-binding site at the C-terminal [29]. FGF23 can bind to multiple FGFRs, including FGFR1c, FGFR3c and FGFR4 [21,30–33]. A recent in vivo study, however, did not show a significant response of FGF23 through FGFR3 or FGFR4 [34]. Furthermore, the interaction between FGF23-FGFRs and subsequent signalling activities required klotho as a cofactor. In the presence of klotho, FGF23 can bind to its receptor complex, with much higher affinity, to activate downstream signalling phosphoproteins, including FGFR substrate-2a, ERK (extracellular-signal-regulated kinase), p38, JNK (c-Jun N-terminal kinase) and AKT proteins [35,36].
The klotho gene encodes a type 1 membrane protein and the full-length 5.2 kb transcript encodes a 130-kDa membrane protein [37]. A disintegrin and metalloproteinases (ADAM-10 and ADAM-17) can cleave klotho from the plasma membrane [38]. Klotho expression is mostly detected in the distal convoluted tubules of the kidney, the parathyroid gland and the epithelium of the choroid plexus in the brain [39]; such restricted expression gives the tissue specificity for FGF23 action. Activation of the FGF23–klotho system can increase urinary phosphate excretion by reducing NaPi-2a and NaPi-2c co-transporter activity [21–23]. Whether the FGF23–klotho system directly reduces renal NaPi co-transporter activity, or such response is mediated through other FGF23 target molecules, is an unsolved issue. The reduction of serum phosphate levels following activation of the FGF23–klotho system is a universal phenomenon detected in both animal and human studies [28,40–45]. For instance, increased serum levels of FGF23 due to gain-of-function mutations of the human FGF23 gene are associated with hypophosphataemia, a condition triggered by excessive urinary phosphate wasting in patients with ADHR (autosomal-dominant hypophosphataemic rickets) [28]. Similarly, hypophosphataemia in patients with ARHR (autosomal-recessive hypophosphataemic rickets), also known as osteomalacia, has been attributed to high serum levels of FGF23 [46]. Likewise, transgenic mice overexpressing human FGF23 develop hypophosphataemia due to severe urinary loss of phosphate, while Fgf23 knockout mice develop hyperphosphataemia due to increased renal uptake of filtrated phosphate. Genetic restoration of the systemic actions of human FGF23 in Fgf23 knockout mice can reverse hyperphosphataemia to hypophosphataemia [47]. As it happens, the phenotype of Fgf23 knockout mice mimics the clinical features of FTC (familial tumoral calcinosis) patients, in which hyperphosphataemia develops due to reduced activity of human FGF23 [44,47].
Recently, genetically modified animal models have convincingly demonstrated the in vivo requirement for klotho in FGF23-mediated regulation of phosphate metabolism. For example, bioactive FGF23 protein is unable to exert phosphate-lowering effects in mice lacking klotho activities (either klotho−/− mice or Fgf23−/−/klotho−/− double knockout mice) [45]. Similarly, the inactivation of the klotho function from phex mutant mice resulted in hyperphosphataemia in phex/klotho double mutant mice, even though double mutant mice have significantly high serum levels of FGF23 [43,48]. In a separate experiment, genetic inactivation of klotho from FGF23 transgenic mice reversed a hypophosphataemic phenotype to a hyperphosphataemic phenotype [49]. In a similar study involving humans, a homozygous loss-of-function mutation in the Klotho gene induced severe hyperphosphataemia, despite high serum levels of FGF23 in the affected patient with tumoral calcinosis [42]. It is clear from previous studies that klotho is indispensable for FGF23-mediated regulation of systemic phosphate homoeostasis [11,50,51] and that suppression of the FGF23–klotho system can induce phosphate toxicity [52,53].
