

Abstract
Phosphorus is an essential nutrient and is routinely assimilated through consumption of food. The body’s need of phosphate is usually fulfilled by intestinal absorption of this element from the consumed food, whereas its serum level is tightly regulated by renal excretion or reabsorption. Sodium-dependent phosphate transporters, located in the luminal side of the proximal tubular epithelial cells, have a molecular control on renal phosphate excretion and reabsorption. The systemic regulation of phosphate metabolism is a complex multiorgan process, and the identification of fibroblast growth factor-23 (FGF23)–Klotho system as a potent phosphatonin has provided new mechanistic insights into the homeostatic control of phosphate. Hypophosphatemia as a result of an increase in urinary phosphate wasting after activation of the FGF23–Klotho system is a common phenomenon, observed in both animal and human studies, whereas suppression of the FGF23–Klotho system leads to the development of hyperphosphatemia. This article will briefly summarize how delicate interactions of the FGF23–Klotho system can regulate systemic phosphate homeostasis.
Keywords: Klotho, FGF23, Vitamin D, Calcium, NaPi, PTH
Phosphate is a widely distributed element, and more than 80% of the total phosphate in the body is present in bones and teeth, whereas the rest is present in the viscera and skeletal muscle, with a very small amount in the extracellular fluids. About 20% of the intracellular phosphate is present in the mitochondria, and influences various essential cellular functions, including oxidative phosphorylation, whereas about 30% of total cellular phosphate is stored in the endoplasmic reticulum and is used for phosphorylation of various proteins.1-3 The remaining cellular phosphate is distributed in other cellular components, including nucleus, Golgi complex, and lysosomes. Maintaining phosphate balance is not only important for structural and functional activities of bones and teeth, but also essential for normal cellular function and survival. Despite clinical importance of maintaining phosphate balance, the molecular events related to its regulation are not yet clearly understood. Recent identification of bone-derived factors in regulating numerous systemic functions has helped us to realize the importance of endocrine regulation of mineral ion homeostasis.4-8
Traditionally, it was a well accepted fact that parathyroid hormone (PTH) and the active form of vitamin D, 1, 25-dihydroxyvitamin D3 [1α,25(OH)2D3] were 2 major endocrine factors that regulated calcium homeostasis. When calcium-sensing receptors in the parathyroid gland detect a decrease in serum calcium levels, PTH is secreted and acts to restore the serum calcium level to normal. PTH stimulates 1α-hydroxylase enzyme expression in the kidneys, and thus increases synthesis of 1α,25(OH)2D3.9 The function of 1α,25(OH)2D3 is to increase the efficiency of calcium and phosphate absorption in the intestine and kidneys (Fig 1). PTH also acts on bones to stimulate osteoclastogenesis and to promote mobilization of calcium and phosphate from the reservoir, thereby increasing blood calcium levels. However, it does not cause phosphate retention as a result of its phosphaturic effect in the kidneys. Therefore, maintenance of phosphate balance was considered to be passively regulated by PTH and 1α,25(OH)2D3.
Figure 1.
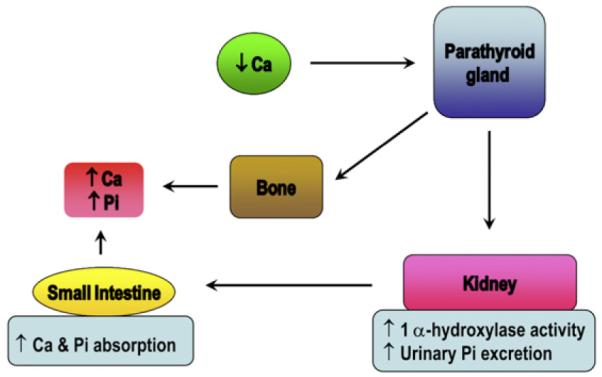
Classical pathway of passively regulated phosphate (Pi) balance. On decrease in serum calcium (Ca2+) level, parathyroid hormone (PTH) is secreted and acts on its cognate G-protein-coupled receptor, PTH1R, at its target tissues (bones and kidneys) and restores the serum Ca2+ level. In kidneys, PTH induces phosphaturia and increases levels of circulating 1α,25(OH)2D3. 1α,25(OH)2D3 in turn acts on the small intestine to increase Ca2+ and Pi absorption. In bones, PTH causes an increase in bone resorption, and thus increases serum Ca2+ and Pi levels. For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.
Recent studies have added new dimensions to this traditional view on the endocrine regulation of phosphate balance. Phosphatonins are the most recently discovered players in phosphate homeostasis. The term “phosphatonin” was originally described as a circulating factor that induced renal inorganic phosphate wasting in patients with tumor-induced osteomalacia (TIO).10 Patients with TIO have mesenchymal tumors and exhibit mineral metabolism derangements, including normal or slightly low serum calcium, normal PTH, inappropriately low serum 1α,25(OH)2D3, and low serum phosphate levels owing to renal phosphate wasting with defective bone mineralization.11 Because removal of tumors restores these mineral metabolism derangements, it was speculated that the tumors secreted a putative endocrine factor (phosphatonin) that induced renal phosphate wasting and suppressed 1α,25(OH)2D3 synthesis. Search for phosphatonin in patients with TIO led to the identification of fibroblast growth factor-23 (FGF23). Many phosphatonin molecules, such as the FGF23, secreted frizzled-related protein-4 (sFRP-4), fibroblast growth factor-7 (FGF-7), and matrix extracellular phosphoglycoprotein (MEPE), have been described in recent studies.12-17 Identification of these phosphatonins has significantly advanced our understanding of the molecular regulation of phosphate homeostasis, and in this brief article, we will present how the FGF23–Klotho system is involved in such regulation.
