

Abstract
Resonance Raman (RR) studies of intermediates generated by cryoreduction of the oxyferrous complex of the D251N mutant of cytochrome P450cam (CYP101) are reported. Owing to the fact that proton delivery to the active site is hindered in this mutant, the unprotonated peroxo-ferric intermediate is observed as the primary species after radiolytic reduction of the oxy-complex in frozen solutions at 77 K. Inasmuch as previous EPR and ENDOR studies have shown that annealing of this species to ~180 K results in protonation of the distal oxygen atom to form the hydroperoxo intermediate, this system has been exploited to permit direct RR interrogation of the changes in the Fe-O and O-O bonds caused by the reduction and subsequent protonation. Our results show that the ν(O-O) mode decreases from a “superoxo-like” frequency near ~1130 cm−1 to 792 cm−1 upon reduction. The latter frequency, as well as its lack of sensitivity to H/D exchange, is consistent with a heme-bound peroxide formulation. This species also exhibits a ν(Fe-O) mode, whose 553 cm−1 frequency is higher than that observed for the non-reduced oxy P450 precursor (537 cm−1), implying a strengthened Fe-O linkage upon reduction. Upon subsequent protonation, the resulting Fe-O-OH fragment exhibits a lowered ν(O-O) mode at 774 cm−1, while the ν(Fe-O) increases to 564 cm−1, both modes exhibiting downshifts upon H/D exchange, as expected for a hydroperoxo-ferric formulation. These experimental RR data are compared with those previously acquired for the wild-type protein and the shifts observed upon reduction and subsequent protonation are discussed with reference to theoretical predictions.
Keywords: Peroxo-ferric heme complex, Hydroperoxo-ferric heme complex, Cytochrome P450, Cryoradiolysis, Oxygen activation
Introduction
Activation of molecular oxygen by the heme enzymes such as the cytochromes P450, nitric oxide synthase and heme oxygenase represents a sequence of coordinated events which involve two electron transfers interspersed by the binding of a dioxygen molecule to the heme iron1–3 (species [2] – [5], Scheme 1). As a result, the transient peroxo-ferric complex [5a] is formed, which is readilyprotonated to form a hydroperoxo -ferric intermediate [5b], often termed Compound 0. The evaluation of electronic structure of these iron-oxygen intermediates and of the pathways for proton delivery is at the heart of understanding how Nature can utilize atmospheric dioxygen for controlled biological oxidations.
Scheme 1.
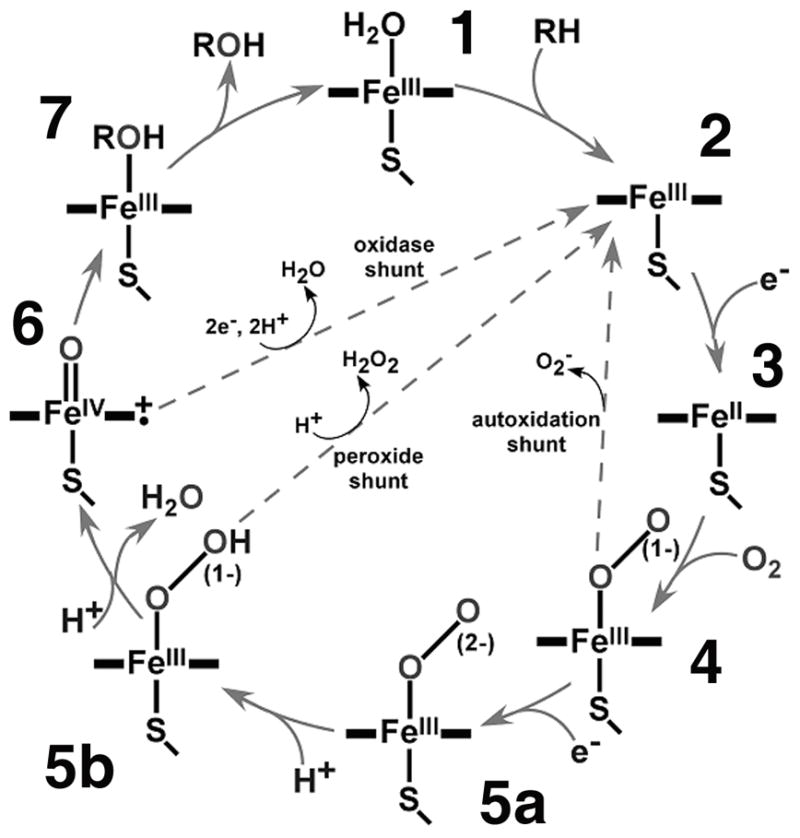
The properties of (hydro)peroxo-ferric intermediates on the path of oxygen activation in heme and non-heme systems are the subject of continuing interest in inorganic chemistry and biochemistry.4–9 Using X-ray crystallography and multiple spectroscopic methods, a great amount of information has been obtained on the series of mononuclear non-heme iron complexes where a (hydro)peroxide is coordinated in an end-on (η1), or side-on (η2) fashion. In particular, the results of resonance Raman spectroscopic studies revealed the essential role played by the spin state of Fe3+ in the strength of Fe-O and O-O bond strengths in the peroxo/hydroperoxo-ferric complexes.4,10–15 Recently, considerable progress has been achieved in characterization of similar intermediates in heme proteins obtained by using radiolytic reduction of the oxy-ferrous precursor frozen at cryogenic temperatures. Application of EPR, ENDOR, and Mössbauer spectroscopy provided detailed information on the structure and stability of these complexes in Hb, Mb, P450, HO and on the variety of protonation pathways and reactivity of these intermediates.16–24 Several high resolution X-ray structures of peroxo and hydroperoxo intermediates were also recently obtained using cryogenic radiolysis, highlighting the importance of active-site details (including solvent) in the transformation of these intermediates. 25–28 Detailed understanding of the mechanism of formation of [5a] and [5b] and their role in heme enzyme catalysis is also supported by many advanced theoretical studies,29–34 which provide a consistent framework for the general picture of oxygen activation in heme enzymes.
