

Abstract
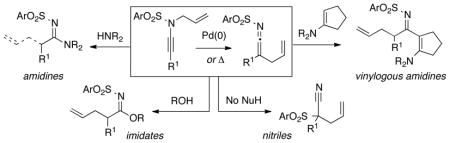
A detailed study of amidine synthesis from N-allyl-N-sulfonyl ynamides is described here. Mechanistically, this is a fascinating reaction consisting of diverging pathways that could lead to deallylation or allyl transfer depending upon the oxidation state of palladium catalysts, the nucleophilicity of amines, and the nature of the ligands. It essentially constitutes a Pd(0)-catalyzed aza-Claisen rearrangement of N-allyl ynamides, which can also be accomplished thermally. An observation of N-to-C 1,3-sulfonyl shift was made when examining these aza-Claisen rearrangements thermally. This represents a useful approach to nitrile synthesis. While attempts to render this 1,3-sulfonyl shift stereoselective failed, we uncovered another set of tandem sigmatropic rearrangements, leading to vinyl imidate formation. Collectively, this work showcases the rich array of chemistry one can discover using these ynamides.
Introduction
Amidines1,2 represent a prolific functional group in medicinal chemistry and an important pharmacophore in drug discovery.3–6 One notable example is the DNA and RNA binding di-amidine Diminazene, which is found in drugs like Azidin, Berenil, or Pirocide to treat the parasitic protozoan that causes Trypanosoniasis for which African sleeping sickness and Chagas disease are a form.7 There are several different variations of amidines depending on the substituents on the nitrogen or the sp2-amidinyl carbon1,2 [Figure 1]. Aliphatic and aromatic amidines are generally prepared in a very similar manner, frequently from amides, nitriles, and thioamides with a nitrogen nucleophile.1,2,8 The Pinner reaction and modified Pinner transformations,9,10 which employ nitriles and go through an imidate, are the most common protocols for synthesizing amidines. However, this method is not good for hindered nitriles and it cannot be used for the synthesis of tertiary amidines. Consequently, amides and thioamides are frequently used.1,2 Activation of the mono-substituted amide by chlorinating agents or alkylation of the alkoxy group can yield amidines when the resulting compound is reacted with an amine. Thioamides can more directly yield an amidine by simply reacting it with an amine and a mercury salt to act as a sulfide scavenger [Figure 1]. Whitby11 recently reported an interesting usage of isonitrile for accessing amidines by reacting with aryl or allyl bromides and an amine using a palladium catalyst. Lastly, there has been extensive development in constructing amidines through decomposing N-sulfonyl triazoles [not shown here].12–15
Figure 1.

Amidines and Their General Preparation.
We recently found that ynamides16–18 could serve as excellent precursors for synthesizing amidines via a Pd(0)-catalyzed N-to-C allyl transfer.19 As shown in Scheme 1, the oxidative addition [O.A.] of N-allyl-ynamides 1 would lead to novel ynamido-π-allyl complexes 2a and ketenimino-π-allyl complexes 2b. The subsequent formation of amidines 4 would occur in the presence of an amine either through trapping of ketenimines 3, which is derived from 2b via reductive elimination [R.E.]; or through π-allyl complex 5, which is a result of the amine addition to complexes 2b prior to R. E. We also found that while amidines 4 represent a successful N-to-C allyl transfer, the reaction did not always lead to transfer of the allyl group. Instead, a diverging pathway would lead to amidines 6 that could be prepared with concomitant de-allylation depending upon the amine and the nature of the palladium catalyst and ligands. Although it is quite reasonable to presume that this de-allylation could have occurred through amine attacking the palladium π-allyl complexes 2b, it could also be envisioned from keteniminium intermediate 7 generated from 1 with the Pd(II) species serving as a π-Lewis acid. We recognized that not only does this signify a de novo transformation of ynamides with an invaluable opportunity to study ynamido-metal complexes,12–16,20,21 but also an excellent method for synthesizing amidines especially given that ynamides are now synthetic readily available substrates.22–24 We report here details of our investigation in developing this amidine synthesis.
Scheme 1.

Amidine Formation from N-Allyl-Ynamides.
Results and Discussion
1. Deallylative Amidine Formation
We made our initial discovery of the deallylation of ynamides when treating N-allyl-ynamide 8a with 5 mol% of PdCl2(PPh3)2 with an original intent to hydroaminate the alkyne [Scheme 2]. Instead, we found that amidine 9 was formed in high yield when using 10.0 equiv of H2N-t-Bu, and that the allyl group was missing. Intriguingly, when lowering the amount of the amine to 3.0 equiv, the allyl resonances visibly reappeared in the proton NMR and no longer on the nitrogen atom but on the formerly the β-carbon of the ynamide after isolating 10 in 37% yield. We recognized this unexpected transformation represents an excellent protocol for synthesizing amidines, and to avoid complication with the allyl group issue, we initially focused on deallylation via using 5.0 equiv of the amine. The generality and the effectiveness of this amidine synthesis could be thoroughly captured in Table 1 through using a wide range of primary amines, and demonstrated in Table 2 via employing a diverse array of secondary amines as well as varying the N-allyl-ynamide substitutions. It is noteworthy that based on NOE experiments, these amidines adopt an E-geometry with respect to the C=N bond.19
Scheme 2.

Amidine Formation with Pd(II) Catalyst.
Table 1.
Deallylative Primary Amidine Formation.
| entry | amines [5.0 equiv]a | amidine products | yield [%]b |
|---|---|---|---|
| 1 |
|
 11 1112 |
87 |
| 2 | 92 | ||
| 3 |
|
14 15 |
50 |
| 4 | 71 | ||
| 5 | 22 | ||
| 6 |
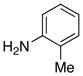
|
 16 16
|
30 |
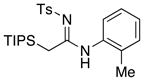
| 7 |
|
 17 1718 |
≥95 |
| 8 | ≥95 |
All entries utilized ynamide 8a except in entry 8, Ph-substituted ynamide 8b was used. All reactions employed 5.0 mol % PdCl2(PPh3)2, 1.0 equiv K2CO3, THF [conc = 0.05 M], 80 °C, 5–8 h.
Isolated yields.
Table 2.
Secondary Amidine Formation.
| entry | ynamides | aminesa | amidine products | yield [%]b |
|---|---|---|---|---|
| 1 |
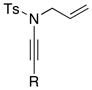 8a 8a8c 8d 8e |
|
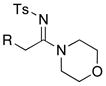 19: R = TIPS 19: R = TIPS20: R = t-Bu 21: R = (CH2)3OTBS 22: R = 2-MeO-Ph |
≥95 |
| 2 | ≥95 | |||
| 3 | 92 | |||
| 4 | 37 | |||
| 5 |
 8f 8f
|
|
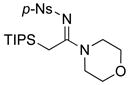 23 23
|
39 |
| 6 |
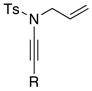 8a 8a8b 8g |
|
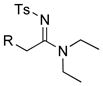 24: R = TIPS 24: R = TIPS25: R = Ph 26: R = n-hex |
≥95 |
| 7 | ≥95 | |||
| 8 | ≥95 | |||
| 9 |
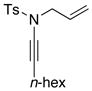 8g 8g
|
|
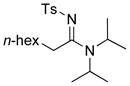 27 27
|
70 |
| 10 |
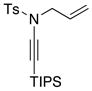 8a 8a
|
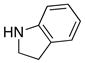
|
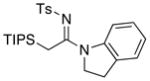 28 28
|
≥95 |
| 11 |
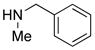
|
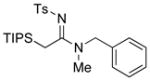 29 29
|
77 | |
| 12 |
|
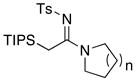 30a: n = 1 30a: n = 130b: n = 2 30c: n = 3 |
≥95 | |
| 13 | ≥95 | |||
| 14 | ≥95 | |||
| 15 |
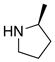
|
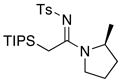 31 31
|
91 | |
| 16 |
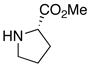
|
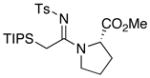 32 32
|
≥95 | |
| 17 |
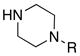
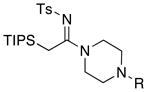
|
 33a: R = CH3 33a: R = CH333b: R = Ph |
≥95 | |
| 18 | ≥95 | |||
| 19 |
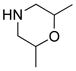
|
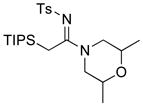 34 34
|
≥95 | |
| 20 |
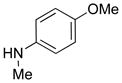
|
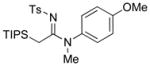 35 35
|
82 |
All reactions utilized 5.0 equiv of the amine, 5.0 mol % PdCl2(PPh3)2, and 1.0 equiv K2CO3; and were run in THF [conc = 0.05 M] at 80 °C over 5–8 h.
Isolated yields.
2. Deallylation Mechanism and Allyl Transfer
While this is an interesting amidine synthesis, we were fascinated by its possible mechanism. Because of their basicity, polarity, and/or volatility, the respective allyl amine byproducts [R2N-CH2CH=CH2] from deallylation were difficult to isolate. Nevertheless, as shown in Scheme 3, a mechanistically revealing experiment involved the use of piperizine, which led to the N-allylated amine 36 in 63% yield. Consequently, deallylation could be either a Pd(0)- or Pd(II)-catalyzed process. The former would require oxidative addition of Pd(0) to N-allyl ynamide 8a, leading to 5 through an N-to-C allyl transfer from ynamido-palladium π–allyl complex 2a; while the latter would involve a palladium(II)-substituted keteniminium ion 7 with Pd(II) serving to activate the alkynyl motif. Deallylation could take place either in an intramolecular manner as shown in 5; or an intermolecular manner as shown in 7. Although attempts were made, these reactions were too fast to allow NMR studies to be revealing of possible ynamido-π-allyl complexes even at 25 °C. Only the starting ynamide, silyl-ketenimine such as 3 and respective amidine product [if the reaction was run in the presence of an amine] were clearly in display spectroscopically.
Scheme 3.

Two Possible Deallylation Pathways.
While both pathways could be operative for the deallylation, we suspected that suppression of the Pd(II)-catalyzed pathway through directly using a Pd(0) source could lead to suppression of the deallylation.25 As shown in Table 3, this predictive assessment turned out to be true. While most Pd(II) sources led to predominantly the deallylation when using 3.0 equiv of c-hex-NH2, Pd(PPh3)4 gave exclusively the allyl transferred amidine 37 [entry 6]. This preservation of the allyl group turned to be general for a number of primary amines, as evident from amidine products 38a–c.
Table 3.
Dependence of Deallylation on Palladium Species.
|
| ||||||
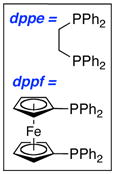
|
|
|||||
| entry | Pd(II) or Pd(0) cat | time [h] | yield [%]a | 12 | 37 | |
|
| ||||||
| 1 | PdCl2(PPh3)2 | 2 | 63 | 24 | ||
| 2 | PdCl2(dppe) | 48 | 30 | <5 | ||
| 3 | PdCl2(dppf) | 24 | 95 | 0 | ||
| 4 | PdCl2 | 20 | 66 | 16 | ||
| 5 | Pd(OAc)2 | 20 | 65 | 8 | ||
| 6 | Pd(PPh3)4 | 3 | 0 | ≥95 | ||
|
| ||||||
All are isolated yields.
However, the use of Pd(PPh3)4 was not sufficient especially in the case of secondary amines such as pyrrolidine or piperidine, which are more nucleophilic than primary amines, leading to exclusively deallylation.26 The nature of the ligand also mattered. Those that favored reductive elimination such as xantphos27,28 allowed the isolation of a range of different allyl transferred amidines such as 39 and 40a–e [Scheme 4]. X-phos29 was also examined and was comparable in terms of yield of allylated amidines, but xantphos allowed shorted reaction times.19
Scheme 4.

The Use of Xantphos Ligand.
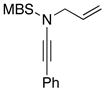
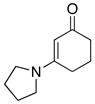
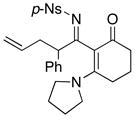
Furthermore, we found that enamines or vinylogous amides could also be used as nucleophiles under these conditions for allyl transfers. As shown in Table 4, vinylogous amidines 45 and 46 [entries 1 and 2] as well as 47 and 51 [entries 3 and 7] could be accessed utilizing enamine 41 and vinylogous amide 42, respectively. For reasons unknown to us at this moment, the allyl group did not transfer for vinylogous amidines 48 and 49 [entries 4 and 5], and primary vinylogous amides such as 43 did not work well [entry 6]. Nevertheless, a general trend for the dichotomy of deallylation versus N-to-C allyl transfer could be summarized as shown in Figure 2. Deallylation is favored when using excess of amine, or Pd(II) sources, and/or more nucleophilic secondary amines whereas N-to-C allyl transfer is favored with less nucleophilic primary amines, Pd(0) sources, and/or R.E. favoring ligands such as xantphos.
Table 4.