Fibroblast Growth Factor-23
FGF23 is a bone-derived phosphatonin that has been most extensively studied. It was originally identified as a gene mutated in patients with autosomal dominant hypophosphatemic rickets (ADHR).12 Patients with ADHR carry missense mutations in the FGF23 gene that confer resistance to proteolysis inactivation of the FGF23 protein. As a result, patients with ADHR have high serum FGF23 levels. FGF23 has an activity that reduces the number of sodium-dependent phosphate (NaPi) cotransporters, type-2a (NaPi-2a), on the brush border membrane of proximal tubules, thereby promoting renal phosphate excretion.18 This reduction in the number of NaPi-2a by FGF23 seems to be independent of PTH.19 When the serum phosphate level is high, FGF23 is secreted from bones and acts on the kidneys to induce phosphaturia and suppress active vitamin D synthesis, thereby inducing a negative phosphate balance to maintain phosphate homeostasis.20 In an animal study, administration of FGF23 intravenously to rodents decreased serum 1α,25(OH)2D3 levels within hours.21 FGF23 downregulates expression of the Cyp27b1 gene, which encodes 1α-hydroxylase, the enzyme that synthesizes the active form of vitamin D [1α,25(OH)2D3]. Furthermore, FGF23 upregulates Cyp24 gene expression that encodes 24-hydroxylase, the enzyme that inactivates 1α,25(OH)2D3. Therefore, FGF23 functions as a counter-regulatory hormone for vitamin D.22 In contrast, 1α,25 (OH)2D3 upregulates FGF23 gene expression and forms a negative feedback loop (Fig 2). Activated 1α,25(OH)2D3 binds to nuclear vitamin D receptor and triggers the formation of a heterodimer with another nuclear receptor RXR. The vitamin D receptor-retinoid X receptor (RXR) heterodimer in turn transactivates the FGF23 gene expression.23 Similarly, elevated serum 1α,25(OH)2D3 levels in klotho knockout mice is associated with extremely high serum FGF23 levels. More importantly, eliminating vitamin D activities from klotho knockout mice resulted in almost undetectable levels of FGF23 in klotho/1α-hydroxylase double knockout mice, clearly suggesting the in vivo role of vitamin D in the induction of FGF23.24,25
Figure 2.
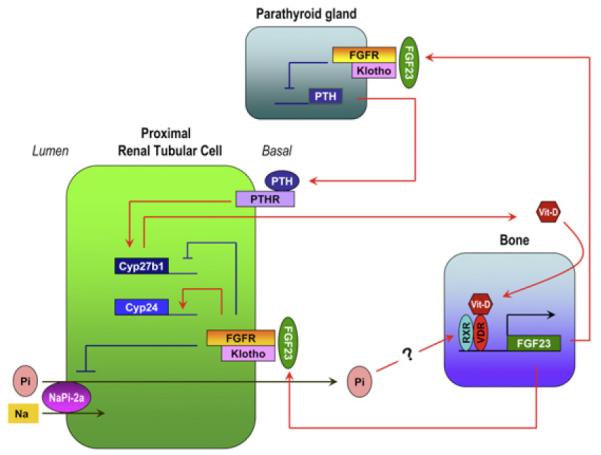
The bone–kidney–parathyroid endocrine axes mediated by fibroblast growth factor-23 (FGF23) and Klotho. High serum and/or dietary Pi increases FGF23 secretion from the bones. FGF23 acts on the Klotho–FGF receptor (FGFR) complex expressed in the kidneys and the parathyroid gland. In the kidneys, FGF23 downregulates expression of the Cyp27b1 gene and upregulates Cyp24 gene, thus suppresses synthesis of active vitamin D. Binding of FGF23 to Klotho–FGFR complex also suppresses Pi reabsorption by inhibiting NaPi-2a activity. In the parathyroid gland, FGF23 suppresses production and secretion of PTH. PTH binds to PTH receptor (PTHR) expressed on the renal tubular cells, in turn, upregulating Cyp27b1 gene expression. For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.
The biological response to FGF23 in its target tissue (kidneys) requires binding and activation of its cognate fibroblast growth factor receptors (FGFRs) that belong to type 1 transmembrane phosphotyrosine kinase receptors. Although FGF23 belongs to the FGF ligand superfamily,26 phylogenetic and sequence analyses have segregated FGF23 and 2 additional FGFs (FGF19 and FGF21) from the other FGF family members.27 These 3 atypical FGFs are collectively called endocrine FGFs.23 These FGFs have low affinity to heparan sulfate (HS). The unique structural feature of reduced affinity of endocrine FGFs to HS allows them to escape from HS-rich extracellular matrices and enter into systemic circulation. Although the low affinity of endocrine FGFs to HS may be advantageous for the endocrine mode of action, it may be disadvantageous for signal transduction through FGFRs. FGF23 cannot activate FGF signaling in most cultured cells, whereas classic FGFs, such as FGF2, can do so,28 thus implicating that endocrine FGFs may require a cofactor(s) other than HS for FGFR activation. Identity of the putative cofactor(s) required for FGF23 to activate FGFRs was not clear until it was realized that the phenotypes of Fgf23-deficient mice are identical to those of Klotho-deficient mice. In fact, the physical, morphological, biochemical, and molecular phenotypes of fgf23 or klotho single knockout mice are indistinguishable from fgf23–klotho double knockout mice.29
Klotho
The klotho gene was originally identified as a gene mutated in the klotho mouse. A defect in klotho gene expression in mice leads to a complex phenotype resembling human premature-aging syndromes, including shorten life span, growth retardation, hypogonadism, skin atrophy, muscle atrophy, premature thymic involution, osteopenia, pulmonary emphysema, and vascular and soft-tissue calcification.30 This gene encodes a 1014-aminoacid-long protein with a long extracellular domain, and a short cytoplasmic region that does not contain signaling capabilities. The extracellular domain is composed of 2 homologous regions, named KL1 and KL2. Klotho is expressed at the cell surface (membrane-bound KL, mKL) and is also present in the plasma as secreted forms (sKL). One form of sKL results from the shedding of mKL from the cell surface. This form of sKL comprises the KL1 and KL2 domains, referred to as “cut mKL.”31 The second sKL is formed by alternative splicing in exon 3 that results in a protein containing only the KL1 domain, but not the transmembrane domain. Both cut mKL and sKL found in the circulation have led to the interpretation that Klotho itself may act as a hormone. However, function of the Klotho protein was not clear until it was realized that phenotypes of Klotho-deficient mice were almost identical to those of mice lacking Fgf23. The fgf23-null mice develop phosphate–retention phenotypes characterized by hyperphosphatemia and extensive soft-tissue calcification.32 Genetic restoration of the systemic actions of human FGF23 in fgf23 knockout mice reversed the hyperphosphatemia to hypophosphatemia and prevented associated complications, including ectopic calcification.33 In addition to these phenotypes, fgf23-null mice also exhibit complex phenotypes, including growth retardation, hypogonadism, premature thymic involution, osteopenia, skin atrophy, muscle atrophy, and pulmonary emphysema, which are reminiscent of the premature aging syndrome in Klotho-deficient mice. These observations revealed an unexpected link between FGF23 and Klotho.