While a great deal of information is available on the role of iron and porphyrin in the stepwise reduction of dioxygen, and on the specific role of sequential protonation steps of the distal oxygen atom of coordinated dioxygen, there is still a considerable lack of information in heme enzymes on the properties of the reduced dioxygen moiety itself, [5a] and [5b], Scheme 1. Theoretical calculations predict a significant lengthening of the O-O bond in [5a] as compared to the oxy-ferrous complex [4] and a further increase in O-O distance in [5b]. 29–34 These predictions were supported qualitatively by the recent structures of [5a] in Mb27,28 and [5b] in CPO.26 Note that the resolution of these X-ray structures was not high enough to distinguish between [5a] and [5b], and the assignment of protonation state of peroxo/hydroperoxo moiety in these works was based on additional quantum chemical modeling. However, these structures cannot be directly compared since they are obtained in different proteins with different axial ligands trans to coordinated peroxide, His in Mb and Cys in CPO.
The use of an active-site mutant of cytochrome P450 CYP101, in which proton delivery is hampered, provides a unique opportunity to isolate the unprotonated peroxo-ferric intermediate. As a result, a definitive comparison of Fe-O-O− and Fe-O-OH fragments is made possible in the present work, wherein we report the first resonance Raman study of a cryoreduced oxyferrous complex prepared in the D251N mutant of CYP101. Because the first proton delivery in this mutant is perturbed,17,35–39 the unprotonated peroxo-ferric intermediate [5a] is observed as the primary species after radiolytic reduction of the oxy-complex in the presence of the substrate camphor at 77 K.17 Since camphor hydroxylation in this mutant is fully coupled, although much slower,35,36 and the hydroperoxo-ferric complex [5b] was identified by EPR and ENDOR after annealing of [5a] at 180 K,17 this system represents an ideal opportunity to monitor directly the changes in Fe-O and O-O bonds caused by the first protonation event and to study both peroxo-ferric [5a] and hydroperoxo-ferric [5b] intermediates using cryogenic resonance Raman spectroscopy.
Methods
Mutation of CYP101, expression of the mutant D251N CYP101 in E. coli and purification were performed as described. 40 Mutagenesis of the CYP101 gene (pCamT7) was performed using the Stratagene Quikchange kit following the manufacturer’s recommendations. Mutations were confirmed through sequencing of the entire CYP101 gene (ACGT, Wheeling, IL). In order to construct the CYP101 gene with the N-terminal hexahistidine tag, (pT7-P450 His), the ends of the gene were modified using PCR to introduce new restriction cites (Nde 1 and Hind III), and the resulting gene was cloned into pET28(a) vector (Novagen, Madison, WI). Protein was expressed in BL21 (DE3) cells (Stratagene, La Jolla, CA) grown overnight in the presence of 0.5 mM δ-aminolevulinic acid. After cell sonication, resuspension in 50 mM KPi pH 7.4, 150 mM KCl (buffer A), and ultracentrifugation the protein extract was loaded on a Ni-NTA column (Amersham, Piscataway, NJ), washed with buffer A in the presence of 50 mM imidazole, and eluted with the same buffer supplemented with 20 mM EDTA. The resulting red fractions were dialyzed in 50 mM KPi, 400 μM D-camphor, 20 mM β-mercaptoethanol, pH 7.4 (designated as RB buffer) overnight at 4 °C. Solid ammonium sulfate was added to 30% saturation over 30 minutes and the protein was subsequently loaded onto a Phenyl-Sepharose CL6B column equilibrated in the same buffer and eluted in a linear gradient from 30% to 0% ammonium sulfate. Resulting fractions with an Rz-value (A391/A280) greater than 1.45 were judged as pure and concentrated using an Amicon cell.
All samples for Raman and UV-Vis spectroscopy contained 100 mM KPi pH 7.4, 100 mM KCl, and 0.6 mM d-camphor. Samples of oxy-ferrous CYP101 for the resonance Raman spectroscopy were prepared as described previously41,42 with minor modifications listed below. Concentrated solutions of ferric CYP101 were deaerated under a stream of Ar gas, mixed with the degassed 9:1 v/v mixture of glycerol and 1 M phosphate buffer in an anaerobic chamber (Coy Lab Products, Grass Lake, MI) to the final glycerol/buffer ratio 3:7 v/v and placed into the 5 mm NMR glass tubes (Wilmad LabGlass, Buena, NJ). CYP101 was reduced by anaerobic addition of a small molar excess of dithionite using a solution of known concentration, determined spectrophotometrically using a molar absorption coefficient Σ315 = 8050 M−1cm−1. Complete reduction was verified by absorption spectroscopy. Oxygenation of the ferrous CYP101 samples was done directly in NMR tubes connected to the Schlenck line at 4o C by sequential application of vacuum, high purity argon, vacuum, and then 16O2 or 18O2 gas followed by quick vortexing to facilitate oxygenation. Oxygenated samples were immediately frozen in liquid nitrogen to minimize autoxidative decomposition of oxy-ferrous CYP101.
Samples of oxy-ferrous CYP101 for the UV-Vis absorption spectroscopy were prepared in UV-enhanced methacrylate semimicro spectroscopic cells (Fisher Scientific, Allentown, PA) as described.43,44 Different concentrations, from 20 μM to 80 μM, and an optical pathlength of 4.3 mm were used for accurate determination of absorption spectra in the Soret and Q-band regions and for the optimal background subtraction (see below). To ensure formation of clear transparent glassy solvent matrix, a high glycerol content, 2:1 v/v was used as described previously.43,44 Samples for absorption spectroscopy were irradiated together with the resonance Raman samples to generate the peroxo-ferric intermediate and illuminated at 77 K for 8 to 12 minutes before measurement in order to bleach the strong absorption from trapped electrons generated during radiolysis. 44 Absorption spectra of all samples were measured before and after irradiation at different temperatures in the range 80 K – 200 K using the home made cryostat as described earlier.43,44 The remaining smooth background due to the radicals generated by radiolysis in the aqueous glycerol solvent matrix was the same in all samples with different CYP101 concentrations, and could be eliminated by subtraction of the spectrum of the sample with the lowest concentration of CYP101 from all other spectra taken at the same temperatures.