Vinylogous Amidine Synthesis.
| entry | ynamides | enaminesa | vinylogous amidines | yield [%]b |
|---|---|---|---|---|
| 1 |
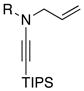 8a: R = Ts 8a: R = Ts8h: R = MBS |
|
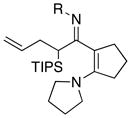 45 4546 |
52c |
| 2 | 71d | |||
| 3 |
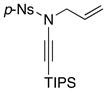 8f 8f
|
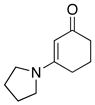 42 42
|
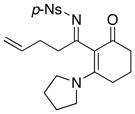 47 47
|
58e |
| 4 |
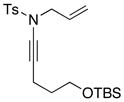 8d 8d
|
|
 48 48
|
54f |
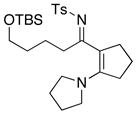
| 5 |
 8i 8i
|
|
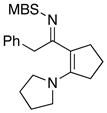 49 49
|
57g |
| 6 |
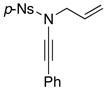 8j 8j
|
 43 43
|
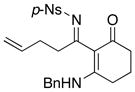 50 50
|
-- |
| 7 |
 42 42
|
 51 51
|
62h |
Unless otherwise noted, all reactions utilized 3.0 equiv of the enamine, 5.0 mol% Pd2(dba)3, and 10.0 mol% of xantphos; and were run in THF [conc = 0.05 M].
Isolated yields.
2 h at 70°C.
2 h at 50 °C with 1.5 equiv of K2CO3.
12 h at 70°C with 1.5 equiv of 42; 2.0 mol % Pd2(dba)3; and 4.0 mol% of xantphos.
30 min at 50 °C.
2 h at RT.
2 h at 75 °C with 1.5 equiv of K2CO3 and 1.5 equiv of 44.
Figure 2.

A Dichotomy of Deallylation and Allyl Transfer.
3. Aza-Claisen Rearrangement and 1,3-Sulfonyl Shift
Our efforts described above allowed us to recognize that these reactions in essence are aza-Claisen rearrangements30–32 promoted by palladium catalysts. In fact, aza-Claisen rearrangements could be carried out thermally without any metal catalysts. As shown in Scheme 5, a non-palladium involved pathway would entail an aza-Claisen transition state [see A], leading to allyl-ketenimine intermediate B. Trapping of B would then lead to many products, and in the case of an alcohol, imidates could be accessed.33,34
Scheme 5.

Thermal Aza-Claisen Rearrangment.
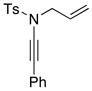
Trapping of the allyl-ketenimine intermediate such as 55 with an external amine could also lead to allyl-transferred amidine 39. This indeed is true not only in the case of 39 but is also highly effective as shown in Table 5 in giving a wide range of amidines 56–60 in good yields.35 However, this pathway requires much higher temperature and longer reaction time. When carried out at 65 °C to 80 °C in THF, the reaction was sluggish and slow,36 thereby suggesting that the palladium catalyst was indeed promoting these transformations.
Table 5.
Amidine Synthesis via Aza-Claisen Rearrangement.
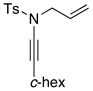
| entry | ynamides | aminesa | amidine products | yield [%]b |
|---|---|---|---|---|
| 1 |
 8b 8b
|
|
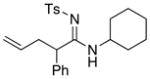 56 56
|
94 |
| 2 |
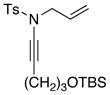 8d 8d
|
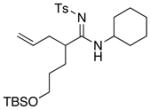 57 57
|
58 | |
| 3 |
 8k 8k
|
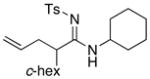 58 58
|
77 | |
| 4 |
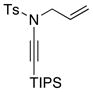 8a 8a
|
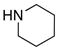
|
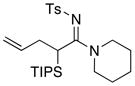 59 59
|
≥95 |
| 5 |
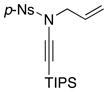 8f 8f
|
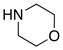
|
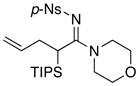 60 60
|
93 |
All reactions utilized 3.0 equiv of the amine and were run in toluene [concn = 0.05 M] at 110 °C over 24 h except it was 48 h for entry 4.
Isolated yields.
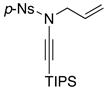
It was during this study that we observed an interesting N-to-C 1,3-sulfonyl shift.37 As shown in Scheme 6, when using ynamides not substituted with TIPS at the terminal position, we observed the formation of quaternary nitriles 61 in the absence of an amine. This observation suggested that allyl ketenimines 3 in which R ≠ TIPS had undergone a N-to-C 1,3-sulfonyl shift; whereas this was not the case when R = TIPS. Silyl ketenimines such as 55 were sufficiently stable38,39 and could be trapped subsequently.
Scheme 6.

An Unexpected N-to-C 1,3-Sulfonyl Shift.
This nitrile formation is very general including a number of different sulfonyl groups as demonstrated in Table 6 [see entries 1–4]. While the 1,3-sulfonyl shift tolerated various substitutions [entries 5 and 6], again when using 8a containing the TIPS substitution [entry 7], the 1,3-sulfonyl shift only took place only when in conjunction with desilylation. It is also noteworthy that given results in Table 5, the N-to-C 1,3-sulfonyl shift most likely took place after the aza-Claisen rearrangement. In addition, monitoring the thermal aza-Claisen rearrangement of ynamide 8b using proton NMR did not reveal any respective ketenimine, thereby suggesting that the 1,3-sulfonyl shift was very fast at 110 °C.
Table 6.
Tandem Aza-Claisen - 1,3-Sulfonyl Shift
| entry | ynamides | nitriles | yielda,b |
|---|---|---|---|
| 1 |
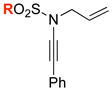 8b: R = Ts 8b: R = Ts8i: R = 4-MeO-Ph 8j: R = 4-NO2-Ph 8l: R = Me |
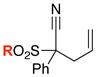 62 6263 64 65 |
48 |
| 2 | 53 | ||
| 3 | 53 | ||
| 4 | 64 | ||
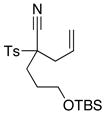
| 5 |
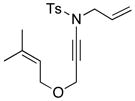 8m 8m
|
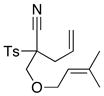 66 66
|
57 |
| 6 |
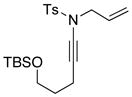 8d 8d
|
 67 67
|
45 |
| 7 |
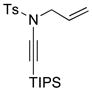 8a 8a
|
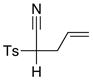 68 68
|
57 |
Conditions: Toluene, 110 °C, and 14 h.
Isolated yields.
We then attempted to extend this interesting 1,3-sulfonyl shift because the formation of tertiary nitriles holds significant merit in synthesis.40 As shown in Scheme 7, we envisioned that using ynamide 69 containing a propargylic stereocenter could lead to a stereoselective 1,3-sulfonyl shift to give 70 or 70′. The level of selectivity would depend upon conformational preference of allyl-ketenimine intermediate 72 and 72′, and in this case, the conformer 72′ could be more preferred given the A1,2-strain present in 72. This preference could lead to a facial selective 1,3-sulfonyl shift to give 70′. In addition, we could vary the P group so that it could lead to the conformational situation shown in 73′ in which anchiomeric assistance could take place, again leading to possible facial selective 1,3-sulfonyl shift.
Scheme 7.

Diastereoselective N-to-C 1,3-Sulfonyl Shift.
Unfortunately, after examining a number of such ynamides [69a–d], the best ratio was 2:1 using 69c [entry 3 in Table 7]. Most intriguingly, when exploring ynamide 69e [P = N-dimethyl carbamoyl] and 69d [P = Piv] with hope of the aforementioned anchiomeric assistance, we obtained α,β-unsaturated imidates 79 and 81, respectively. The double bond geometry was assured using NOE experiments. In addition, when we went back to crude proton NMR of the reaction using 69b in which P = Ac, we also saw ~10% of the respective imidate. Intriguingly, no nitrile was found in the case of 69e, due to the increased electron density in the carbamate. The isolation of these imidates implied a sequence of tandem sigmatropic rearrangement: an aza-Claisen followed by another [3,3]-sigmatropic rearrangement through the respective allyl-ketenimine intermediates 78 and 80.
Table 7.
Possible Diasteoselective 1,3-Sulfonyl Shift.
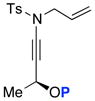 (±)-69a: P = TIPS
(±)-69a: P = TIPS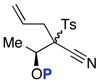 74
74 (±)-69c: P = TIPS
(±)-69c: P = TIPS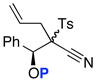 76
76CONCLUSION
We have described here a detailed study of amidine synthesis from N-allyl-N-sulfonyl ynamides. Mechanistically, this is a fascinating reaction consisting of diverging pathways that could lead to deallylation or allyl transfer depending upon the oxidation state of palladium catalysts, the nucleophilicity of amines, and the nature of the ligands. It essentially constitutes a Pd(0)-catalyzed aza-Claisen rearrangement of N-allyl ynamides, which can also be accomplished thermally. An observation of N-to-C 1,3-sulfonyl shift was made when examining these aza-Claisen rearrangements thermally, thereby representing a useful approach to nitrile synthesis. While attempts to render this 1,3-sulfonyl shift stereoselective failed, we uncovered another set of tandem sigmatropic arrangements, leading to vinyl imidate formation, and thereby suggesting rich chemistry one can develop using these ynamides.
EXPERIMENTAL SECTION
General Procedure for the Preparation of Des-Allyl Amidines from N-allyl Ynamides
To a flame dried screw-cap vial was added ynamide 8a (117.0 mg, 0.300 mmol), PdCl2(PPh3)2 (10.5 mg, 0.015 mmol), THF (6 mL), and tert-butyl amine (158.0 μL, 1.50 mmol), then sealed under a dry nitrogen atmosphere and heated to 80 °C for 2 h. After the reaction was judged to be complete by TLC, the solvent was removed in vacuo and the resulting crude residue was purified via silica gel flash column chromatography (isocratic eluent: 6:1 hexane/EtOAc) to afford amidine 9 as a white solid (119.0 mg, 0.282 mmol, 94%).
9: Rf = 0.27 [4:1 hexanes:EtOAc]; white solid; mp = 134 – 135 °C; 1H NMR (400 MHz, CDCl3) δ 1.11 (d, 18H, J = 8.8 Hz), 1.28 (sept, 3H, J = 8.8 Hz), 1.31 (s, 9H), 2.39 (s, 3H), 2.45 (s, 2H), 4.91 (brs, 1H), 7.25 (d, 2H, J = 10.0 Hz), 7.79 (d, 2H, J = 10.0 Hz); 13C NMR (100 MHz, CDCl3) δ 11.5, 18.7, 18.8, 21.6, 28.7, 53.4, 126.3, 129.2, 141.7, 141.8, 167.1; IR (film) cm−1 3328m, 2942m, 2867m, 1570m, 1530s, 1341m; mass spectrum (ESI): m/e (% relative intensity) 447 (100) (M+Na)+, 425 (40) (M+H)+; HRMS (ESI) m/e calcd for C22H40N2O2SSiNa 447.2472, found 447.2476.
11 (87%): Rf = 0.31 [4:1 hexanes:EtOAc]; white solid; mp = 73 – 76 °C; 1H NMR (400 MHz, CDCl3) (showing as two rotamers in 2.3:1 ratio) major rotamer δ 0.96 (d, 18H, J = 6.0 Hz), 0.96 – 1.02 (m, 1H), 1.10 (s, 3H), 1.20 – 1.35 (m, 2H), 1.35 – 1.50 (m, 2H), 1.60 (pent, 2H, J = 7.2 Hz), 1.85 (s, 2H), 2.39 (s, 3H), 3.27 (t, 2H, J = 7.2 Hz), 7.22 (d, 2H, J = 8.0 Hz), 7.76 (d, 2H, J = 8.0 Hz), 8.28 (brs, 1H); 13C NMR (100 MHz, CDCl3) major rotamer δ 11.7, 13.9, 18.0, 18.7, 20.1, 21.7, 31.8, 44.6, 126.5, 129.1, 140.4, 142.4, 169.7; IR (film) cm−1 3322m, 2942m, 2869m, 1532s, 1465m; mass spectrum (APCI): m/e (% relative intensity) 425 (50) (M+H)+; HRMS (ESI) m/e calcd for C22H40N2O2SSiH 425.2653, found 425.2640.
13 (41%): Rf = 0.20 [4:1 hexanes:EtOAc]; white solid; mp = 84 – 87 °C; 1H NMR (400 MHz, CDCl3) δ 0.86 (d, 18H, J = 6.4 Hz), 0.90 (sept, 3H, J = 6.4 Hz), 1.88 (s, 2H), 2.42 (s, 3H), 3.82 (s, 3H), 6.90 (d, 2H, J = 8.8 Hz), 7.08 (d, 2H, J = 8.8 Hz), 7.26 (d, 2H, J = 8.0 Hz), 7.83 (d, 2H, J = 8.0 Hz), 9.80 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 11.5, 18.3, 18.6, 21.7, 55.8, 114.8, 126.6, 128.2, 129.2, 130.3, 140.1, 142.7, 159.1, 169.3; IR (film) cm−1 3272m, 2943m, 2869m, 1610m, 1573s, 1513m; mass spectrum (ESI): m/e (% relative intensity) 497 (100) (M+Na)+, 475 (40) (M+H)+; HRMS (ESI) m/e calcd for C25H38N2O3SSiNa 497.2265, found 497.2258.