FGF23–Klotho System
Klotho protein forms a binary complex with several FGFR isoforms and significantly enhances their affinity for FGF23.28,34 Co-immunoprecipitation studies indicated that Klotho binds to FGFR1c, 3c, and 4 with higher affinity than to the other FGFR isoforms. Without Klotho, these FGFRs cannot bind to FGF23, indicating that FGF23 requires Klotho as a coreceptor. This explains why Klotho-deficient mice have extremely high levels of FGF23. Also, kidney-specific expression of Klotho explains why FGF23 can identify kidneys as its target organ among many other tissues that express multiple FGFR isoforms. Thus, Klotho is required for FGF23 to induce negative phosphate balance by increasing renal phosphate excretion as well as reduce intestinal phosphate absorption.
Recent mouse genetic studies have convincingly demonstrated the in vivo importance of Klotho in FGF23-mediated regulation of phosphate homeostasis.35-37 For instance, functionally active FGF23 recombinant protein is unable to reduce serum phosphate levels in mice lacking Klotho activities (either Klotho-deficient mice or fgf23/klotho double knockout mice).29,35 Similarly, phex mutant mice have hypophosphatemia because of high serum levels of FGF23, whereas inactivation of the Klotho function from the phex mutant mice resulted in hyperphosphatemia in phex/klotho double mutant mice, even though these mice have significantly higher serum levels of FGF23.29,35,38 Likewise, genetically ablating Klotho function from FGF23 transgenic mice reversed the hypophosphatemic phenotype in these mice to hyperphosphatemic.39 In a similar study in human beings, despite high serum levels of FGF23, a homozygous loss-of-function mutation in the KLOTHO gene induced severe hyperphosphatemia in the affected patient with tumoral calcinosis.40 Together, these human and experimental evidences clearly suggest that Klotho is indispensable for the FGF23-mediated regulation of systemic phosphate homeostasis, and that dysregulation of the FGF23–Klotho system, either alone or in combination, can induce phosphate imbalance.
Klotho and NaPi System
Regulation of phosphate homeostasis in the kidneys occurs primarily in the proximal convoluted tubule and is controlled mainly by PTH and the FGF23–Klotho endocrine axis. Approximately 80% of filtered phosphate is reabsorbed along with sodium through NaPi cotransporters, NaPi-2a and NaPi-2c, located in the proximal tubule. In fact, molecular and biochemical analyses suggest that increased renal activity of NaPi-2a leads to severe hyperphosphatemia in Klotho knockout mice.41,42 More importantly, the extensive physical, morphological, and molecular phenotypes in klotho knockout mice can be suppressed by genetically reducing serum phosphate levels in NaPi-2a/klotho double knockout mice to extend their survival. For instance, both male and female hyperphosphatemic klotho knockout mice have hypogonadism that is associated with premature infertility in these mutant mice, whereas genetically reducing serum phosphate levels can regain fertility in the NaPi2a/klotho double knockout mice.41,42 A similar trend is also noted in hyperphosphatemic fgf23 knockout mice, where suppressing NaPi-2a activity can reduce serum phosphate levels in NaPi2a/fgf23 double knockout mice, and reverse associated complications, including fertility.43 These genetics studies in mice clearly suggest that the NaPi system is actively involved in FGF23–Klotho-mediated regulation of phosphate homeostasis.41,42,44,45 Whether the FGF23–Klotho system directly reduces the renal NaPi cotransporter activity or whether such a response is mediated through other FGF23 target molecules is an unsolved issue; however, recent studies have provided possible molecular insights.46
The bone–kidney endocrine axis mediated by FGF23 and Klotho reduces the number of NaPi-2a on the brush border membrane of the proximal tubules, thereby promoting renal phosphate excretion. It should be noted that Klotho protein is expressed much more abundantly in distal convoluted tubules than in proximal tubules. This has raised the question as to how Klotho protein expressed in distal tubules controls the function of proximal tubules. A recent animal study reported that activation of the FGF signaling pathway was detectable only in the distal convoluted tubules after injection of FGF23 into mice.47 This study supported the possibility that FGF23 might first act on distal convoluted tubules and then generate a paracrine factor that acts on the adjacent proximal tubules to reduce phosphate reabsorption and vitamin D synthesis. A candidate of this putative paracrine factor might be sKL protein because it functions as a phosphaturic substance independent of FGF23. Injection of sKL protein induces phosphaturia in fgf23 knockout mice. This study showed that sKL mediates deglycosylation of N-linked glycans on the NaPi2a protein through its putative glycosidase activity. Deglycosylation of N-linked glycans renders NaPi-2a protein more susceptible to proteases residing in the proximal tubular brush border membrane, resulting in reduction in the number and activity of NaPi-2a.46 Therefore, Klotho can induce phosphaturia by 2 ways: by functioning as a coreceptor for FGF23 and by functioning as a phosphaturic substance independent of FGF23.