Frozen samples of oxy-ferrous CYP101 were irradiated with 4 Mrad of 60 Co gamma rays at 77 K and a dose rate 0.95 Mrad/hour. X-Band EPR experiments were performed at the University of Illinois EPR Resource Center on a Varian E-122 X-Band (9.03 GHz) spectrometer, using microwave power of 0.5 mW and modulation amplitude of 5 Gauss at 100 kHz. An Air Products (Allentown, PA) liquid helium flow system was used for measurements at 15 K.
The resonance Raman spectra were acquired using a Spex 1269 spectrometer equipped with a Newton EMCCD detector (Model DU971, Andor Technologies). The 413.1 nm line from a Kr+ laser (Coherent Innova Model 100-K3) was used to measure spectra of oxygenated samples before irradiation, while the 441.6 nm line provided by a He-Cd laser (Liconix Model 4240) was used for probing the irradiated and annealed samples. The RR spectra were collected using a back scattering (180°) geometry with the laser beam (power of 1.0 mW or less) being focused by a cylindrical lens to form a line image on the sample. All measurements were taken at 77 K. The frozen samples were contained in 5 mm O.D. NMR tubes (WG-5 ECONOMY, Wilmad). The NMR tubes were positioned into a double-walled quartz low temperature cell of in-house design filled with liquid nitrogen. The sample tubes were spun to avoid local heating by the laser beam. Spectra were calibrated with fenchone and processed with Grams/32 AI software (ThermoGalactic, Salem, NH).
Results
In order to document the changes of the Fe-O-O moiety in the key iron-oxygen intermediates of P450 catalysis, we have prepared intermediates [4], [5a], and [5b] in the D251N mutant of CYP101 and studied these complexes using resonance Raman spectroscopy. As described earlier, 17,18 the cryogenic radiolytic reduction of oxy-ferrous complex [4] in this mutant CYP101 results in formation of the unprotonated state [5a], which is stable at 77 K and undergoes protonation to afford [5b] after brief annealing above 180 K. Using cryogenic Raman spectroscopy, we have measured the resonance Raman spectra of these intermediates before and after protonation, and assigned the positions of ν(O-O) and ν(Fe-O) modes. The protonation state of both intermediates [5a] and [5b] was confirmed by the EPR spectroscopic signatures of these states before and after annealing at 185 K, correspondingly. Both EPR and Raman data sets were obtained using the same samples, to ensure the proper assignment of the spectroscopic results. In addition, the UV-Visible absorption spectra of complexes [4], [5a], and [5b] are also reported herein.
A. Electronic and EPR Spectroscopy
We measured EPR spectra on the same samples used in the Raman experiments, both before and after Raman measurements, to confirm formation of the peroxo-ferric complex [5a] in CYP101 immediately after irradiation at 77 K, and to monitor protonation of this intermediate and formation of the hydroperoxo-ferric complex [5b] after annealing at 185 K. As shown in Figure 1A, X-band EPR spectra of the cryoreduced oxy-ferrous D251N mutant of CYP101 measured after irradiation displayed a low-spin rhombic species with g-values [2.245, 2.16, 1.95] diagnostic of an unprotonated ferric-peroxo moiety with end-on (η1) geometry, in agreement with the earlier Q-band EPR studies of the same mutant in CYP101.17 After annealing to 185 K, an almost complete conversion to the protonated ferric peroxo species with g-values [2.29, 2.17, 1.95] is observed (Figure 1A), again confirmingearlier results .17
Figure 1.


(A) EPR spectra of the reduced oxy-ferrous complex of D251N CYP101. Data measured after irradiation at 77 K, and after all resonance Raman experiments and annealing at 185 K. The hydrogen atom doublet is marked by the star sign. Vertical bars indicate the features of peroxo-ferric (upper line) and hydroperoxo-ferric (bottom line) complexes with the corresponding g-values. Frozen samples are in 100 mM phosphate (H2O or D2O, as indicated), 100 mM KCl, 30% glycerol v/v, T = 15 K. Other details of EPR spectroscopy are described in Materials and Methods. (B) Absorption spectra of D251N CYP101: (1) Oxy-ferrous complex, T=85 K, before irradiation; (2) peroxo-ferric complex, 90 K, after irradiation at 77 K; (3) hydroperoxo-ferric complex, 180 K, after irradiation at 77 K and annealing at 185 K. Frozen samples are in 100 mM phosphate (H2O), 100 mM KCl, pH 7.4, 65% glycerol.
The transitions in protonation state of the cryoreduced oxy P450 mutant are similarly reflected in low-temperature optical spectroscopy experiments (Figure 1B), with the spectrum of the parent, non-reduced, dioxygen adduct being shown for reference. The features of the absorption spectrum of the reduced oxy-ferrous complex of the mutant protein at 90 – 120 K in both the split Soret band (main maximum at 439 nm with the second one at 361 nm) and Q-band (α, β bands at 561 nm and 530 nm) are similar to those obtained in analogous experiments with the CYP101 reported earlier.43,44 Upon annealing from 90 to 180 K, the Soret band shifts to 437 nm, and the second band shifts to 365 nm, indicating the decreased splitting in [5b] as compared to [5a] with only minor changes observable in intensities of the Q bands. These small changes are consistent with the protonation of [5a] and formation of the hydroperoxo-ferric intermediate [5b]. 17
B. Resonance Raman Spectroscopy
Resonance Raman spectroscopy is a most powerful method for providing direct information about Fe-X-Y bond strengths and, through the observation of hydrogen-deuterium isotope shifts of corresponding vibrational modes, reveals important details concerning the protonation state of H/D-exchangeable groups, such as the peroxo-ferric fragments of interest here. In this work we present the RR characterization of the precursor dioxygen adduct [4] and each of the cryoreduced forms [5a] and [5b].
1. The Dioxygen Adduct(s) of D251N
Two series of isotopically (16O2 and 18O2) labeled samples of D251N were prepared in H2O and D2O buffers and their RR spectra were collected before irradiation in order to establish the vibrational signature of this oxygenated precursor and to document successful isotopic labeling. The high frequency RR spectrum of oxygenated D251N is shown in trace A of Figure 2. This spectrum resembles that already published earlier by Sjodin et al.,45 with a strong, slightly broadened, band centered near 1134 cm−1 that is attributable to bands associated with the ν(16O-16O) stretching mode. As was pointed out in earlierwork, 45 this broadened band is most reasonably interpreted to reflect the existence of two or three structural conformers of the Fe-O-O fragment. It was concluded 45 that for both the WT and D251N oxygenated proteins there is one major conformer exhibiting its ν(16O-16O) at 1138 cm−1 or 1136 cm−1, respectively, with both proteins having two minor conformers detected at 1146 cm−1 and near 1130 cm−1. Interestingly, it was also shown that upon binding of its reduced electron transfer partner putidaredoxin (Pd) to the D251N mutant, the 1129 cm−1 component was populated to an extent nearly comparable to the 1137 cm−1 species; i.e., two nearly equally intense bands were observed.45
Figure 2.