14 (22%): Rf = 0.31 [4:1 hexanes:EtOAc]; white solid; mp = 125 – 128 °C; 1H NMR (500 MHz, CDCl3) δ 0.85 (d, 18H, J = 6.0 Hz), 0.85 – 0.95 (m, 3H), 1.94 (s, 2H), 2.42 (s, 3H), 7.17 (d, 2H, J = 7.5 Hz), 7.27 (d, 2H, J = 8.0 Hz), 7.31 (t, 1H, J = 7.5 Hz), 7.40 (t, 2H, J = 7.5 Hz), 7.84 (d, 2H, J = 8.0 Hz), 9.96 (s, 1H); 13C NMR (125 MHz, CDCl3) δ 11.5, 18.4, 18.6, 21.8, 126.7, 126.8, 127.9, 129.3, 129.7, 137.6, 140.0,142.8, 168.9; IR (film) cm−1 3338w, 2962m, 2867m, 1608m, 1569m, 1527s, 1441m; mass spectrum (ESI): m/e (% relative intensity) 467 (100) (M+Na)+, 445 (35) (M+H)+; HRMS (ESI) m/e calcd for C24H36N2O2SSiNa 467.2159, found 467.2173.
15 (22%): Rf = 0.30 [4:1 hexanes:EtOAc]; white solid; mp = 147 – 150 °C; 1H NMR (400 MHz, CDCl3) δ 0.87 (s, 18H), 0.90 – 1.20 (m, 3H), 1.90 (brs, 2H), 2.42 (s, 3H), 7.05 – 7.17 (m, 2H), 7.27 (d, 2H, J = 8.0 Hz), 7.32 – 7.42 (m, 2H), 7.81 (d, 2H, J = 8.0 Hz), 9.91 (brs, 1H); 13C NMR (125 MHz, CDCl3) δ 11.6, 18.6, 21.7, 29.9, 126.6, 128.1, 129.3, 129.9, 133.7, 136.2, 139.7, 142.9, 168.7; IR (film) cm−1 3314m, 2943m, 2867m, 1600m, 1569s, 1517s, 1493s; mass spectrum (ESI): m/e (% relative intensity) 501 (100) (M+Na)+, 479 (45) (M+H)+; HRMS (ESI) m/e calcd for C24H35ClN2O2SSiNa 501.1770, found 501.1766.
16 (30%): Rf = 0.33 [4:1 hexanes:EtOAc]; white solid; mp = 74 – 76 °C; 1H NMR (500 MHz, CDCl3) δ 0.87 (d, 18H, J = 7.0 Hz), 0.94 – 0.98 (m, 3H), 1.82 (s, 2H), 2.26 (s, 3H), 2.42 (s, 3H), 7.10 – 7.17 (m, 1H), 7.20 – 7.26 (m, 3H), 7.27 (d, 2H, J = 8.5 Hz), 7.84 (d, 2H, J = 8.5 Hz), 9.79 (brs, 1H); 13C NMR (125 MHz, CDCl3) δ 11.6, 18.2, 18.2, 18.6, 21.8, 126.7, 127.1, 127.5, 128.2, 129.3, 131.5, 134.7, 136.3, 140.0, 142.7, 169.2; IR (film) cm−1 3256m, 2941m, 2866m, 1566s, 1461m; 1369m, mass spectrum (ESI): m/e (% relative intensity) 481 (100) (M+Na)+, 459 (35) (M+H)+; HRMS (ESI) m/e calcd for C25H38N2O2SSi 481.2316, found 481.2306.
17 (≥95%): Rf = 0.19 [4:1 hexanes:EtOAc], colorless oil; 1H NMR (500 MHz, CDCl3) [showing as two rotamers in a 3:2 ratio] major rotamer δ 0.97 (d, 18H, J = 6.5 Hz), 1.02 (sept, 3H, J = 6.5 Hz), 2.41 (s, 2H), 2.52 (s, 3H), 4.48 (d, 2H, J = 6.0 Hz), 7.24 (d, 2H, J = 7.0 Hz), 7.39 – 7.19 (m, 5H), 7.77 (d, 2H, J = 8.0 Hz), 8.65 (s, 1H); 13C NMR (125 MHz, CDCl3) major rotamer δ 11.7, 18.3, 18.7, 21.7, 48.5, 126.6, 127.2, 128.4, 129.2, 129.3, 136.3, 140.2, 142.6, 170.0; IR (film) cm−1 3326brs, 2942m, 2866m, 1537s, 1496m, 1270m; mass spectrum (APCI): m/e (% relative intensity) 459 (100) (M+H)+; HRMS (ESI) m/e calcd for C25H38N2O2SSiH 459.2496, found 459.2506.
19 (≥95%): Rf = 0.36 [2:1 hexanes:EtOAc]; white solid; mp = 143 – 145 °C; 1H NMR (300 MHz, CDCl3) δ 1.15 (d, 18H, J = 7.5 Hz), 1.38 (sept, 3H, J = 7.5 Hz), 2.40 (s, 3H), 2.70 (s, 2h), 3.57 (brs, 2H), 3.69 (brs, 6H), 7.25 (d, 2H, J = 8.1 Hz), 7.80 (d, 2H, J = 8.1 Hz); 13C NMR (75 MHz, CDCl3) δ 12.3, 16.3, 18.9, 21.6, 44.1, 47.1, 66.4, 77.6, 126.1, 129.2, 141.7, 142.1, 168.7; IR (film) cm−1 2966m, 2945m, 2868m, 1519s, 1444m, 1271s, 1089s; mass spectrum (APCI): m/e (% relative intensity) 439 (100) (M+H)+; HRMS (ESI) m/e calcd for C22H38N2O3SSiNa 461.2265, found 461.2275.
20 (≥95%): Rf = 0.28 [1:1 hexanes:EtOAc]; white solid; mp = 125 – 132 °C; 1H NMR (400 MHz, CDCl3) δ 1.16 (s, 9H), 2.39 (s, 3H), 3.09 (s, 2H), 3.66 (brs, 8H), 7.23 (d, 2H, J = 8.4 Hz), 7.78 (d, 2H, J = 8.4 Hz); 13C NMR (100 MHz, CDCl3) δ 21.6, 31.0, 31.4, 39.9, 66.8, 126.1, 129.3, 141.9, 142.3, 165.8 [missing one sp3 carbon due to overlap]; IR (film) cm−1 2960w, 2868w, 1599s, 1459m, 1398m, 1293s, 1141m, 1065s; mass spectrum (APCI): m/e (% relative intensity) 339 (100) (M+H)+; HRMS (ESI) m/e calcd for C17H26N2O3SNa 361.1556, found 361.1574.
21 (92%): Rf = 0.36 [1:1 hexanes:EtOAc]; brown oil; 1H NMR (400 MHz, CDCl3) δ 0.04 (s, 6H), 0.88 (s, 9H), 1.57 – 1.72 (m, 4H), 2.40 (s, 3H), 2.93 – 2.98 (m, 2H), 3.51 – 3.53 (m, 2H), 3.62 – 3.72 (m, 8H), 7.25 (d, 2H, J = 8.4 Hz), 7.81 (d, 2H, J = 8.4 Hz); 13C NMR (100 MHz, CDCl3) δ −5.2, 14.3, 18.4, 21.2, 21.6, 23.5, 26.1, 30.4, 32.3, 60.5, 62.1, 66.5, 126.4, 128.6, 129.3, 132.3, 142.1; IR (film) cm−1 3058m, 2929m, 2856m, 1536s, 1438m, 1388w, 1359w, 1268s, 1143s, 1117s, 1087s; mass spectrum (APCI): m/e (% relative intensity) 455 (100) (M+H)+; HRMS (ESI) m/e calcd for C22H38N2O4SSiNa 477.2214, found 477.2222.
22 (37%): Rf = 0.21 [1:1 hexanes:EtOAc]; yellow foam; mp = 54 – 60 °C; 1H NMR (400 MHz, CDCl3) δ 2.37 (s, 3H), 3.24 (t, 2H, J = 4.8 Hz), 3.33 (t, 2H, J = 4.8 Hz), 3.63 (t, 2H, J = 5.2 Hz), 3.81 (t, 2H, J = 5.2 Hz), 3.83 (s, 3H), 4.35 (s, 2H), 6.84 (d, 1H, J = 8.0 Hz), 6.85 (td, 1H, J = 1.2, 7.6 Hz), 7.04 (dd, 1H, J = 1.2, 7.6 Hz), 7.20 (d, 2H, J = 8.0 Hz), 7.21 – 7.23 (m, 1H), 7.80 (d, 2H, J = 8.0 Hz); 13C NMR (125 MHz, CDCl3) δ 21.7, 30.4, 45.2, 46.9, 55.7, 66.5, 66.5, 110.6, 121.2, 122.5, 126.7, 128.5, 128.7, 129.3, 141.1, 142.2, 156.1, 166.4; IR (film) cm−1 2966w, 2858w, 1540s, 1463m, 1271s, 1069s, 998s; mass spectrum (APCI): m/e (% relative intensity) 389 (100) (M+H)+; HRMS (ESI) m/e calcd for C20H24N2O4SH 389.1530, found 389.1523.
23 (39%): Rf = 0.50 [1:1 hexanes:EtOAc]; pale yellow solid; mp = 159 – 161 °C; 1H NMR (400 MHz, CDCl3) δ 1.15 (d, 18H, J = 7.2 Hz), 1.37 (sept, 3H, J = 7.2 Hz), 2.71 (s, 2H), 3.53 (brs, 2H), 3.65 (brs, 2H), 3.68 (brs, 2H), 3.73 (brs, 2H), 8.08 (d, 2H, J = 9.2 Hz), 8.29 (d, 2H, J = 9.2 Hz); 13C NMR (100 MHz, CDCl3) δ 12.3, 17.0, 18.9, 45.5, 47.3, 66.4, 124.1, 127.4, 149.4, 150.4, 169.2; IR (film) cm−1 2983w, 2360w, 1740s, 1526m, 1444m, 1374m, 1242s, 1047s; mass spectrum (APCI): m/e (% relative intensity) 470 (100) (M+H)+; HRMS (ESI) m/e calcd for C21H35N3O5SSiH 470.2140, found 470.2150.
24 (≥95%): Rf = 0.32 [4:1 hexanes:EtOAc]; white solid; mp = 129 – 130 °C; 1H NMR (500 MHz, CDCl3) δ 1.08 (t, 3H, J = 7.5 Hz), 1.18 (d, 18H, J = 7.5 Hz), 1.19 (t, 3H, J = 7.0 Hz), 1.39 (sept, 3H, J = 7.5 Hz), 2.36 (s, 3H), 2.62 (s, 2H), 3.32 (q, 2H, J = 7.0 Hz) 3.42 (q, 2H, J = 7.0 Hz), 7.20 (d, 2H, J = 8.0 Hz), 7.79 (d, 2H, J = 8.0 Hz); 13C NMR (125 MHz, CDCl3) δ 12.3, 12.4, 13.6, 16.4, 18.9, 21.5, 43.5, 43.6, 125.9, 129.0, 141.2, 142.7, 167.7; IR (film) cm−1 2941s, 2868m, 2361w, 1548s, 1469s, 1362m, 1261m, 1395s; mass spectrum (APCI): m/e (% relative intensity) 425 (100) (M+H)+; HRMS (ESI) m/e calcd for C22H40N2O2SSiNa 447.2472, found 447.2479.
25 (≥95%): Rf = 0.29 [2:1 hexanes:EtOAc]; pale yellow oil; 1H NMR (300 MHz, CDCl3) δ 0.94 (t, 3H, J = 7.2 Hz), 1.16 (t, 3H, J = 7.2 Hz), 2.36 (s, 3H), 3.20 (q, 2H, J = 7.2 Hz), 3.50 (q, 2H, J = 7.2 Hz), 4.38 (s, 2H), 7.09 – 7.27 (m, 7H), 7.76 (d, 2H, J = 8.1 Hz); 13C NMR (75 MHz, CDCl3) δ 12.0, 13.5, 21.5, 36.6, 43.3, 43.4, 126.2, 126.7, 127.8, 128.8, 129.0, 134.3, 141.4, 141.6, 164.5; IR (film) cm−1 2978w, 2937w, 2359w, 1650w, 1549s, 1475m, 1274m, 1143m; mass spectrum (APCI): m/e (% relative intensity) 345 (100) (M+H)+; HRMS (ESI) m/e calcd for C19H24N2O2SNa 367.1451, found 367.1438.
26 (≥95%): Rf = 0.09 [4:1 hexanes:EtOAc]; waxy white solid; mp = 30 – 31 °C; 1H NMR (500 MHz, CDCl3) δ 0.88 (t, 3H, J = 7.0 Hz), 1.10 (t, 3H, J = 7.0 Hz), 1.22 (t, 3H, J = 7.0 Hz), 1.24 – 1.32 (m, 6H), 1.35 – 1.40 (m, 2H), 1.58 – 1.63 (m, 2H), 2.39 (s, 3H), 2.81 – 2.86 (m, 2H), 3.34 (q, 2H, J = 7.0 Hz), 3.44 (q, 2H, J = 7.0 Hz), 7.23 (d, 2H, J = 7.5 Hz), 7.82 (d, 2H, J = 7.5 Hz); 13C NMR (125 MHz, CDCl3) δ 12.3, 14.2, 14.4, 21.6, 22.7, 27.6, 29.0, 30.1, 31.1, 31.8, 43.3, 126.2, 129.1, 141.6, 142.1, 167.8; IR (film) cm−1 2920m, 2855m, 1550s, 1474s, 1453s, 1434s; mass spectrum (ESI): m/e (% relative intensity) 375 (100) (M+Na)+, 353 (30) (M+H)+; HRMS (ESI) m/e calcd for C19H32N2O2SNa 375.2077, found 375.2075.