Phosphate-independent Effects of Klotho
Although FGF23-mediated functions are mostly Klotho-dependent, Klotho also has numerous FGF23-independent functions, including adipogenesis.48 Studies have shown Klotho as a regulator of oxidative stress and senescence.49 The other functional role of Klotho in the kidneys is its involvement in renal calcium handling. Klotho may regulate calcium homeostasis through direct regulation of renal ion transport in addition to modulation of PTH and 1α,25(OH)2D3 levels.50,51 sKL activates the transient receptor potential v-5 (TRPV5) by entrapping the channel on the plasma membrane and inhibiting its endocytosis, leading to enhanced calcium reabsorption in the distal nephron.52,53 A similar Klotho effect was also found on renal outer medullary potassium channel (ROMK1).54 Treatment with sKL removes terminal sialic acids from N-linked glycans of TRPV5 and ROMK1. Removal of terminal sialic acids exposes underlying galactose residues, which are good ligands for galectin-1 in extracellular matrices. Therefore, Klotho-mediated removal of sialic acids triggers interaction between galectin-1 and glycans of these ion channels, preventing them from endocytosis. As a result, sKL promotes calcium absorption and potassium excretion. The in vivo ability of sKL to activate TRPV5 and ROMK1 to renal physiology and pathophysiology remains to be determined.
Other Phosphatonins
Several other phosphatonins, including MEPE, sFRP-4, dentin matrix protein-1 (DMP-1), and FGF-7, have not been studied as extensively as FGF23 (Table 1). Similar to FGF23, sFRP-4 has been shown to decrease renal phosphate reabsorption by reducing the number of NaPi cotransporters in the renal proximal tubules and by inhibiting synthesis of 1α,25(OH)2D3.15 FGF-7 inhibits NaPi transport in opossum kidney cells and reportedly has a phosphaturic activity in vivo.58 Similarly, MEPE has been shown to increase the fractional excretion of phosphate and induce hypophosphatemia in vivo.17 In contrast to FGF23, MEPE does not inhibit 1α,25(OH)2D3 formation. As such, one phosphatonin can influence the functionality of the others. For instance, mutations in the DMP-1 gene, found in patients with autosomal recessive hypophosphatemic rickets/osteomalacia,59 resulted in increased circulating levels of FGF23, leading to hypophosphatemia. Similarly, dmp-1 knockout mice developed hypophosphatemia because of increased bioactivities of FGF23.57 One of the areas that will need additional studies includes determining how numerous phosphatonins molecularly interact with each other to regulate physiologic phosphate balance, and how these molecules facilitate cross-organ talks among bones, kidneys, parathyroid gland, and intestine. Such understanding will help us to determine the cause of abnormal regulation of phosphate homeostasis in a wide range of diseases, including in patients with CKD.
Table 1.
Phosphatonins Other than FGF23
| Phosphatonins | Associated Diseases | References |
|---|---|---|
| MEPE | Increased expression in tumors associated with TIO | 13 |
| Increased serum levels in XLH | 55 | |
| Induced phosphaturia when injected into rats | 56 | |
| sFRP-4 | Increased expression in tumors associated with TIO | 13 |
| Induced phosphaturia when injected into rats | 15 | |
| DMP-1 | Increased expression in tumors associated with TIO | 13 |
| Loss-of-function causes ARHR, increased serum FGF23 | 57 | |
| FGF-7 | Increased expression in tumors associated with TIO | 13 |
| Suppression of phosphate uptake in cultured cells | 58 |
Abbreviations: MEPE, matrix extracellular phosphoglycoprotein; sFRP-4, secreted frizzled-related protein 4; DMP-1, dentin matrix protein 1; FGF-7, fibroblast growth factor 7; TIO, tumor-induced osteomalacia; XLH, X-linked hypophosphatemia; ARHR, autosomal recessive hypophosphatemic rickets.