The high frequency resonance Raman spectra of oxy D251N CYP101 measured at 77 K, and the difference spectra before irradiation (excitation at 413 nm). Inset shows low frequency difference spectra of 16O2-18O2in H 2O (A) and in D 2O (B) buffer.
As will be discussed, our new data acquiredat 77 Knot only support this previous suggestion of multiple conformers, but reveal the fact that the difference in the two major conformers appearing at 1136 and 1125 cm−1 at 77 K is associated with changes in H-bonding. Thus, careful inspection of the spectra (traces B and C of Figure 2) reveals the existence of two isotope-sensitive modes, whose vibrational parameters can be extracted by fitting the difference traces with 50/50 Gaussian / Lorentzian profiles. It was found that the broad positive feature centered at around 1134 cm−1 , as well as negative one centered at around 1065 cm−1 in trace B, had apparent bandwidths of 22–23 cm−1, while the relatively isolated heme mode, ν4 , appearing at 1373 cm−1 had a bandwidth of 12 cm−1. Not surprisingly, the best fit of those isotope sensitive features is achieved using two functions that had 11–12 cm−1 bandwidth, yielding two modes at 1136 cm−1 and 1125 cm−1 exhibiting 66 and 65 cm−1 shifts upon 18O2 substitution, respectively, in agreement with that predicted by Hooke’s law. Moreover, the results obtained with the samples prepared in deuterated solvent show that the lower frequency ν(O-O) stretch exhibits a clear 2 cm−1 upshift upon D2O exchange, which suggests that the lower frequency conformer is in fact participating in a hydrogen bonding interaction with a distal side residue apparently different than that corresponding to the higher frequency conformer. It is important to emphasize that in the spectra of oxygenated wild type CYP101 at 77 K only one strong ν(O-O) stretch was detected at 1139 cm−1, which also failed to exhibit an H/D shift.42 Thus, the present work shows that the population of this type of second conformer actually occurs merely by lowering the temperature of the oxygenated D251N mutant to 77 K and does not require binding of Pd.45
The lower frequency of the proposed H-bonded form is consistent with early work with model compounds where, comparing ν(O-O) stretching frequencies of dioxygen adducts of pyridine ligated CoTPP with those of corresponding “picket fence” derivatives, it was demonstrated that the picket fence adducts exhibited significantly (up to 5 cm−1) lower frequencies, a shift that was reasonably attributed to H-bonding interactions with the amide linkages of the picket fence fragments. 46 Furthermore, in recent RR studies of dioxygen ligated NOS isoforms, shifts of precisely this magnitude have been attributed to H-bonding differences; i.e., the substrate-free dioxygen adduct exhibited a ν(O-O) at 1133 cm−1, while that of the arginine-bound form was observed at 1126 cm−1.47
Referring to the sensitivity to deuterium substitution, while H/D induced 2-5 cm−1 shifts to higher frequency of the ν(O-O) modes of dioxygen adducts of cobalt-substituted globins have been plausibly attributed to D- vs H-bonding interactions 48 it has been demonstrated that such interpretations are potentially compromised by complications arising from unusually strong vibrational coupling interactions between the ν(O-O) mode and internal modes of the trans-axial histidyl imidazole, the coupling patterns changing as the latter modes shift in response to H/D exchange at the imidazole H-N fragment. 49–51 In the present case, however, the absence of the trans-axial imidazole obviates consideration of such complications and the 2 cm−1 upshift can reasonably be attributed to H-bonding; in fact, a recent RR study of dioxygen adducts of cytochrome P450 model compounds, in which hydroxyl groups were incorporated into the distal pocket to serve as H-bond donors to the bound dioxygen, the ν(O-O) band indeed displayed an identical 2 cm−1shift to higherfrequency upon H/D exchange of the distal pocket hydroxyl groups .52
The low frequency spectra of oxygenated D251N samples (Figure 2, inset) exhibit a feature in the ν(Fe-O2) stretching region at 537 cm−1 that shifts down by 30 cm−1 upon 18O2 substitution. Based on the observation of two ν(O-O) modes in the high frequency region, it might have been expected that two ν(Fe-O) modes would be observed in the low frequency region. Owing to the relatively low S/N ratio seen here, however, no significant broadening is discernible and no clear H/D shift detected (Table 1). Such H/D shifts, if present, are indeed expected to be quite small and are not normally observed for these low frequency ν(M-O2) modes. 45,53–56 While we have obtained no clear evidence for separation of the ν(Fe-O) region into two distinct features, similar to earlier results, 45 we note the presence of the positive correlation between the ν(Fe-O) and ν(O-O) modes. The presence of a lower frequency ν(O-O) mode at 1125 cm−1 is correlated with an apparent shift to lower frequency for the observed ν(Fe-O) mode(s) from 546 to 537 cm−1.
Table 1.