27 (70%): Rf = 0.68 [1:1 hexanes:EtOAc]; yellow solid; mp = 78 – 80 °C; 1H NMR (400 MHz, CDCl3) δ 0.89 (t, 3H, J = 8.0 Hz), 1.22 – 1.49 (m, 20H), 1.56 – 1.68 (m, 2H), 2.39 (s, 3H), 2.85 – 2.89 (m, 2H), 3.51 (brs, 1H), 4.03 (sept, 1H, J = 6.4 Hz), 7.24 (d, 2H, J = 8.0 Hz), 7.81 (d, 2H, J = 8.0 Hz); 13C NMR (100 MHz, CDCl3) δ 14.3, 20.3, 20.9, 21.6, 22.8, 27.5, 29.1, 30.0, 31.9, 33.0, 48.1, 50.1, 126.3, 129.2, 141.6, 142.2, 166.7; IR (film) cm−1 3479w, 2970w, 2932w, 1539s, 1494m, 1449m, 1267m, 1086s; mass spectrum (APCI): m/e (% relative intensity) 381 (100) (M+H)+; HRMS (ESI) m/e calcd for C21H36N2O2SH 381.2570, found 381.2561.
28 (≥95%): Rf = 0.45 [4:1 hexanes:EtOAc]; brown solid; mp = 182 – 184 °C; 1H NMR (400 MHz, CDCl3) δ 1.15 (d, 18H, J = 7.2 Hz), 1.46 (sept, 3H, J = 7.2 Hz), 2.39 (s, 3H), 2.84 (brs, 2H), 3.14 (t, 2H, J = 8.4 Hz), 4.11 (t, 2H, J = 8.4 Hz), 7.00 (td, 1H, J = 0.8, 7.2 Hz), 7.06 (t, 1H, J = 7.2 Hz), 7.17 (d, 1H, J = 7.2 Hz), 7.25 (dd, 2H, J = 0.4, 8.8 Hz), 7.85 (d, 2H, J = 8.8 Hz), 8.07, (brs, 1H); 13C NMR (100 MHz, CDCl3) δ 12.8, 19.0, 20.1, 21.7, 27.7, 50.4, 119.8, 124.8, 124.8, 126.5, 127.7, 129.3, 132.9, 141.6, 141.8, 142.6, 166.3; IR (film) cm−1 3055w, 2941w, 2867w, 1541m, 1481m, 1265s, 1143m, 1089m; mass spectrum (APCI): m/e (% relative intensity) 471 (100) (M+H)+; HRMS (ESI) m/e calcd for C26H38N2O2SSiNa 493.2316, found 493.2340.
29 (77%): Rf = 0.67 [1:1 hexanes:EtOAc]; white solid; mp = 89 – 90 °C; 1H NMR (400 MHz, CDCl3) [showing as two rotamers in 1.7:1 ratio] major rotamer δ 1.13 (d, 18H, J = 7.6 Hz), 1.40 (sept, 3H, J = 7.6 Hz), 2.37 (s, 3H), 2.75 (s, 2H), 3.01 (s, 3H), 4.68 (s, 2H), 7.06 – 7.11 (m, 3H), 7.17 (d, 2H, J = 8.4 Hz), 7.22 – 7.26 (m, 2H), 7.74 (d, 2H, J = 8.4 Hz); 13C NMR (125 MHz, CDCl3) major rotamer δ 12.6, 17.0, 18.9, 21.6, 36.9, 54.2, 126.2, 127.8, 128.5, 128.7, 129.1, 136.1, 141.4, 142.3, 169.4; IR (film) cm−1 3060w, 2944m, 2867m, 2362w, 1530s, 1454m, 1266s, 1142s, 1088s, 1017m; mass spectrum (APCI): m/e (% relative intensity) 473 (100) (M+H)+; HRMS (ESI) m/e calcd for C26H40N2O2SSiH 473.2653, found 473.2676.
30a (≥95%): Rf = 0.30 [3:1 hexanes:EtOAc]; white solid; mp = 161 – 163 °0C; 1H NMR (300 MHz, CDCl3) δ 1.15 (d, 18H, J = 7.2 Hz), 1.41 (sept, 3H, J = 7.2 Hz), 1.87 (quint, 2H, J = 6.6 Hz), 1.98 (quint, 2H, J = 6.6 Hz), 2.40 (s, 3H), 2.63 (s, 2H), 3.51 (q, 4H, J = 8.7 Hz), 7.23 (d, 2H, J = 8.4 Hz), 7.85 (d, 2H, J = 8.4 Hz); 13C NMR (75 MHz, CDCl3) δ 12.6 18.3, 18.9, 21.5, 24.4, 26.1, 48.3, 49.0, 126.1, 129.0, 141.3, 142.7, 167.2; IR (film) cm−1 2945w, 2868m, 1530s, 1463m, 1421m, 1337w, 1268m, 1139m, 1089s; mass spectrum (APCI): m/e (% relative intensity) 423 (100) (M+H)+; HRMS (ESI) m/e calcd for C22H38N2O2SSiNa 445.2316, found 445.2329.
30b (≥95%): Rf = 0.28 [4:1 hexanes:EtOAc]; white solid; mp = 121 – 122 °C; 1H NMR (300 MHz, CDCl3) δ 1.15 (d, 18H, J = 7.5 Hz), 1.39 (sept, 3H, J = 7.5 Hz), 1.54 (brs, 2H), 1.64 (brs, 4H), 2.39 (s, 3H), 2.69 (s, 2H), 3.45 (brs, 2H), 3.70 (brs, 2H), 7.28 (d, 2H, J = 8.4 Hz), 7.80 (d, 2H, J = 8.4 Hz); 13C NMR (75 MHz, CDCl3) δ 12.3, 16.5, 18.9, 21.5, 24.3, 25.5, 26.4, 46.3, 48.1, 126.0, 129.0, 141.3, 142.7, 167.9; IR (film) cm−1 2943m, 2867m, 1526s, 1468m, 1445m, 1271m, 1144s, 1089s; mass spectrum (APCI): m/e (% relative intensity) 437 (100) (M+H)+; HRMS (ESI) m/e calcd for C23H40N2O2SSiNa 459.2472, found 459.2461.
30c (≥95%): Rf = 0.34 [4:1 hexanes:EtOAc]; white solid; mp = 150 – 152 °C; 1H NMR (300 MHz, CDCl3) δ 1.15 (d, 18H, J = 7.2 Hz), 1.39 – 1.73 (m, 11H), 2.40 (s, 3H), 2.68 (s, 2H), 3.49 (t, 2H, J = 6.0 Hz), 3.61 (t, 2H, J = 6.0 Hz), 7.23 (d, 2H, J = 7.8 Hz), 7.81 (d, 2H, J = 8.1 Hz); 13C NMR (75 MHz, CDCl3) δ 12.4, 16.4, 18.9, 21.5, 26.3, 26.4, 26.7, 29.0, 50.0, 50.1, 125.9, 129.0, 141.2, 142.6, 168.4; IR (film) cm−1 2939m, 2866m, 1533s, 1473m, 1370w, 1271m, 1143m, 1089m; mass spectrum (APCI): m/e (% relative intensity) 451 (100) (M+H)+; HRMS (ESI) m/e calcd for C24H42N2O2SSiNa 473.2629, found 473.2643.
31 (91%): Rf = 0.64 [1:1 hexanes:EtOAc]; pale solid; mp = 92 – 96 °C; 1H NMR (400 MHz, CDCl3) [showing as two rotamers in a 2.0:1 ratio] major rotamer δ 1.10 – 1.24 (m, 21H), 1.33 – 1.40 (m, 3H), 1.86 – 2.04 (m, 4H), 2.36 (s, 3H), 2.43 (d, 1H, J = 12.4 Hz), 2.73 (d, 1H, J = 12.4 Hz), 3.41 – 3.53 (m, 2H), 4.27 – 4.30 (m, 1H), 7.20 (d, 2H, J = 8.4 Hz), 7.79 (d, 2H, J = 8.4 Hz); 13C NMR (100 MHz, CDCl3) major rotamer δ 12.7, 18.4, 19.0, 19.0, 21.6, 23.8, 31.8, 48.6, 55.8, 126.1, 129.1, 141.3, 142.7, 166.9; IR (film) cm−1 2972m, 2868m, 2839m, 2361m, 1781w, 1738s, 1517s, 1461m, 1240s; mass spectrum (APCI): m/e (% relative intensity) 437 (100) (M+H)+; HRMS (ESI) m/e calcd for C23H40N2O2SSiNa 459.2472, found 459.2471.
32 (≥95%): Rf = 0.80 [1:1 hexanes:EtOAc]; [α]D23 = − 63.5 (c 0.43, dichloromethane); pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 1.13 – 1.16 (m, 18H), 1.40 (sept, 3H, J = 6.8 Hz), 1.92 – 1.99 (m, 2H), 2.09 – 2.16 (m, 2H), 2.39 (s, 3H), 2.43 (d, 1H, J = 12.4 Hz), 2.95 (d, 1H, J = 12.4 Hz), 3.34 (s, 3H), 3.59 – 3.60 (m, 1H), 3.66 – 3.72 (m, 1H), 4.45 (dd, 1H, J = 4.0, 8.4 Hz), 7.21 (d, 2H, J = 8.4 Hz), 7.74 (d, 2H, J = 8.4 Hz); 13C NMR (100 MHz, CDCl3) δ 12.6, 18.0, 19.0, 21.6, 24.8, 29.5, 48.7, 52.0, 61.5, 126.4, 129.0, 141.6, 142.0, 167.6, 172.3; IR (film) cm−1 3058w, 2949m, 2871m, 2364w, 1747s, 1519s, 1459m, 1367w, 1267s, 1142m; mass spectrum (APCI): m/e (% relative intensity) 481 (100) (M+H)+; HRMS (ESI) m/e calcd for C29H40N2O4SSiNa 503.2370, found 503.2348.
33a (≥95%): Rf = 0.46 [95:5 CH2Cl2:MeOH]; pale yellow oil; 1H NMR (300 MHz, CDCl3) δ 1.15 (d, 18H, J = 7.5 Hz), 1.38 (sept, 3H, J = 7.5 Hz), 2.30 (s, 3H), 2.39 (brs, 4H), 2.40 (s, 3H), 2.70 (s, 2H), 3.51 (brs, 2H), 3.74 (brs, 2H), 7.24 (d, 2H, J = 8.4 Hz), 7.81 (d, 2H, J = 8.4 Hz); 13C NMR (75 MHz, CDCl3) δ 12.2, 16.5, 18.9, 21.6, 44.8, 45.9, 46.6, 54.4, 54.9, 77.2, 128.6, 133.2, 141.5, 142.3, 168.4; IR (film) cm−1 2943m, 2868m, 2796m, 1525s, 1452m, 1363w, 1271m, 1143m, 1092m; mass spectrum (APCI): m/e (% relative intensity) 452 (100) (M+H)+; HRMS (ESI) m/e calcd for C23H41N3O2SSiH 452.2672, found 452.2668.
33b (≥95%): Rf = 0.50 [2:1 hexanes:EtOAc]; white solid; mp = 126 – 127 °C; 1H NMR (300 MHz, CDCl3) δ 1.19 (d, 18H, J = 7.2 Hz), 1.43 (sept, 3H, J = 7.2 Hz), 2.42 (s, 3H), 2.77 (s, 2H), 3.17 (brs, 2H), 3.24 (brs, 2H), 3.71 (brs, 2H), 3.92 (brs, 2H), 6.92 – 6.98 (m, 3H), 7.26 – 7.35 (m, 4H), 7.85 (d, 2H, J = 8.1 Hz); 13C NMR (75 MHz, CDCl3) δ 12.0, 16.2, 18.6, 21.3, 44.4, 46.3, 48.5, 49.2, 116.3, 120.6, 125.9, 128.9, 129.2, 141.4, 141.9, 150.3, 168.3; IR (film) cm−1 3029w, 2945m, 2868m, 1600m, 1525s, 1453m, 1277m, 1143m, 1091m; mass spectrum (APCI): m/e (% relative intensity) 514 (100) (M+H)+; HRMS (ESI) m/e calcd for C28H43N3O2SSiNa 536.2738, found 536.2737.
34 (≥95%): Rf = 0.59 [1:1 hexanes:EtOAc]; yellow solid; mp = 125 – 126 °C; 1H NMR (400 MHz, CDCl3) δ 1.12 (d, 18H, J = 7.2 Hz), 1.10 – 1.20 (m, 6H), 1.34 (sept, 3H, J = 7.2 Hz), 2.28 – 2.31 (m, 1H), 2.37 (s, 3H), 2.62 – 2.74 (m, 2H), 3.47 – 3.68 (m, 3H), 3.37 (sept, 1H, J = 4.8 Hz), 4.62 (d, 1H, J = 13.2 Hz), 7.20 (d, 2H, J = 7.6 Hz), 7.75 (d, 2H, J = 7.6 Hz); 13C NMR (75 MHz, CDCl3) δ 12.3, 12.4, 16.5, 18.9, 19.0, 21.6, 71.6, 126.1, 129.2, 141.7, 142.2, 168.4; IR (film) cm−1 2973m, 2870m, 1523s, 1463m, 1271m, 1148s, 1093s; mass spectrum (APCI): m/e (% relative intensity) 467 (100) (M+H)+; HRMS (ESI) m/e calcd for C24H42N2O3SSiH 467.2758, found 467.2747.