FGF23–Klotho System and CKD
In patients with CKD, circulating levels of FGF23 are progressively increased as kidney function declines,60 whereas Klotho expression in the kidneys is significantly decreased in such patients.61 Therefore, CKD may be viewed as a state of FGF23 resistance caused by Klotho deficiency. Increases in FGF23 precede increases in serum phosphate (hyperphosphatemia) during CKD progression, suggesting that patients with early-stage CKD maintain normal serum phosphate levels by increasing FGF23 to compensate for increased renal resistance to FGF23. However, this is at the expense of 1α,25(OH)2D3, as FGF23 induces phosphaturia and suppresses 1α,25(OH)2D3 synthesis at the same time. Low serum levels of 1α,25(OH)2D3 not only cause secondary hyperparathyroidism but also reduce Klotho expression further, because 1α,25(OH)2D3 is a potent inducer of Klotho expression.51 Therefore, low serum levels of 1α,25(OH)2D3 induced by compensatory increase in serum FGF23 levels can trigger deterioration spiral of Klotho expression. The benefit of calcitriol therapy may be partly attributed to interruption of this vicious cycle. A recent study showed that high serum FGF23 levels were associated with resistance to calcitriol treatment, and concluded that FGF23 could be a predictor for calcitriol therapy resistance.62
Dietary phosphorus load as well as serum phosphate levels can influence FGF23 levels in human beings. For instance, serum FGF23 levels increased during high dietary phosphate/calcium intake, whereas serum PTH levels declined; 1α,25(OH)2D3 levels showed an inverse relation with FGF23 levels.63,64 In a similar study, effects of providing various amount of phosphate-containing food (400, 800, and 1200 mg) on serum FGF23 showed that its levels remained either similar or slightly decreased by 6 hours after consumption of a high phosphate meal. In contrast, a significantly increased serum FGF23 level was detected in approximately 8 hours after intake of 1200 mg of phosphate, suggesting that high amount of phosphate consumption may induce FGF23 to restore phosphate balance.65
Recently, an increased serum level of FGF23 has been shown to affect mortality of incident hemodialysis patients.66 Furthermore, an association between increased serum levels of FGF23 and left ventricular hypertrophy has found in patients with CKD undergoing hemodialysis treatment.67 If the serum FGF23 level is a surrogate marker for the renal Klotho expression level, high FGF23 may suggest low Klotho expression in kidneys and severe loss of functional nephron mass.66 When functional renal mass is decreased to a level that fails to maintain phosphate excretion in response to increased FGF23 and PTH, hyperphosphatemia ensues.
The molecular interaction of FGF23–Klotho system and PTH is a complex process, and whether FGF23–Klotho system is involved in the genesis of secondary hyperparathyroidism in patients with CKD is still not clear. Studies have found decreased expression of Klotho and FGFR1 in the hyperplastic parathyroid glands of uremic patients. This observation implicates that intrinsic changes in the parathyroid gland may provoke FGF23 resistance or unresponsiveness toward the gland.68 Contrary to human observation, the experimental studies found increased parathyroid expression of Klotho and FGFR1 in uremic animals.69 Furthermore, exogenous FGF23 failed to inhibit development of hyperparathyroidism in experimental uremic animals.70 It remains to be determined whether decline in renal Klotho expression precedes increase in serum FGF23 during CKD progression. Of relevance, experimental studies have shown that eliminating Klotho function from mice is associated with extremely high serum levels of FGF23.14,35
Conclusions
Our knowledge of the mechanisms that regulate phosphate homeostasis has greatly improved during the past few years.19,20,37 Seminal discoveries on the FGF23–Klotho endocrine system have been a major driving force for this progress. It has become increasingly clear that hyperphosphatemia should be aggressively treated to improve life expectancy of patients with CKD. Within this context, the manipulation of the FGF23–Klotho axis is expected to be a novel target of therapeutic intervention in diseases associated with abnormal mineral ion metabolism. However, it is worth mentioning that, at present, there is no direct evidence that can establish a positive link between accelerated progression of CKD and FGF23 toxicity.71 Any strategy of therapeutic lowering of FGF23 in patients with CKD needs additional studies. A controlled therapeutic suppression of FGF23 might be of therapeutic benefit for patients with excessive urinary phosphate wasting diseases, including ADHR, autosomal recessive hypophosphatemic rickets/osteomalacia, and X-linked hypophosphatemic rickets. As such, the current treatment for hypophosphatemic genetic diseases is mostly symptomatic, including oral phosphate replacement. The prolonged use of such replacement therapy can lead to the development of secondary hyperparathyroidism. Again, whether patients with CKD might also benefit from the therapeutic lowering of FGF23 is an area that needs additional studies. In contrast to anti-FGF23 therapy, providing exogenous bioactive FGF23 or Klotho protein might help restore the phosphate balance and reduce abnormal calcifications in patients with familial tumoral calcinosis (FTC), which is usually caused by the reduced activity of FGF23. Our challenge will be to use experimental observations to fine-tune the existing therapeutic options by manipulating the FGF23–Klotho system to treat patients suffering from the complications of abnormal mineral ion metabolism, including CKD.14,36,72
Acknowledgments
Some of the original research that formed the basis of this review article is supported by grants R01-AG019712 (to M.K.) and R01-DK077276 (to M.S.R.) from the National Institute of Health.