Frequencies of isotopically sensitive modes of wild-type (WT) and D251N mutant of CYP101 and their isotopic shifts. Spectra were measured at 77 K.
| Frequency and isotopic shift [cm−1] of | ||
|---|---|---|
| ν(Fe-O) Δ[16O2/18O2 ; H2O/D2O] | ν(O-O) Δ[16O2/18O2 ; H2O/D2O] | |
| WTa | ||
| Fe(II)-O2 [4] | 546 [31 ; 0] | 1139 [66 ; 0] |
| Fe(III)-O22− [5a] | - | - |
| Fe(III)-O2H−[5b] | 559 [27 ; −3] | 799 [40 ; −3] |
| D251N | ||
| Fe(II)-O2 [4] | 537 [30 ; 0] | 1136 [66 ; 0] 1125 [65 ; +2] |
| Fe(III)-O22− [5a] | 553 [27 ; 0] | 792 [38 ; 0] |
| Fe(III)-O2H− [5b] | 564 [28 ; −2] | 774 [37 ; −4] |
data from ref. 42
2. Irradiated and annealed samples of oxyD251N
The RR spectra of irradiated oxyD251N samples show two isotopically sensitive species (Figure 3). The mode observed in the spectra of 16O2 in H2O at 553 cm−1 shows a 27 cm−1 down shift upon 18O2 substitution. The other isotopically sensitive mode at 792 cm−1 exhibits the expected 38 cm−1 down shift upon 16O2/18O2 exchange (Table 1). Most importantly, neither of these modes exhibit H2O / D2O sensitivity, an observation most consistent with assignment of these features to the ν(Fe-O) (at 553 cm−1) and ν(O-O) (at 792 cm−1) modes of a peroxo- species. Note that distinct H/D shifts have been documented for these modes for the hydroperoxo intermediate of the WT protein.42 This is the first direct observation of the vibrational frequencies of the non-protonated peroxo intermediate [5a] of a member of the cytochrome P450 protein family, and its assignment is consistent with earlier EPR and absorption spectroscopicstudies. 17,18
Figure 3.

Theresonance Raman spectra of irradiated P450 D251N samples in H 2O buffer: spectra A (16O2) and B (18O2) and in the D2O buffer: spectra C (16O2) and D (18O2). The two bottom traces shows the difference spectra of 16O2-18O2 in H2O and 16O2-18O2 in D2O buffer (excitation line 442 nm, temperature 77 K).
Earlier it was shown that annealing of such irradiated samples to higher temperatures (above 170 K) allows protonation of this peroxo species to yield the hydroperoxo form. 17,18 Consistent with this picture, the spectra of our samples annealed to 185 K and then cooled to 77 K for RR measurements are presented in Figure 4 and document a difference pattern of isotopically sensitive modes that is similar to that observed before annealing, but which reveals distinctly different component frequencies. The mode appearing at 564 cm−1, seen in the trace A of Figure 4, exhibits a 28 cm−1 downshift upon 18O2 substitution as well as a 2 cm−1 downshift in D2O buffer. Moreover, the higher frequency mode at 774 cm−1 shifts down by 37 cm−1 and 4 cm−1 upon 18O2 and H/D exchange, respectively (Table 1). Those H/D shifts are in a good agreement with previously published RR data for known metallo-hydroperoxo species, 4,41,42,57 and are consistent the hydroperoxo formulation derived from EPR and UV-Vis data for annealed samples of irradiated D251N. 17 Clearly, the modes at 564 cm−1 and 774 cm−1 are most reasonably assigned, respectively, as the ν(Fe-O) and ν(O-O) modes of the hydroperoxo-ferricspecies [5b]. Further annealing to higher temperatures (195 K and 205 K) did not lead to the observation of any new isotopically sensitive modes, at least when employing the 442 nm excitation line used here for the peroxo / hydroperoxo forms (Figure S1, Supporting Information).
Figure 4.

The resonance Raman spectra of irradiated and annealed at 185 K samples of P450 D251N in H2O buffer: spectra A (16O2) and B (18O2) and in the D2O buffer: spectra C (16O2) and D (18O2). The two bottom traces shows the difference spectra of 16O2-18O2 in H2O and 16O2-18O2in D 2O buffer (excitation at 442 nm, temperature 77 K).
Future work, employing higher concentrations, different excitation lines and other solution conditions, including the use of different substrates, will be conducted in order to search for these later, highly reactive intermediates.
Discussion
In this study we document the changes in the Fe-O-O moiety monitored by resonance Raman spectroscopy in the key intermediates of dioxygen activation in the cytochromes P450. The isolation and separate spectroscopic studies of unprotonated peroxo-ferric and protonated hydroperoxo-ferric complexes [5a] and [5b] have been made possible by using the D251N mutant of CYP101, which yields exclusively [5a] as a results of cryogenic radiolytic reduction of [4]. 17,18 The protonation states of [5a] as prepared at 77 K, and of [5b] after annealing at 185 K have been assigned using characteristic EPR signature spectra, measured on the same samples used for the Raman spectroscopy. As a result, we can compare for the first time the vibrational modes ν(O-O) and ν(Fe-O) measured for [4], [5a] and [5b] on the same sample to evaluate the effects of the one-electron reduction and of protonation on the structure and stability of these intermediates of dioxygen activation.