35 (82%): Rf = 0.64 [1:1 hexanes:EtOAc]; brown oil; 1H NMR (400 MHz, CDCl3) δ 0.91 (d, 18H, J = 7.6 Hz), 1.18 (sept, 3H, J = 7.6 Hz), 2.42 (s, 3H), 2.73 (s, 2H), 3.27 (s, 3H), 3.82 (s, 3H), 6.92 (d, 2H, J = 8.4 Hz), 7.12 (d, 2H, J = 8.4 Hz), 7.25 (d, 2H, J = 7.6 Hz), 7.88 (d, 2H, J = 7.6 Hz); 13C NMR (100 MHz, CDCl3) δ 12.2, 17.3, 18.6, 18.8, 21.6, 55.9, 113.8, 115.1, 115.2, 126.2, 128.9, 129.2, 141.6, 159.3, 169.9; IR (film) cm−1 2945m, 2868m, 2252w, 1608m, 1509s, 1463m, 1406m; mass spectrum (APCI): m/e (% relative intensity) 498 (100) (M+H)+; HRMS (ESI) m/e calcd for C26H40N2O3SSiNa 511.2421, found 511.2411.
General Procedure for the Preparation of α-Allyl Amidines from Secondary Amines using Pd2(dba)3 and Xantphos.3
To a flame-dried vial filled with nitrogen was added Pd2(dba)3 (8.90 mg, 0.010 mmol), xantphos (11.2 mg, 0.020 mmol), and anhyd THF (4 mL). The resulting solution was stirred at rt for 10 min. Subsequently, a respective ynamide (79.0 mg, 0.20 mmol), K2CO3 (27.8 mg, 0.20 mmol), and pyrollidine (49.0 μL, 0.60 mmol) were added. The reaction mixture was stirred under nitrogen at 65 °C for 6 h. The progress of the reaction was monitored by TLC. After complete consumption of the starting ynamide, the crude reaction mixture was filtered through Celite™ and concentrated in vacuo. Purification of the crude residue via silica gel flash column chromatography [isocratic eluent: 5:1 hexanes/EtOAc] afforded amidine 39 (95.0 mg, 0.20 mmol, ≥95%).
39: Rf = 0.22 [5:1 hexanes:EtOAc]; white solid; mp 75 – 76 °C; 1H NMR (400 MHz, CDCl3) delta; 0.98 (d, 18H, J = 6.0 Hz), 1.16 (m, 3H), 1.76 – 1.82 (m, 1H), 1.92 – 2.08 (m, 3H), 2.17 – 2.22 (m, 1H), 2.26 – 2.29 (m, 1H), 2.38 (s, 3H), 2.44 – 2.52 (m, 1H), 3.56 (q, 1H, J = 8.0 Hz), 3.68 (td, 1H, J = 2.4, 8.0 Hz), 4.22 (brs, 2H), 4.97 (d, 1H, J = 10.8 Hz), 5.02 (d, 1H, J = 17.6 Hz), 5.81 (m, 1H), 7.20 (d, 2H, J = 8.4 Hz), 7.78 (d, 2H, J = 8.4 Hz); 13C NMR (125 MHz, CDCl3) δ 11.5, 19.1, 21.5, 25.4, 25.8, 35.8, 35.9, 51.8, 53.2, 116.0, 126.1, 128.8, 137.8, 140.9, 143.2, 167.1; IR (neat) cm−1 2924ms, 2863m, 1547s, 1414s, 1274s, 1139s; mass spectrum (APCI): m/e (% relative intensity) 463 (100) (M+H)+. HRMS (ESI) m/e calcd for C25H42N2O2SSiNa 485.2628, found 486.2630.
General Procedure for the Preparation of Vinylogous Amidines
To a flame dried screw-cap vial was added ynamide 8a (75.0 mg, 0.192 mmol), Pd2(dba)3 (8.8 mg, 0.0096 mmol), xantphos (11.1 mg, 0.019 mmol), THF (2.0 mL), and enamine 41 (77.0 μL, 0.576 mmol), then sealed under a dry nitrogen atmosphere and heated to 70 °C for 2 h. After the reaction was judged to be complete by TLC, the solvent was removed in vacuo and the resulting crude residue was purified via silica gel flash column chromatography (isocratic eluent: 1:1 hexane/EtOAc + 5% NEt3 buffer) to afford the vinylogous amidine 45 as an orange solid (52.4 mg, 0.099 mmol, 52%).
45: Rf = 0.23 [1:1 hexanes:EtOAc]; orange solid, mp = 95 – 97 °C; 1H NMR (400 MHz, CDCl3) [spectrum complicated by rotamers] δ 1.07 (d, 9H, J = 6.4 Hz), 1.11 (d, 9H, J = 5.2 Hz), 1.10 – 1.20 (m, 3H), 1.83 – 1.90 (m, 2H), 1.90 – 2.01 (m, 3H), 2.01 – 2.15 (m, 2H), 2.25 – 2.35 (m, 1H), 2.34 (s, 3H), 2.50 (d, 1H, J = 11.2 Hz), 2.57 – 2.72 (m, 2H), 2.80 (t, 1H, J = 4.8 Hz), 2.85 – 2.92 (m, 1H), 3.18 (brs, 2H), 3.89 (brs, 2H), 4.66 (d, 1H, J = 18.8 Hz), 4.67 (d, 1H, J = 9.2 Hz), 5.51 – 5.62 (m, 1H), 7.14 (d, 2H, J = 8.4 Hz), 7.76 (d, 2H, J = 8.4 Hz); 13C NMR (125 MHz, CDCl3) δ 12.3, 19.3, 19.4, 21.6, 21.7, 25.4, 29.9, 35.2, 36.2, 40.4, 53.0, 111.4, 113.6, 126.0, 128.7, 131.1, 140.0, 140.2, 143.7, 148.1; IR (film) cm−1 2944m, 2865m, 1736m, 1579m, 1469s, 1445s, 1342m; mass spectrum (ESI): m/e (% relative intensity) 529 (100) (M+H)+; HRMS (ESI) m/e calcd for C30H48N2O2SSiH 529.3279, found 529.3269.
46 (71%): Rf = 0.18 [1:1 hexanes:EtOAc], orange oil; 1H NMR (400 MHz, CDCl3) [spectrum complicated by rotamers] δ 1.05 (d, 9H, J = 6.4 Hz), 1.09 (d, 9H, J = 5.2 Hz), 1.09 – 1.15 (m, 3H), 1.70 – 2.10 (m, 9H), 2.32 (brs, 1H), 2.48 (d, 1H, J = 11.2 Hz), 2.57 – 2.70 (m, 2H), 2.76 (brs, 1H), 2.80 – 2.90 (m, 1H), 3.13 (brs, 2H), 3.80 (s, 3H), 3.83 (brs, 2H), 4.63 (d, 1H, J = 5.6 Hz), 4.65 (d, 1H, J = 10.8 Hz), 5.48 – 5.60 (m, 1H), 6.81 (d, 2H, J = 8.8 Hz), 7.79 (d, 2H, J = 8.8 Hz); 13C NMR (100 MHz, CDCl3) δ 12.3, 19.3, 19.4, 21.7, 25.4, 35.1, 36.2, 40.3, 52.9, 55.5, 113.2, 113.6,127.9, 139.0, 139.8, 139.9, 160.9 [missing two sp2 signals due to rotamers]; IR (film) cm−1 2943m, 2866m, 1596m, 1590m, 1468s, 1249s; mass spectrum (APCI): m/e (% relative intensity) 545 (100) (M+H)+; HRMS (ESI) m/e calcd for C30H48N2O3SSiH 545.3228, found 545.3240.
47 (58%): Rf = 0.12 [1:2 hexanes:EtOAc]; orange solid, mp = 69 – 72 °C; 1H NMR (500 MHz, CDCl3) δ 1.98 (pent, 2H, J = 6.5 Hz), 2.02 (brs, 4H), 2.32 (t, 2H, J = 8.5 Hz), 2.34 (t, 2H, J = 8.5 Hz), 2.71 (t, 2H, J = 6.5 Hz), 3.07 (t, 2H, J = 6.5 Hz), 3.30 – 3.55 (m, 4H), 4.92 (dq, 1H, J = 1.5, 10.0 Hz), 5.00 (dq, 1H, J = 1.5, 17.0 Hz), 5.81 (ddt, 1H, J = 6.5, 10.5, 17.0 Hz), 8.30 (d, 2H, J = 9.0 Hz), 8.05 (d, 2H, J = 9.0 Hz); 13C NMR (125 MHz, CDCl3) δ 19.3, 25.3, 31.2, 32.1, 38.1, 41.8, 52.5, 109.7, 115.2, 124.1, 128.0, 137.7, 148.6, 149.7, 169.5, 186.5, 193.0; IR (film) cm−1 3062m, 2954m, 2877m, 1634m, 1608m, 1527s, 1436s, 1349s; mass spectrum (ESI): m/e (% relative intensity) 454 (100) (M+Na)+, 432 (15) (M+H)+; HRMS (ESI) m/e calcd for C21H25N3O5SNa 454.1408, found 454.1402.
48 (54%): Rf = 0.14 [1:1 hexanes:EtOAc]; orange solid; mp = 104 – 107 °C; 1H NMR (500 MHz, CDCl3) δ 0.00 (s, 6H), 0.84 (s, 9H), 1.22 (s, 2H), 1.51 – 1.56 (m, 4H), 1.71 – 1.79 (m, 6H), 2.35 (s, 3H), 2.57 (t, 2H, J = 7.5 Hz), 2.61 (t, 2H, J = 7.5 Hz), 2.75 – 2.78 (m, 2H), 3.32 (t, 4H, J = 6.5 Hz), 3.55 (t, 2H, J = 6.5 Hz), 7.19 (d, 2H, J = 8.0 Hz), 7.78 (d, 2H, J = 8.0 Hz); 13C NMR (125 MHz, CDCl3) δ –5.1, 18.5, 21.3, 21.6, 25.0, 25.6, 26.2, 32.8, 33.3, 36.6, 37.1, 53.8, 62.9, 107.0, 126.8, 129.2, 141.6, 142.4, 165.8, 176.9; IR (film) cm−1 2928m, 2855m, 1569s, 1470s, 1416s; mass spectrum (ESI): m/e (% relative intensity) 505 (100) (M+H)+; HRMS (ESI) m/e calcd for C27H44N2O3SSiH 505.2915, found 505.2894.
49 (57%): Rf = 0.17 [1:2 hexanes:EtOAc]; orange solid; mp = 79 – 83 °C; 1H NMR (500 MHz, CDCl3) δ 1.69 (pent, 2H, J = 7.5 Hz), 1.82 – 1.85 (m, 4H), 2.40 (t, 2H, J = 7.5 Hz), 2.53 (t, 2H, J = 7.5 Hz), 3.43 (t, 4H, J = 7.5 Hz), 3.81 (s, 3H), 4.24 (s, 2H), 6.85 (d, 2H, J = 9.0 Hz), 7.13 – 7.22 (m, 5H), 7.84 (d, 2H, J = 9.0 Hz); 13C NMR (125 MHz, CDCl3) δ 21.2, 25.6, 32.8, 36.4, 42.6, 53.8, 55.7, 108.2, 113.7, 126.1, 128.4, 128.7, 129.2, 137.0, 137.6, 161.9, 166.9, 172.6; IR (film) cm−1 2953m, 2870m, 1665m, 1594m, 1494m, 1412s; mass spectrum (ESI): m/e (% relative intensity) 425 (100) (M+H)+; HRMS (ESI) m/e calcd for C24H28N2O3SH 425.1894, found 425.1897.
51 (62%): Rf = 0.15 [1:1 hexanes:EtOAc]; yellow solid; mp = 177 – 179 °C; 1H NMR (500 MHz, d8-toluene, 90 °C) spectrum not resolved due to rotamers δ 1.00 – 1.30 (m, 8H), 1.30 – 1.60 (m, 6H), 1.75 – 1.85 (m, 2H), 1.95 – 2.10 (m, 2H), 2.32 (brs, 4H), 2.55 – 2.62 (m, 3H), 2.83 (brs, 6H), 3.05 – 3.13 (m, 1H), 3.31 (brs, 1H), 3.79 (dd, 1H, J = 6.5, 15.0 Hz), 4.65 – 4.75 (m, 2H), 4.76 – 4.85 (m, 2H), 5.52 (brs, 1H), 5.60 – 5.65 (m, 1H), 6.80 – 7.20 (m, 10H), 7.40 (brs, 2H), 7.52 (d, 2H, J = 7.5 Hz), 7.65 (d, 2H, J = 6.5 Hz), 7.65 – 7.80 (m, 2H). 13C NMR (125 MHz, CDCl3) could not obtain spectrum due to rotamers; IR (film) cm−1 2970m, 2924w, 1738s, 1621m, 1527s, 1432s, 1350s; mass spectrum (APCI): m/e (% relative intensity) 508 (100) (M+H)+; HRMS (ESI) m/e calcd for C27H29N3O5SH 508.1901, found 508.1900.
General Procedure for the Preparation of α-Allyl Amidines via Thermal Aza-Claisen Rearrangement
To a flame dried screw-cap vial was added ynamide 8b (62.0 mg, 0.20 mmol), cyclohexylamine (69 μL, 0.60 mmol) and anhyd toluene (2.0 mL). The vial was flushed with nitrogen and heated to 110 °C overnight. When the reaction was judged to be complete by TLC, removal of the solvent in vacuo followed by purification via silica gel flash column chromatography (isocratic eluent: 5:1 hexanes/EtOAc) afforded the amidine 56 (77.4 mg, 0.19 mmol, 94% yield).