PHOSPHATE TOXICITY
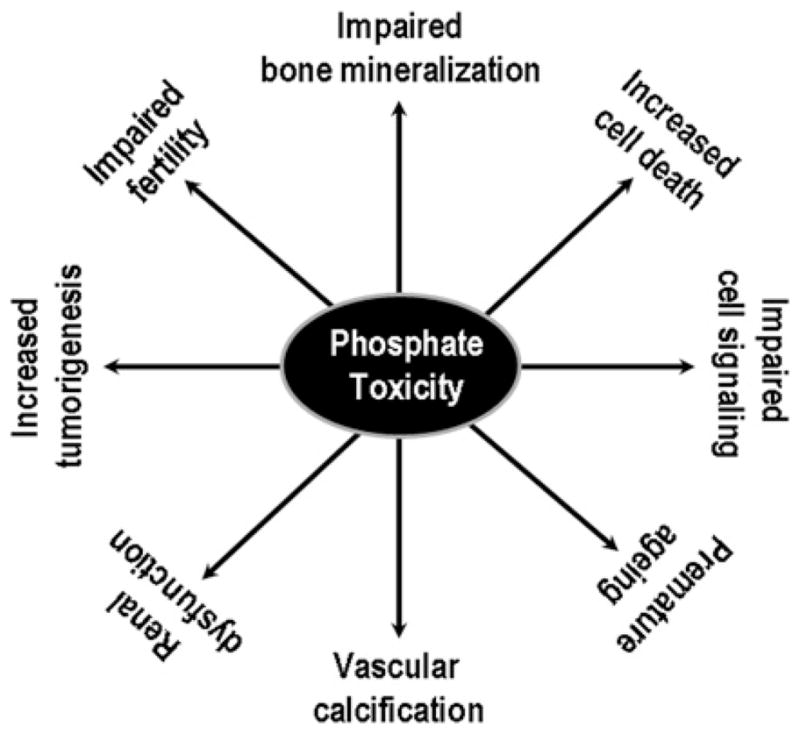
Phosphate toxicity due to excessive retention of phosphate in the body can cause a wide range of cellular and tissue injuries (Figure 2). For instance, higher occurrence of vascular calcification, encountered in patients with CKD (chronic kidney disease), is related to the increased retention of phosphate in the body [1,48]. Genetic studies with mice have shown that phosphate toxicity is closely associated with cardiovascular calcification in klotho knockout mice [22]. More importantly, lowering serum phosphate levels in klotho knockout mice can markedly reduce vascular calcification, despite the presence of significantly higher serum calcium and 1,25-dihydroxyvitamin D levels [22,54]. A similar trend of phosphate toxicity and ectopic calcification is also noted in Fgf23 knockout mice [55–58]. In humans, phosphate toxicity and low serum vitamin D levels have been implicated as independent risk factors for high mortality in CKD patients [59–61].
Figure 2. Partial list of pathological events related to phosphate toxicity as documented in both human and animal studies.
Complications of phosphate toxicity are also encountered in patients treated with phosphate-containing laxatives or enemas. Serum analysis of 14 elderly patients exposed to a phosphate-containing enema has shown significantly elevated serum levels of phosphate within an hour and a significant decrease in serum calcium levels by 12 h [62]. In addition, administration of hypertonic phosphate-containing enemas to a paediatric group of patients resulted in a wide range of complications, including tetany, dehydration, hypotension, tachycardia, hyperpyrexia, cardiac arrest and coma [63,64]. Some of these complications can be attributed to abnormal mineral ion and electrolyte balance induced by phosphate toxicity, which can also serve to increase anion gaps. For instance, Domico et al. [65] found a total anion gap of 29 mmol/l in a patient with phosphate toxicity (38.3 mg/dl), which was normalized after therapeutically reducing serum phosphate [65]. In a separate study, rectally administered hypertonic phosphate solution to a 4-year-old chronically constipated girl with normal renal function was reported to have developed phosphate toxicity (23 mg/dl) with breathing difficulties and a depressed level of consciousness [66]. The girl experienced a generalized seizure 16 h following a phosphate-containing enema and was unresponsive to multiple doses of lorazepam. The patient only responded to 100 mg of intravenous calcium chloride, suggesting deleterious effects of phosphate toxicity on other mineral ions and electrolyte levels that provoked subsequent complications, even without the presence of predisposing risk factors [66]. In an animal study, phosphate toxicity (7–20-fold increase over control by 4 h) induced by a commercially available phosphate-containing enema (30–50 ml/kg of body weight) has been shown to induce 100 % mortality [67].