The RR spectra of the dioxygen adduct [4] of the D251N protein show small but distinct differences from the corresponding spectra of the WT protein.42 The D251N adduct exhibits two relatively strong ν(O-O) stretching modes at 1136 cm−1 and 1125 cm−1 , while the WT shows only one intense mode near 1139 cm−1. Neither the 1136 (D251N) nor the 1139 cm−1 (WT) features are sensitive to H/D exchange, but the 1125 cm−1 mode of the mutant exhibits a distinct 2 cm−1 shift to higher frequency in the deuterated solutions (Figure 2), implying that one effect of the D251N mutation is to facilitate substantial population of a second conformer, a result in agreement with more recent crystallographic data.58 While early crystallographic studies of the oxygenated complex of the D251N mutant did not show multiple conformers, 59 more recent work indicates that two conformations of the Asn251-Thr252 region of the protein are present in crystals of the dioxygen adduct of D251NCYP1 01. 58
The frequencies of the ν(O-O) and ν(Fe-O) modes for all species detected here and in the previous study of the hydroperoxo form of the WT protein are collected in Table 1. As mentioned, cryoradiolysis of the oxygenated WT protein at 77 K yields directly the hydroperoxo derivative [5a], whose ν(O-O) and ν(Fe-O) modes appear at 799 cm−1 and 559 cm−1, both exhibiting telltale shifts to lower frequency in D2O solutions.42 Given that the most significant effect of the D251N mutation is to restrict proton delivery to the initially formed ferri-peroxo fragment, it is not surprising that, as shown previously17,18 , this species is trapped at 77 K and, as shown here, exhibits ν(O-O) and ν(Fe-O) modes at 792 cm−1 and 553 cm−1, neither of which shows any sensitivity to H/D exchange. This strongly suggests that these modes refer to the peroxo-ferric intermediate [5a]. The hydroperoxo derivative of D251N [5b], formed upon annealing to higher temperatures, exhibits a substantially lower frequency ν(O-O) mode at 774 cm−1 and a significantly higher ν(Fe-O) mode (i.e., 564 cm−1) compared to its non-protonated precursor. Assuming a similar (18 cm−1) shift of ν(O-O) for transition between [5a] and [5b] occurs for the WT protein, the estimated frequency for the unobservable reactive ferri-peroxo [5a] derivative of WT CYP101 would be ~817 cm−1; i.e., 799+18 cm−1. Based on this estimated frequency for the [5a] form of the WT protein, the shift in ν(O-O) upon conversion from peroxo- to hydroperoxo forms for both proteins would be 18 cm−1 , and the observed shifts in comparing similar forms of both proteins are 25 cm−1. For both the [5a] and [5b] states, ν(O-O) in the mutant is 25 cm−1 lower than in the WT protein. Thus, not only does the mutation retard proton delivery to the Fe-O-O− fragment, but the replacement of Asp251 acid side chain with amide also leads to substantial active site differences that significantly affect the disposition of the Fe-O-O structure, as reflected in the downshift of ν(O-O) vibrational mode. These shifts to lower frequency for the reduced forms of the D251N mutant relative to the WT protein are also consistent with the increased stabilization of the lower frequency conformer of the parent form [4], whose 1125 cm−1component was shown to be much more highly populated in the mutant, compared to the WT CYP101, at least at 77 K. While it had been shown previously that at 4° C, the higher frequency conformer dominates the spectra for both the WT and D251N mutant,45 the results obtained here clearly show that the lower frequency conformer is substantially populated at 77 K, but only in the case of the mutant. Thus, while the H-bonded conformer is more easily accessed in the case of the mutant, its population is significant only at low temperatures. Such structural changes at cryogenic temperatures have been previously documented in EPR studies of single crystals of oxygenated cobalt myoglobin60 and in RR studies of deoxy hemoglobin, where significant increases in the frequency of the ν(Fe-Nhis) stretching mode, associated with the linkage to the proximal histidine, were reasonably attributed to thermal contraction that stabilizes certain key interactions. 61
Collectively, the data presented in Table 1 provide the first clear opportunity to directly compare experimental vibrational data for these superoxo / peroxo / hydroperoxo heme derivatives in similar environments. In CYP101, upon reduction from [4] to [5a], there is a large shift of ν(O-O) from ~ 1135 cm−1, characteristic of bound superoxide, to ~800 cm−1, a frequency indicative of a bound peroxo-fragment. At the same time the observed ν(Fe-O) mode shifts to higher frequencies by ~ 15 cm−1; e.g., 537 to 553 cm−1 . Protonation to yield the hydroperoxo-derivative causes the ν(O-O) to decrease by 18 cm−1, while the ν(Fe-O) simultaneously increases by 11 cm−1 to 564 cm−1. As can be seen by inspection of Table S1 (Supporting Information), where the results of several computational studies are summarized, the predicted changes in vibrational parameters in proceeding from the parent [4] through the [5a] and [5b] intermediates are generally consistent with those observed here. An exception is the behavior of the ν(Fe-O) modes in proceeding from [4] to [5a], where strengthening of this bond is implied from the vibrational data, while a weakening (longer bond) is predicted from all of the calculations. However, it is particularly satisfying to note that virtually all computational work correctly predicts the anti-correlation, involving increased Fe-O and decreased O-O bond strengths, upon formation of the hydroperoxo-intermediate. The weaker O-O bond in [5b] can be rationalized in terms of the higher electron density on the dioxygen moiety filling the anti-bonding π* orbital of hydroperoxide in [5b], as compared to unprotonated [5a]. This is consistent with the overall lower spin density on the distal and proximal oxygen atoms in [5b] than in [5a] as suggested based on the proton ENDOR measurements, 17 as well as with the similar predictions of DFT theoretical studies .29–34
In conclusion, we have documented the vibrational characteristics of the Fe-O-O moiety in the main intermediate states of oxygen activation by the cytochromes P450. Using the D251N mutant of CYP101, we have prepared the oxy-ferrous complex [4], and used cryogenic radiolytic reduction at 77 K followed by annealing at 185 K to isolate peroxo-ferric and hydroperoxo-ferric intermediates [5a] and [5b]. Using cryogenic Raman spectroscopy, we have identified for the first time both the ν(Fe-O) and ν(O-O) stretching modes in all three iron-dioxygen intermediates, and evaluated experimentally the influence of reduction and protonation on the changes in Fe-O and O-O bond strength. Reduction of [4] and the concomitant formation of peroxo-ferric complex [5a] result in significant weakening of the O-O bond, indicating transition from superoxo- to peroxo- electronic structure. Further weakening of O-O bond is caused by protonation of the distal oxygen atom in [5b], consistent with the predictions of theoretical studies. Contrary, the Fe-O bond is becoming stronger during the transition from [4] through [5a] and to [5b], revealing the presence of the negative correlation between ν(Fe-O) and ν(O-O). As the first available set of vibrational characteristics of Fe-O-O intermediates, our data provide further insight into the mechanism of oxygen activation in heme enzymes, as well as the convenient benchmark for the refinement of the detailed quantum chemical calculations.
Supplementary Material
Acknowledgments
We thank Dr. Na Ke for the help in constructing of the plasmid with PT7-P450 His gene, Dr. S.Toshkov for irradiation of the samples at the 60 Co source, and Dr. M. Nilges for the help in EPR measurements. This work was supported by National Institutes of Health grants GM 31756 to S.G.S, and DK 35153 to J.R.K.