56: Rf = 0.33 [5:1 hexanes:EtOAc]; white solid; mp = 106 – 108 °C; 1H NMR (400 MHz, CDCl3) [showing as two rotamers in 2.5:1 ratio] major rotamer δ 0.80 – 1.40 (m, 6H), 1.40 – 1.70 (m, 3H), 1.70 – 1.95 (m, 2H), 2.41 (s, 3H), 2.82 (pent, 1H, J = 7.2 Hz), 3.47 (brs, 1H), 3.64 (t, 1H, J = 7.2 Hz), 4.89 (d, 1H, J = 11.2 Hz), 4.93 (d, 1H, J = 19.2 Hz), 5.55 – 5.68 (m, 1H), 7.21 (brs, 5H), 7.25 (d, 2H, J = 7.6 Hz), 7.78 (d, 2H, J = 8.4 Hz), 8.34 (d, 1H, J = 8.4 Hz); 13C NMR (125 MHz, CDCl3) major rotamer δ 21.7, 24.6, 24.7, 25.1, 33.3, 34.3, 39.3, 48.7, 52.6, 117.3, 126.5, 127.7, 128.0, 129.0, 129.3, 135.7, 139.5, 140.0, 142.7, 166.9; IR (film) cm−1 3320brs, 2933m, 2856m, 1532s, 1451m; mass spectrum (ESI): m/e (% relative intensity) 844 (30) (2M+Na+H), 433 (100) (M+Na)+; HRMS (ESI) m/e calcd for C48H60N4O4S2Na (2M+Na) 843.3949, found 843.3962.
60 (93%): Rf = 0.12 [4:1 hexanes:EtOAc]; colorless oil; 1H NMR (400 MHz, CDCl3) δ 1.03 (s, 18H), 1.15 – 1.30 (m, 3H), 2.18 – 2.32 (m, 2H), 2.32 – 2.61 (m, 1H), 3.45 – 4.20 (m, 8H), 4.99 (d, 1H, J = 16.8 Hz), 5.00 (d, 1H, J = 10.0 Hz), 5.65 – 5.85 (m, 1H), 8.06 (d, 2H, J = 9.2 Hz), 8.30 (d, 2H, J = 9.2 Hz); 13C NMR (125 MHz, CDCl3) δ 11.6, 19.1, 34.0, 35.0, 51.6, 66.7, 116.7, 123.9, 127.3, 137.2, 149.2, 150.6 170.7; IR (film) cm−12947m, 2869m, 1640w, 1529s, 1425m, 1350m; mass spectrum (ESI): m/e (% relative intensity) 532 (100) (M+Na)+; HRMS (ESI) m/e calcd for C24H39N3O5SSiNa 532.2272, found 532.2264.
General Procedure for the Preparation of Nitriles and Unsaturated Imidates via Tandem Thermal Rearrangements
To a flame dried screw-cap vial was added ynamide 8i (100.0 mg, 0.306 mmol) and anhyd toluene (2.5 mL). The vial was flushed with nitrogen and heated to 110 °C overnight. When the reaction was judged to be complete by TLC, removal of the solvent in vacuo followed by purification via silica gel flash column chromatography (isocratic eluent: 10:1 hexanes/EtOAc) afforded the nitrile 63 (52.7 mg, 0.161 mmol, 53% yield).
63: Rf = 0.32 [4:1 hexanes:EtOAc]; waxy white solid; mp = 50 – 53 °C; 1H NMR (400 MHz, CDCl3) δ 3.32 (dd, 1H, J = 7.2, 14.0 Hz), 3.45 (dd, 1H, J = 6.8, 14.0 Hz), 3.85 (s, 3H), 5.18 (dd, 1H, J = 1.2, 10.0 Hz), 5.30 (dd, 1H, J = 1.2, 16.8 Hz), 5.55 (dddd, 1H, J = 6.8, 7.2, 10.0, 16.8 Hz), 6.84 (d, 2H, J = 9.2 Hz), 7.33 – 7.35 (m, 2H), 7.38 – 7.41 (m, 3H), 7.44 (d, 2H, J = 9.2 Hz); 13C NMR (100 MHz, CDCl3) δ 36.1, 56.0, 72.2, 114.2, 116.6, 122.2, 125.1, 128.8, 128.9, 129.0, 129.3, 130.2, 133.2, 164.9; IR (film) cm−1 2946m, 2843m, 2238w, 1641s, 1592s, 1495s, 1365s; mass spectrum (ESI): m/e (% relative intensity 350 (100) (M+Na)+; HRMS (ESI) m/e calcd for C18H17NO3SNa 350.0822, found 350.0818.
64 (53%): Rf = 0.21 [4:1 hexanes:EtOAc]; yellow solid; mp = 125 – 128 °C; 1H NMR (400 MHz, CDCl3) δ 3.39 (dd, 1H, J = 7.2, 14.0 Hz,), 3.49 (dd, 1H, J = 6.4, 14.0 Hz), 5.24 (d, 1H, J = 10.0 Hz), 5.35 (d, 1H, J = 16.8 Hz), 5.60 – 5.50 (m, 1H), 7.44 – 7.34 (m, 5H), 7.71 (d, 2H, J = 8.4 Hz), 8.22 (d, 2H, J = 8.4 Hz); 13C NMR (100 MHz, CDCl3) δ 35.7, 72.6, 115.8, 123.1, 123.9, 127.8, 128.4, 128.9, 129.4, 130.9, 132.3, 139.5, 151.6; IR (film) cm−1 3104m, 2932m, 2850m, 2240w, 1606m, 1530s, 1450m; mass spectrum (ESI): m/e (% relative intensity) 365 (100) (M+Na)+; HRMS (ESI) m/e calcd for C17H14N2O4SNa 365.0566, found 365.0559.
65 (64%): Rf = 0.22 [4:1 hexanes:EtOAc]; white solid; mp = 48 – 50 °C; 1H NMR (400 MHz, CDCl3) δ 2.83 (s, 3H), 3.28 – 3.30 (m, 3H), 5.22 (dd, 1H, J = 1.2, 10.0 Hz), 5.31 (dd, 1H, J = 1.2, 16.8 Hz), 5.55 (ddt, 1H, J = 7.2, 10.0, 16.8 Hz), 7.48 – 7.53 (m, 3H), 7.69 – 7.73 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 35.9, 37.1, 70.8, 116.1, 122.8, 128.4, 128.7, 128.7, 129.7, 130.8; IR (film) cm−1 3008m, 2930m, 2241w, 1642m, 1599m, 1450s; mass spectrum (ESI): m/e (% relative intensity) 258 (100) (M+Na)+; HRMS (ESI) m/e calcd for C12H13NO2SNa 258.0559, found 258.0552.
74 (91%, 1.5:1 dr): Rf = 0.41 [6:1 hexanes:EtOAc], colorless oil; 1H NMR (400 MHz, CDCl3) major isomer δ 1.06 (s, 18H), 1.05 – 1.08 (m, 3H), 1.50 (d, 3H, J = 6.0 Hz), 2.48 (s, 3H), 2.82 – 3.00 (m, 2H), 4.50 (q, 1H, J = 6.0 Hz), 5.12 – 5.22 (m, 1H), 5.28 (dd, 1H, J = 1.2, 17.6 Hz), 5.90 (ddtd, 1H, J = 1.2, 7.2, 10.0, 17.2 Hz), 7.41 (d, 2H, J = 8.4 Hz), 7.90 (d, 2H, J = 8.4 Hz); minor isomer δ 0.98 – 1.02 (m, 21H), 1.60 (d, 3H, J = 6.4 Hz), 2.48 (s, 3H), 2.82 – 3.00 (m, 2H), 4.73 (q, 1H, J = 6.4 Hz), 5.12 – 5.22 (m, 2H), 5.68 – 5.78 (m, 1H), 7.38 (d, 2H, J = 8.4 Hz), 7.90 (d, 2H, J = 8.4 Hz); 13C NMR (125 MHz, CDCl3) both isomers δ 12.9, 13.1, 18.2, 18.3, 18.3, 18.4, 19.2, 21.6, 22.0, 22.0, 34.8, 69.5, 71.9, 116.4, 116.7, 120.3, 121.0, 130.0, 130.1, 130.6, 131.0, 131.1, 131.3, 132.5, 133.7, 146.4, 146.8; IR (film) cm−1 2945m, 2868m, 2256w, 1596m, 1493m, 1330m; mass spectrum (ESI): m/e (% relative intensity) 458 (100) (M+Na)+; HRMS (ESI) m/e calcd for C23H37NO3SSiNa 458.2156, found 458.2171.
75 (50%, 1.3:1 dr): Rf = 0.18 [4:1 hexanes:EtOAc]; colorless oil; 1H NMR (400 MHz, CDCl3) major diastereomer δ 1.47 (d, 3H, J = 6.4 Hz), 1.90 (s, 3H), 2.49 (s, 3H), 2.81 – 3.04 (m, 2H), 5.25 – 5.30 (m, 2H), 5.46 (q, 1H, J = 6.4 Hz), 5.85 – 5.98 (m, 1H), 7.42 (d, 2H, J = 8.4 Hz), 7.89 (d, 2H, J = 8.4 Hz); minor diastereomer δ 1.50 (d, 3H, J = 6.4 Hz), 1.97 (s, 3H), 2.49 (s, 3H), 2.81 – 3.04 (m, 2H), 5.25 – 5.30 (m, 2H), 5.46 (q, 1H, J = 6.4 Hz), 5.85 – 5.98 (m, 1H), 7.43 (d, 2H, J = 8.4 Hz), 7.91 (d, 2H, J = 8.4 Hz); 13C NMR (125 MHz, CDCl3) both diastereomers δ 16.6, 17.8, 21.0, 21.1, 22.1, 31.8, 34.3, 34.7, 68.6, 69.3, 71.6, 115.4, 121.7, 121.7, 129.8, 130.0, 130.2, 130.3, 130.8, 131.2, 132.4, 133.2, 147.0, 147.1, 169.1, 169.2 [missing one sp2 carbon from minor]; IR (film) cm−1 2923m, 2243w, 1752s, 1642m, 1597m, 1374, 1335s; mass spectrum (ESI): m/e (% relative intensity) 344 (100) (M+Na)+; HRMS (ESI) m/e calcd for C16H19NO4SNa 344.0927, found 344.0914.
76 (≥95%, 2.0:1 dr): Rf = 0.23 [8:1 hexanes:EtOAc]; colorless oil; 1H NMR (500 MHz, CDCl3) major diastereomer δ 0.99 (s, 18H), 1.02 (s, 3H), 2.40 (s, 3H), 3.08 (ddd, 1H, J = 1.0, 7.0, 15.0 Hz), 3.18 (ddd, 1H, J = 1.0, 7.0, 15.0 Hz), 5.13 (d, 1H, J = 9.0 Hz), 5.16 (d, 1H, J = 16.0 Hz), 5.54 (s, 1H), 5.74 (ddt, 1H, J = 7.0, 9.0, 16.0 Hz), 7.21 (d, 2H, J = 8.0 Hz), 7.25 – 7.30 (m, 2H), 7.34 – 7.37 (m, 1H), 7.50 (d, 2H, J = 7.0 Hz), 7.63 (d, 2H, J = 8.0 Hz); minor diastereomer δ 0.99 (s, 18H), 1.02 (s, 3H), 2.44 (ddd, 1H, J =1.0, 6.5, 15.5 Hz), 2.45 (s, 3H), 2.64 (ddd, 1H, J = 1.0, 6.5, 15.5 Hz), 4.81 (d, 1H, J = 17.0 Hz), 4.93 (d, 1H, J = 10.0 Hz); 5.45 (ddt, 1H, J = 6.5, 10.0, 17.0 Hz), 5.81 (s, 1H), 7.25 – 7.30 (m, 2H), 7.31 (d, 2H, J = 8.0 Hz), 7.34 – 7.37 (m, 1H), 7.54 – 7.59 (m, 2H), 7.89 (d, 2H, J = 8.0 Hz); 13C NMR (125 MHz, CDCl3) both diastereomers δ 13.02, 13.12, 18.19, 18.23, 21.95, 22.02, 34.99, 35.38, 72.86, 72.87, 75.50, 75.66, 116.02, 120.06, 120.84, 128.29, 129.14, 129.24, 129.26, 129.43, 129.56, 129.62, 129.65, 129.89, 130.52, 130.89, 130.93, 131.25, 131.31, 133.47, 138.58, 145.94 [missing two sp2 signals from minor]; IR (film) cm−1 2945m, 2893m, 2867m, 2240w, 1639w, 1596m, 1494m, 1365m; mass spectrum (ESI): m/e (% relative intensity) 520 (100) (M+Na)+; HRMS (ESI) m/e calcd for C28H39NO3SSiNa 520.2313, found 520.2322.
77-major (69% for both, 1.5:1 dr): Rf = 0.28 [6:1 hexanes:EtOAc]; colorless oil; 1H NMR (500 MHz, CDCl3) δ 1.04 (s, 9H), 2.38 (dd, 1H, J = 7.0, 15.5 Hz), 2.48 (s, 3H), 2.64 (dd, 1H, J = 7.0, 15.5 Hz), 4.76 (dd, 1H, J = 1.5, 17.0), 4.93 (dd, 1H, J = 1.5, 10.0 Hz), 5.27 (ddt, 1H, J = 7.0, 10.0, 17.0 Hz), 6.30 (s, 1H), 7.35 – 7.38 (m, 3H), 7.41 (d, 2H, J = 8.5 Hz), 7.46 – 7.49 (m, 2H), 7.93 (d, 2H, J = 8.5 Hz); 13C NMR (125 MHz, CDCl3) δ 22.0, 26.8, 36.1, 38.9, 69.8, 73.6, 114.9, 120.8, 128.5, 129.1, 129.2, 130.1, 130.3, 131.1, 133.3, 134.8, 146.9, 176.0; IR (film) cm−1 3068w, 2974m, 2934m, 2873w, 2242w, 1739s, 1595m, 1494m, 1336s; mass spectrum (ESI): m/e (% relative intensity) 448 (100) (M+Na)+; HRMS (ESI) m/e calcd for C24H27NO4SNa 448.1553, found 448.1558.