Phosphate toxicity has recently been found to accelerate the mammalian aging process by inflicting tissue damage and reducing survival [40]. Genetically engineered klotho-null mice developed phosphate toxicity as early as 3 weeks of age that induced premature aging. The effects of premature aging in genetically engineered mice included, but were not limited to, loss of body weight, kyphosis, hypogonadism, infertility, generalized tissue atrophy and reduced life span [40,41,43,68–70]. Some of these changes in klotho knockout mice bear similarities to human aging. Molecular and biochemical analysis suggests that increased renal activity of NaPi-2a leads to phosphate toxicity in klotho knockout mice. In fact, the extensive aging phenotypes in short-lived klotho knockout mice can be suppressed by genetically reducing phosphate toxicity in NaPi2a/klotho double knockout mice to extend their survival [40]. The genital organs of hyperphosphataemic klotho knockout mice of both sexes are severely atrophic and such hypogonadism is associated with premature infertility, which is a major consequence of accelerated aging in both humans and experimental animals [68,69]. Notably, the hyperphosphataemic klotho knockout mice regained fertility by genetically reducing serum phosphate levels, as evidenced in the NaPi2a/klotho double knockout mice. More importantly, the NaPi2a/klotho double knockout mice lost their fertility when fed a high-phosphate diet, clearly suggesting that phosphate toxicity can affect fertility and thereby influence the aging process. A similar premature aging trend is exhibited in klotho knockout mice where widespread tissue atrophy in the spleen, skeletal muscle, intestine and skin is present [71]. However, reducing serum phosphate levels in klotho knockout mice rescues tissue atrophy in NaPi2a/klotho double knockout mice. When phosphate toxicity is induced in NaPi2a/klotho double knockout mice by feeding on a high-+phosphate diet (1.2 %), premature aging, including generalized tissue atrophy, reappeared and all the mice died by 15 weeks. Of relevance, none of the NaPi2a/klotho double knockout mice fed on a normal-phosphate diet (0.6 %) died until 20 weeks of the observational period [40]. These in vivo genetic and dietary observations clearly suggest that phosphate toxicity can accelerate the mammalian aging process.
In addition to accelerating the aging process, increased dietary phosphate intake has been shown to stimulate the growth and size of lung tumours [72]. A large number of processed foods, including meats, cheeses, beverages and bakery products, use phosphate-containing food additives. Consumption of these items could significantly increase phosphorus intake and impair the normal homoeostatic balance of calcium and phosphate. For instance, rats fed with phosphoric-acid-containing soft drinks developed significant hypercalciuria and hyperphosphaturia, along with dysregulation of serum PTH and vitamin D, provoking reduced bone mineralization [73,74]. In a related human study, consumption of phosphoric acid-containing soft drinks was found to be associated with hypocalcaemia in postmenopausal women [75].
The mechanism by which phosphate toxicity accelerates the aging process is not clear. Phosphate toxicity can exert cytotoxic effects to compromise the functional ability of various organ systems. Phosphate toxicity can induce an increased rate of apoptosis in various tissues that can be suppressed by reducing the phosphate burden [40]. In an experimental study, when rabbits were injected intradermally with hypertonic phosphate solution, a pronounced erythema and indurations were noted by 24 h, which progressed to central necrosis and full thickness tissue loss by 5–7 days [76]. In fact, patients exposed to a phosphate-containing enema developed necrotic changes of the abdominal tissues, including loss of internal and external sphincters due to extensive tissue necrosis [76]. As mentioned above, phosphate is essential for cell signalling activities and altered phosphate balance may impair the homoeostatic control of signalling activities leading to cellular and tissue damage. Phosphorylation of inositol to phosphatidylinositol and cleavage of inositol triphosphate represent a major intracellular regulation of phosphate metabolism that also affects intracellular calcium metabolism. Recently, dietary phosphate has been shown to stimulate the Akt-mediated signalling network and provoke an increase in lung tumorigenesis [72].
CONCLUSIONS
Common causes of phosphate toxicity in humans include impaired renal function, rhabdomyolysis and tumour lysis syndrome. In addition, exogenous phosphate toxicity is also documented in patients with Hirschsprung disease when exposed to hypertonic phosphate enemas [77]. Of relevance, phosphate toxicity induced by excessive exogenous phosphate administration can be fatal [66,67,78,79]. Although the lethal dose of phosphate in humans is unknown, Martin et al. [67] reported that the lethal dose of phosphate in pigs was 35 mmol/kg of body weight. As for mice, 14–16 mg/dl of phosphate serum level could cause 100 % mortality by 15 weeks of age [40]. Overall, human and animal studies have convincingly demonstrated the toxic effects of phosphate in accelerating various pathologies, ranging from vascular calcification to tumour formation and aging. Acute phosphate toxicity can provoke hypocalcaemia and associated symptoms including tetany, hypotension and tachycardia. Moderate phosphate toxicity that takes longer to develop can lead to the deposition of calcium phosphate crystals in various tissues, including often fatal cardiovascular calcification. An abnormal deposit of calcium phosphate crystals due to phosphate toxicity is usually an irreversible process. Although, without pre-existing renal or gastrointestinal diseases, acute phosphate toxicity is relatively rare, the deleterious effects of chronic ingestion of unrestricted amounts of phosphate in individuals is not clear, and needs to be studied in more depth. Particularly, the effects of chronic unrestricted ingestion of high phosphate-containing processed foods and soft drinks on functionality of various organ systems require careful analysis. Finally, maintaining the phosphate balance in the diet may be important for a healthy life and longevity, as phosphate imbalance can induce serious debilitating complications.