References
- 1.Iotti S, Lodi R, Gottardi G, Zaniol P, Barbiroli B. Inorganic phosphate is transported into mitochondria in the absence of ATP biosynthesis: an in vivo 31P NMR study in the human skeletal muscle. Biochem Biophys Res Commun. 1996;225:191–194. doi: 10.1006/bbrc.1996.1152. [DOI] [PubMed] [Google Scholar]
- 2.Hutson SM, Williams GD, Berkich DA, LaNoue KF, Briggs RW. A 31P NMR study of mitochondrial inorganic phosphate visibility: effects of Ca2+, Mn2+, and the pH gradient. Biochemistry. 1992;31:1322–1330. doi: 10.1021/bi00120a007. [DOI] [PubMed] [Google Scholar]
- 3.Gaasbeek A, Meinders AE. Hypophosphatemia: an update on its etiology and treatment. Am J Med. 2005;118:1094–1101. doi: 10.1016/j.amjmed.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 4.Schaefer B, Schlosser K, Wuhl E, et al. Long-term control of parathyroid hormone and calcium-phosphate metabolism after parathyroidectomy in children with chronic kidney disease. Nephrol Dial Transplant. 2010;25:2590–2595. doi: 10.1093/ndt/gfq074. [DOI] [PubMed] [Google Scholar]
- 5.Carter JL, O’Riordan SE, Eaglestone GL, Delaney MP, Lamb EJ. Bone mineral metabolism and its relationship to kidney disease in a residential care home population: a cross-sectional study. Nephrol Dial Transplant. 2008;23:3554–3565. doi: 10.1093/ndt/gfn302. [DOI] [PubMed] [Google Scholar]
- 6.Razzaque MS. Osteocalcin: a pivotal mediator or an innocent bystander in energy metabolism? Nephrol Dial Transplant. doi: 10.1093/ndt/gfq721. In press. doi:10.1093/ndt/gfq1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fukagawa M, Kazama JJ, Kurokawa K. Renal osteodystrophy and secondary hyperparathyroidism. Nephrol Dial Transplant. 2002;17(suppl 10):2–5. doi: 10.1093/ndt/17.suppl_10.2. [DOI] [PubMed] [Google Scholar]
- 8.Fulzele K, Riddle RC, Digirolamo DJ, et al. Insulin receptor signaling in osteoblasts regulates postnatal bone acquisition and body composition. Cell. 2010;142:309–319. doi: 10.1016/j.cell.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berndt TJ, Schiavi S, Kumar R. “Phosphatonins” and the regulation of phosphorus homeostasis. Am J Physiol Renal Physiol. 2005;289:F1170–F1182. doi: 10.1152/ajprenal.00072.2005. [DOI] [PubMed] [Google Scholar]
- 10.Cai Q, Hodgson SF, Kao PC, et al. Brief report: inhibition of renal phosphate transport by a tumor product in a patient with oncogenic osteomalacia. N Engl J Med. 1994;330:1645–1649. doi: 10.1056/NEJM199406093302304. [DOI] [PubMed] [Google Scholar]
- 11.Kumar R. Tumor-induced osteomalacia and the regulation of phosphate homeostasis. Bone. 2000;27:333–338. doi: 10.1016/s8756-3282(00)00334-3. [DOI] [PubMed] [Google Scholar]
- 12.White KE, Evans WE, O’Rlordan JLH, et al. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet. 2000;26:345–348. doi: 10.1038/81664. [DOI] [PubMed] [Google Scholar]
- 13.De Beur SM, Finnegan RB, Vassiliadis J, et al. Tumors associated with oncogenic osteomalacia express genes important in bone and mineral metabolism. J Bone Miner Res. 2002;17:1102–1110. doi: 10.1359/jbmr.2002.17.6.1102. [DOI] [PubMed] [Google Scholar]
- 14.Razzaque MS. The FGF23-Klotho axis: endocrine regulation of phosphate homeostasis. Nat Rev Endocrinol. 2009;5:611–619. doi: 10.1038/nrendo.2009.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berndt T, Craig TA, Bowe AE, et al. Secreted frizzled-related protein 4 is a potent tumor-derived phosphaturic agent. J Clin Invest. 2003;112:785–794. doi: 10.1172/JCI18563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Razzaque MS, Lanske B. The emerging role of the fibroblast growth factor-23-klotho axis in renal regulation of phosphate homeostasis. J Endocrinol. 2007;194:1–10. doi: 10.1677/JOE-07-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rowe PS, Kumagai Y, Gutierrez G, et al. MEPE has the properties of an osteoblastic phosphatonin and minhibin. Bone. 2004;34:303–319. doi: 10.1016/j.bone.2003.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shimada T, Urakawa I, Yamazaki Y, et al. FGF-23 transgenic mice demonstrate hypophosphatemic rickets with reduced expression of sodium phosphate cotransporter type IIa. Biochem Biophys Res Commun. 2004;314:409–414. doi: 10.1016/j.bbrc.2003.12.102. [DOI] [PubMed] [Google Scholar]
- 19.Kuro-o M. Klotho as a regulator of fibroblast growth factor signaling and phosphate/calcium metabolism. Curr Opin Nephrol Hypertens. 2006;15:437–441. doi: 10.1097/01.mnh.0000232885.81142.83. [DOI] [PubMed] [Google Scholar]
- 20.Quarles LD. Endocrine functions of bone in mineral metabolism regulation. J Clin Invest. 2008;118:3820–3828. doi: 10.1172/JCI36479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shimada T, Hasegawa H, Yamazaki Y, et al. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004;19:429–435. doi: 10.1359/JBMR.0301264. [DOI] [PubMed] [Google Scholar]
- 22.Liu S, Tang W, Zhou J, et al. Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin D. J Am Soc Nephrol. 2006;17:1305–1315. doi: 10.1681/ASN.2005111185. [DOI] [PubMed] [Google Scholar]
- 23.Kuro-o M. Endocrine FGFs and Klothos: emerging concepts. Trends Endocrinol Metab. 2008;19:239–245. doi: 10.1016/j.tem.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 24.Ohnishi M, Nakatani T, Lanske B, Razzaque MS. Reversal of mineral ion homeostasis and soft-tissue calcification of klotho knockout mice by deletion of vitamin D 1alpha-hydroxylase. Kidney Int. 2009;75:1166–1172. doi: 10.1038/ki.2009.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Razzaque MS. The dualistic role of vitamin D in vascular calcifications. Kidney Int. doi: 10.1038/ki.2010.432. In press. doi:10.1038/ki.2010.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamashita T, Yoshioka M, Itoh N. Identification of a novel fibroblast growth factor, FGF-23, preferentially expressed in the ventrolateral thalamic nucleus of the brain. Biochem Biophys Res Commun. 2000;277:494–498. doi: 10.1006/bbrc.2000.3696. [DOI] [PubMed] [Google Scholar]