77-minor: Rf = 0.35 [6:1 hexanes:EtOAc]; colorless oil; 1H NMR (500 MHz, CDCl3) δ 1.27 (s, 9H), 2.46 (s, 3H), 3.03 (dd, 1H, J = 7.0, 15.0 Hz), 3.16 (dd, 1H, J = 7.0, 15.0 Hz), 5.22 (d, 1H, J = 9.0 Hz), 5.23 (d, 1H, J = 17.5 Hz), 5.81 – 5.84 (m, 1H), 6.22 (s, 1H), 7.30 – 7.37 (m, 7H), 7.75 (d, 2H, J = 8.5 Hz); 13C NMR (125 MHz, CDCl3) δ 22.1, 27.2, 34.7, 39.2, 70.0, 73.0, 115.4, 121.5, 128.5, 128.7, 129.8, 130.4, 131.2, 132.2, 134.4, 146.7, 175.8; IR (film) cm−1 3069w, 2978m, 2937m, 2874w, 2244w, 1746s, 1597m, 1495m, 1338s; mass spectrum (ESI): m/e (% relative intensity) 448 (100) (M+Na)+; HRMS (ESI) m/e calcd for C24H27NO4SNa 448.1553, found 448.1566.
79 (74%): Rf = 0.28 [2:1 hexanes:EtOAc]; white solid; mp = 80 – 84 °C; 1H NMR (400 MHz, CDCl3) δ 1.70 (d, 3H, J = 7.2 Hz), 2.43 (s, 3H), 2.93 (d, 2H, J = 6.4 Hz), 3.18 (s, 6H), 4.92 (dq, 1H, J = 2.0, 10.2 Hz), 4.95 (dq, 1H, J = 2.0, 16.8 Hz), 5.64 (ddt, 1H, J = 6.4, 10.2, 16.8 Hz), 6.06 (q, 1H, J = 7.2 Hz), 7.31 (d, 2H, J = 8.4 Hz), 7.98 (d, 2H, J = 8.4 Hz); 13C NMR (100 MHz, CDCl3) δ 14.2, 22.0, 32.3, 116.4, 129.4, 129.6, 133.4, 134.1, 134.3, 136.3, 145.3, 169.0 [missing NMe2 carbon signal]; IR (film) cm−1 2927m, 2251w, 1703s, 1641m, 1598m, 1495m, 1448m, 1359s; mass spectrum (ESI): m/e (% relative intensity) 373 (100) (M+Na)+; HRMS (ESI) m/e calcd for C17H22N2O4SNa 373.1193, found 373.1188. mass spectrum (ESI): m/e (% relative intensity) 373 (100) (M+Na)+; HRMS (ESI) m/e calcd for C17H22N2O4SNa 373.1193, found 373.1188.
81 (30%): Rf = 0.41 [6:1 hexanes:EtOAc]; white solid; mp = 65 – 67 °C; 1H NMR (400 MHz, CDCl3) δ 1.46 (s, 9H), 2.43 (s, 3H), 3.28 (d, 2H, J = 5.6 Hz), 5.08 (d, 1H, J = 10.4 Hz), 5.11 (d, 1H, J = 17.2 Hz), 5.89 (ddt, 1H, J = 5.6, 10.4, 17.2 Hz), 7.31 (d, 2H, J = 8.4 Hz), 7.40 (s, 6H), 7.54 (s, 1H), 7.85 (d, 2H, J = 8.4 Hz); 13C NMR (125 MHz, CDCl3) δ 21.9, 27.4, 32.2, 39.9, 116.9, 127.4, 128.9, 129.6, 130.0, 130.0, 132.2, 134.4, 134.9, 138.3, 143.7, 144.0, 162.1, 174.4; IR (film) cm−1 2966m, 1765s, 1740s, 1601s, 1481m, 1326s; mass spectrum (ESI): m/e (% relative intensity) 448 (100) (M+Na)+; HRMS (ESI) m/e calcd for C24H27NO4SNa 448.1553, found 448.1543.
Supplementary Material
Scheme 8.

A Tandem Sigmatropic Rearrangement.
Acknowledgments
Authors thank the NIH [GM066055] for financial support. KAD thanks American Chemical Society for a Division of Medical Chemistry Predoctoral Fellowship. HD thanks China Scholarship Council for a Visiting Scholar Fellowship.
Footnotes
SUPPORTING INFORMATION AVAILABLE. Experimental procedures, characterization data for all new compounds, and 1H/13C NMR spectra are available free of charge via Internet at http://pubs.acs.org.
References
- 1.Boyd GV. In: In The Chemistry of Amidines and Imidates. Patai S, Rappoport Z, editors. 2 Chapter 8 Wiley; New York: 1991. [Google Scholar]
- 2.Dunn PJ. In: Compreh Org Funct Group Transform II, Amidines and N-Substituted Amidines. Katritzky Alan R, Taylor Richard JK., editors. Vol. 5. Pfizer Global Research and Development; Sandwich, UK: 2005. pp. 655–699. [Google Scholar]
- 3.Greenhill JV, Lue P. Prog Med Chem. 1993;30:203. doi: 10.1016/s0079-6468(08)70378-3. [DOI] [PubMed] [Google Scholar]
- 4.For a leading review on amidine derivatives serving as selective muscarinic agonists in the treatment of Alzheimer’s diseases, see: Messer WS, Jr, Dunbar PG. Muscarinic Agonists Treat Alzheimer’s Dis. 1996. pp. 131–153.
- 5.(a) Traveras AG, Chao J, Biju PJ, Yu Y, Fine JS, Hipkin W, Aki CJ, Merritt JR, Li G, Baldwin JJ, Lai G, Wu M, Hecker EA. 2004033440. WO. 2004; (b) Varghese J, Maillard M, Jagodzinska B, Beck JP, Gailunas A, Fang L, Sealy J, Tenbrink R, Freskos J, Mickelson J, Samala L, Hom R. 2003040096. WO. 2003; (c) Ito K, Spears GW, Yamada A, Toshima M, Kato M. 2001220375. JP. 2001
- 6.(a) Edwards PD, Albert JS, Sylvester M, Aharony D, Andisik D, Callaghan O, Campbell JB, Carr RA, Chessari G, Congreve M, Frederickson M, Folmer RHA, Geschwindner S, Koether G, Kolmodin K, Krumrine J, Mauger RC, Murray CW, Olsson LL, Patel S, Spear N, Tian G. J Med Chem. 2007;50:5912. doi: 10.1021/jm070829p. [DOI] [PubMed] [Google Scholar]; (b) Peterlin-Masic L, Kikelj D. Tetrahedron. 2001;57:7073. [Google Scholar]
- 7.Clement B, Immel M, Raether W. Arzneimittel-Forschung. 1992;42:1497. [PubMed] [Google Scholar]
- 8.For some examples of amidine synthesis, see: Shang Y, He X, Hu J, Wu J, Zhang M, Yu S, Zhang Q. Adv Synth Catal. 2009;351:2709.She J, Jiang Z, Wang Y. Synlett. 2009;12:2023.Yu RT, Rovis T. J Am Chem Soc. 2008;130:3262. doi: 10.1021/ja710065h.Wang J, Xu F, Cai T, Shen Q. Org Lett. 2008;10:445. doi: 10.1021/ol702739c.Malik H, Frederic B, Alexandre M, Jean-Jacques B. Org Biom Chem. 2006;4:3142.Katritzky AR, Cai C, Singh SK. J Org Chem. 2006;71:3375. doi: 10.1021/jo052443x.Kumagai N, Matsunaga S, Shibasaki M. Angew Chem, Int Ed. 2004;43:478. doi: 10.1002/anie.200352750.
- 9.Pinner A, Klein F. Ber. 1877;10:1889, 1878, 11, 1475. [Google Scholar]
- 10.Caron S, Wei L, Douville J, Ghosh A. J Org Chem. 2010;75:945. doi: 10.1021/jo902159z. [DOI] [PubMed] [Google Scholar]
- 11.(a) Tetala KKR, Whitby RJ, Light ME, Hurtshouse MB. Tetrahedron Lett. 2004;45:6991. [Google Scholar]; (b) Saluste CG, Whitby RJ, Furber M. Angew Chem Int Ed. 2000;39:4156. [PubMed] [Google Scholar]
- 12.Yoo EJ, Ahlquist M, Bae I, Sharpless BK, Fokin VV, Chang S. J Org Chem. 2008;73:5520. doi: 10.1021/jo800733p. [DOI] [PubMed] [Google Scholar]
- 13.Bae I, Han H, Chang S. J Am Chem Soc. 2005;127:2038. doi: 10.1021/ja0432968.Cho SH, Yoo EJ, Bae I, Chang S. J Am Chem Soc. 2005;127:16046. doi: 10.1021/ja056399e.Yoo EJ, Bae I, Cho SH, Han H, Chang S. Org Lett. 2006;8:1347. doi: 10.1021/ol060056j.Kim SH, Jung DY, Chang S. J Org Chem. 2007;72:9769. doi: 10.1021/jo7016247.Cho SH, Chang S. Angew Chem, Int Ed. 2008;47:2836. doi: 10.1002/anie.200705940.Kim J, Lee SY, Lee J, Do Y, Chang S. J Org Chem. 2008;73:9454. doi: 10.1021/jo802014g.For an intramolecular addition, see: Chang S, Lee M, Jung DY, Yoo EJ, Cho SH, Han SK. J Am Chem Soc. 2006;128:12366. doi: 10.1021/ja064788i.
- 14.For a study using ynamides, see: Kim JY, Kim SH, Chang S. Tetrahedron Lett. 2008;49:1745.
- 15.For some more recent examples, see: Yavari I, Ahmadian S, Ghazanfarpur-Darjani M, Solgi Y. Tetrahedron Lett. 2011;52:668.Kim J, Lee SJ, Zhu S. Synthesis. 2011:1142.
- 16.For current leading reviews on chemistry of ynamides, see: Evano G, Coste A, Jouvin K. Angew Chem Int Ed. 2010;49:2840. doi: 10.1002/anie.200905817.DeKorver KA, Li H, Lohse AG, Hayashi R, Lu Z, Zhang Y, Hsung RP. Chem Rev. 2010;110:5064. doi: 10.1021/cr100003s.
- 17.For other reviews on ynamides, see: Zificsak CA, Mulder JA, Hsung RP, Rameshkumar C, Wei LL. Tetrahedron. 2001;57:7575.Katritzky AR, Jiang R, Singh SK. Heterocycles. 2004;63:1455.For a special issue dedicated to the chemistry of ynamides, see: Tetrahedron-Symposium-In-Print: “Chemistry of Electron-Deficient Ynamines and Ynamides”. Tetrahedron. 2006;62(16)Ackermann L, Potukuchi HK. Org Biomol Chem. 2010;8:4503. doi: 10.1039/c0ob00212g.