Acknowledgments
Some of the original research that formed the basis of this review was performed by Dr Teruyo Nakatani, Dr Mutsuko Ohnishi, Dr Shigeko Kato and Dr Kazuyoshi Uchihashi of the Department of Oral Medicine, Infection and Immunity at the Harvard School of Dental Medicine, Boston, MA, U.S.A.
FUNDING
The author’s work was supported by the National Institute of Diabetes and Digestive and Kidney Diseases [grant number R01-DK077276].
Abbreviations
- ADAM
a disintegrin and metalloproteinase
- ADHR
autosomal-dominant hypophosphataemic rickets
- CKD
chronic kidney disease
- FGF23
fibroblast growth factor 23
- FGFR
fibroblast growth factor receptor
- NaPi
sodium-dependent phosphate transporter
- PTH
parathyroid hormone
References
- 1.Fukagawa M, Hamada Y, Nakanishi S, Tanaka M. The kidney and bone metabolism: nephrologists’ point of view. J Bone Miner Metab. 2006;24:434–438. doi: 10.1007/s00774-006-0719-7. [DOI] [PubMed] [Google Scholar]
- 2.Laroche M, Boyer JF. Phosphate diabetes, tubular phosphate reabsorption and phosphatonins. J Bone Spine. 2005;72:376–381. doi: 10.1016/j.jbspin.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 3.Huttunen MM, Pietila PE, Viljakainen HT, Lamberg-Allardt CJ. Prolonged increase in dietary phosphate intake alters bone mineralization in adult male rats. J Nutr Biochem. 2006;17:479–484. doi: 10.1016/j.jnutbio.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 4.Heilberg IP, Weisinger JR. Bone disease in idiopathic hypercalciuria. Curr Opin Nephrol Hypertens. 2006;15:394–402. doi: 10.1097/01.mnh.0000232880.58340.0c. [DOI] [PubMed] [Google Scholar]
- 5.Gaasbeek A, Meinders AE. Hypophosphatemia: an update on its etiology and treatment. Am J Med. 2005;118:1094–1101. doi: 10.1016/j.amjmed.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 6.Razzaque MS. The FGF23-Klotho axis: endocrine regulation of phosphate homeostasis. Nat Rev Endocrinol. 2009;5:611–619. doi: 10.1038/nrendo.2009.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iotti S, Lodi R, Gottardi G, Zaniol P, Barbiroli B. Inorganic phosphate is transported into mitochondria in the absence of ATP biosynthesis: an in vivo31P NMR study in the human skeletal muscle. Biochem Biophys Res Commun. 1996;225:191–194. doi: 10.1006/bbrc.1996.1152. [DOI] [PubMed] [Google Scholar]
- 8.Hutson SM, Williams GD, Berkich DA, LaNoue KF, Briggs RW. A 31P NMR study of mitochondrial inorganic phosphate visibility: effects of Ca2+, Mn2+, and the pH gradient. Biochemistry. 1992;31:1322–1330. doi: 10.1021/bi00120a007. [DOI] [PubMed] [Google Scholar]
- 9.KEGG (Kyoto Encyclopedia of Genes and Genomes) http://www.genome.jp/
- 10.Drezner M. Phosphorus homeostasis and related disorders. In: Bilezikian J, Raisz L, Rodan G, editors. Principles in Bone Biology. Academic Press; New York: 2002. pp. 321–338. [Google Scholar]
- 11.Razzaque MS. FGF23-mediated regulation of systemic phosphate homeostasis: is Klotho an essential player? Am J Physiol Renal Physiol. 2009;296:F470–F476. doi: 10.1152/ajprenal.90538.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Econs MJ. New insights into the pathogenesis of inherited phosphate wasting disorders. Bone. 1999;25:131–135. doi: 10.1016/s8756-3282(99)00108-8. [DOI] [PubMed] [Google Scholar]
- 13.Miyamoto K, Ito M, Segawa H, Kuwahata M. Molecular targets of hyperphosphataemia in chronic renal failure. Nephrol Dial Transplant. 2003;18(Suppl 3):iii79–iii80. doi: 10.1093/ndt/gfg1020. [DOI] [PubMed] [Google Scholar]
- 14.Quarles LD. FGF23, PHEX, and MEPE regulation of phosphate homeostasis and skeletal mineralization. Am J Physiol Endocrinol Metab. 2003;285:E1–E9. doi: 10.1152/ajpendo.00016.2003. [DOI] [PubMed] [Google Scholar]
- 15.Marks J, Debnam ES, Unwin RJ. Phosphate homeostasis and the renal–gastrointestinal axis. Am J Physiol Renal Physiol. 2010;299:F285–F296. doi: 10.1152/ajprenal.00508.2009. [DOI] [PubMed] [Google Scholar]