- 27.Itoh N, Ornitz DM. Functional evolutionary history of the mouse Fgf gene family. Dev Dyn. 2008;237:18–27. doi: 10.1002/dvdy.21388. [DOI] [PubMed] [Google Scholar]
- 28.Kurosu H, Ogawa Y, Miyoshi M, et al. Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem. 2006;281:6120–6123. doi: 10.1074/jbc.C500457200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakatani T, Sarraj B, Ohnishi M, et al. In vivo genetic evidence for klotho-dependent, fibroblast growth factor 23 (Fgf23) -mediated regulation of systemic phosphate homeostasis. FASEB J. 2009;23:433–441. doi: 10.1096/fj.08-114397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuro-o M, Matsumura Y, Aizawa H, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- 31.Imura A, Iwano A, Tohyama O, et al. Secreted Klotho protein in sera and CSF: implication for post-translational cleavage in release of Klotho protein from cell membrane. FEBS Lett. 565:143–147. doi: 10.1016/j.febslet.2004.03.090. [DOI] [PubMed] [Google Scholar]
- 32.Shimada T, Kakitani M, Yamazaki Y, et al. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest. 2004;113:561–568. doi: 10.1172/JCI19081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DeLuca S, Sitara D, Kang K, et al. Amelioration of the premature aging-like features of Fgf-23 knockout mice by genetically restoring the systemic actions of FGF-23. J Pathol. 2008;216:345–355. doi: 10.1002/path.2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Urakawa I, Yamazaki Y, Shimada T, et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006;444:770–774. doi: 10.1038/nature05315. [DOI] [PubMed] [Google Scholar]
- 35.Nakatani T, Ohnishi M, Razzaque MS. Inactivation of klotho function induces hyperphosphatemia even in presence of high serum fibroblast growth factor 23 levels in a genetically engineered hypophosphatemic (Hyp) mouse model. FASEB J. 2009;23:3702–3711. doi: 10.1096/fj.08-123992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Razzaque MS. FGF23-mediated regulation of systemic phosphate homeostasis: is Klotho an essential player? Am J Physiol Renal Physiol. 2009;296:F470–F476. doi: 10.1152/ajprenal.90538.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Razzaque MS. Phosphate toxicity: new insights into an old problem. Clin Sci (Lond) 2011;120:91–97. doi: 10.1042/CS20100377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Razzaque MS. Therapeutic potential of klotho-FGF23 fusion polypeptides: WO2009095372. Expert Opin Ther Pat. 2010;20:981–985. doi: 10.1517/13543771003774100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bai X, Dinghong Q, Miao D, Goltzman D, Karaplis AC. Klotho ablation converts the biochemical and skeletal alterations in FGF23 (R176Q) transgenic mice to a Klotho-deficient phenotype. Am J Physiol Endocrinol Metab. 2009;296:E79–E88. doi: 10.1152/ajpendo.90539.2008. [DOI] [PubMed] [Google Scholar]
- 40.Ichikawa S, Imel EA, Kreiter ML, et al. A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J Clin Invest. 2007;117:2684–2691. doi: 10.1172/JCI31330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ohnishi M, Razzaque MS. Dietary and genetic evidence for phosphate toxicity accelerating mammalian aging. FASEB J. 2010;24:3562–3571. doi: 10.1096/fj.09-152488. 10.1096/fj.1009-152488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ohnishi M, Nakatani T, Lanske B, Razzaque MS. In vivo genetic evidence for suppressing vascular and soft-tissue calcification through the reduction of serum phosphate levels, even in the presence of high serum calcium and 1,25-Dihydroxyvitamin D levels. Circ Cardiovasc Genet. 2009;2:583–590. doi: 10.1161/CIRCGENETICS.108.847814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sitara D, Kim S, Razzaque MS, et al. Genetic evidence of serum phosphate-independent functions of FGF-23 on bone. PLoS Genet. 2008;4:e1000154. doi: 10.1371/journal.pgen.1000154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Razzaque MS, Sitara D, Taguchi T, St-Arnaud R, Lanske B. Premature ageing-like phenotype in fibroblast growth factor 23 null mice is a vitamin-D mediated process. FASEB J. 2006;20:720–722. doi: 10.1096/fj.05-5432fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Razzaque MS, Lanske B. Hypervitaminosis D and premature aging: lessons learned from Fgf23 and Klotho mutant mice. Trends Mol Med. 2006;12:298–305. doi: 10.1016/j.molmed.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 46.Hu MC, Shi M, Zhang J, et al. Klotho: a novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. FASEB J. 2010;24:3438–3450. doi: 10.1096/fj.10-154765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Farrow EG, Davis SI, Summers LJ, White KE. Initial FGF23-mediated signaling occurs in the distal convoluted tubule. J Am Soc Nephrol. 2009;20:955–960. doi: 10.1681/ASN.2008070783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chihara Y, Rakugi H, Ishikawa K, et al. Klotho protein promotes adipocyte differentiation. Endocrinology. 2006;147:3835–3842. doi: 10.1210/en.2005-1529. [DOI] [PubMed] [Google Scholar]
- 49.Kuro-o M. Klotho as a regulator of oxidative stress and senescence. Biol Chem. 2008;389:233–241. doi: 10.1515/BC.2008.028. [DOI] [PubMed] [Google Scholar]