The abbreviations used are
- CPO
chloroperoxidase
- H/D
hydrogen-deuterium
- Hb
hemoglobin
- HO
heme oxygenase
- KPi
potassium phosphate
- Mb
myoglobin
- Pd
putidaredoxin
- WT
wild-type
Footnotes
Supporting Information. One figure representing resonance Raman spectra of irradiated and annealed at different temperatures samples and one table with the calculated parameters of Fe-O-O bonds in [4], [5a] and [5b] taken from literature. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Makris TM, Davydov R, Denisov IG, Hoffman BM, Sligar SG. Drug Metab Rev. 2002;34:691–708. doi: 10.1081/dmr-120015691. [DOI] [PubMed] [Google Scholar]
- 2.Makris TM, Denisov IG, Schlichting I, Sligar SG. Activation of Molecular Oxygen by Cytochrome P450. In: Ortiz de Montellano PR, editor. Cytochrome P450: Structure, Function, Genetics. 3. Kluwer Academic/Plenum Publishers; New York: 2005. pp. 149–182. [Google Scholar]
- 3.Denisov IG, Makris TM, Sligar SG, Schlichting I. Chem Rev. 2005;105:2253–2277. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- 4.Que L, Jr, Ho RYN. Chem Rev. 1996;96:2607–2624. doi: 10.1021/cr960039f. [DOI] [PubMed] [Google Scholar]
- 5.Bukowski MR, Halfen HL, van den Berg TA, Halfen JA, Que L., Jr Angew Chemie, Int Ed. 2005;44:584–587. doi: 10.1002/anie.200461527. [DOI] [PubMed] [Google Scholar]
- 6.Decker A, Solomon EI. Curr Opin Chem Biol. 2005;9:152–163. doi: 10.1016/j.cbpa.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 7.Neidig ML, Solomon EI. Chem Commun. 2005:5843–5863. doi: 10.1039/b510233m. [DOI] [PubMed] [Google Scholar]
- 8.Bakac A. Adv Inorg Chem. 2004;55:1–59. [Google Scholar]
- 9.Bakac A. Coord Chem Rev. 2006;250:2046–2058. [Google Scholar]
- 10.Neese F, Zaleski JM, Zaleski KL, Solomon EI. J Am Chem Soc. 2000;122:11703–11724. [Google Scholar]
- 11.Roelfes G, Vrajmasu V, Chen K, Ho RYN, Rohde JU, Zondervan C, la Crois RM, Schudde EP, Lutz M, Spek AL, Hage R, Feringa BL, Muenck E, Que L., Jr Inorg Chem. 2003;42:2639–2653. doi: 10.1021/ic034065p. [DOI] [PubMed] [Google Scholar]
- 12.Solomon EI, Decker A, Lehnert N. Proc Natl Acad Sci U S A. 2003;100:3589–3594. doi: 10.1073/pnas.0336792100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee SK, Lehnert N, Neese F, Skulan AJ, Yang YS, Zhou J. Chem Rev. 2000;100:235–349. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- 14.Costas M, Mehn MP, Jensen MP, Que L., Jr Chem Rev. 2004;104:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 15.Girerd JJ, Banse F, Simaan AJ. Structure and Bonding. 2000;97:145–177. [Google Scholar]
- 16.Davydov RM, Yoshida T, Ikeda-Saito M, Hoffman BM. J Am Chem Soc. 1999;121:10656–10657. [Google Scholar]
- 17.Davydov R, Makris TM, Kofman V, Werst DE, Sligar SG, Hoffman BM. J Am Chem Soc. 2001;123:1403–1415. doi: 10.1021/ja003583l. [DOI] [PubMed] [Google Scholar]
- 18.Davydov R, Macdonald IDG, Makris TM, Sligar SG, Hoffman BM. J Am Chem Soc. 1999;121:10654–10655. [Google Scholar]
- 19.Davydov R, Ledbetter-Rogers A, Martasek P, Larukhin M, Sono M, Dawson JH, Masters BS, Hoffman BM. Biochemistry. 2002;41:10375–10381. doi: 10.1021/bi0260637. [DOI] [PubMed] [Google Scholar]
- 20.Davydov R, Kofman V, Fujii H, Yoshida T, Ikeda-Saito M, Hoffman BM. J Am Chem Soc. 2002;124:1798–1808. doi: 10.1021/ja0122391. [DOI] [PubMed] [Google Scholar]
- 21.Davydov R, Kofman V, Nocek JM, Noble RW, Hui H, Hoffman BM. Biochemistry. 2004;43:6330–6338. doi: 10.1021/bi036273z. [DOI] [PubMed] [Google Scholar]
- 22.Davydov R, Perera R, Jin S, Yang TC, Bryson TA, Sono M, Dawson JH, Hoffman BM. J Am Chem Soc. 2005;127:1403–1413. doi: 10.1021/ja045351i. [DOI] [PubMed] [Google Scholar]
- 23.Kim SH, Yang TC, Perera R, Jin S, Bryson TA, Sono M, Davydov R, Dawson JH, Hoffman BM. Dalton Trans. 2005:3464–3469. doi: 10.1039/b506764m. [DOI] [PubMed] [Google Scholar]
- 24.Garcia-Serres R, Davydov RM, Matsui T, Ikeda-Saito M, Hoffman BM, Huynh BH. J Am Chem Soc. 2007;129:1402–1412. doi: 10.1021/ja067209i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beitlich T, Kuhnel K, Schulze-Briese C, Shoeman RL, Schlichting I. J Synchrotron Radiat. 2007;14:11–23. doi: 10.1107/S0909049506049806. [DOI] [PubMed] [Google Scholar]
- 26.Kuhnel K, Derat E, Terner J, Shaik S, Schlichting I. Proc Natl Acad Sci U S A. 2007;104:99–104. doi: 10.1073/pnas.0606285103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Unno M, Chen H, Kusama S, Shaik S, Ikeda-Saito M. J Am Chem Soc. 2007;129:13394–13395. doi: 10.1021/ja076108x. [DOI] [PubMed] [Google Scholar]
- 28.Hersleth HP, Hsiao YW, Ryde U, Gorbitz CH, Andersson KK. Biochem J. 2008 doi: 10.1042/BJ20070921. [DOI] [PubMed] [Google Scholar]