- 16.Hattenhauer O, Traebert M, Murer H, Biber J. Regulation of small intestinal Na-Pi type IIb cotransporter by dietary phosphate intake. Am J Physiol Gastrointest Liver Physiol. 1999;277:G756–G762. doi: 10.1152/ajpgi.1999.277.4.G756. [DOI] [PubMed] [Google Scholar]
- 17.Sabbagh Y, O’Brien SP, Song W, Boulanger JH, Stockmann A, Arbeeny C, Schiavi SC. Intestinal Npt2b plays a major role in phosphate absorption and homeostasis. J Am Soc Nephrol. 2009;20:2348–2358. doi: 10.1681/ASN.2009050559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Corut A, Senyigit A, Ugur SA, Altin S, Ozcelik U, Calisir H, Yildirim Z, Gocmen A, Tolun A. Mutations in SLC34A2 cause pulmonary alveolar microlithiasis and are possibly associated with testicular microlithiasis. Am J Hum Genet. 2006;79:650–656. doi: 10.1086/508263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tenenhouse HS. Regulation of phosphorus homeostasis by the type IIa Na/phosphate cotransporter. Annu Rev Nutr. 2005;25:197–214. doi: 10.1146/annurev.nutr.25.050304.092642. [DOI] [PubMed] [Google Scholar]
- 20.Murer H, Forster I, Hilfiker H, Pfister M, Kaissling B, Lotscher M, Biber J. Cellular/molecular control of renal Na/Pi-cotransport. Kidney Int Suppl. 1998;65:S2–S10. [PubMed] [Google Scholar]
- 21.Gattineni J, Bates C, Twombley K, Dwarakanath V, Robinson ML, Goetz R, Mohammadi M, Baum M. FGF23 decreases renal NaPi-2a and NaPi-2c expression and induces hypophosphatemia in vivo predominantly via FGF receptor 1. Am J Physiol Renal Physiol. 2009;297:F282–F291. doi: 10.1152/ajprenal.90742.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ohnishi M, Nakatani T, Lanske B, Razzaque MS. In vivo genetic evidence for suppressing vascular and soft-tissue calcification through the reduction of serum phosphate levels, even in the presence of high serum calcium and 1,25-dihydroxyvitamin D levels. Circ Cardiovasc Genet. 2009;2:583–590. doi: 10.1161/CIRCGENETICS.108.847814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miyamoto K, Ito M, Kuwahata M, Kato S, Segawa H. Inhibition of intestinal sodium-dependent inorganic phosphate transport by fibroblast growth factor 23. Ther Apher Dial. 2005;9:331–335. doi: 10.1111/j.1744-9987.2005.00292.x. [DOI] [PubMed] [Google Scholar]
- 24.Hu MC, Shi M, Zhang J, Pastor J, Nakatani T, Lanske B, Shawkat Razzaque M, Rosenblatt KP, Baum MG, Kuro-o M, Moe OW. Klotho: a novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. FASEB J. 2010;24:3438–3450. doi: 10.1096/fj.10-154765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keusch I, Traebert M, Lotscher M, Kaissling B, Murer H, Biber J. Parathyroid hormone and dietary phosphate provoke a lysosomal routing of the proximal tubular Na/Pi-cotransporter type II. Kidney Int. 1998;54:1224–1232. doi: 10.1046/j.1523-1755.1998.00115.x. [DOI] [PubMed] [Google Scholar]
- 26.Lotscher M, Kaissling B, Biber J, Murer H, Levi M. Role of microtubules in the rapid regulation of renal phosphate transport in response to acute alterations in dietary phosphate content. J Clin Invest. 1997;99:1302–1312. doi: 10.1172/JCI119289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamashita T, Yoshioka M, Itoh N. Identification of a novel fibroblast growth factor, FGF-23, preferentially expressed in the ventrolateral thalamic nucleus of the brain. Biochem Biophys Res Commun. 2000;277:494–498. doi: 10.1006/bbrc.2000.3696. [DOI] [PubMed] [Google Scholar]
- 28.White KE, Evons WE, O’Riordan JLH, Speer MC, Econs MJ, Lorenz-Depiereux B, Grabowski M, Meitinger T, Strom JM. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet. 2000;26:345–348. doi: 10.1038/81664. [DOI] [PubMed] [Google Scholar]
- 29.Yamashita T. Structural and biochemical properties of fibroblast growth factor 23. Ther Apher Dial. 2005;9:313–318. doi: 10.1111/j.1744-9987.2005.00288.x. [DOI] [PubMed] [Google Scholar]