- 18.For reports in 2010–2011 that appeared after the above reviews in reference 16, see: Balieu S, Toutah K, Carro L, Chamoreau L-M, Rouselière H, Courillon C. Tetrahedron Lett. 2011;52:2876.Fadel A, Legrand F, Evano G, Rabasso N. Adv Synth Catal. 2011;353:263.Schotes C, Mezzetti A. Angew Chem. 2011;123:3128. doi: 10.1002/anie.201007753.Barbazanges M, Meyer C, Cossy J, Turner P. Chem-Eur J. 2011;17:4480. doi: 10.1002/chem.201003265.Pizzetti M, Russo A, Petricci E. Chem–Eur J. 2011;17:4523. doi: 10.1002/chem.201100447.Garcia P, Evanno Y, George P, Sevrin M, Ricci G, Malacria M, Aubert C, Gandon V. Org Lett. 2011;13:2030. doi: 10.1021/ol200417p.Kramer S, Friis SD, Xin Z, Odabachian Y, Skrydstrup T. Org Lett. 2011;13:1750. doi: 10.1021/ol200269h.Li C, Zhang L. Org Lett. 2011;13:1738. doi: 10.1021/ol2002607.Wang YP, Danheiser RL. Tetrahedron Lett. 2011;52:2111. doi: 10.1016/j.tetlet.2010.11.002.Mak XY, Crombie AL, Danheiser RL. J Org Chem. 2011;76:1852. doi: 10.1021/jo2000308.Davies PW, Cremonesi A, Martin N. Chem Commun. 2011:379. doi: 10.1039/c0cc02736g.Xu C-F, Mei Xu M, Jia YX, Li C-Y. Org Lett. 2011;13:1556. doi: 10.1021/ol200270t.Chen Z, Zheng D, Wu J. Org Lett. 2011;13:848. doi: 10.1021/ol102775s.Shindoh N, Takemoto Y, Taksu K. Heterocycles. 2011;82:1133.Saito N, Katayama T, Sato K. Heterocycles. 2011;82:1181.Wakamatsu H, Takeshita M. Synlett. 2010:2322.Valenta P, Carroll PJ, Walsh PJ. J Am Chem Soc. 2010;132:14179. doi: 10.1021/ja105435y.Lu B, Li C, Zhang L. J Am Chem Soc. 2010;132:14070. doi: 10.1021/ja1072614.Zhang C, Jiao N. J Am Chem Soc. 2010;132:28. doi: 10.1021/ja908911n.Li H, Antoline JE, Yang JH, Al-Rashid ZF, Hsung RP. New J Chem. 2010;34:1309. doi: 10.1039/c0nj00063a.Li H, Hsung RP, De Korver KA, Wei Y. Org Lett. 2010;12:3780. doi: 10.1021/ol101418d.Xu H, Zhang Y, Huang J, Chen W. Org Lett. 2010;12:3704. doi: 10.1021/ol101563f.Kramer S, Madsen JLH, Rottländer M, Skrydstrup T. Org Lett. 2010;12:2758. doi: 10.1021/ol1008685.Banerjee B, Litvinov DN, Kang J, Bettale JD, Castle SL. Org Lett. 2010;12:2650. doi: 10.1021/ol1008679.Gourdet B, Rudkin ME, Lam HW. Org Lett. 2010;12:2554. doi: 10.1021/ol100769p.Jia W, Jiao N. Org Lett. 2010;12:2000. doi: 10.1021/ol1004615.Li CW, Pati K, Lin GY, Sohel SMA, Hung HH, Liu RS. Angew Chem Int Ed. 2010;49:9891. doi: 10.1002/anie.201004647.Boyce GR, Johnson JS. Angew Chem Int Ed. 2010;49:8930. doi: 10.1002/anie.201003470.Wakamatsu H, Sakagami M, Hanata M, Takeshita M, Mori M. Macromol Symp. 2010;293:5.Sato A, Yorimitsu H, Oshima K. Bull Korean Chem Soc. 2010;31:570.Poloukhtine A, Rassadin V, Kuzmin A, Popik VV. J Org Chem. 2010;75:5953. doi: 10.1021/jo101238x.Burley GA, Davies DL, Griffith GA, Lee M, Singh K. J Org Chem. 2010;75:980. doi: 10.1021/jo902466f.Yamasaki R, Terashima N, Sotome I, Komagawa S, Saito S. J Org Chem. 2010;75 doi: 10.1021/jo902251m.Grimster NP, Wilton DAA, Chan LKM, Godfrey CRA, Green C, Owen DR, Gaunt MJ. Tetrahedron. 2010;66:6429.Gourdet B, Smith DL, Lam HW. Tetrahedron. 2010;66:6026.Jouvin K, Couty F, Evano G. Org Lett. 2010;12:3272. doi: 10.1021/ol101322k.Gourdet B, Lam HW. Angew Chem Int Ed. 2010;122:8915. doi: 10.1002/anie.201004328.Basheer A, Marek I. Beilstein J Org Chem. 2010;6:77. doi: 10.3762/bjoc.6.77.Felpin FX, Fouquet E. Chem-Eur J. 2010;16:12440. doi: 10.1002/chem.201001377.Hashmi ASK, Pankajakshan S, Rudolph M, Enns E, Bander T, Rominger F, Frey W. Adv Synth Catal. 2009;351:2855.Garcia P, Harrak Y, Diab L, Cordier P, Ollivier C, Gandon V, Malacria M, Fensterbank L, Aubert C. Organic Lett. ASAP; 2011.
- 19.Zhang Y, DeKorver KA, Lohse AG, Zhang YS, Huang J, Hsung RP. Org Lett. 2009;11:899. doi: 10.1021/ol802844z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.For leading references on related Cu-complexes generated from triazolyl copper intermediates, see: Gerard B, Ryan J, Beeler AB, Porco JA., Jr Tetrahedron. 2006;62:6405.Wu YM, Deng J, Li Y, Chen QY. Synthesis. 2005:1314.Cassidy MP, Raushel J, Fokin VV. Angew Chem, Int Ed. 2006;45:3154. doi: 10.1002/anie.200503805.and see footnote 11.
- 21.For leading examples of trapping ynamido-metal complexes, see: (a) See references 12–14. Cui SL, Lin XF, Wang YG. Org Lett. 2006;8:4517. doi: 10.1021/ol061685w.Xu X, Cheng D, Li J, Guo H, Yan J. Org Lett. 2007;9:1585. doi: 10.1021/ol070485x.Jin Y, Fu H, Yin Y, Jiang Y, Zhao Y. Synlett. 2007:901.Cui SL, Wang J, Wang YG. Org Lett. 2007;9:5023. doi: 10.1021/ol702241e.Cui SL, Wang J, Wang YG. Org Lett. 2008;10:1267. doi: 10.1021/ol800160q.For an earlier study on trapping of ynamido-lithium complexes, see: Fromont C, Masson S. Tetrahedron. 1999;55:5405.
- 22.For leading reviews on synthesis of ynamides, see: Tracey MR, Hsung RP, Antoline JA, Kurtz KCM, Shen L, Slafer BW, Zhang Y. Science of Synthesis, Houben-Weyl Methods of Molecular Transformations. In: Weinreb Steve M., editor. Vol. 21.4. Georg Thieme Verlag KG; Stuttgart, Germany: 2005. Mulder JA, Kurtz KCM, Hsung RP. Synlett. 2003:1379.
- 23.For org syn procedures, see: Kohnen AL, Dunetz JR, Danheiser RL. Organic Syn. 2007;84:88.Sagamanova IK, Kurtz KCM, Hsung RP. Organic Syn. 2007;84:359.
- 24.For reports on synthesis of ynamides, see: Frederick MO, Mulder JA, Tracey MR, Hsung RP, Huang J, Kurtz KCM, Shen L, Douglas CJ. J Am Chem Soc. 2003;125:2368. doi: 10.1021/ja021304j.Dunetz JR, Danheiser RL. Org Lett. 2003;5:4011. doi: 10.1021/ol035647d.Zhang Y, Hsung RP, Tracey MR, Kurtz KCM, Vera EL. Org Lett. 2004;6:1151. doi: 10.1021/ol049827e.Rodríguez D, Castedo L, Saá C. Synlett. 2004:377.Rodríguez D, Castedo L, Saá C. Synlett. 2004:783.Riddell N, Villeneuve K, Tam W. Org Lett. 2005;7:3681. doi: 10.1021/ol0512841.Couty S, Barbazanges M, Meyer C, Cossy J. Synlett. 2005:905.Zhang X, Zhang Y, Huang J, Hsung RP, Kurtz KCM, Oppenheimer J, Petersen ME, Sagamanova IK, Tracey MR. J Org Chem. 2006;71:4170. doi: 10.1021/jo060230h.Rodríguez D, Martinez-Esperon MF, Castedo L, Saá C. Synlett. 2007:1963.Hamada T, Ye X, Stahl SS. J Am Chem Soc. 2008;130:833. doi: 10.1021/ja077406x.Dooleweerdt K, Birkedal H, Ruhland T, Skrydstrup T. J Org Chem. 2008;73:9447. doi: 10.1021/jo801935b.Coste A, Karthikeyan G, Couty F, Evano G. Angew Chem In Ed. 2009;48:4381. doi: 10.1002/anie.200901099.Yao B, Liang Z, Niu T, Zhang Y. J Org Chem. 2009;74:4630. doi: 10.1021/jo900595c.Coste A, Couty F, Evano G. Org Lett. 2009;11:4454. doi: 10.1021/ol901831s.Jouvin K, Couty F, Evano G. Org Lett. 2010;12:3272. doi: 10.1021/ol101322k.
- 25.DeKorver KA, Hsung RP, Lohse AG, Zhang Y. Org Lett. 2010;12:1840. doi: 10.1021/ol100446p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.For a leading reference on relative nucleophilicity of amines, see: Brotzel F, Chu YC, Mayr H. J Org Chem. 2007;72:3679. doi: 10.1021/jo062586z.
- 27.For a leading reference on xantphos, see: Kranenburg M, van der Burgt YEM, Kamer PCJ, van Leeuwen PWNM. Organometallics. 1995;14:3081.
- 28.For a rationale on the usage of bidentate xantphos in promoting reductive elimination with its unique bite angle, see: Hartwig observed that in comparison with mono-dentate phosphine ligands, the usage of bidentate ligands such as xantphos leads to a much faster amidative cross-coupling. This is presumably due to the ability of amido-type carbonyl groups to engage in tight complexation with the palladium metal. As a result, when using xantphos, its unique bite angle promotes reductive elimination. See: Fujita KI, Yamashita M, Puschmann F, Alvarez-Falcon MM, Incarvito CD, Hartwig JF. J Am Chem Soc. 2006;128:9044. doi: 10.1021/ja062333n.
- 29.For leading references on X-phos, see: Huang X, Anderson KW, Zim D, Jiang L, Klapars A, Buchwald SL. J Am Chem Soc. 2003;125:6653. doi: 10.1021/ja035483w.Barder TE, Buchwald SL. J Am Chem Soc. 2007;129:12003. doi: 10.1021/ja073747z.
- 30.For leading reviews on aza-Claisen rearrangements, see: Majumdar KC, Bhayyacharyya T, Chattopadhyay B, Nandi RK. Synthesis. 2009:2117.Nubbemeyer U. Top Curr Chem. 2005;244:149.
- 31.For general reviews on Claisen rearrangements, see: Castro AMM. Chem Rev. 2004;104:2939. doi: 10.1021/cr020703u.Ito H, Taguchi T. Chem Soc Rev. 1999;28:43.Enders D, Knopp M, Schiffers R. Tetrahedron Asym. 1996;7:1847.Wipf P. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, editors. Vol. 5. Pergamon Press; Oxford: 1991. p. 827.Hill RK. In: Asymmetric Synthesis. Morrison JD, editor. Academic Press; New York: 1984.
- 32.For some examples of aza-Claisen rearrangements, see: Majumdar KC, Samanta S, Chattopadhyay B, Nandi RK. Synthesis. 2010:863.Cheung LLW, Yudin AK. Org Lett. 2009;11:1281. doi: 10.1021/ol900118d.Cant AA, Bertrand GHV, Henderson JL, Roberts L, Greaney MF. Angew Chem Int Ed. 2009;48:5199. doi: 10.1002/anie.200901410.Yasui H, Yorimitsu H, Oshima K. Chem Lett. 2008;37:40.Walters MA. J Am Chem Soc. 1994;116:11618.Jolidon S, Hansen HJ. Helv Chim Acta. 1977;60:978.For some earlier work of palladium promoted aza-Claisen-type rearrangements using N-allylthioamide, allylimidates, or allyl carbamates, see: Overman LE. Acc Chem Res. 1980;13:218.Overman LE. Angew Chem, Int Ed Engl. 1984;23:579.Tamaru Y, Kagotani M, Yoshida Z. J Org Chem. 1985;50:764.Overman LE, Donde Y. J Am Chem Soc. 1999;121:2933.Lei A, Lu X. Org Lett. 2000;2:2357. doi: 10.1021/ol006130u.
- 33.DeKorver KA, North TD, Hsung RP. Synlett. 2010:2397. doi: 10.1055/s-0030-1258544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.For examples of imidates serving as prodrugs, see: Maryanoff BE, McComsey DF, Costanzo MJ, Yabut SC, Lu T, Player MR, Giardino EC, Damiano BP. Chem Biol Drug Des. 2006;68:29. doi: 10.1111/j.1747-0285.2006.00408.x.Poon SF, Stock N, Payne JE, McGuire AR, Stearns B, Yang X, Chen W, Munoz B, Smith ND. Bioorg Med Chem Lett. 2005;15:2259. doi: 10.1016/j.bmcl.2005.03.009.
-
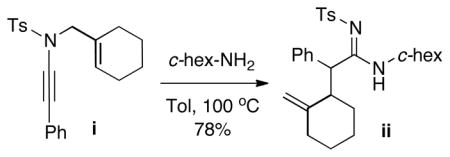
35.Ynamide i with substitution along the allyl group was also tolerated, but due to the complication of diastereomers and rotamers of the amidine product ii, this effort was not further pursued.
- 36.For a recent account on a related thermal transformation using ynol ethers, see: Sosa JR, Tudjarian AA, Minehan TG. Org Lett. 2008;10:5091. doi: 10.1021/ol802147h.Tudjarian AA, Minehan TG. J Org Chem. 2011;76:3576. doi: 10.1021/jo200271s.
- 37.For a related 1,3-sulfonyl shift, see: Bendikov M, Duong HM, Bolanos E, Wudl F. Org Lett. 2005;7:783. doi: 10.1021/ol0477327.
- 38.For documentation of silicon-stabilization of ketenes and ketenimines, see: Brady WT, Saidi K. J Org Chem. 1990;55:4215.
- 39.For reviews on the chemistry of ketenimines, see: Krow GR. Angew Chem, Int Ed. 1971;10:435.Gambaryan NP. Usp Khim. 1976;45:1251.Dondoni A. Heterocycles. 1980;14:1547.Barker MW, McHenry WE. In: In The Chemistry of Ketenes, Allenes and Related Compounds. Patai S, editor. Part 2. Wiley-Interscience; Chichester, UK: 1980. pp. 701–720.Alajarin M, Vidal A, Tovar F. Targets Heterocycl Syst. 2000;4:293.
- 40.For leading references on synthetic applications of nitriles, see: Fleming FF, Liu W. Eur J Org Chem. 2009:699.Fleming FF, Wei G, Steward OW. J Org Chem. 2008;73:3674. doi: 10.1021/jo8001062.Mermerian AH, Fu GC. Angew Chem Int Ed. 2005;44:949. doi: 10.1002/anie.200461886.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.