- 50.Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V, et al. The parathyroid is a target organ for FGF23 in rats. J Clin Invest. 2007;117:4003–4008. doi: 10.1172/JCI32409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsujikawa H, Kurotaki Y, Fujimori T, Fukuda K, Nabeshima Y. Klotho, a gene related to a syndrome resembling human premature aging, functions in a negative regulatory circuit of vitamin D endocrine system. Mol Endocrinol. 2003;17:2393–2403. doi: 10.1210/me.2003-0048. [DOI] [PubMed] [Google Scholar]
- 52.Chang Q, Hoefs S, van der Kemp AW, Topala CN, Bindels RJ, Hoenderop JG. The beta-glucuronidase klotho hydrolyzes and activates the TRPV5 channel. Science. 2005;310:490–493. doi: 10.1126/science.1114245. [DOI] [PubMed] [Google Scholar]
- 53.Cha SK, Ortega B, Kurosu H, Rosenblatt KP, Kuro-o M, Huang CL. Removal of sialic acid involving Klotho causes cell-surface retention of TRPV5 channel via binding to galectin-1. Proc Natl Acad Sci U S A. 2008;105:9805–9810. doi: 10.1073/pnas.0803223105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cha SK, Hu MC, Kurosu H, Kuro-o M, Moe O, Huang CL. Regulation of ROMK1 channel and renal K+ excretion by Klotho. Mol Pharmacol. 2009;76:38–46. doi: 10.1124/mol.109.055780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bresler D, Bruder J, Mohnike K, Fraser WD, Rowe PS. Serum MEPE-ASARM-peptides are elevated in X-linked rickets (HYP): implications for phosphaturia and rickets. J Endocrinol. 2004;183:R1–R9. doi: 10.1677/joe.1.05989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dobbie H, Unwin RJ, Faria NJ, Shirley DG. Matrix extracellular phosphoglycoprotein causes phosphaturia in rats by inhibiting tubular phosphate reabsorption. Nephrol Dial Transplant. 2008;23:730–733. doi: 10.1093/ndt/gfm535. [DOI] [PubMed] [Google Scholar]
- 57.Feng JQ, Ward LM, Liu S, et al. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet. 2006;38:1310–1315. doi: 10.1038/ng1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Carpenter TO, Ellis BK, Insogna KL, Philbrick WM, Sterpka J, Shimkets R. Fibroblast growth factor 7: an inhibitor of phosphate transport derived from oncogenic osteomalacia-causing tumors. J Clin Endocrinol Metab. 2005;90:1012–1020. doi: 10.1210/jc.2004-0357. [DOI] [PubMed] [Google Scholar]
- 59.Lorenz-Depiereux B, Bastepe M, Benet-Pages A, et al. DMP1 mutations in autosomal recessive hypophosphatemia implicate a bone matrix protein in the regulation of phosphate homeostasis. Nat Genet. 2006;38:1248–1250. doi: 10.1038/ng1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Larsson T, Nisbeth U, Ljunggren O, Juppner H, Jonsson KB. Circulating concentration of FGF-23 increases as renal function declines in patients with chronic kidney disease, but does not change in response to variation in phosphate intake in healthy volunteers. Kidney Int. 2003;64:2272–2279. doi: 10.1046/j.1523-1755.2003.00328.x. [DOI] [PubMed] [Google Scholar]
- 61.Koh N, Fujimori T, Nishiguchi S, et al. Severely reduced production of klotho in human chronic renal failure kidney. Biochem Biophys Res Commun. 2001;280:1015–1020. doi: 10.1006/bbrc.2000.4226. [DOI] [PubMed] [Google Scholar]
- 62.Kazama JJ, Sato F, Omori K, et al. Pretreatment serum FGF-23 levels predict the efficacy of calcitriol therapy in dialysis patients. Kidney Int. 2005;67:1120–1125. doi: 10.1111/j.1523-1755.2005.00178.x. [DOI] [PubMed] [Google Scholar]
- 63.Vervloet MG, van Ittersum FJ, Buttler RM, Heijboer A, Blankenstein MA, Ter Wee PM. Effects of dietary phosphate and calcium intake on fibroblast growth factor-23. Clin J Am Soc Nephrol. doi: 10.2215/CJN.04730510. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Antoniucci DM, Yamashita T, Portale AA. Dietary phosphorus regulates serum fibroblast growth factor-23 concentrations in healthy men. J Clin Endocrinol Metab. 2006;91:3144–3149. doi: 10.1210/jc.2006-0021. [DOI] [PubMed] [Google Scholar]
- 65.Nishida Y, Taketani Y, Yamanaka-Okumura H, et al. Acute effect of oral phosphate loading on serum fibroblast growth factor 23 levels in healthy men. Kidney Int. 2006;70:2141–2147. doi: 10.1038/sj.ki.5002000. [DOI] [PubMed] [Google Scholar]
- 66.Gutierrez OM, Mannstadt M, Isakova T, et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med. 2008;359:584–592. doi: 10.1056/NEJMoa0706130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hsu HJ, Wu MS. Fibroblast growth factor 23: a possible cause of left ventricular hypertrophy in hemodialysis patients. Am J Med Sci. 2009;337:116–122. doi: 10.1097/MAJ.0b013e3181815498. [DOI] [PubMed] [Google Scholar]
- 68.Komaba H, Goto S, Fujii H, et al. Depressed expression of Klotho and FGF receptor 1 in hyperplastic parathyroid glands from uremic patients. Kidney Int. 2010;77:232–238. doi: 10.1038/ki.2009.414. [DOI] [PubMed] [Google Scholar]
- 69.Hofman-Bang J, Martuseviciene G, Santini MA, Olgaard K, Lewin E. Increased parathyroid expression of klotho in uremic rats. Kidney Int. 2010;78:1119–1127. doi: 10.1038/ki.2010.215. [DOI] [PubMed] [Google Scholar]
- 70.Canalejo R, Canalejo A, Martinez-Moreno JM, et al. FGF23 fails to inhibit uremic parathyroid glands. J Am Soc Nephrol. 2010;21:1125–1135. doi: 10.1681/ASN.2009040427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Razzaque MS. Does FGF23 toxicity influence the outcome of chronic kidney disease? Nephrol Dial Transplant. 2009;24:4–7. doi: 10.1093/ndt/gfn620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Razzaque MS. Can fibroblast growth factor 23 fine-tune therapies for diseases of abnormal mineral ion metabolism? Nat Clin Pract Endocrinol Metab. 2007;3:788–789. doi: 10.1038/ncpendmet0667. [DOI] [PubMed] [Google Scholar]