- 30.Eswarakumar VP, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005;16:139–149. doi: 10.1016/j.cytogfr.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 31.Mohammadi M, Olsen SK, Ibrahimi OA. Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev. 2005;16:107–137. doi: 10.1016/j.cytogfr.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 32.Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP, Baum MG, Schiavi S, Hu MC, Moe OW, Kuro-o M. Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem. 2006;281:6120–6123. doi: 10.1074/jbc.C500457200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, Fujita T, Fukumoto S, Yamashita T. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006;444:770–774. [Google Scholar]
- 34.Liu S, Vierthaler L, Tang W, Zhou J, Quarles LD. FGFR3 and FGFR4 do not mediate renal effects of FGF23. J Am Soc Nephrol. 2008;19:2342–2350. doi: 10.1681/ASN.2007121301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuro-o M. Klotho as a regulator of fibroblast growth factor signaling and phosphate/calcium metabolism. Curr Opin Nephrol Hypertens. 2006;15:437–441. doi: 10.1097/01.mnh.0000232885.81142.83. [DOI] [PubMed] [Google Scholar]
- 36.Medici D, Razzaque MS, Deluca S, Rector TL, Hou B, Kang K, Goetz R, Mohammadi M, Kuro OM, Olsen BR, Lanske B. FGF-23–Klotho signaling stimulates proliferation and prevents vitamin D-induced apoptosis. J Cell Biol. 2008;182:459–465. doi: 10.1083/jcb.200803024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nabeshima Y. The discovery of α-Klotho and FGF23 unveiled new insight into calcium and phosphate homeostasis. Cell Mol Life Sci. 2008;65:3218–3230. doi: 10.1007/s00018-008-8177-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen CD, Podvin S, Gillespie E, Leeman SE, Abraham CR. Insulin stimulates the cleavage and release of the extracellular domain of Klotho by ADAM10 and ADAM17. Proc Natl Acad Sci USA. 2007;104:19796–19801. doi: 10.1073/pnas.0709805104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matsumura Y, Aizawa H, Shiraki-Iida T, Nagai R, Kuro-o M, Nabeshima Y. Identification of the human klotho gene and its two transcripts encoding membrane and secreted klotho protein. Biochem Biophys Res Commun. 1998;242:626–630. doi: 10.1006/bbrc.1997.8019. [DOI] [PubMed] [Google Scholar]
- 40.Ohnishi M, Razzaque MS. Dietary and genetic evidence for phosphate toxicity accelerating mammalian aging. FASEB J. 2010;24:3562–3571. doi: 10.1096/fj.09-152488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ohnishi M, Nakatani T, Lanske B, Razzaque MS. Reversal of mineral ion homeostasis and soft-tissue calcification of klotho knockout mice by deletion of vitamin D 1α-hydroxylase. Kidney Int. 2009;75:1166–1172. doi: 10.1038/ki.2009.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ichikawa S, Imel EA, Kreiter ML, Yu X, Mackenzie DS, Sorenson AH, Goetz R, Mohammadi M, White KE, Econs MJ. A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J Clin Invest. 2007;117:2684–2691. doi: 10.1172/JCI31330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nakatani T, Ohnishi M, Razzaque MS. Inactivation of klotho function induces hyperphosphatemia even in presence of high serum fibroblast growth factor 23 levels in a genetically engineered hypophosphatemic (Hyp) mouse model. FASEB J. 2009;23:3702–3711. doi: 10.1096/fj.08-123992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Benet-Pages A, Orlik P, Strom TM, Lorenz-Depiereux B. An FGF23 missense mutation causes familial tumoral calcinosis with hyperphosphatemia. Hum Mol Genet. 2005;14:385–390. doi: 10.1093/hmg/ddi034. [DOI] [PubMed] [Google Scholar]
- 45.Nakatani T, Bara S, Ohnishi M, Densmore MJ, Taguchi T, Goetz R, Mohammadi M, Lanske B, Razzaque MS. In vivo genetic evidence of klotho-dependent functions of FGF23 in regulation of systemic phosphate homeostasis. FASEB J. 2009;23:433–441. doi: 10.1096/fj.08-114397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lorenz-Depiereux B, Bastepe M, Benet-Pages A, Amyere M, Wagenstaller J, Muller-Barth U, Badenhoop K, Kaiser SM, Rittmaster RS, Shlossberg AH, et al. DMP1 mutations in autosomal recessive hypophosphatemia implicate a bone matrix protein in the regulation of phosphate homeostasis. Nat Genet. 