- 29.Loew GH, Harris DL. Chem Rev. 2000;100:407–419. doi: 10.1021/cr980389x. [DOI] [PubMed] [Google Scholar]
- 30.Ogliaro F, de Visser Sam P, Cohen S, Sharma Pankaz K, Shaik S. J Am Chem Soc. 2002;124:2806–2817. doi: 10.1021/ja0171963. [DOI] [PubMed] [Google Scholar]
- 31.Rydberg P, Sigfridsson E, Ryde U. J Biol Inorg Chem. 2004;9:203– 223. doi: 10.1007/s00775-003-0515-y. [DOI] [PubMed] [Google Scholar]
- 32.Shaik S, Kumar D, de Visser SP, Altun A, Thiel W. Chem Rev. 2005;105:2279–2328. doi: 10.1021/cr030722j. [DOI] [PubMed] [Google Scholar]
- 33.Kumar D, Hirao H, De Visser SP, Zheng J, Wang D, Thiel W, Shaik S. J Phys Chem B. 2005;109:19946–19951. doi: 10.1021/jp054754h. [DOI] [PubMed] [Google Scholar]
- 34.Hackett JC, Brueggemeier RW, Hadad CM. J Am Chem Soc. 2005;127:5224–5237. doi: 10.1021/ja044716w. [DOI] [PubMed] [Google Scholar]
- 35.Gerber NC, Sligar SG. J Am Chem Soc. 1992;114:8742–8743. [Google Scholar]
- 36.Gerber NC, Sligar SG. J Biol Chem. 1994;269:4260–4266. [PubMed] [Google Scholar]
- 37.Vidakovic M, Sligar SG, Li H, Poulos TL. Biochemistry. 1998;37:9211–9219. doi: 10.1021/bi980189f. [DOI] [PubMed] [Google Scholar]
- 38.Schlichting I, Berendzen J, Chu K, Stock AM, Maves SA, Benson DE, Sweet RM, Ringe D, Petsko GA, Sligar SG. Science. 2000;287:1615–1622. doi: 10.1126/science.287.5458.1615. [DOI] [PubMed] [Google Scholar]
- 39.Taraphder S, Hummer G. J Am Chem Soc. 2003;125:3931–3940. doi: 10.1021/ja016860c. [DOI] [PubMed] [Google Scholar]
- 40.Makris TM, von Koenig K, Schlichting I, Sligar SG. Biochemistry. 2007;46:14129–14140. doi: 10.1021/bi7013695. [DOI] [PubMed] [Google Scholar]
- 41.Ibrahim M, Denisov IG, Makris TM, Kincaid JR, Sligar SG. J Am Chem Soc. 2003;125:13714–13718. doi: 10.1021/ja036949d. [DOI] [PubMed] [Google Scholar]
- 42.Mak PJ, Denisov IG, Victoria D, Makris TM, Deng T, Sligar SG, Kincaid JR. J Am Chem Soc. 2007;129:6382–6383. doi: 10.1021/ja071426h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Denisov IG, Makris TM, Sligar SG. J Biol Chem. 2001;276:11648–11652. doi: 10.1074/jbc.M010219200. [DOI] [PubMed] [Google Scholar]
- 44.Denisov IG, Makris TM, Sligar SG. Meth Enzymol. 2002;357:103–115. doi: 10.1016/s0076-6879(02)57670-9. [DOI] [PubMed] [Google Scholar]
- 45.Sjodin T, Christian JF, Macdonald IDG, Davydov R, Unno M, Sligar SG, Hoffman BM, Champion PM. Biochemistry. 2001;40:6852–6859. doi: 10.1021/bi002510b. [DOI] [PubMed] [Google Scholar]
- 46.Bajdor K, Kincaid JR, Nakamoto K. J Am Chem Soc. 1984;106:7741–7747. [Google Scholar]
- 47.Li D, Kabir M, Stuehr DJ, Rousseau DL, Yeh SR. J Am Chem Soc. 2007;129:6943–6951. doi: 10.1021/ja070683j. [DOI] [PubMed] [Google Scholar]
- 48.Kitagawa T, Ondrias MR, Rousseau DL, Ikeda-Saito M, Yonetani T. Nature. 1982;298:869–871. doi: 10.1038/298869a0. [DOI] [PubMed] [Google Scholar]
- 49.Bruha A, Kincaid JR. J Am Chem Soc. 1988;110:6006–6014. doi: 10.1021/ja00226a014. [DOI] [PubMed] [Google Scholar]
- 50.Proniewicz LM, Kincaid JR. J Am Chem Soc. 1990;112:675–681. [Google Scholar]
- 51.Proniewicz LM, Kincaid JR. Coord Chem Rev. 1997;161:81–127. [Google Scholar]
- 52.Tani F, Matsuura M, Nakayama S, Ichimura M, Nakamura N, Naruta Y. J Am Chem Soc. 2001;123:1133–1142. doi: 10.1021/ja003430y. [DOI] [PubMed] [Google Scholar]
- 53.Macdonald IDG, Sligar SG, Christian JF, Unno M, Champion PM. J Am Chem Soc. 1999;121:376–380. [Google Scholar]
- 54.Hirota S, Ogura T, Appelman EH, Shinzawa-Itoh K, Yoshikawa S, Kitagawa T. J Am Chem Soc. 1994;116:10564–10570. [Google Scholar]
- 55.Das TK, Couture M, Ouellet Y, Guertin M, Rousseau DL. Proc Natl Acad Sci U S A. 2001;98:479–484. doi: 10.1073/pnas.98.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jeyarajah S, Proniewicz LM, Bronder H, Kincaid JR. J Biol Chem. 1994;269:31047–31050. [PubMed] [Google Scholar]
- 57.Rajani C, Kincaid JR, Petering DH. J Am Chem Soc. 2004;126:3829–3836. doi: 10.1021/ja030622v. [DOI] [PubMed] [Google Scholar]
- 58.von Koenig K, Schlichting I. Cytochromes P450 - structural basis for binding and catalysis. In: Siegel A, Siegel H, Siegel RKO, editors. Ubiquitous Roles of Cytochrome P450 Proteins. Vol. 3. Wiley and Sons; 2007. pp. 235–265. [Google Scholar]
- 59.Nagano S, Poulos TL. J Biol Chem. 2005;280:31659–31663. doi: 10.1074/jbc.M505261200. [DOI] [PubMed] [Google Scholar]
- 60.Hori H, Ikeda-Saito M, Yonetani T. Nature. 1980;288:501–502. doi: 10.1038/288501a0. [DOI] [PubMed] [Google Scholar]
- 61.Rousseau DL, Friedman JM. Transient and cryogenic studies of photodissociated hemoglobin and myoglobin. In: Spiro TG, editor. Biological Applications of Raman Spectroscopy. Vol. 3. Wiley and Sons; New York: 1988. pp. 133–216. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.