2006;38:1248–1250. doi: 10.1038/ng1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.DeLuca S, Sitara D, Kang K, Marsell R, Jonsson K, Taguchi T, Erben RG, Razzaque MS, Lanske B. Amelioration of the premature aging-like features of Fgf-23 knockout mice by genetically restoring the systemic actions of FGF-23. J Pathol. 2008;216:345–355. doi: 10.1002/path.2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Razzaque MS. Therapeutic potential of klotho-FGF23 fusion polypeptides: WO2009095372. Expert Opin Ther Pat. 2010;20:981–985. doi: 10.1517/13543771003774100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bai X, Dinghong Q, Miao D, Goltzman D, Karaplis AC. Klotho ablation converts the biochemical and skeletal alterations in FGF23 (R176Q) transgenic mice to a Klotho-deficient phenotype. Am J Physiol Endocrinol Metab. 2009;296:E79–E88. doi: 10.1152/ajpendo.90539.2008. [DOI] [PubMed] [Google Scholar]
- 50.Goetz R, Nakada Y, Hu MC, Kurosu H, Wang L, Nakatani T, Shi M, Eliseenkova AV, Razzaque MS, Moe OW, et al. Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23–FGFR–Klotho complex formation. Proc Natl Acad Sci U S A. 2010;107:407–412. doi: 10.1073/pnas.0902006107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goetz R, Beenken A, Ibrahimi OA, Kalinina J, Olsen SK, Eliseenkova AV, Xu C, Neubert TA, Zhang F, Linhardt RJ, et al. Molecular insights into the klotho-dependent, endocrine mode of action of fibroblast growth factor 19 subfamily members. Mol Cell Biol. 2007;27:3417–3428. doi: 10.1128/MCB.02249-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lanske B, Razzaque MS. Premature aging in klotho mutant mice: cause or consequence? Ageing Res Rev. 2007;6:73–79. doi: 10.1016/j.arr.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Razzaque MS. Does FGF23 toxicity influence the outcome of chronic kidney disease? Nephrol Dial Transplant. 2009;24:4–7. doi: 10.1093/ndt/gfn620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Razzaque MS. The dualistic role of vitamin D in vascular calcifications. Kidney Int. 2010 doi: 10.1038/ki.2010.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Razzaque MS, Lanske B. Hypervitaminosis D and premature aging: lessons learned from Fgf23 and Klotho mutant mice. Trends Mol Med. 2006;12:298–305. doi: 10.1016/j.molmed.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 56.Razzaque MS, Lanske B. The emerging role of the fibroblast growth factor-23–klotho axis in renal regulation of phosphate homeostasis. J Endocrinol. 2007;194:1–10. doi: 10.1677/JOE-07-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Razzaque MS, Sitara D, Taguchi T, St-Arnaud R, Lanske B. Premature ageing-like phenotype in fibroblast growth factor 23 null mice is a vitamin-D mediated process. FASEB J. 2006;20:720–722. doi: 10.1096/fj.05-5432fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Razzaque MS, St-Arnaud R, Taguchi T, Lanske B. FGF-23, vitamin D and calcification: the unholy triad. Nephrol Dial Transplant. 2005;20:2032–2035. doi: 10.1093/ndt/gfh991. [DOI] [PubMed] [Google Scholar]
- 59.Voormolen N, Noordzij M, Grootendorst DC, Beetz I, Sijpkens YW, van Manen JG, Boeschoten EW, Huisman RM, Krediet RT, Dekker FW. High plasma phosphate as a risk factor for decline in renal function and mortality in pre-dialysis patients. Nephrol Dial Transplant. 2007;22:2909–2916. doi: 10.1093/ndt/gfm286. [DOI] [PubMed] [Google Scholar]
- 60.Wang TJ, Pencina MJ, Booth SL, Jacques PF, Ingelsson E, Lanier K, Benjamin EJ, D’Agostino RB, Wolf M, et al. Vitamin D deficiency and risk of cardiovascular disease. Circulation. 2008;117:503–511. doi: 10.1161/CIRCULATIONAHA.107.706127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shoji T, Shinohara K, Kimoto E, Emoto M, Tahara H, Koyama H, Inaba M, Fukumoto S, Ishimura E, Miki T, et al. Lower risk for cardiovascular mortality in oral 1alpha-hydroxy vitamin D3 users in a haemodialysis population. Nephrol Dial Transplant. 2004;19:179–184. doi: 10.1093/ndt/gfg513. [DOI] [PubMed] [Google Scholar]
- 62.Grosskopf I, Graff E, Charach G, Binyamin G, Spinrad S, Blum I. Hyperphosphataemia and hypocalcaemia induced by hypertonic phosphate enema: an experimental study and review of the literature. Hum Exp Toxicol. 1991;10:351–355. doi: 10.1177/096032719101000509. [DOI] [PubMed] [Google Scholar]
- 63.Sotos JF, Cutler EA, Finkel MA, Doody D. Hypocalcemic coma following two pediatric phosphate enemas. Pediatrics. 1977;60:305–307. [PubMed] [Google Scholar]
- 64.Loughnan P, Mullins GC. Brain damage following a hypertonic phosphate enema. Am J Dis Child. 1977;131:1032. doi: 10.1001/archpedi.1977.02120220098018. [DOI] [PubMed] [Google Scholar]
- 65.Domico MB, Huynh V, Anand SK, Mink R. Severe hyperphosphatemia and hypocalcemic tetany after oral laxative administration in a 3-month-old infant. Pediatrics. 2006;118:e1580–1583. doi: 10.1542/peds.2006-1249. [DOI] [PubMed] [Google Scholar]
- 66.Marraffa JM, Hui A, Stork CM. Severe hyperphosphatemia and hypocalcemia following the rectal administration of a phosphate-containing Fleet pediatric enema. Pediatr Emerg Care. 2004;20:453–456. doi: 10.1097/01.pec.0000132217.65600.52. [DOI] [PubMed] [Google Scholar]
- 67.Martin RR, Lisehora GR, Braxton M, Jr, Barcia PJ. Fatal poisoning from sodium phosphate enema. Case report and experimental study. JAMA, J Am Med Assoc. 1987;257:2190–2192. [PubMed] [Google Scholar]
- 68.Nabeshima Y. Klotho: a fundamental regulator of aging. Ageing Res Rev. 2002;1:627–638. doi: 10.1016/s1568-1637(02)00027-2. [DOI] [PubMed] [Google Scholar]
- 69.Kuro-o M. Disease model: human aging. Trends Mol Med. 2001;7:179–181. doi: 10.1016/s1471-4914(01)01921-9. [DOI] [PubMed] [Google Scholar]
- 70.Nabeshima Y. Toward a better understanding of Klotho. Sci Aging Knowledge Environ. 2006:pe11. doi: 10.1126/sageke.2006.8.pe11. [DOI] [PubMed] [Google Scholar]
- 71.Kuro-o M. A potential link between phosphate and aging: lessons from Klotho-deficient mice. Mech Ageing Dev. 2010;131:270–275. doi: 10.1016/j.mad.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jin H, Xu CX, Lim HT, Park SJ, Shin JY, Chung YS, Park SC, Chang SH, Youn HJ, Lee KH, et al. High dietary inorganic phosphate increases lung tumorigenesis and alters Akt signaling. Am J Respir Crit Care Med. 2009;179:59–68. doi: 10.1164/rccm.200802-306OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Amato D, Maravilla A, Montoya C, Gaja O, Revilla C, Guerra R, Paniagua R. Acute effects of soft drink intake on calcium and phosphate metabolism in immature and adult rats. Rev Invest Clin. 1998;50:185–189. [PubMed] [Google Scholar]
- 74.Garcia-Contreras F, Paniagua R, Avila-Diaz M, Cabrera-Munoz L, Martinez-Muniz I, Foyo-Niembro E, Amato D. Cola beverage consumption induces bone mineralization reduction in ovariectomized rats. Arch Med Res. 2000;31:360–365. doi: 10.1016/s0188-4409(00)00090-4. [DOI] [PubMed] [Google Scholar]
- 75.Fernando GR, Martha RM, Evangelina R. Consumption of soft drinks with phosphoric acid as a risk factor for the development of hypocalcemia in postmenopausal women. J Clin Epidemiol. 1999;52:1007–1010. doi: 10.1016/s0895-4356(99)00097-9. [DOI] [PubMed] [Google Scholar]
- 76.Pietsch JB, Shizgal HM, Meakins JL. Injury by hypertonic phosphate enema. Can Med Assoc J. 1977;116:1169–1170. [PMC free article] [PubMed] [Google Scholar]
- 77.Moseley PK, Segar WE. Fluid and serum electrolyte disturbances as a complication of enemas in Hirschsprung’s disease. Am J Dis Child. 1968;115:714–718. doi: 10.1001/archpedi.1968.02100010716013. [DOI] [PubMed] [Google Scholar]
- 78.Ismail EA, Al-Mutairi G, Al-Anzy H. A fatal small dose of phosphate enema in a young child with no renal or gastrointestinal abnormality. J Pediatr Gastroenterol Nutr. 2000;30:220–221. doi: 10.1097/00005176-200002000-00025. [DOI] [PubMed] [Google Scholar]
- 79.Perlman JM. Fatal hyperphosphatemia after oral phosphate overdose in a premature infant. Am J Health Syst Pharm. 1997;54:2488–2490. doi: 10.1093/ajhp/54.21.2488. [DOI] [PubMed] [Google Scholar]